DOI:
10.1039/D3SC03936F
(Edge Article)
Chem. Sci., 2023,
14, 10508-10514
Deaminative ring contraction for the synthesis of polycyclic heteroaromatics: a concise total synthesis of toddaquinoline†
Received
29th July 2023
, Accepted 7th September 2023
First published on 8th September 2023
Abstract
A concise strategy to prepare polycyclic heteroaromatics involving a deaminative contraction cascade is detailed. The efficient deaminative ring contraction involves the in situ methylation of a biaryl-linked dihydroazepine to form a cyclic ammonium cation that undergoes a base-induced [1,2]-Stevens rearrangement/dehydroamination sequence. The presence of pseudosymmetry guides the retrosynthetic analysis of pyridyl-containing polycyclic heteroaromatics, enabling their construction by the reductive cyclization and deaminative contraction of tertiary amine precursors.
Introduction
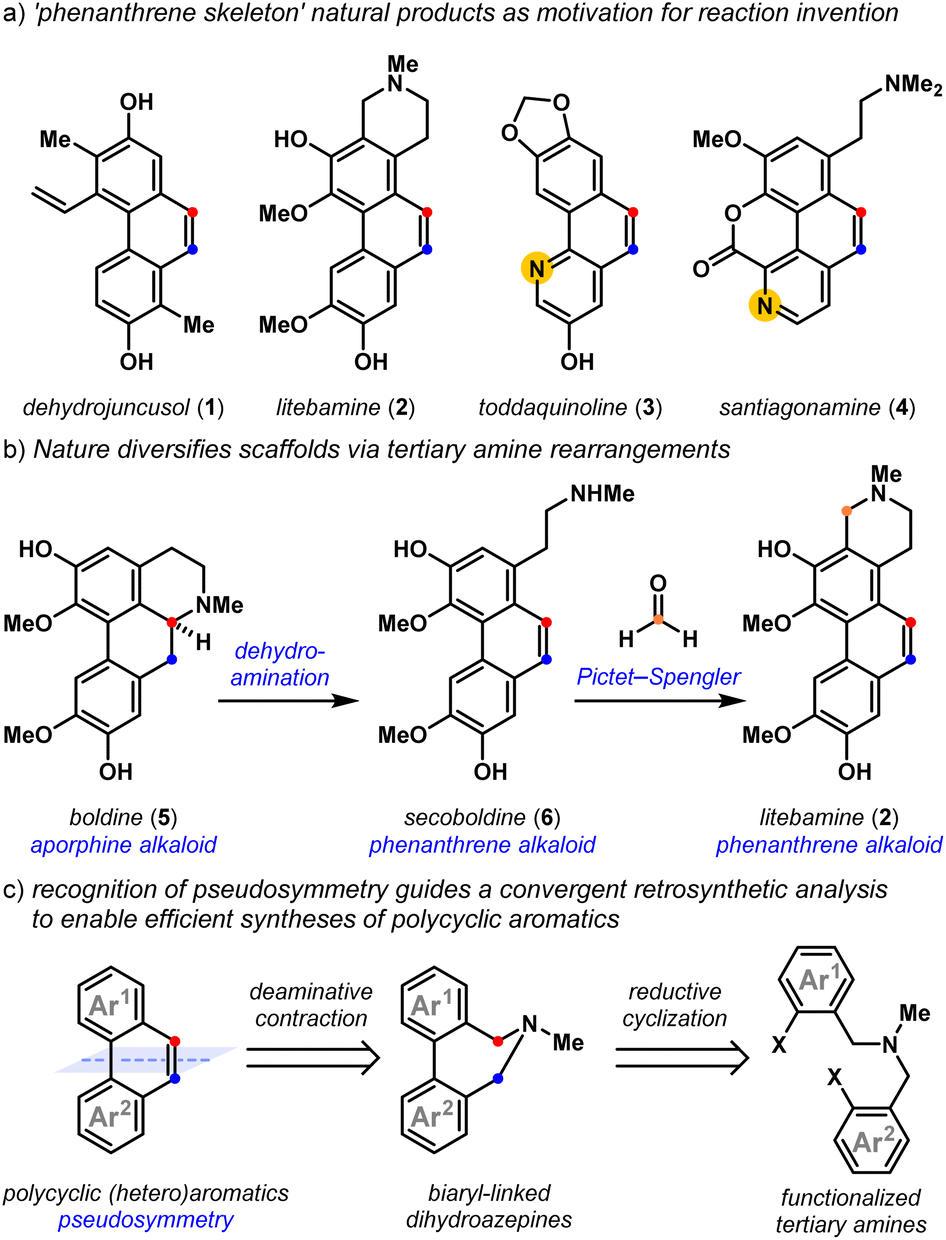
Polycyclic aromatic natural products exhibit a host of exciting biological properties,1,2 and their highly varied arene substitution patterns have motivated creative strategies3 for their assembly. Dehydrojuncusol 1,4 litebamine 2,5 toddaquinoline 3,6 and santiagonamine 4,7 serve to emphasize the diverse substitution patterns displayed by biologically active secondary plant metabolites that share the ‘phenanthrene skeleton’ (Fig. 1a). Biosynthetic rearrangements of the embedded tertiary amine lead to scaffold diversification, as exemplified by the dehydroaminative conversion of the aporphine alkaloid boldine 5, into phenanthrene alkaloids secoboldine 6 and litebamine 2 (Fig. 1b).8 The experimental replication of this biogenic pathway8c reveals the potential of dehydroamination toward family-level access to these phenanthrenes from tertiary amine precursors,8,9 and highlights the utility of amine-derived molecular rearrangements10,11 for the convergent synthesis of densely substituted polycyclic aromatics.
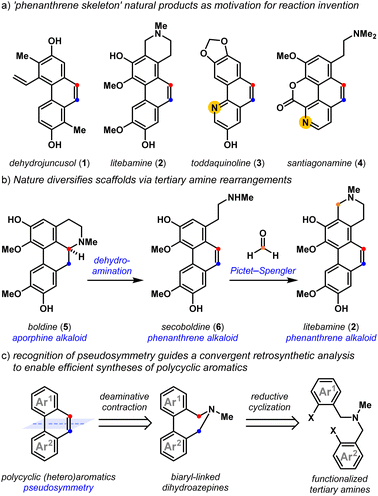 |
| | Fig. 1 Polycyclic aromatic natural products as motivation for reaction invention: (a) ‘phenanthrene skeleton’ natural products as motivation for reaction invention; (b) nature diversifies scaffolds via tertiary amine rearrangements; (c) recognition of pseudosymmetry guides a convergent retrosynthetic analysis. | |
