
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rh(III)-catalyzed highly site- and regio-selective alkenyl C–H activation/annulation of 4-amino-2-quinolones with alkynes via reversible alkyne insertion†
Naohiro
Hirako
,
Takeshi
Yasui
 * and
Yoshihiko
Yamamoto
*
* and
Yoshihiko
Yamamoto
*
Department of Basic Medicinal Sciences, Graduate School of Pharmaceutical Sciences, Nagoya University, Furo-cho Chikusa, Nagoya 464-8603, Japan. E-mail: t-yasui@ps.nagoya-u.ac.jp; yamamoto-yoshi@ps.nagoya-u.ac.jp
First published on 14th September 2023
Abstract
3,4-Fused 2-quinolone frameworks are important structural motifs found in natural products and biologically active compounds. Intermolecular alkenyl C–H activation/annulation of 4-amino-2-quinolone substrates with alkynes is one of the most efficient methods for accessing such structural motifs. However, this is a formidable challenge because 4-amino-2-quinolones have two cleavable C–H bonds: an alkenyl C–H bond at the C3-position and an aromatic C–H bond at the C5-position. Herein, we report the Rh(III)-catalyzed highly site-selective alkenyl C–H functionalization of 4-amino-2-quinolones to afford 3,4-fused 2-quinolones. This method has a wide substrate scope, including unsymmetrical internal alkynes, with complete regioselectivity. Several control experiments using an isolated key intermediate analog suggested that the annulation reaction proceeds via reversible alkyne insertion involving a binuclear Rh complex although alkyne insertion is generally recognized as an irreversible process due to the high activation barrier of the reverse process.
Introduction
Transition-metal-catalyzed site-selective C–H functionalization with the aid of directing groups is one of the most powerful methods of accessing complex molecules from readily available starting materials with high efficiency.1 The Rh(III)-catalyzed oxidative annulation of aromatic compounds with alkynes via C–H activation has been recognized as an efficient method of synthesizing fused carbo- and heterocycles such as naphthalenes, isoquinolones, isoquinolines, indoles, and isocoumarins.2 However, the site-selective C–H functionalization of aromatic compounds with multiple cleavable C–H bonds, such as the ortho and peri C–H bonds of the naphthalene ring, remains a challenge. Several research groups have addressed this issue by developing Rh(III)-catalyzed site-selective functionalizations of such aromatic compounds.3 For example, Jin et al. reported the Rh(III)-catalyzed site-selective C–H functionalization of 1-naphthylcarbamates (Scheme 1a).4 In this reaction, a neutral Rh(III) catalyst enabled peri C–H activation via selective coordination to the carbamate nitrogen, while a cationic Rh(III) catalyst selectively activated the ortho C–H bond via coordination to the carbamate oxygen in preference to the nitrogen due to its Lewis acidity. In contrast, the C–H functionalization of aromatic heterocycles is more complicated because these heterocycles usually have a greater electronic bias than aromatic carbocycles such as benzene and naphthalene.5 For example, Huckins, Bercot, et al. reported that the C–H functionalization of pyridine N-oxides proceeds selectively at the C2-position, whereas the reaction of the corresponding pyridine analog proceeds with poor site-selectivity (Scheme 1b).6a The computational mechanistic study conducted by Huckins, Thiel, Houk, et al. elucidated that the electrostatic interaction between the substrates and alkynes is the pivotal factor affecting the selectivity, whereas the selectivity-determining step can be either C–H activation or alkyne insertion depending on alkynes used.6b However, mechanistic studies to gain insight into the principles behind such selectivity in the C–H functionalization of aromatic heterocycles have rarely been conducted.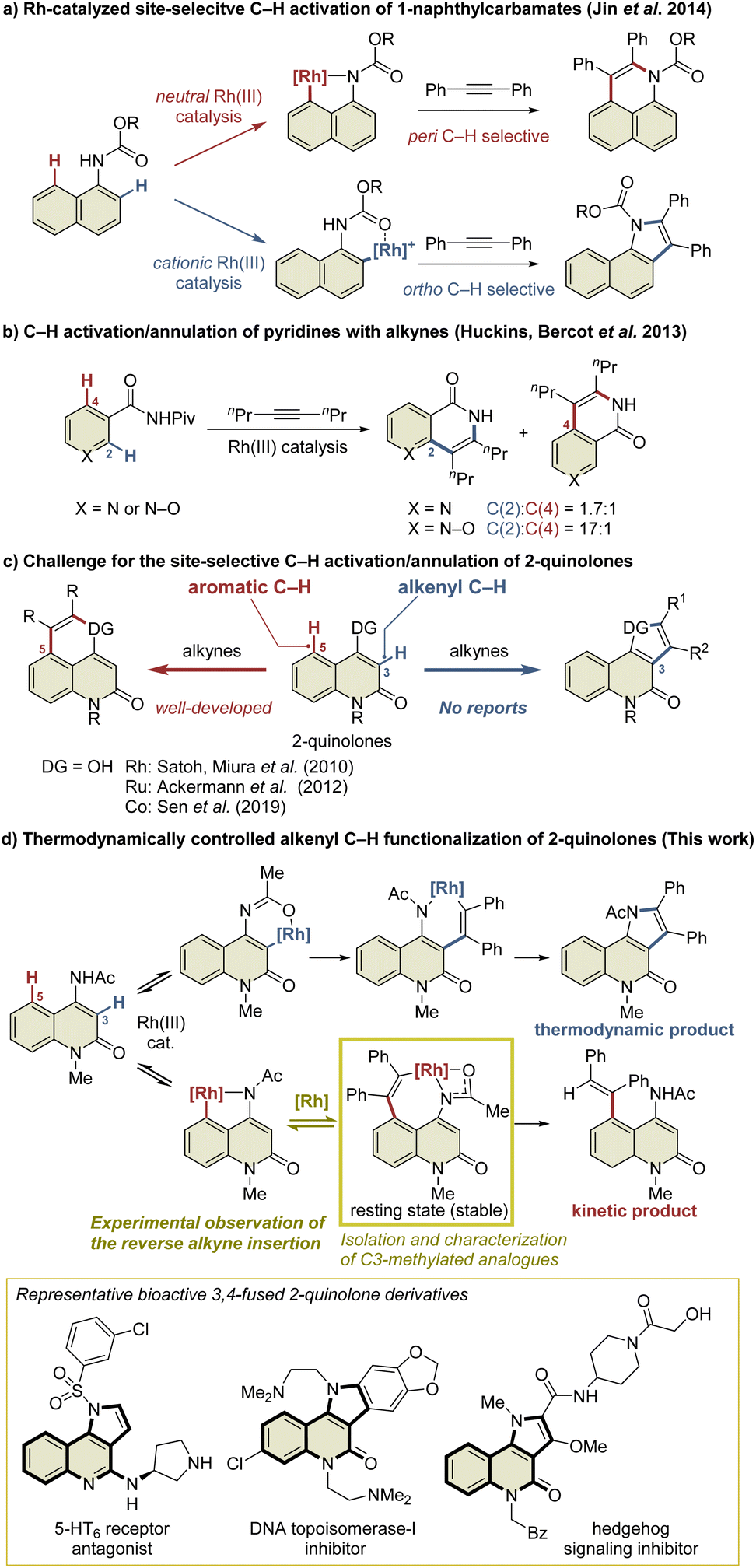 | ||
