
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA five-carbon unsaturated Criegee intermediate: synthesis, spectroscopic identification, and theoretical study of 3-penten-2-one oxide†
Tarun Kumar
Roy‡
 ,
Tianlin
Liu‡
,
Yujie
Qian
,
Christopher A.
Sojdak
,
Marisa C.
Kozlowski
and
Marsha I.
Lester
*
,
Tianlin
Liu‡
,
Yujie
Qian
,
Christopher A.
Sojdak
,
Marisa C.
Kozlowski
and
Marsha I.
Lester
*
Department of Chemistry, University of Pennsylvania, Philadelphia, PA 19104-6323, USA. E-mail: milester@sas.upenn.edu
First published on 5th September 2023
Abstract
Biogenic alkenes, such as isoprene and α-pinene, are the predominant source of volatile organic compounds (VOCs) emitted into the atmosphere. Atmospheric processing of alkenes via reaction with ozone leads to formation of zwitterionic reactive intermediates with a carbonyl oxide functional group, known as Criegee intermediates (CIs). CIs are known to exhibit a strong absorption (π* ← π) in the near ultraviolet and visible (UV-vis) region due to the carbonyl oxide moiety. This study focuses on the laboratory identification of a five-carbon CI with an unsaturated substituent, 3-penten-2-one oxide, which can be produced upon atmospheric ozonolysis of substituted isoprenes. 3-Penten-2-one oxide is generated in the laboratory by photolysis of a newly synthesized precursor, (Z)-2,4-diiodopent-2-ene, in the presence of oxygen. The electronic spectrum of 3-penten-2-one oxide was recorded by UV-vis induced depletion of the VUV photoionization signal on the parent m/z 100 mass channel using a time-of-flight mass spectrometer. The resultant electronic spectrum is broad and unstructured with peak absorption at ca. 375 nm. To complement the experimental findings, electronic structure calculations are performed at the CASPT2(12,10)/aug-cc-pVDZ level of theory. The experimental spectrum shows good agreement with the calculated electronic spectrum and vertical excitation energy obtained for the lowest energy conformer of 3-penten-2-one oxide. In addition, OH radical products resulting from unimolecular decay of energized 3-penten-2-oxide CIs are detected by UV laser-induced fluorescence. Finally, the experimental electronic spectrum is compared with that of a four-carbon, isoprene-derived CI, methyl vinyl ketone oxide, to understand the effects of an additional methyl group on the associated electronic properties.
Introduction
Criegee intermediates (CIs) are zwitterionic carbonyl oxides, produced by ozonolysis of alkenes emitted into the troposphere from biogenic and anthropogenic sources.1 Owing to the large exothermic nature of alkene ozonolysis reactions (ca. 50 kcal mol−1), the CIs are initially produced with high excess energy and may promptly decay to OH radicals and other products.2 Alternatively, the CIs can be collisionally stabilized and undergo thermal unimolecular decay, again leading to OH radicals, thereby enhancing the oxidizing capacity of the troposphere.3–6 In addition, CIs can react with trace atmospheric species, often producing highly oxidized compounds that can lead to secondary organic aerosol (SOA) formation.7,8The fate of CIs in the atmosphere is governed by the nature of the substituents and their conformations.5 Simple alkyl-substituted CIs, such as syn-CH3CHOO, (CH3)2COO, and (CH3)(CH3CH2)COO, generally undergo unimolecular decay via an alkyl 1,4 H-atom transfer mechanism to form OH radicals.6 In contrast, CIs with alkenyl substituents (hereafter abbreviated as unsaturated CIs) can result in extended conjugation, e.g. across vinyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and carbonyl oxide (CO+O−) groups, that stabilize the CIs. Notably, these unsaturated CIs are anticipated in the ozonolysis of biogenic alkenes or conjugated dienes.9 For example, methyl vinyl ketone oxide [(CH3)(CH2CH)COO, MVK-oxide] and methacrolein oxide [(CH2C(CH3))CHOO, MACR-oxide] are four-carbon unsaturated CIs formed in the ozonolysis of isoprene, the most abundant alkene emitted into the Earth's atmosphere.10 Ozonolysis of 1,3-pentadiene and branched diolefins lead to another four-carbon CI, 2-butenal oxide [CH3CHCHCHOO], an isomer of MVK-oxide and MACR-oxide, with similar extended conjugation across the vinyl and carbonyl oxide groups.11,12 This laboratory has made significant advances in studying the electronic spectroscopy and resultant dissociation dynamics12–18 as well as the infrared (IR) action spectroscopy and unimolecular decay dynamics of CIs under jet-cooled conditions.11,19–21 These studies indicate that extended π conjugation across the carbonyl oxide and vinyl group in CIs can strongly impact their electronic spectra, photochemistry, and unimolecular decay pathways compared to simple alkyl-substituted CIs.