Classic approaches12 to build phenanthrenes from stilbene precursors include the Pschorr,13 and Mallory reactions,14,15 which have seen impressive procedural updates.16,17 Unsurprisingly, the advent of transition metal-catalyzed cross-coupling reactions makes construction of the biaryl bond a natural entry point to these scaffolds.18 Recent approaches toward polycyclic aromatics have relied on these methods, using transition metal-,19 oxidation-,20 Lewis acid-,21,22 and base-promoted23 alkene-forming reactions. With some exceptions, these methods demonstrate limited access to polycyclic heteroaromatics containing important pyridyl motifs24 and while useful, Friedländer annulation strategies25 are less convergent. Recently, Lambert and coworkers elegantly addressed this through the development of a hydrazine-catalyzed ring-closing carbonyl–olefin metathesis reaction that is compatible with Lewis basic functionality.26 We drew inspiration from strategies that are also tolerant of Lewis basic arenes that use a Z-selective Wittig olefination to build the alkene, followed by a transition-metal catalyzed cyclization to forge the biaryl bond.27 The recognition of pseudosymmetry and knowing that tertiary amines can react to form alkenes guided our retrosynthetic analyses,11,28 leading to the development of a convergent approach to polycyclic (hetero)aromatics by the reductive cyclization and deaminative contraction of appropriately halogenated tertiary amine precursors (Fig. 1c).
In an earlier effort,28 we demonstrated that functionalized tertiary amines can be converted into substituted benzo[h]quinolines following reductive cyclization29–33 and deaminative contraction reactions (Fig. 2a). We were able to show that the deaminative contraction of 7 comprises a cascade reaction sequence wherein amine methylation, a [1,2]-Stevens rearrangement of the resultant ammonium cation, and a dehydroamination of the rearranged tertiary amine ensued to yield 8. Yet, the multievent transformation suffered from low efficiency and system-dependent side reactions. Herein, we detail our use of these mechanistic insights to develop a general and high yielding reductive cyclization/deaminative contraction strategy to prepare a diverse array of these polycyclic heteroaromatics. We show that appropriately halogenated tertiary amines28 are simple to prepare and cyclize. Readily accessed biaryl-linked dihydroazepines undergo efficient deaminative contraction, providing substituted polycyclic (hetero)aromatic products as highlighted by a concise total synthesis of toddaquinoline 3.