| Scheme 1 Site-selective C–H functionalization of aromatic compounds with two cleavable C–H bonds. | ||
2-Quinolone derivatives fused at the C3- and C4-positions are important motifs found in natural products and biologically active substances.7 The annulation reaction of 2-quinolones involving C–H activation is one of the most efficient methods of preparing 3,4-fused 2-quinolones such as indoloquinolones.8 In particular, the intermolecular annulation of 2-quinolones is highly desirable, as substrates are readily available compared to those required for intramolecular reactions. However, 2-quinolones bearing a directing group at the C4-position possess two cleavable C–H bonds: an alkenyl C–H bond at the C3-position and an aromatic C–H bond at the C5-position (Scheme 1c). For selective access to 3,4-fused 2-quinolones, C3-selective C–H functionalization is required. As an effective method of managing this issue, Pd/norbornene-mediated C3-selective C–H functionalizations (the Catellani reaction) of 4-iodo-2-quinolones have recently been developed by the present authors and other research groups.9,10 However, the related annulation of 4-iodo-2-quinolones with activated alkynes, such as dimethyl acetylenedicarboxylate, can potentially afford both 3,4- and 4,5-fused 2-quinolone products.11 Thus, the C3-selective annulation of 2-quinolones with alkynes is a formidable challenge, whereas the C5-selective annulation has been well developed.12
Herein, we report the Rh(III)-catalyzed highly selective alkenyl C–H functionalization of 4-amino-2-quinolones, providing access to 3,4-fused 2-quinolones (Scheme 1d). The mechanistic study suggested that the present C3-selective annulation reaction proceeds via an unprecedented reversible alkyne insertion. In general, the reverse process of alkyne insertion is recognized as an unfavorable process. In fact, only a few examples of β-carbon elimination of alkenylmetal species have been reported to date.13 Ishii et al. reported the β-carbon elimination of Ru or Rh complexes, and indicated that the β-carbon elimination was thermodynamically unfavorable.13b,c On the other hand, in the previously reported Rh(III)-catalyzed C–H activation/annulation reactions, the DFT calculations indicated that the β-carbon elimination of alkenylrhodium intermediates required relatively high activation energy, which is estimated to be more than +27 kcal mol−1 in most cases.14 Thus, subsequent processes such as reductive elimination are generally more facile to occur, and the β-carbon elimination of an alkenylrhodium complex is hard to observe, even if possible. Nevertheless, in this study, we have succeeded in the preparation of a key alkenylrhodium intermediate analog and the observation of the β-carbon elimination involving a binuclear Rh complex.