C) and carbonyl oxide (CO+O−) groups, that stabilize the CIs. Notably, these unsaturated CIs are anticipated in the ozonolysis of biogenic alkenes or conjugated dienes.9 For example, methyl vinyl ketone oxide [(CH3)(CH2CH)COO, MVK-oxide] and methacrolein oxide [(CH2C(CH3))CHOO, MACR-oxide] are four-carbon unsaturated CIs formed in the ozonolysis of isoprene, the most abundant alkene emitted into the Earth's atmosphere.10 Ozonolysis of 1,3-pentadiene and branched diolefins lead to another four-carbon CI, 2-butenal oxide [CH3CHCHCHOO], an isomer of MVK-oxide and MACR-oxide, with similar extended conjugation across the vinyl and carbonyl oxide groups.11,12 This laboratory has made significant advances in studying the electronic spectroscopy and resultant dissociation dynamics12–18 as well as the infrared (IR) action spectroscopy and unimolecular decay dynamics of CIs under jet-cooled conditions.11,19–21 These studies indicate that extended π conjugation across the carbonyl oxide and vinyl group in CIs can strongly impact their electronic spectra, photochemistry, and unimolecular decay pathways compared to simple alkyl-substituted CIs.
The present study focuses on the laboratory identification of a five-carbon, unsaturated CI, 3-penten-2-one oxide [(CH3CHCH)(CH3)COO], which can be produced upon atmospheric ozonolysis of substituted isoprenes, e.g., 2-methylpenta-1,3-diene (ESI, Scheme S1†) or other branched unsaturated alkenes, and thus is also of interest from an atmospheric standpoint. To best of our knowledge, 3-penten-2-one oxide is the largest CI stabilized under gas-phase conditions to date. The structure of this five-carbon CI is analogous to MVK-oxide aside from an additional methyl group (Scheme 1). In 3-penten-2-one oxide, the extra methyl group is attached to the β site of the vinyl group compared to MVK-oxide. The characteristic electronic absorption spectra of CIs are strong π* ← π transitions associated with the carbonyl oxide group. Prior studies on MVK-oxide and MACR-oxide with extended π conjugation have shown that the experimental electronic absorption maxima associated with their first π* ← π transition are shifted to significantly longer wavelengths (ca. 50 nm) compared to those of simple alkyl substituted CIs, such as CH3CHOO.15,16 3-Penten-2-one oxide has analogous extended π conjugation and thus its first π* ← π transition is expected to be similarly shifted to longer wavelengths.
 | ||
| Scheme 1 Structural comparison of the five-carbon Criegee intermediate, 3-penten-2-one oxide, with the four-carbon Criegee intermediate MVK-oxide in their lowest energy conformations. | ||
Here, we investigate the electronic spectroscopy of a five-carbon unsaturated CI, 3-penten-2-one oxide, under jet-cooled conditions. This CI is generated in the laboratory by 248 nm photolysis of a newly synthesized 2,4-diiodopent-2-ene precursor and subsequent reaction with O2 in a quartz capillary reactor tube. The electronic spectrum of 3-penten-2-one oxide is recorded by UV-vis induced depletion of its VUV photoionization signal (10.5 eV) on the m/z 100 mass channel using a time-of-flight mass spectrometer (TOF-MS). The experimental results are complemented by theoretical calculations of the relative stability of its eight conformers, their vertical excitation energies, and the absorption spectra of the lowest energy conformers. In addition, the experimentally observed UV-vis spectrum of this five-carbon unsaturated CI is compared with a structurally similar four-carbon CI, MVK-oxide. Finally, OH radical products are observed that arise from unimolecular decay of energized 3-penten-2-one oxide CIs.
Synthetic methods
In this study, a new 1,3-diiodoalkene precursor, (Z)-2,4-diiodopent-2-ene [CH3(I)CCHCH(I)CH3], is synthesized and utilized to generate the 3-penten-2-one oxide CI by UV photolysis and subsequent reaction with O2. Similar precursors were used previously to generate MVK-oxide and MACR-oxide,15,20 which are the unsaturated four-carbon CIs derived from isoprene ozonolysis.