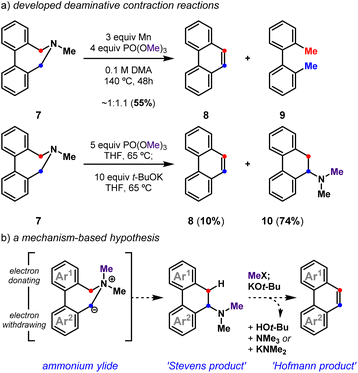 |
| | Fig. 2 Deaminative contraction for the synthesis of polycyclic (hetero)aromatics: (a) developed deaminative contraction reactions; (b) a mechanism-based hypothesis. | |
Results and discussion
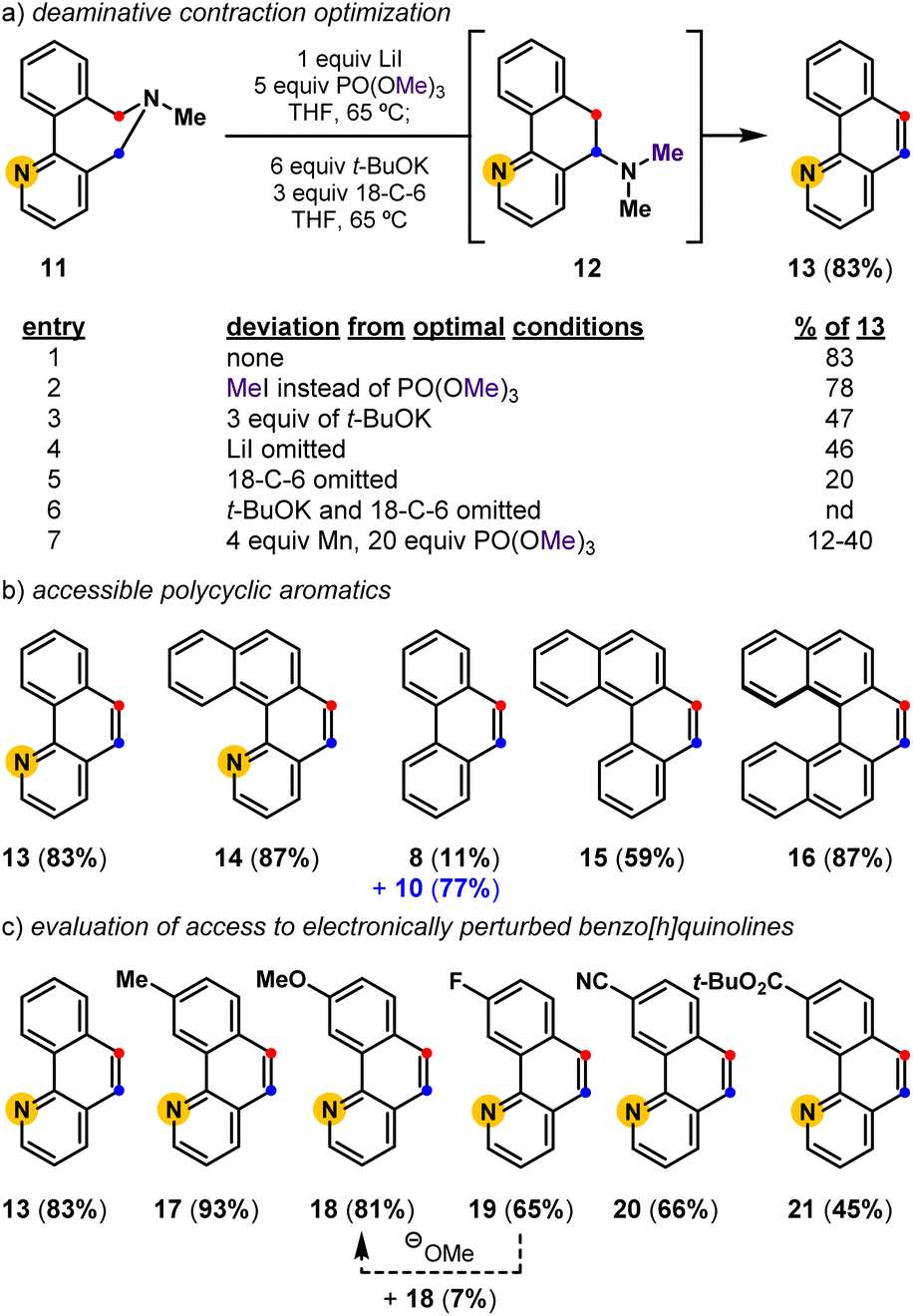
We previously demonstrated that biaryl-linked dihydroazepine 7 can form phenanthrene 8 and 2,2′-dimethylbiaryl 9 upon treatment with Mn and trimethyl phosphate, PO(OMe)3 (Fig. 2).28 The Mn additive served no obvious mechanistic role, but its presence improved the yield for some benzo[h]quinoline-forming examples. Excess Mn led to reductive deamination without contraction to favor 2,2′-dimethylbiaryls (e.g., 9), especially when the substrate lacked a pyridyl motif. With a better mechanistic level understanding of this cascade reaction,28 we developed one-pot conditions that leverage PO(OMe)3 as an amine-methylating reagent34 and t-BuOK as a Brønsted base. Here we presume Stevens product 10 forms en route to phenanthrene 8, as 10 can be resubjected to the reaction conditions to exclusively form 8. Závada and coworkers previously noted that biaryl-linked dihydroazepinium salts are susceptible to a [1,2]-Stevens rearrangement/Hofmann elimination process upon treatment with excess base (3.1 equiv n-BuLi).35 They noted that binaphthyl dihydroazepeniums undergo the [1,2]-Stevens rearrangement more efficiently than the corresponding biphenyl derivatives by several orders of magnitude.35,36 We reasoned the net deaminative ring contraction process involving amine methylation, [1,2]-Stevens rearrangement, and Hofmann elimination events could be made general by improving the