The resulting product has a pyrroloquinolone scaffold, which is found in a number of bioactive compounds such as hedgehog signaling inhibitors,7b 5-HT6 receptor antagonists,7d and topoisomerase-I inhibitors.7e As an efficient method for the construction of this scaffold, Cai et al. have reported the Cu-catalyzed tandem [3 + 2] cycloaddition/C–C coupling of N-methyl-N-(2-iodophenyl) propiolamides with isocyanides,15a although preparation methods of pyrroloquinolone derivatives are still scarce and are mostly based on linear synthesis.15 However, the preparation of isocyanides generally requires several steps, most of which still rely on the dehydration of formamides with highly electrophilic dehydration reagents such as POCl3 and p-TsCl.16 In addition, this method is unsuitable for the collective synthesis of diverse products because lengthy synthetic manipulations are required for the introduction of different substituents on the pyrrole ring. Compared with this method, in the present Rh-catalyzed annulation reaction, numerous alkynes can be used as stable and readily available reagents, allowing quick access to diverse pyrroloquinolone derivatives from 4-amino-2-quinolones.
Results and discussion
Reaction development
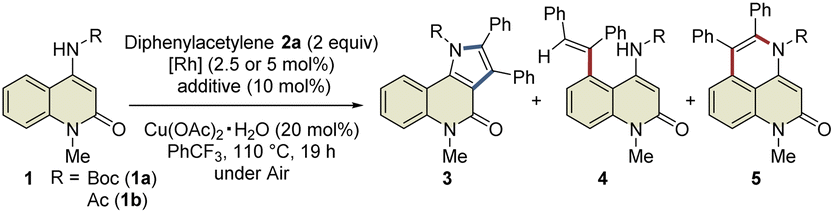
This study was commenced with the reaction of 4-amino-N-Boc-2-quinolone (1a) using a cationic Rh(III) catalyst ([Cp*RhCl2]2/AgSbF6) to realize C3-selective annulation with diphenylacetylene, based on Jin's report.4 However, the undesired C5-annulated product 5aa was selectively obtained in 65% yield, along with several unidentified products (Table 1, entry 1). The use of the neutral Rh(III) catalyst ([Cp*RhCl2]2) also resulted in the selective formation of 5aa (entry 2). These results indicate that the principle for the site-selective annulation of 1-naphthalenylcarbamates with alkynes is not applicable to the present reaction. Surprisingly, however, the reaction of 4-amino-N-acetyl-2-quinolone (1b) using the neutral Rh(III) catalyst afforded the desired C3-annulated product 3ba in 63% yield without the formation of C5-functionalized products (entry 3). An optimization study revealed that the use of benzotrifluoride as the solvent gave the highest yield of 3ba (entry 4). Notably, when Cp*Rh(OAc)2·H2O was used as the catalyst, 3ba was obtained in 65% yield, along with C5-functionalized product 4ba in 5% yield (entry 5); the latter might be generated via protodemetalation associated with increasing the amount of acetate derived from the catalyst. However, the reaction did not proceed when one equivalent of acetic acid was added to promote the formation of 4ba (entry 6).|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1 | Additive | 3 (%) | 4 (%) | 5 (%) | RSM (%) |
| a Reaction conditions: 1 (0.2 mmol), 2a (0.4 mmol), [Cp*RhCl2]2 (2.5 mol%) or Cp*Rh(OAc)2·H2O (5 mol%), Cu(OAc)2·H2O (20 mol%), PhCF3 (2 mL). For details of the optimization study, see Tables S1 and S2. The isolated yields are reported. n.d. = not detected. b Yields were determined by 1H NMR spectroscopy of the crude mixture. c 1.2-Dichloroethane was used as the solvent instead of PhCF3. d 2 equiv. of Cu(OAc)2·H2O was used under Ar atmosphere. e Cp*Rh(OAc)2·H2O was used instead of [Cp*RhCl2]2. f AcOH (1 equiv.) was added. | ||||||
| 1b | 1a | AgSbF6 | <10 | n.d. | 65 | 0 |
| 2c | 1a | — | n.d. | n.d. | 78 | 0 |
| 3c,d | 1b | — | 63 | n.d. | n.d. | 0 |
| 4 | 1b | — | 68 | n.d. | n.d. | 0 |
| 5e | 1b | — | 65 | 5 | n.d. | 0 |