Synthesis of (Z/E)-2,4-diiodopent-2-ene ((Z/E)-4) can be accomplished in three steps starting from propargylic alcohol 1 in 15% overall yield. Scheme 2 shows an overview of the synthesis; further information are provided in the ESI (see Section S1 and Fig. S1–S6†). To avoid the use of acetaldehyde due to its propensity for polymerization, an alternative approach was sought to improve reproducibility. n-BuLi is used to exhaustively deprotonate the alkyne C–H and alcohol O–H of propargylic alcohol 1; subsequent addition of methyl iodide leads to selective alkyne methylation. Isolation of internal alkyne 2via distillation leads to a fair yield of 47%. Low yields arise from mixed fractions of 1 and 2; however, these fractions can be added to subsequent batches to increase product recovery. Alcohol directed hydroalumination of alkyne 2 followed by trapping with iodine yielded alkenyl iodide 3 exclusively as the Z isomer.22 Finally, displacement of alcohol via an Appel reaction provides final product (Z/E)-4. Trace amounts of isomerization in this step led to the formation of (E)-4 as the minor isomer. With this procedure in hand (Z/E)-4 could be rapidly and reliably synthesized and, with appropriate precautions, stored for several weeks with negligible decomposition.
 | ||
| Scheme 2 Synthetic route to generate the diiodoalkene precursor (Z)-2,4-diiodopent-2-ene (4). | ||
The 3-penten-2-one oxide CI is generated in the laboratory by photolysis of the (Z)-2,4-diiodopent-2-ene precursor and subsequent reaction with O2 as shown in Scheme 3.20,23,24 Photolysis of the precursor at 248 nm leads to preferential dissociation of the weaker allylic C–I bond (sp3-hybridized), rather than the vinylic C–I bond, which is sp2-hybridized. The resultant monoiodoalkene radical intermediate, Int(1), is resonantly stabilized with delocalization leading to the preferred radical site on the tertiary carbon. The subsequent reaction of Int(1) with O2 produces an iodoalkenyl peroxy radical, Int(2), which can readily rotate about the C–C and C–O bonds before dissociation of the second I atom to generate 3-penten-2-one oxide in up to eight low-energy conformational forms.
 | ||
| Scheme 3 Synthetic route to generate 3-penten-2-one oxide. | ||
Results and discussion
3-Penten-2-one oxide CIs are generated in a quartz capillary reactor tube, collisionally stabilized, and subsequently cooled in a pulsed jet expansion. A detailed description of the experimental methods is provided in Section S2 of the ESI† as well as in preceding literature.13,14,25,26 The precursor is initially detected by photoionization with 118 nm (10.5 eV) vacuum ultraviolet (VUV) radiation using a time-of-flight (TOF) mass spectrometer. Fig. 1a (gray trace) shows the TOF mass spectrum of the (Z)-2,4-diiodo-pent-2-ene precursor, where mass channel m/z 322 corresponds to the precursor mass (m/z 322; C5H8I2). Other masses are observed, for example, m/z 195 (C5H8I), which originates from the loss of an I-atom from the precursor upon photoionization (see Fig. S7† for full mass spectrum). The precursor was then photolyzed at 248 nm in a 20% O2/Ar carrier gas to generate 3-penten-2-one oxide. Upon photoionization, a new mass channel at m/z 100, consistent with the chemical composition of C5H8O2, has been identified as 3-penten-2-one oxide CI (Fig. 1a, blue trace); the structure of the lowest energy tEE conformer of 3-penten-2-one oxide is also shown. | ||
| Fig. 1 (a) Mass spectra recorded with 118 nm photoionization of the (Z)-2,4-diiodo-pent-2-ene precursor (gray). Upon photolysis of the precursor at 248 nm in a 20% O2/Ar carrier gas, the m/z 322 mass peak arising from the precursor (C5H8I2) is decreased and a new mass peak appears at m/z 100 with C5H8O2 composition (blue), which is ascribed to the 3-penten-2-one oxide CI. (b) The m/z 100 photoionization signal (blue) decreases upon 410 nm (ca. 4 mJ per pulse) excitation (red) with the difference trace (violet) illustrating the depletion signal. Also shown is the lowest energy tEE conformer of 3-penten-2-one oxide. | ||
Two types of experiments are performed to characterize the newly formed 3-penten-2-one oxide CI. In first set of experiments, UV-vis excitation resonant on the first π* ← π transition of 3-penten-2-one oxide readily depletes the ground state population of one or more of its conformers. The ground state depletion is detected after a short time delay (Δt ∼ 50 ns) as a reduced VUV photoionization signal on m/z 100. Fig. 1b shows a significant depletion of ∼35% of the m/z 100 photoionization signal upon excitation at 410 nm (ca. 4 mJ per pulse). The percentage depletion can be expressed as (N0 − N)/N0 × 100%, where N and N0 represent the ground state abundances with and without UV-vis radiation, respectively. A large depletion of 35% can be reached (Fig. S8†), indicative of a strong electronic transition and rapid decay from the excited ππ* state. The corresponding absorbance, −ln(N/N0), exhibits a linear dependence with the laser power up to ∼4 mJ per pulse, indicating a one-photon process (Fig. S8†). The magnitude of the depletion (∼35%) and associated absorbance remain almost constant at higher power, as seen previously for MVK-oxide and MACR-oxide.15,16 This suggests that the m/z 100 photoionization signal may have contributions from an isomer of 3-penten-2-oxide. Several potential isomers that can be ionized at 10.5 eV are considered in Section S4;† however, none are predicted to have significant absorption in the 320–460 nm region. Rather, their computed vertical excitation energies (VEE, Table S4†) suggest that the isomers will absorb at significantly shorter wavelengths (<265 nm).