methylation and base-catalyzed events. We proposed that in situ formed biaryl-linked azepeniums would undergo more efficient [1,2]-Stevens rearrangement and Hofmann elimination events. These hypotheses, and our interest in pyridyl-containing natural products such as 3 and 4, directed us to develop a general deaminative reaction for access to substituted and pyridyl-containing polycyclic heteroaromatics (Fig. 3).
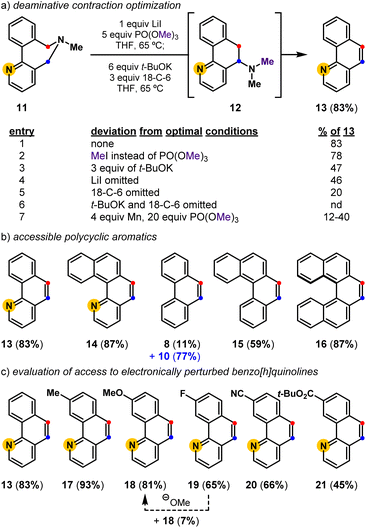 |
| | Fig. 3 Deaminative contractions for the convergent synthesis of polycyclic aromatics and substituted benzo[h]quinolines: (a) deaminative contraction optimization; (b) accessible polycyclic aromatics; (c) evaluation of electronically perturbed benzo[h]quinolines. nd = not detected. | |
Deaminative contraction for the convergent synthesis of substituted benzo[h]quinolines and other polycyclic aromatics
Optimization revealed that the reagent combination of LiI and PO(OMe)3 facilitates tertiary amine methylation,37 while subsequently added t-BuOK38 and 18-crown-6 (18-C-6) affects [1,2]-Stevens rearrangement and concomitant Hofmann elimination events to convert biaryl-linked dihydroazepine 11, via Stevens product 12, into benzo[h]quinoline 13 in 83% isolated yield (Fig. 3). Toward substituted benzo[h]quinolines, the Stevens product is only observed in reactions that are stopped prematurely. Mechanistic studies have shown the combination of LiI and PO(OMe)3 to be useful for the metered generation of MeI, especially when substrates harbor Lewis basic sites (e.g., pyridines) that are susceptible to methylation (entry 2).37 Other deviations from the optimal conditions such as less t-BuOK (entry 3), the