| 6f | 1b | — | n.d. | n.d. | n.d. | 92 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 regioisomeric ratio. In addition, 4-octyne and phenylacetylene were not tolerated in this transformation. Regarding the substituents on the benzene ring of the 2-quinolone scaffold, methyl, trifluoromethyl, fluoro, bromo, and methoxy groups at the C6- or C7-positions were compatible with this reaction, affording the desired products 3ca–ga in 57–74% yields with excellent site-selectivity. Remarkably, the introduction of a pivaloyl group instead of the acetyl group in 1b led to the formation of 3b′g in 95% yield with excellent regioselectivity (>20:1 rr), which is probably due to the steric hindrance between the tert-butyl and phenyl groups. To evaluate the potential utility of this reaction, we performed a scale-up experiment, which produced 1.16 g of 3ba without a significant decrease in the yield (63%).
:1 regioisomeric ratio. In addition, 4-octyne and phenylacetylene were not tolerated in this transformation. Regarding the substituents on the benzene ring of the 2-quinolone scaffold, methyl, trifluoromethyl, fluoro, bromo, and methoxy groups at the C6- or C7-positions were compatible with this reaction, affording the desired products 3ca–ga in 57–74% yields with excellent site-selectivity. Remarkably, the introduction of a pivaloyl group instead of the acetyl group in 1b led to the formation of 3b′g in 95% yield with excellent regioselectivity (>20:1 rr), which is probably due to the steric hindrance between the tert-butyl and phenyl groups. To evaluate the potential utility of this reaction, we performed a scale-up experiment, which produced 1.16 g of 3ba without a significant decrease in the yield (63%).
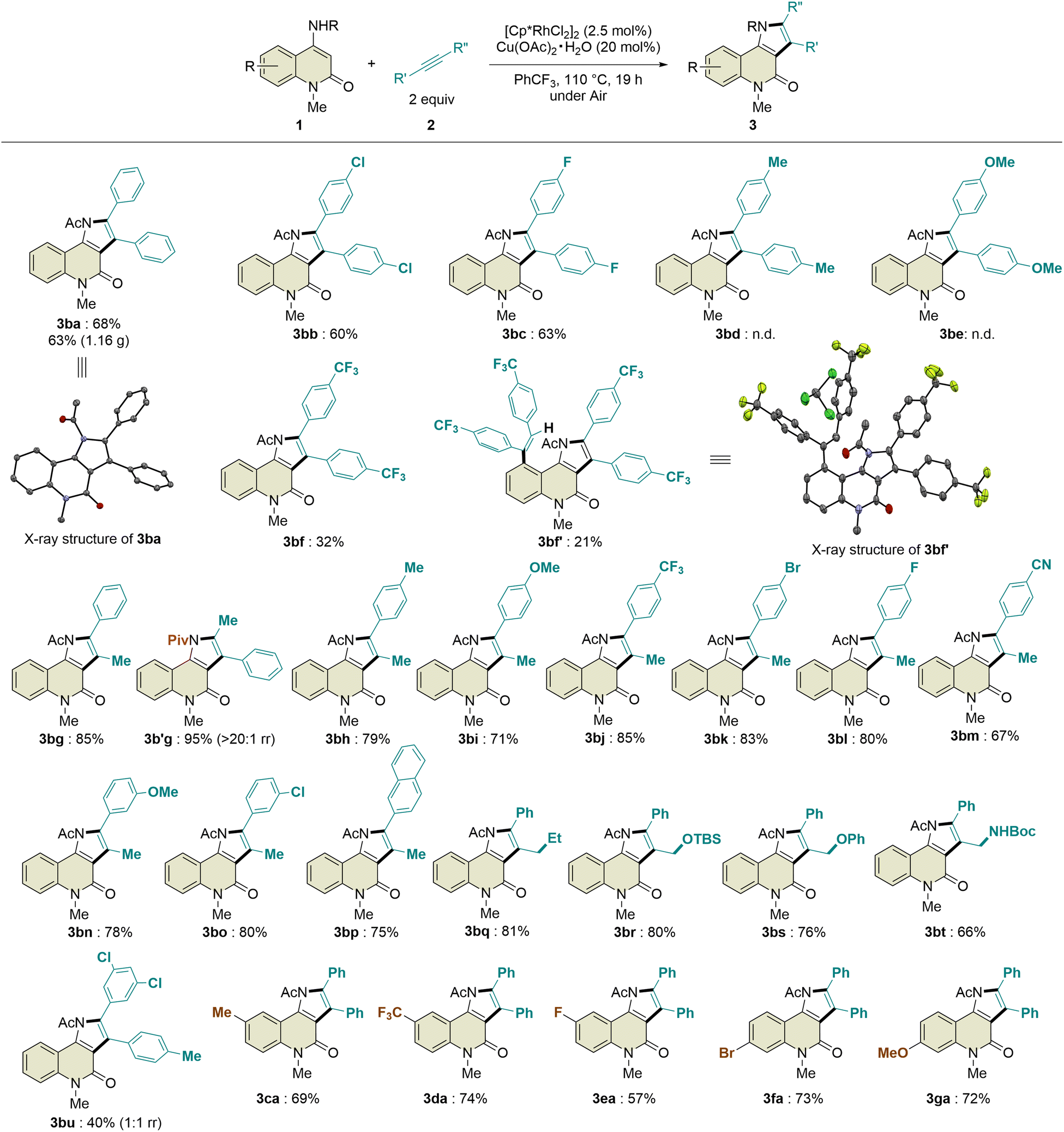 | ||
| Scheme 2 Substrate scope. | ||
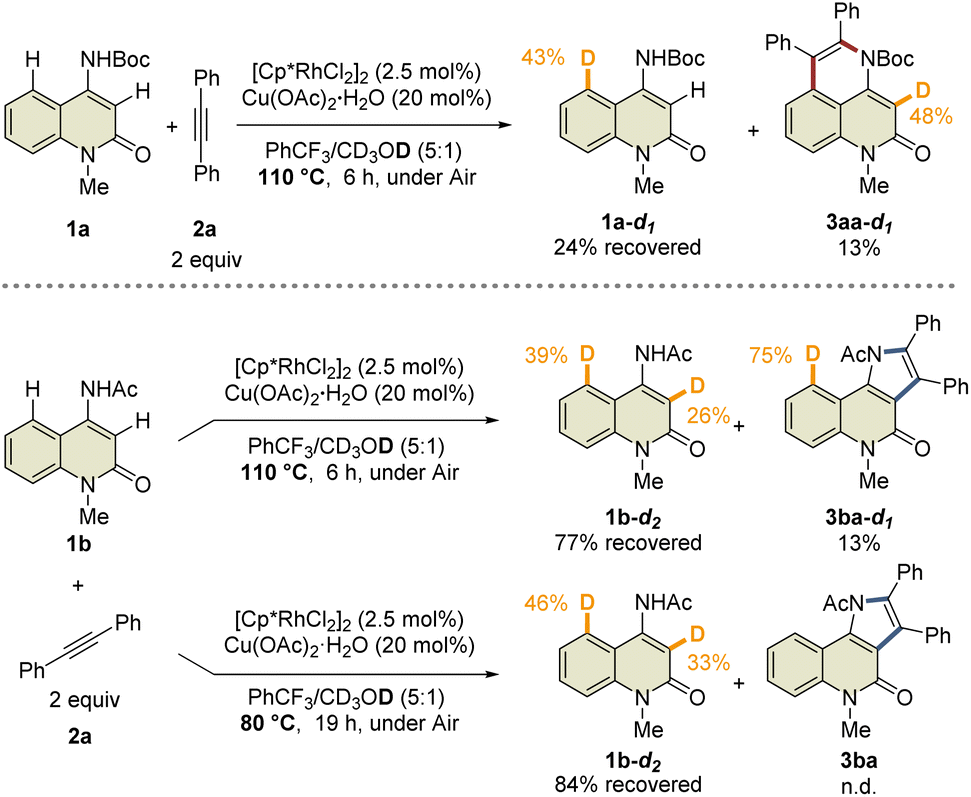 | ||
| Scheme 3 Analysis of hydrogen/deuterium exchange. | ||
During the reaction optimization study, we found that not only C3-functionalized product 3ba but also C5-functionalized product 4ba were obtained when Cp*Rh(OAc)2·H2O was used as the catalyst (Table 1, entry 5), indicating that alkyne insertion can also occur at the C5-position. Interestingly, lowering the reaction temperature to 100 °C led to an increase in the ratio of 4ba to 3ba (Fig. 1a). Furthermore, the same reaction conducted at 70 °C for 6 d afforded 3ba and 4ba in 29% and 23% NMR yields, respectively. These results clearly show that 3ba was formed under thermodynamic control, whereas 4ba was formed under kinetic control. Based on these results, we assumed that the alkyne insertion into the five-membered rhodacycle intermediate generated by C5–H activation is a reversible process (Fig. 1b). Although the β-carbon elimination from an alkenylmetal complex is very rare, Ishii et al. recently reported that a related seven-membered rhodacycle undergoes an exchange reaction of the alkenyl unit through β-carbon elimination.13c Similar alkyne deinsertion could also occur in the present reaction system.
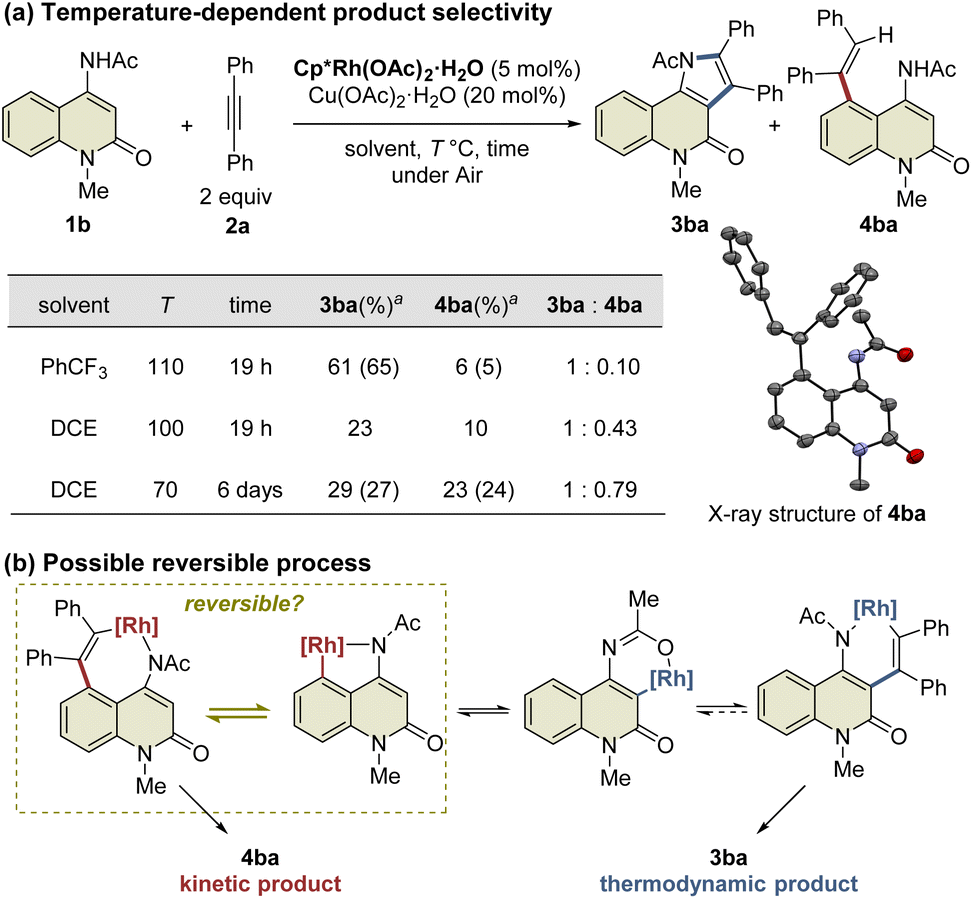 | ||
| Fig. 1 Temperature-dependence of the reaction. aYields are determined by 1H NMR. Isolated yields are shown in parentheses. | ||
To prove this hypothesis, we performed an NMR experiment using 1b and 2a-d10 in the presence of a stoichiometric amount of Cp*Rh(OAc)2·H2O as a preliminary investigation. Triethylamine was used as an additive to suppress the protodemetalation. When we monitored the progress of the reaction at 40 °C in CDCl3, two different species, which might be rhodacycle intermediates, other than 3ba-d10 and 4ba-d10, were observed immediately after initiation of the reaction (Fig. 2a). To our delight, one of these intermediates was isolated by silica gel column chromatography, and