The electronic spectrum of 3-penten-2-one oxide is recorded on its first π* ← π transition from 325 to 460 nm on the parent mass channel of the CI (m/z 100) as shown in Fig. 2. The electronic spectrum is obtained by UV-vis induced depletion of the associated VUV photoionization signal in 5 nm steps using the signal (410–460 nm) and sum frequency generated (325–405 nm) outputs of a BBO–OPO. Here, the UV-vis spectrum is normalized to OPO power, which is maintained at ca. 2.5 mJ per pulse from 325 to 405 nm and ca. 3.5 mJ per pulse from 410 to 460 nm. The experimental data points shown in Fig. 2 are averages of repeated measurements (∼4000 UV-vis pulses per point) with ±1σ uncertainty represented by the shaded region. The solid trace is obtained through smoothing (5-point) with a binomial method through the experimental data points. As evident from Fig. 2, the UV-vis spectrum of 3-penten-2-oxide shows a broad spectral profile, spanning across the entire 325–460 nm range with a maximum at ca. 375 nm. The experimental spectrum declines to half-maximum at ca. 420 nm on the long wavelength side and further drops at longer wavelengths. On the shorter wavelength side, the absorption initially decreases, showing a dip at ca. 340 nm, before starting to rise again at shorter wavelengths.
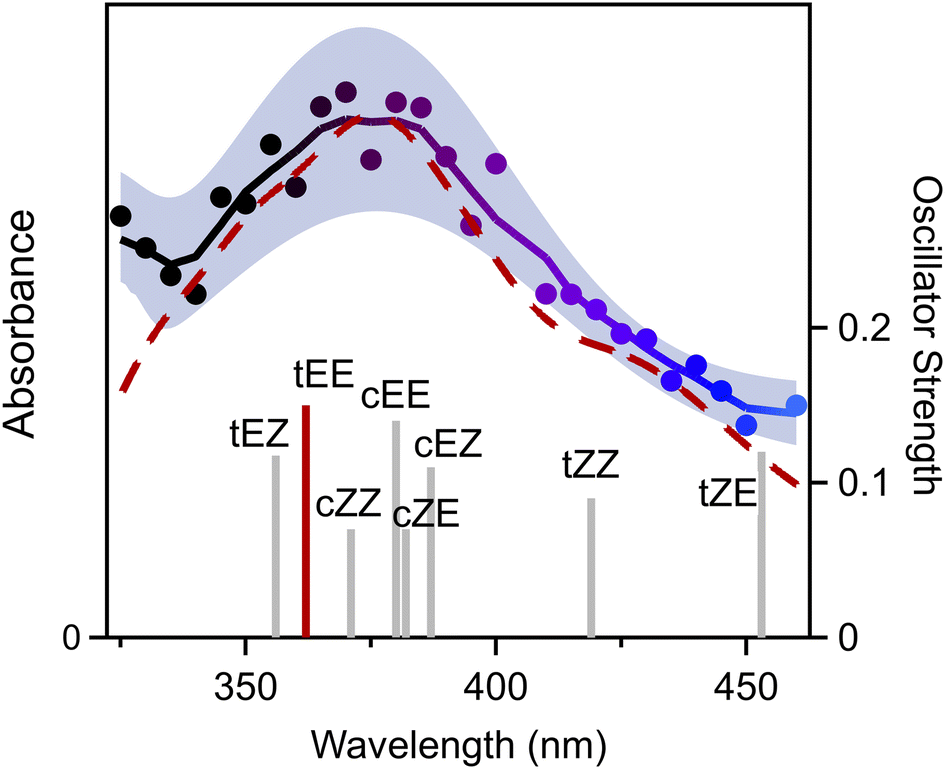 | ||
| Fig. 2 Electronic spectrum of 3-penten-2-one oxide obtained using the UV-vis induced depletion method with photoionization detection at m/z 100. The solid trace depicts a smoothed curve through the experimental data. Experimental uncertainties from repeated measurements are shown as the shaded region (±1σ). The red dashed line is the computed absorption spectrum for the most stable tEE conformer of 3-penten-2-one oxide on the first π* ← π transition. The bars indicate the vertical excitation energies (VEE) and associated oscillator strengths (f) for the eight conformers. | ||
Electronic structure calculations are carried out to characterize the relative energies of 3-penten-2-oxide conformers, relevant transition states (TS), and torsional barriers (see Section S3 of the ESI† for detailed description). Eight low energy conformations are predicted for 3-penten-2-one oxide (Fig. S9†) with ground state stabilities given in Table 1. The conformers are labelled according to the orientation of carbonyl oxide group (first Z or E) and the 1-propenyl [–CHCHCH3] group (second Z or E), with each pair of E/Z conformers separated by high barriers associated with rotation about the CO or CC bond. In addition, each E/Z pair has cis and trans (c/t) conformers separated by relatively low barriers associated with internal rotation about the C–C bond. Notably, the second lowest energy conformer, cEE, can undergo c ↔ t isomerization with a low torsional barrier (<7 kcal mol−1; Table S1†) to the lowest energy conformer, tEE. Such isomerization is likely feasible within the capillary and at the early stage of jet-expansion, increasing the population of the lowest energy tEE conformer.27,28
| Conformer | Relative energies (kcal mol−1) | VEE/λ (eV nm−1) | f |
|---|---|---|---|
| tEE | 0.00 | 3.42/362 | 0.15 |
| cEE | 1.65 | 3.25/380 | 0.14 |
| cZE | 1.98 | 3.24/382 | 0.07 |
| tZE | 2.15 | 2.73/453 | 0.12 |
| cEZ | 2.84 | 3.20/387 | 0.11 |
| tEZ | 4.07 | 3.48/356 | 0.12 |
| tZZ | 6.26 | 2.96/419 | 0.09 |
| cZZ | 7.67 | 3.34/371 | 0.07 |