omission of LiI (entry 4), and the omission of 18-C-6 (entry 5), result in diminished yields. Finally, a control reaction (entry 6) indirectly shows that lithium dimethyl phosphate is an ineffective base at 65 °C, suggesting that base-promoted rearrangements, and eliminations do not occur until t-BuOK and 18-C-6 are added. Relative to previously developed conditions (entry 7), this deaminative contraction method showed general applicability toward polycyclic aromatics. Readily accessible biaryl-linked dihydroazepines undergo deaminative contraction to afford 14, 8, 15, and 16 in good yields (Fig. 3b). Interestingly, Stevens product 10 is halted en route to phenanthrene 8, supporting the prior observation made by Závada and coworkers.35 Our interest in pyridyl-containing polycyclic aromatic natural products directed us to further evaluate the impact of system electronics. Electron-rich (17, 18) and electron-deficient (19–21) benzo[h]quinolines are all accessed in good yields (Fig. 3c). Notably, during the preparation of 19 (65%), we observed the formation of a minor amount of 18 (7%). We speculate that methoxide is a potential byproduct of the one-pot conditions and may convert 19 into 18via nucleophilic aromatic substitution (SNAr).39 While unoptimized, this observation suggests that other benzo[h]quinoline substitution patterns could be accessible via SNAr of an appropriately halogenated precursor. Due to the preliminary efficiency of this deaminative contraction method and its potential orthogonality to recent oxidative deaminative ring contraction methods,40,41 we set out to develop a general build-cyclize-contract strategy to prepare polycyclic heteroaromatics (Fig. 4).