X-ray crystallographic analysis revealed the structure to be the rhodium sandwich complex A-d10 with an η4-pyrrole ligand. However, another intermediate B-d10 could not be isolated because of its instability. In the NMR experiment, a distinctive 1H NMR signal of the alkenyl proton derived from intermediate B-d10 was observed at δ 6.35 ppm as a singlet (Fig. S7†). In addition, the corresponding signal of the Cp* methyl groups was also observed at δ 1.32 ppm as a singlet. Based on these observations, we assumed that intermediate B-d10 might be a five-membered rhodacycle generated by C5–H activation or a seven-membered rhodacycle formed by subsequent alkyne insertion. Reaction monitoring by 1H NMR revealed the formation of a small amount of rhodacycle B-d10 over a long period of time, suggesting that this intermediate might be in a resting state and gradually evolved into 4ba-d10via protodemetalation or 3ba-d10 through the regeneration of 1b in equilibrium. To obtain further information regarding intermediate B, we designed a C3-methylated 2-quinolone 1h to prevent the C3-metalation, in order to selectively prepare the corresponding rhodacycle intermediate (Fig. 2b). The treatment of 1h with Cp*Rh(OAc)2·H2O (1 equiv.) and 2a (2 equiv.) at 40 °C in CDCl3 led to a rapid and quantitative formation of rhodacycle B-Me, for which the 1H NMR signals are similar to those of intermediate B observed in the NMR experiment (Fig. S7†). Complex B-Me was isolated in 66% yield and X-ray crystallographic analysis unambiguously revealed the structure to be a seven-membered rhodacycle. This rhodium complex adopted a three-legged piano stool geometry, in which the amidate moiety coordinates to the Rh center as a κ2-N,O bidentate ligand.
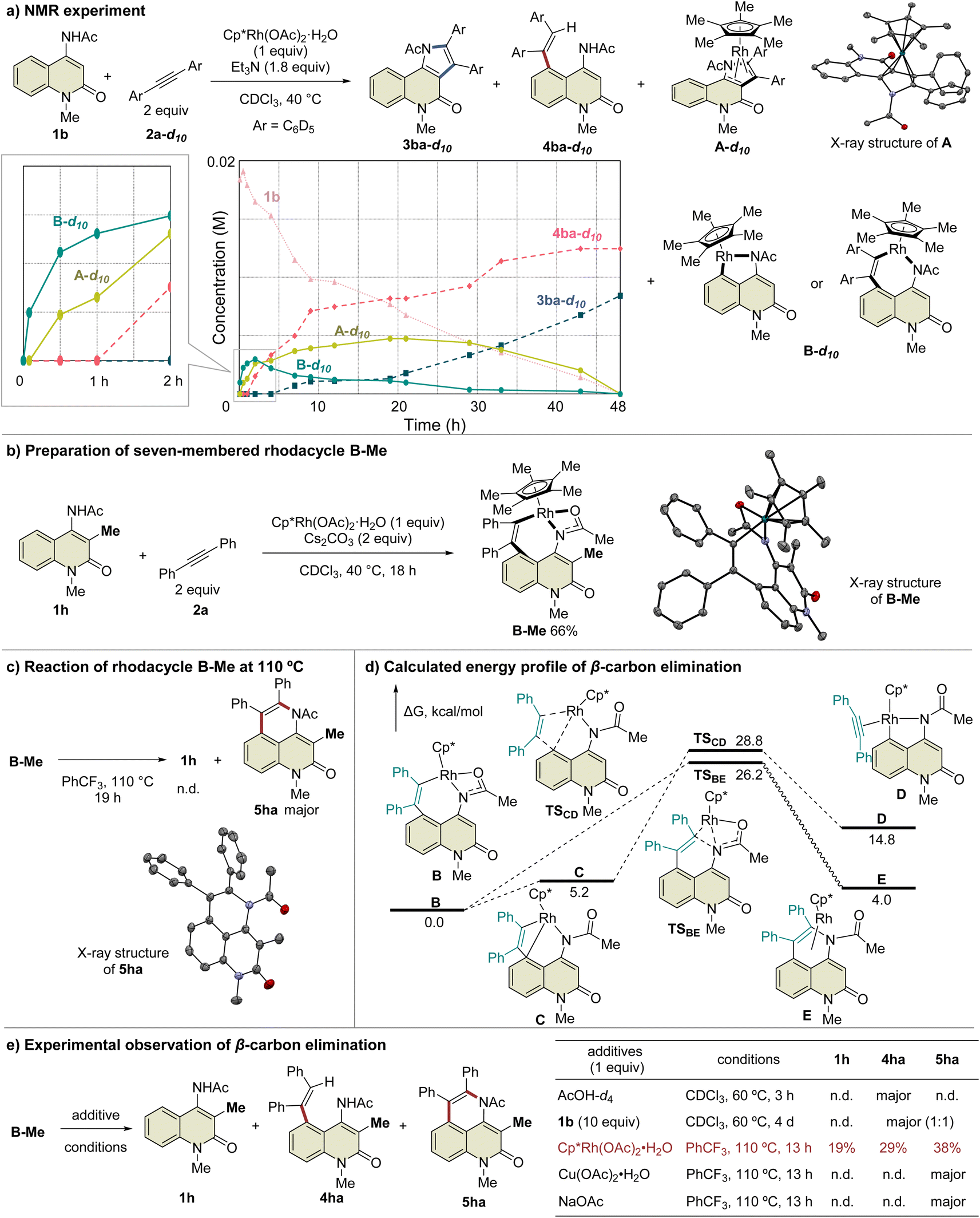 | ||
| Fig. 2 Mechanistic study involving rhodacycle intermediates. | ||
With the isolated key intermediate analog B-Me in hand, we sought to observe the β-carbon elimination experimentally. First, complex B-Me was heated to 110 °C in benzotrifluoride; however, 1h was not obtained, and reductive elimination proceeded to afford 5ha as the main product (Fig. 2c). To estimate the activation energy for the β-carbon elimination process, DFT calculations were performed at the SMD (DCE) B3LYP-D3(BJ)/6-311++G(d,p)-SDD//B3LYP-D3(BJ)/6-31G(d)-LanL2DZ level of theory. The calculated activation energy for the β-carbon elimination was 28.8 kcal mol−1, whereas that for reductive elimination was 26.2 kcal mol−1, indicating that it is difficult for β-carbon elimination to proceed before reductive elimination (Fig. 2d). Accordingly, we hypothesized that a mediator is required to promote the β-carbon elimination. Several additives utilized in the present catalytic reaction were examined (Fig. 2e and Table S3†). The addition of AcOH-d4 resulted in protodemetalation to afford only 4ha. When an excess amount of 1b was used, 1:1 mixture of 4ha and 5ha were observed while 1h was not detected. Finally, we found that the addition of Cp*Rh(OAc)2·H2O (1 equiv.) promoted β-carbon elimination from B-Me to afford 1h in 19% yield, along with the formation of 4ha and 5ha, although the addition of Cu(OAc)2 or NaOAc as an acetate source resulted in the formation of only 5ha. This suggests that β-carbon elimination from B-Me involves a binuclear (or polynuclear) Rh complex, although further investigations are required to elucidate the details of the mechanism of β-carbon elimination in the catalytic reaction process.