The vertical excitation energies (VEE) and oscillator strengths (f) computed for the first π* ← π transition of the eight conformers of 3-penten-2-one oxide are given in Table 1 and Fig. 2. The VEE for the lowest energy conformer, tEE, is computed at 362 nm with significant oscillator strength, which agrees well with the observed absorption maximum at ca. 375 nm. The VEEs computed for the higher energy conformers are consistent with the experimental spectral range of 325–460 nm, indicating that higher energy conformers may contribute to the overall absorption. To aid in comparison, the electronic spectrum of the lowest energy tEE conformer on the first π* ← π transition is theoretically computed using the nuclear ensemble method (red dashed line, Fig. 2),29,30 which shows excellent agreement with the experimental spectrum in terms of peak position and breadth. The simulation yields a peak absorption cross-section of 1.2 × 10−17 cm2, comparable to previously studied CIs.31 Electronic spectra have also been simulated for four other conformers with energies within 3 kcal mol−1 (ESI Section S6.1 and Fig. S11†). Of these, only the cZE conformer (ca. 2 kcal mol−1) gives rise to an electronic absorption spectrum similar to that observed experimentally. However, the cZE conformer can undergo rapid 1,5 ring closure to a dioxole isomer,17 which is expected to significantly reduce the amount of stabilized cZE. As a result, the lowest energy tEE conformer is likely to be most populated and make the largest contribution to the overall absorption spectrum of 3-penten-2-one oxide. Similar to other alkenyl-substituted CIs, a second π* ← π transition, mainly involving the vinyl group, is predicted to occur at shorter wavelengths with calculated VEE below 300 nm.15,16,18 This suggests that the increase in absorption below 340 nm in the 3-penten-2-oxide spectrum may originate from the second π* ← π transition.
In a second set of experiments, OH radical products resulting from the unimolecular decay of energized 3-penten-2-one oxide intermediates and subsequent cooling in the supersonic jet are detected by UV laser-induced fluorescence (LIF) on the OH A–X (1, 0) transition. The photolysis of (Z)-2,4-diiodopent-2-ene precursor leads to allylic C–I bond fission and generates an allylic monoiodoalkene radical (Scheme 3). Typical C–I bond dissociation energy is ca. 50 kcal mol−1; therefore, a large amount of excess energy is partitioned into the resulting monoiodo-radical intermediate.32,33 Prior theoretical studies predicted that subsequent reaction of the monoiodoalkene radical with O2 is nearly thermoneutral,34 thus the significant portion of the energy is anticipated to be retained as internal excitation of the nascent CI.32 As a result, we expect that 3-penten-2-one oxide CI has sufficient internal excitation to undergo prompt unimolecular decay prior to thermalization in the capillary and collisional cooling in the supersonic jet expansion. Two unimolecular decay pathways are anticipated as the primary sources of OH radical products from specific conformers of 3-penten-2-one oxide: 1,4 H-atom transfer from the methyl-substituent to carbonyl oxide group and allylic 1,6 H-atom transfer from methyl group in the 1-propenyl substituent to the carbonyl oxide group. The vinyl hydroperoxide intermediates generated following H-atom transfer in both pathways will undergo rapid O–O fission to yield OH radicals. The 1,4 and 1,6 H-atom transfer pathways for the tEE and cZZ conformers of 3-penten-2-one oxide are illustrated in Schemes S2 and S3.† The four lowest energy conformers of 3-penten-2-one oxide can undergo 1,4 H-atom transfer, while two the highest energy conformers, tZZ and cZZ, can undergo facile 1,6 H-atom transfer to produce OH products (Table S2†).11 The four lowest energy conformers are expected to have a higher population and contribute most significantly to the formation of the OH products. Vinyl 1,4 H-atom transfer also possible for the tZE and tZZ conformers, but this is expected to have a significantly higher TS barrier5,11 and is unlikely to be a significant source of OH products.