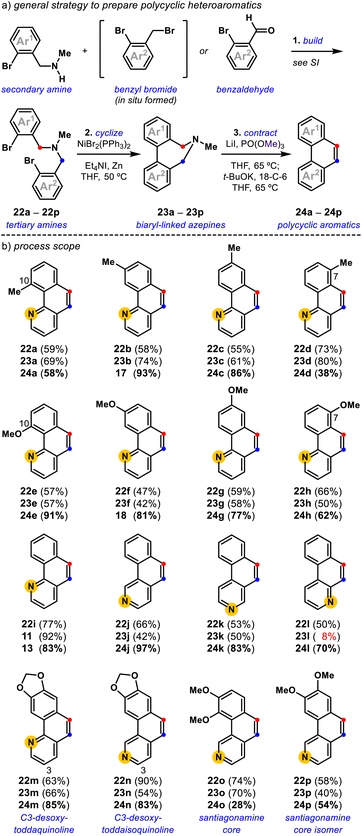 |
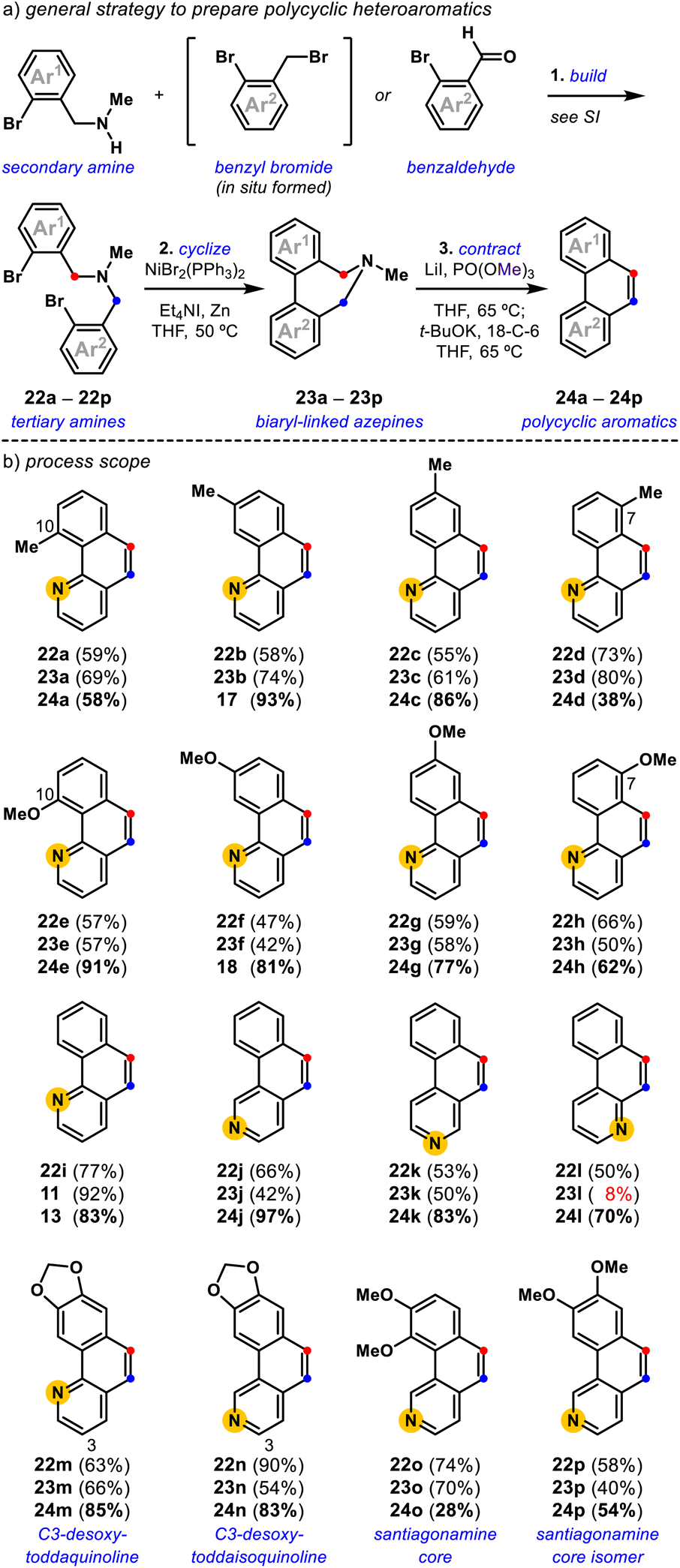
| | Fig. 4 Synthesis of polycyclic aromatics (a) a general strategy to prepare polycyclic heteroaromatics; (b) process scope: the final polycyclic heteroaromatics are shown for each three-step synthesis, the yields for each step are shown in order: build (step 1)-cyclize (step 2)-contract (step 3). | |
A general strategy to prepare polycyclic heteroaromatics
In the build step, synthetic secondary amines are advanced to tertiary amines using either radical halogenation/alkylation or reductive amination procedures, dependent on the availability of the respective methyl aryl and benzaldehyde derivatives (Fig. 4a, see the ESI†). Tertiary amines synthesized by the one-pot radical halogenation/alkylation method proceeded with a yield range of 23–74% for the 18 examples (Fig. 4b, see the ESI†). Tertiary amines prepared by reductive amination are accessible with a yield range of 53–90% for the six examples (Fig. 4b, see the ESI†). Following the build step, synthetic tertiary amines were cyclized using a Ni-promoted reaction28,30,32,33 to access the corresponding biaryl-linked dihydroazepines in moderate to good yields. Notably, the biaryl bond-forming cyclization is tolerant of ortho-substitution (23a, 23e, and 23o) and allows access to all possible pyridyl isomers (11, 23j, 23k, and 23l).42 In some cases, the Ni-mediated reaction conditions give lower conversion, which we attribute to the chelating 1,2-diamine nature of certain tertiary amine substrates.40d This was most evident for the conversion of pyridyl variant 22l into 23l (8%).