A plausible mechanism based on these results is illustrated in Scheme 4. Initially, complex I′ is formed by the coordination of the Rh catalyst to the amide nitrogen of 1b, followed by the formation of five-membered rhodacycle II′via C5–H activation. Subsequent alkyne insertion provides seven-membered rhodacycle B, which is a reversible process that involves another Rh catalyst. On the other hand, C3–H activation proceeds via the coordination of the Rh catalyst to the amide oxygen to form the six-membered rhodacycle II. All these processes are reversible, and complex B is in a resting state. A catalytic amount of acetic acid generated in situ can protonate complex B (or the binuclear Rh complex) to afford 4ba under kinetic control, while alkyne insertion and reductive elimination from complex II proceed to afford the thermodynamic product 3ba. In the above-mentioned NMR experiment, complex B was formed much faster than the 3ba-Rh(I) complex (complex A) in the initial period of the reaction (Fig. 2a), and no NMR signals corresponding to intermediates III or IV were detected during the experiment. These results indicate that alkyne insertion from intermediate II may be a relatively slow process while reductive elimination from intermediate IV may occur more easily. Moreover, because of the low concentration of acetic acid in the catalytic process, protodemetalation from complex B is probably much slower than alkyne insertion and reductive elimination from intermediate II, allowing the smooth progress of C3-selective functionalization at the elevated temperature.
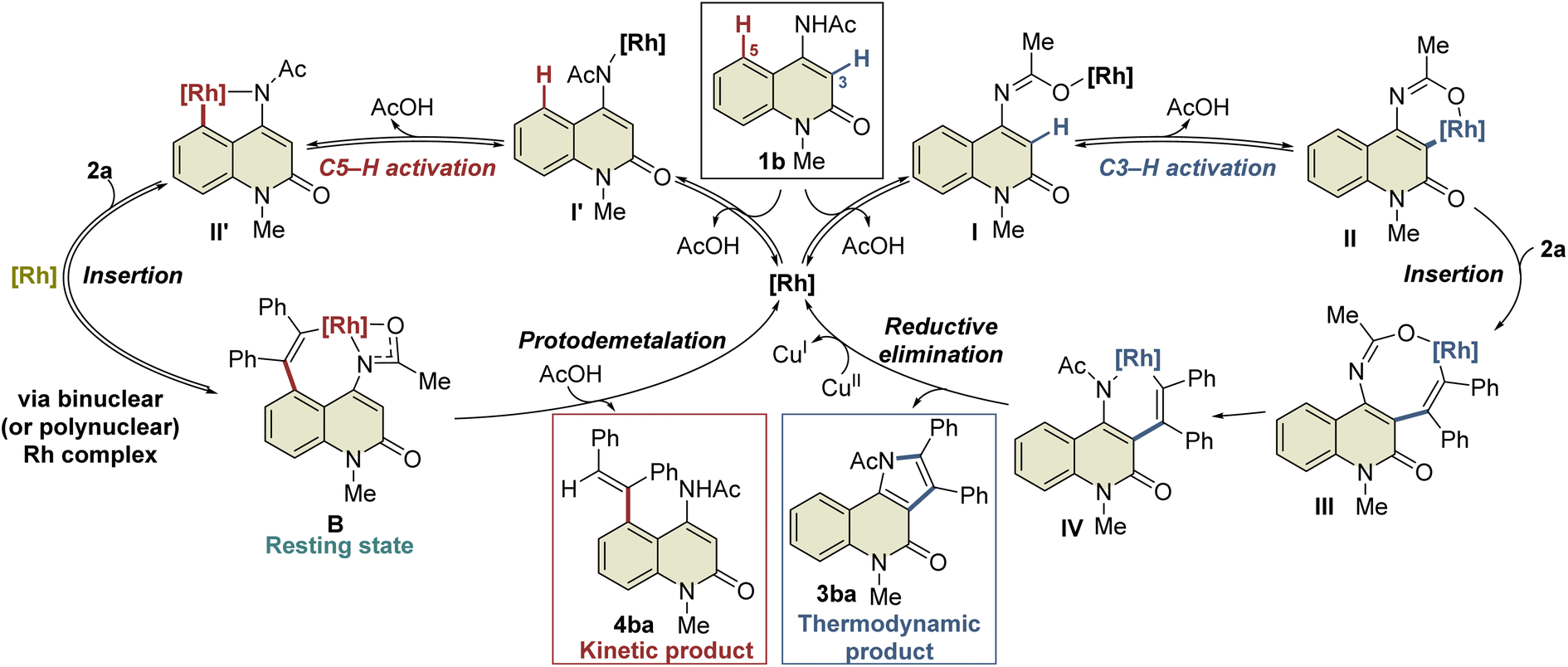 | ||
| Scheme 4 Proposed reaction mechanism. | ||
Conclusions
In summary, we established an efficient method for preparing 3,4-fused 2-quinolones via the Rh(III)-catalyzed selective alkenyl C–H functionalization of 4-amino-2-quinolones bearing an N-acetyl group as a directing group. This protocol has a wide substrate scope, including unsymmetrical internal alkynes, with complete regioselectivity. Several control experiments using an isolated key intermediate analog suggested that the reaction process involves an unprecedented reversible alkyne insertion process, leading to the simultaneous existence of key intermediates for both C3- and C5-functionalization in equilibrium. Alkyne insertion and reductive elimination from the C3-metalated intermediate may occur more readily than protodemetalation (or reductive elimination) from the seven-membered rhodacycle intermediate leading to C5-functionalization, allowing selective functionalization of the alkenyl C3–H bond over the aromatic C5–H bond. Further studies, including theoretical calculations, to elucidate the details of the reaction process are underway in our laboratory.Data availability
Data for the crystal structure reported in this paper have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under the deposition number 2278040 (for 3ba), 2278041 (for 3bf′), 2278042 (for 4ba), 2278043 (for 5ha), 2278044 (for A), and 2278045 (for B-Me). All other data including experimental procedures, compound characterization, NMR spectra, theoretical calculations, and supporting figures and tables are recorded in the ESI.†Author contributions