Here, OH X2Π3/2 (v = 0, N) radicals arising from unimolecular decay of energized 3-penten-2-one oxide intermediates in the capillary tube and subsequent cooling in the free jet expansion are probed via UV excitation in the A2Σ+–X2Π3/2 (1, 0) region with LIF detection. The OH A–X (1, 0) rovibronic transitions exhibit main branch (P1, Q1, R1) and satellite branch (P21, Q21, R21) lines. Fig. 3 shows the OH A–X (1, 0) Q1(1) and Q21(1) transitions originating from the lowest OH X2Π3/2 (v = 0, N = 1) level.35 The detection of OH radicals provides further evidence that the 3-penten-2-one oxide CI has been generated in the capillary reactor tube and undergoes unimolecular decay to OH products.
 | ||
| Fig. 3 UV laser-induced fluorescence (LIF) detection of OH radicals resulting from unimolecular decay of energized 3-penten-2-one oxide and subsequent cooling in the supersonic jet expansion. The OH A2Σ+–X2Π3/2 (1, 0) Q1(1) and Q21(1) transitions originating from the lowest rovibrational level (v = 0, N = 1) are shown. | ||
The present study demonstrates the production of a five-carbon, unsaturated CI, 3-penten-2-one oxide, based on a combination of experimental and theoretical results. 3-penten-2-one oxide is structurally similar to the isoprene-derived four carbon CI, MVK-oxide (Scheme 1), both of which exhibit extended conjugation across the carbonyl oxide and vinyl groups, apart from an additional methyl group attached to the vinyl group in the five-carbon CI (Scheme 1). The electronic spectrum of MVK-oxide has been reported by UV-vis depletion spectroscopy under jet-cooled conditions and direct UV-vis absorption spectroscopy under thermal conditions.15,36 In Fig. 4, the electronic spectrum of 3-penten-2-one oxide obtained in this study is compared with the absorption spectrum of MVK-oxide.36 Both spectra are broad and predominantly associated with the first π* ← π transition; furthermore, the absorption maxima of these spectra are shifted by 50 nm to longer wavelengths than those of simple alkyl substituted CIs due to extended conjugation between the carbonyl oxide and vinyl groups.13,14,25,26 The 3-penten-2-one oxide spectrum is remarkably similar to that of MVK-oxide, peaked at 375 nm and 370 nm, respectively, as shown in Fig. 4. MVK-oxide has four conformational forms,20 although its electronic spectrum originates primarily from the lowest energy (syn–trans or tE) conformer shown in Scheme 1.36 Higher energy (anti or Z) conformers rapidly decay by a 1,5-ring closure mechanism on the timescale of thermal measurements.31,36 In the present study, the electronic spectrum of 3-penten-2-one oxide has predominate contribution from the lowest energy tEE conformer. As shown in Scheme 1, the tEE conformer of 3-penten-2-one oxide and tE conformer of MVK-oxide have the same configuration, consistent with the similarity observed in their electronic spectra. The slight red-shift observed in the electronic spectrum of the five-carbon CI could result from hyperconjugation due to the additional methyl group. Overall, the present study has revealed that the additional methyl group appended to the vinyl group in 3-penten-2-one oxide has a minimal effect on its electronic spectrum compared to MVK-oxide.