With various biaryl-linked dihydroazepines in hand, we evaluated their conversion into polycyclic heteroaromatics by deaminative contraction (Fig. 4). We find that methyl-substituted benzo[h]quinolines, 24a (58%), 17 (93%), 24c (86%), and 24d (38%), are all accessible, with a trend toward slightly diminished conversion for 10-methyl (24a) and 7-methyl (24d) variants. Toward 24a, we attribute the lower yield to the 10-methyl substituent-induced torsional strain,36 likely rendering the [1,2]-Stevens rearrangement difficult. Toward 24d, we attribute the overall lower conversion to the steric encumbrance induced by the amine-neighboring C7-methyl substituent that may preclude the requisite methylation and [1,2]-Stevens rearrangement events. In this case, the Stevens product can be observed. A similar, albeit less pronounced trend is noted for the corresponding methoxy-substituted benzo[h]quinolines, 24e (91%), 18 (81%), 24g (77%), and 24h (62%). The comparatively better yields for 24e (C10-OMe) vs.24a (C10-Me) and 24h (C7-OMe) vs.24d (C7-Me) are consistent with the smaller B-value steric measure for methoxy (B = 5.6 kcal mol−1) vs. methyl (B = 7.4 kcal mol−1) substituents,43 but cannot be decoupled from differences due to electronic effects. Intriguingly, we find that isomers of the parent benzo[h]quinoline system 13 (83%), 24j (97%), 24k (83%), and 24l (70%) are accessible in comparable yields, with 24j produced in exceptional yield. Consistent with our initial substrate polarization hypothesis, we reason that resonance-stabilized ammonium ylide intermediates, undergo more efficient [1,2]-Stevens rearrangement and Hofmann elimination events en route to their corresponding polycyclic heteroaromatics. The ability to access several pyridyl variations of the polycyclic aromatic core enables general access to the benzo[h]quinoline core of toddaquinoline 3 (ref. 6) and the benzo[h]isoquinoline core of santiagonamine 4 (ref. 7) (Fig. 1a). To highlight the efficiency and flexibility of the developed strategy, both natural and isomeric variations are straightforward to access. The build-cyclize-contract strategy provides efficient access to C3-desoxy-toddaquinoline 24m (85%),28,44 C3-desoxy-toddaisoquinoline 24n (83%),26 santiagonamine core 24o (28%),7 and an isomeric santiagonamine core 24p (54%). This general efficiency prompted us to evaluate the build-cyclize-contract strategy to realize a concise total synthesis of toddaquinoline 3.
Total synthesis of toddaquinoline
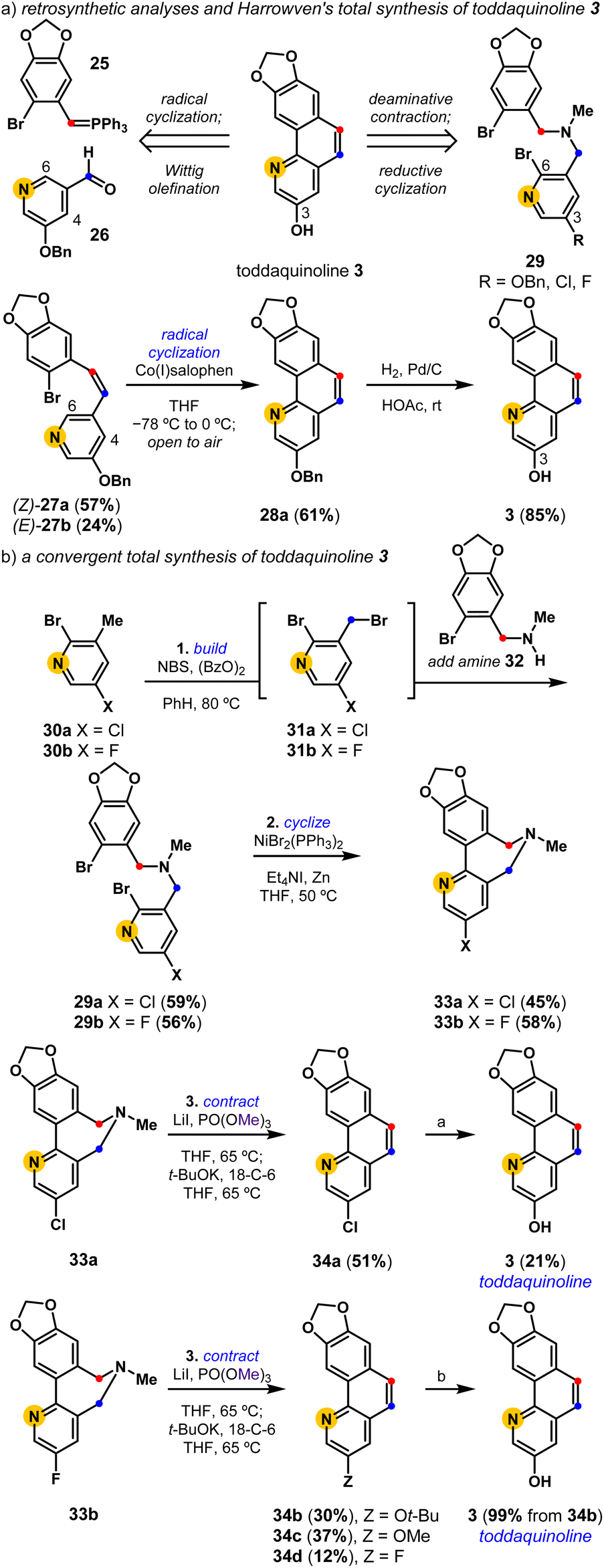
Harrowven and coworkers completed the first and only total synthesis of toddaquinoline 3 in three steps from accessible precursors, 25 and 26 (Scheme 1).45,46 Their concise assembly of the oxygenated benzo[h]quinoline natural product leveraged a partly Z-selective Wittig olefination ((Z)-27a and (E)-27b, 2.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), followed by a cobalt(I) salophen-mediated C6-regiocontrolled radical cyclization of the pendent pyridine. While olefination followed by cyclization sequences have since become a reliable means to build polycyclic aromatics,27,47 these strategies can suffer from alkene isomer selectivity and cyclization regiocontrol issues. We set out to develop an alternative total synthesis of toddaquinoline 3 from the reductive cyclization and deaminative contraction of a functionalized