N. H. performed experiments, computational studies, and X-ray crystal structure analysis. N. H. and T. Y. wrote the manuscript. T. Y. and Y. Y designed, advised, and directed the project. All authors edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was partially supported by the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS) from AMED under Grant Number JP23ama121044) and JSPS KAKENHI (Grant Number JP21K05051). The computations were carried out on the supercomputer of the Research Center for Computational Science, Okazaki, Japan (Project: 23-IMS-C125). We thank Dr Shuhei Ohmura for carrying out the X-ray crystallographic analysis.Notes and references
- For recent reviews, see: (a) R. Zhu, M. E. Farmer, Y. Chen and J. Yu, Angew. Chem., Int. Ed., 2016, 55, 10578–10599 CrossRef CAS; (b) T. Brandhofer and O. García Mancheño, Eur. J. Org Chem., 2018, 2018, 6050–6067 CrossRef CAS; (c) A. Baccalini, G. Faita, G. Zanoni and D. Maiti, Chem. - Eur. J., 2020, 26, 9749–9783 CrossRef CAS; (d) O. Baudoin, Angew. Chem., Int. Ed., 2020, 59, 17798–17809 CrossRef CAS; (e) Y. Zhang and M. Szostak, Chem. - Eur. J., 2022, 28, e202104278 CrossRef CAS.
- For selected reviews, see: (a) R. He, Z.-T. Huang, Q.-Y. Zheng and C. Wang, Tetrahedron Lett., 2014, 55, 5705–5713 CrossRef CAS; (b) X. Cui, J. Mo, L. Wang and Y. Liu, Synthesis, 2015, 47, 439–459 CrossRef; (c) P. Gandeepan and C.-H. Cheng, Chem.–Asian J., 2016, 11, 448–460 CrossRef CAS PubMed; (d) Y. Yang, K. Li, Y. Cheng, D. Wan, M. Li and J. You, Chem. Commun., 2016, 52, 2872–2884 RSC; (e) A. Peneau, C. Guillou and L. Chabaud, Eur. J. Org Chem., 2018, 2018, 5777–5794 CrossRef CAS; (f) C. Wang, F. Chen, P. Qian and J. Cheng, Org. Biomol. Chem., 2021, 19, 1705–1721 RSC; (g) A. Saha, M. Shankar, S. Sau and A. K. Sahoo, Chem. Commun., 2022, 58, 4561–4587 RSC.
- (a) Á. M. Martínez, J. Echavarren, I. Alonso, N. Rodríguez, R. G. Arrayás and J. C. Carretero, Chem. Sci., 2015, 6, 5802–5814 RSC; (b) K. Elumalai and W. K. Leong, Tetrahedron Lett., 2018, 59, 113 CrossRef CAS; (c) T. P. Pabst, J. V. Obligacion, É. Rochette, I. Pappas and P. J. Chirik, J. Am. Chem. Soc., 2019, 141, 15378–15389 CrossRef CAS PubMed; (d) R. A. Alharis, C. L. Mcmullin, D. L. Davies, K. Singh and S. A. Macgregor, J. Am. Chem. Soc., 2019, 141, 8896–8906 CrossRef CAS; (e) D. Prim and B. Large, Synthesis, 2020, 52, 2600–2612 CrossRef.
- X. Zhang, W. Si, M. Bao, N. Asao, Y. Yamamoto and T. Jin, Org. Lett., 2014, 16, 4830–4833 CrossRef CAS.
- (a) R. B. Bedford, S. J. Durrant and M. Montgomery, Angew. Chem., Int. Ed., 2015, 54, 8787–8790 CrossRef CAS PubMed; (b) J. A. Leitch, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 5618–5627 CrossRef CAS; (c) D. Kang, K. Ahn and S. Hong, Asian J. Org. Chem., 2018, 7, 1136–1150 CrossRef CAS; (d) A. Biswas, S. Maity, S. Pan and R. Samanta, Chem.–Asian J., 2020, 15, 2092–2109 CrossRef CAS; (e) A. Corio, C. Gravier-Pelletier and P. Busca, Molecules, 2021, 26, 5467 CrossRef CAS PubMed; (f) Y. Yamamoto in Handbook of CH-Functionalization, ed. D. Maiti, Wiley, 2022 Search PubMed.
- (a) J. R. Huckins, E. A. Bercot, O. R. Thiel, T.-L. Hwang and M. M. Bio, J. Am. Chem. Soc., 2013, 135, 14492–14495 CrossRef CAS PubMed; (b) S. R. Neufeldt, G. Jiménez-Osés, J. R. Huckins, O. R. Thiel and K. N. Houk, J. Am. Chem. Soc., 2015, 137, 9843–9854 CrossRef CAS.