 | ||
| Fig. 4 Electronic spectrum of 3-penten-2-one oxide from this work (blue) compared with that previously reported for MVK-oxide (cyan). The electronic spectrum of MVK-oxide was adapted from ref. 36 with permission. | ||
Conclusions
This work reports the synthesis of (Z)-2,4-diiodopent-2-ene and the first identification of a five-carbon unsaturated CI, 3-penten-2-one oxide, generated photolytically from a diiodoalkene precursor. Employing VUV photoionization and TOF mass spectrometry, the 3-penten-2-one oxide CI is identified on its parent mass channel of m/z 100; moreover, depletion of the m/z 100 photoionization signal is observed when UV-vis laser radiation is introduced prior to the VUV photoionization. The electronic spectrum of 3-penten-2-one oxide is recorded in the wavelength range of 325 to 460 nm by the depletion method, exhibiting broad absorption peaking at ca. 375 nm. Based on computed VEE and simulated absorption spectrum for the first π* ← π transition, the observed spectrum is predominantly attributed to the most stable tEE conformer of 3-penten-2-one oxide. The broad electronic spectrum observed for 3-penten-2-one oxide peaked at ca. 375 nm is analogous to that previously reported for MVK-oxide at ca. 370 nm. The lowest energy conformer of 3-penten-2-one oxide has a similar structure to that of MVK-oxide with an additional methyl group. Furthermore, OH radical products arising from prompt unimolecular decay of energized 3-penten-2-oxide CIs are observed following jet-cooling by UV LIF. Overall, this study advances our efforts to characterize larger CIs as well as understand their electronic spectroscopy.Data availability
All experimental supporting data and methods are available in the ESI.†Author contributions
M. I. L. designed the research project. C. A. S. and M. C. K. synthesized the precursor. T. K. R., T. L. and Y. Q. conducted the experiments and analyzed the experimental data. T. L., Y. Q. and T. K. R. carried out the theoretical calculations. All authors discussed the results and contributed to writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Science Foundation under grants CHE-1955068 and CHE-2301298 (M. I. L.). T. K. R. was primarily supported by a Walter-Benjamin Scholarship funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project number 508074809. M. C. K. is grateful to the NSF (CHE 2102626) for financial support of this research. This work used the Advanced Cyberinfrastructure Coordination Ecosystem: Service & Support (ACCESS) program, which is supported by National Science Foundation grants #2138259, #2138286, #2138307, #2137603, and #2138296, through the allocation TG-CHE190088.References
- D. Johnson and G. Marston, Chem. Soc. Rev., 2008, 37, 699–716 RSC.
- D. L. Osborn and C. A. Taatjes, Int. Rev. Phys. Chem., 2015, 34, 309–360 Search PubMed.
- J. H. Kroll, S. R. Sahay, J. G. Anderson, K. L. Demerjian and N. M. Donahue, J. Phys. Chem. A, 2001, 105, 4446–4457 CrossRef CAS.
- J. H. Kroll, J. S. Clarke, N. M. Donahue, J. G. Anderson and K. L. Demerjian, J. Phys. Chem. A, 2001, 105, 1554–1560 CrossRef CAS PubMed.
- L. Vereecken, A. Novelli and D. Taraborrelli, Phys. Chem. Chem. Phys., 2017, 19, 31599–31612 RSC.
- T. A. Stephenson and M. I. Lester, Int. Rev. Phys. Chem., 2020, 39, 1–33 Search PubMed.
- C. A. Taatjes, D. E. Shallcross and C. J. Percival, Phys. Chem. Chem. Phys., 2014, 16, 1704–1718 RSC.
- M. A. H. Khan, C. J. Percival, R. L. Caravan, C. A. Taatjes and D. E. Shallcross, Environ. Sci.: Processes Impacts, 2018, 20, 437–453 RSC.
- P. O. Wennberg, K. H. Bates, J. D. Crounse, L. G. Dodson, R. C. McVay, L. A. Mertens, T. B. Nguyen, E. Praske, R. H. Schwantes, M. D. Smarte, J. M. St Clair, A. P. Teng, X. Zhang and J. H. Seinfeld, Chem. Rev., 2018, 118, 3337–3390 CrossRef CAS PubMed.
- T. B. Nguyen, G. S. Tyndall, J. D. Crounse, A. P. Teng, K. H. Bates, R. H. Schwantes, M. M. Coggon, L. Zhang, P. Feiner, D. O. Milller, K. M. Skog, J. C. Rivera-Rios, M. Dorris, K. F. Olson, A. Koss, R. J. Wild, S. S. Brown, A. H. Goldstein, J. A. de Gouw, W. H. Brune, F. N. Keutsch, J. H. Seinfeld and P. O. Wennberg, Phys. Chem. Chem. Phys., 2016, 18, 10241–10254 RSC.
- A. S. Hansen, Y. Qian, C. A. Sojdak, M. C. Kozlowski, V. J. Esposito, J. S. Francisco, S. J. Klippenstein and M. I. Lester, J. Am. Chem. Soc., 2022, 144, 5945–5955 CrossRef CAS PubMed.
- G. Wang, T. Liu, M. Zou, C. A. Sojdak, M. C. Kozlowski, T. N. V. Karsili and M. I. Lester, J. Phys. Chem. A, 2023, 127, 203–215 CrossRef CAS PubMed.
- J. M. Beames, F. Liu, L. Lu and M. I. Lester, J. Am. Chem. Soc., 2012, 134, 20045–20048 CrossRef CAS PubMed.
- J. M. Beames, F. Liu, L. Lu and M. I. Lester, J. Chem. Phys., 2013, 138, 244307 CrossRef PubMed.
- M. F. Vansco, B. Marchetti and M. I. Lester, J. Chem. Phys., 2018, 149, 244309 CrossRef PubMed.
- M. F. Vansco, B. Marchetti, N. Trongsiriwat, T. Bhagde, G. Wang, P. J. Walsh, S. J. Klippenstein and M. I. Lester, J. Am. Chem. Soc., 2019, 141, 15058–15069 CrossRef CAS PubMed.
- M. F. Vansco, R. L. Caravan, K. Zuraski, F. A. F. Winiberg, K. Au, N. Trongsiriwat, P. J. Walsh, D. L. Osborn, C. J. Percival, M. A. H. Khan, D. E. Shallcross, C. A. Taatjes and M. I. Lester, J. Phys. Chem. A, 2020, 124, 3542–3554 CrossRef CAS.
- G. Wang, T. Liu, A. Caracciolo, M. F. Vansco, N. Trongsiriwat, P. J. Walsh, B. Marchetti, T. N. V. Karsili and M. I. Lester, J. Chem. Phys., 2021, 155, 174305 CrossRef CAS PubMed.
- F. Liu, J. M. Beames, A. S. Petit, A. B. McCoy and M. I. Lester, Science, 2014, 345, 1596–1598 CrossRef CAS PubMed.
- V. P. Barber, S. Pandit, A. M. Green, N. Trongsiriwat, P. J. Walsh, S. J. Klippenstein and M. I. Lester, J. Am. Chem. Soc., 2018, 140, 10866–10880 CrossRef CAS PubMed.
- V. P. Barber, S. Pandit, V. J. Esposito, A. B. McCoy and M. I. Lester, J. Phys. Chem. A, 2019, 123, 2559–2569 CrossRef CAS PubMed.
- V. Y. Chen and O. Kwon, Angew. Chem., Int. Ed., 2021, 60, 8874–8881 CrossRef CAS PubMed.
- C.-A. Chung and Y.-P. Lee, Commun. Chem., 2021, 4, 8 CrossRef CAS PubMed.
- J.-R. Cai, J.-H. Su and Y.-P. Lee, Commun. Chem., 2022, 5, 26 CrossRef CAS PubMed.
- F. Liu, J. M. Beames, A. M. Green and M. I. Lester, J. Phys. Chem. A, 2014, 118, 2298–2306 CrossRef CAS PubMed.
- T. Liu, M. Zou, A. Caracciolo, C. A. Sojdak and M. I. Lester, J. Phys. Chem. A, 2022, 126, 6734–6741 CrossRef CAS PubMed.
- T. S. Zwier, Annu. Rev. Phys. Chem., 1996, 47, 205–241 CrossRef CAS.
- E. Gloaguen, M. Mons, K. Schwing and M. Gerhards, Chem. Rev., 2020, 120, 12490–12562 CrossRef CAS PubMed.
- J. C. McCoy, B. Marchetti, M. Thodika and T. N. V. Karsili, J. Phys. Chem. A, 2021, 125, 4089–4097 CrossRef CAS PubMed.
- J. C. McCoy, S. J. Léger, C. F. Frey, M. F. Vansco, B. Marchetti and T. N. V. Karsili, J. Phys. Chem. A, 2022, 126, 485–496 CrossRef CAS PubMed.
- T. N. V. Karsili, B. Marchetti, M. I. Lester and M. N. R. Ashfold, Photochem. Photobiol., 2023, 99, 4–18 CrossRef CAS PubMed.
- J. H. Lehman, H. Li and M. I. Lester, Chem. Phys. Lett., 2013, 590, 16–21 CrossRef CAS.
- B. W. Toulson, J. P. Alaniz, J. G. Hill and C. Murray, Phys. Chem. Chem. Phys., 2016, 18, 11091–11103 RSC.
- E. P. F. Lee, D. K. W. Mok, D. E. Shallcross, C. J. Percival, D. L. Osborn, C. A. Taatjes and J. M. Dyke, Chem.–Eur. J., 2012, 18, 12411–12423 CrossRef CAS PubMed.
- J. Luque and D. R. Crosley, LIFBASE: Database and Spectral Simulation Program, 1.5, SRI International Report MP 99-009, SRI International, 1999 Search PubMed.
- R. L. Caravan, M. F. Vansco, K. Au, M. A. H. Khan, Y.-L. Li, F. A. F. Winiberg, K. Zuraski, Y.-H. Lin, W. Chao, N. Trongsiriwat, P. J. Walsh, D. L. Osborn, C. J. Percival, J. J.-M. Lin, D. E. Shallcross, L. Sheps, S. J. Klippenstein, C. A. Taatjes and M. I. Lester, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 9733–9740 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03993e |
| ‡ Equal contributions. |
| This journal is © The Royal Society of Chemistry 2023 |