- (a) C. A. Leach, T. H. Brown, R. J. Ife, D. J. Keeling, S. M. Laing, M. E. Parsons, C. A. Price and K. J. Wiggall, J. Med. Chem., 1992, 35, 1845–1852 CrossRef CAS; (b) T. Ohashi, Y. Oguro, T. Tanaka, Z. Shiokawa, S. Shibata, Y. Sato, H. Yamakawa, H. Hattori, Y. Yamamoto, S. Kondo, M. Miyamoto, H. Tojo, A. Baba and S. Sasaki, Bioorg. Med. Chem., 2012, 20, 5496–5506 CrossRef CAS; (c) Z. Fu, K. Jiang, T. Zhu, J. Torres and Y. R. Chi, Angew. Chem., Int. Ed., 2014, 53, 6506–6510 CrossRef CAS; (d) K. Grychowska, G. Satała, T. Koz, A. Partyka, E. Colacino, S. Chaumont-Dubel, X. Bantreil, A. Wesołowska, M. Pawłowski, J. Martinez, P. Marin, G. Subra, A. J. Bojarski, F. Lamaty, P. Popik and P. Zajdel, ACS Chem. Neurosci., 2016, 7, 972–983 CrossRef CAS PubMed; (e) W.-Y. Hsueh, Y.-S. E. Lee, M.-S. Huang, C.-H. Lai, Y.-S. Gao, J.-C. Lin, Y.-F. Chen, C.-L. Chang, S.-Y. Chou, S.-F. Chen, Y.-Y. Lu, L.-H. Chang, S. F. Lin, Y.-H. Lin, P.-C. Hsu, W.-Y. Wei, Y.-C. Huang, Y.-F. Kao, L.-W. Teng, H.-H. Liu, Y.-C. Chen, T.-T. Yuan, Y.-W. Chan, P.-H. Huang, Y.-T. Chao, S.-Y. Huang, B.-H. Jian, H.-Y. Huang, S.-C. Yang, T.-H. Lo, G.-R. Huang, S.-Y. Wang, H.-S. Lin, S.-H. Chuang and J.-J. Huang, J. Med. Chem., 2021, 64, 1435–1453 CrossRef CAS PubMed.
- (a) L. M. Pardo, A. M. Prendergast, M.-T. Nolan, E. Ó Muim-hneacháin and G. P. Mcglacken, Eur. J. Org Chem., 2015, 2015, 3540–3550 CrossRef CAS; (b) K. Mackey, L. M. Pardo, A. M. Prendergast, M.-T. Nolan, L. M. Bateman and G. P. Mcglacken, Org. Lett., 2016, 18, 2540–2543 CrossRef CAS PubMed; (c) C. Cheng, W.-W. Chen, B. Xu and M.-H. Xu, J. Org. Chem., 2016, 81, 11501–11507 CrossRef CAS PubMed; (d) X. Li, L. Hu, S. Ma, H. Yu, G. Lu and T. Xu, ACS Catal., 2023, 13, 4873–4881 CrossRef CAS.
- (a) Y. Yamamoto, T. Murayama, J. Jiang, T. Yasui and M. Shibuya, Chem. Sci., 2018, 9, 1191–1199 RSC; (b) X.-M. Chen, C. Zhu, D.-F. Chen and L.-Z. Gong, Angew. Chem., Int. Ed., 2021, 60, 24844–24848 CrossRef CAS PubMed.
- For related examples, see: (a) C. Blaszykowski, E. Aktoudianakis, C. Bressy, D. Alberico and M. Lautens, Org. Lett., 2006, 8, 2043–2045 CrossRef CAS PubMed; (b) Y. Shang, C. Wu, Q. Gao, C. Liu, L. Li, X. Zhang, H.-G. Cheng, S. Liu and Q. Zhou, Nat. Commun., 2021, 12, 2988–2998 CrossRef CAS PubMed.
- Y. Yamamoto, J. Jiang and T. Yasui, Chem. - Eur. J., 2020, 26, 3749–3757 CrossRef CAS PubMed.
- (a) S. Mochida, M. Shimizu, K. Hirano, T. Satoh and M. Miura, Chem.–Asian J., 2010, 5, 847–851 CrossRef CAS PubMed; (b) V. S. Thirunavukkarasu, M. Donati and L. Ackermann, Org. Lett., 2012, 14, 3416–3419 CrossRef CAS PubMed; (c) P. K. Dutta, M. K. Ravva and S. Sen, J. Org. Chem., 2019, 84, 1176–1184 CrossRef CAS PubMed.
- (a) M. Etienne, R. Mathieu and B. Donnadieu, J. Am. Chem. Soc., 1997, 119, 3218–3228 CrossRef CAS; (b) Y. Ikeda, Y. Mutoh, K. Imai, N. Tsuchida, K. Takano and Y. Ishii, Organometallics, 2013, 32, 4353–4358 CrossRef CAS; (c) T. Iwamoto, K. Shibuya, T. Takakuwa, T. Kuwabara and Y. Ishii, Organometallics, 2022, 41, 182–186 CrossRef CAS.
- (a) W. Wu, Y. Liu and S. Bi, Org. Biomol. Chem., 2015, 13, 8251–8260 RSC; (b) W.-J. Chen and Z. Lin, Organometallics, 2015, 34, 309–318 CrossRef CAS; (c) R. Thenarukandiyil, S. K. Gupta and J. Choudhury, ACS Catal., 2016, 6, 5132–5137 CrossRef CAS; (d) M. Zhang and G. Huang, Chem. - Eur. J., 2016, 22, 9356–9365 CrossRef CAS; (e) Y.-F. Yang, K. N. Houk and Y.-D. Wu, J. Am. Chem. Soc., 2016, 138, 6861–6868 CrossRef CAS; (f) L. Han, X. Zhang, X. Wang, F. Zhao, S. Liu and T. Liu, Org. Biomol. Chem., 2017, 15, 3938–3946 RSC.
- (a) F. Zhou, J. Liu, K. D. Jinsong and Q. Cai, J. Org. Chem., 2011, 76, 5346–5353 CrossRef CAS PubMed; (b) T. C. T. Ho and K. Jones, Tetrahedron, 1997, 53, 8278–8294 CrossRef; (c) G. T. Manh, H. Bakkali, L. Maingot, M. Pipelier, U. Joshi, J. P. Pradére, S. Sabelle, R. Tuloup and D. Dubreuil, Tetrahedron Lett., 2004, 45, 5913–5916 CrossRef CAS; (d) X. Zhang, L. Huang, H. Peng, F. Ji, X. Li and B. Yin, Tetrahedron, 2014, 70, 5242–5248 CrossRef CAS.
- K. A. Waibel, R. Nickisch, N. Möhl, R. Selim and M. A. R. Meier, Green Chem., 2020, 22, 933–941 RSC , and the references therein..
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization, spectroscopic data, theoretical calculations, and supporting figures and tables. CCDC 2278040–2278045. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03987k |
| This journal is © The Royal Society of Chemistry 2023 |