tertiary amine. The efficient synthesis of 29 (R = OBn) proved difficult as the electron-rich O-benzylated pyridyl derivative is prone to overhalogenation (see the ESI†). We therefore pursued an alternative strategy to complete the total synthesis of 3, leveraging a one-pot radical halogenation/alkylation procedure from halopyridines 30a (X = Cl) and 30b (X = F) to access respective tertiary amines 29a and 29b. Both tertiary amines (29) undergo chemoselective reductive cyclizations to provide biaryl-linked dihydroazepines (33). Their subsequent deaminative contractions provide intriguingly varied results, with both biaryl-linked dihydroazepines enabling access to toddaquinoline 3. The deaminative contraction of 3-Cl dihydroazepine (33a) provides a 3-Cl derivative of toddaquinoline (34a) which is readily converted into 3 (21%) using an unoptimized Pd-catalyzed hydroxylation reaction.48 Alternatively, the deaminative contraction of 3-F dihydroazepine (33b) provides a separable mixture of three ring-contracted products, 34b (Z = Ot-Bu), 34c (Z = OMe), and 34d (Z = F), where we presume SNAr reactivity39 gives rise to substituted variants 34b and 34c. The tert-butyl ether variant (34b) is quantitatively converted into toddaquinoline 3 (99%) using acidolysis conditions.49 Overall, each three-step process yields a polysubstituted benzo[h]quinoline core (34) while completely avoiding alkene isomer and cyclization regiocontrol challenges.
:1), followed by a cobalt(I) salophen-mediated C6-regiocontrolled radical cyclization of the pendent pyridine. While olefination followed by cyclization sequences have since become a reliable means to build polycyclic aromatics,27,47 these strategies can suffer from alkene isomer selectivity and cyclization regiocontrol issues. We set out to develop an alternative total synthesis of toddaquinoline 3 from the reductive cyclization and deaminative contraction of a functionalized tertiary amine. The efficient synthesis of 29 (R = OBn) proved difficult as the electron-rich O-benzylated pyridyl derivative is prone to overhalogenation (see the ESI†). We therefore pursued an alternative strategy to complete the total synthesis of 3, leveraging a one-pot radical halogenation/alkylation procedure from halopyridines 30a (X = Cl) and 30b (X = F) to access respective tertiary amines 29a and 29b. Both tertiary amines (29) undergo chemoselective reductive cyclizations to provide biaryl-linked dihydroazepines (33). Their subsequent deaminative contractions provide intriguingly varied results, with both biaryl-linked dihydroazepines enabling access to toddaquinoline 3. The deaminative contraction of 3-Cl dihydroazepine (33a) provides a 3-Cl derivative of toddaquinoline (34a) which is readily converted into 3 (21%) using an unoptimized Pd-catalyzed hydroxylation reaction.48 Alternatively, the deaminative contraction of 3-F dihydroazepine (33b) provides a separable mixture of three ring-contracted products, 34b (Z = Ot-Bu), 34c (Z = OMe), and 34d (Z = F), where we presume SNAr reactivity39 gives rise to substituted variants 34b and 34c. The tert-butyl ether variant (34b) is quantitatively converted into toddaquinoline 3 (99%) using acidolysis conditions.49 Overall, each three-step process yields a polysubstituted benzo[h]quinoline core (34) while completely avoiding alkene isomer and cyclization regiocontrol challenges.
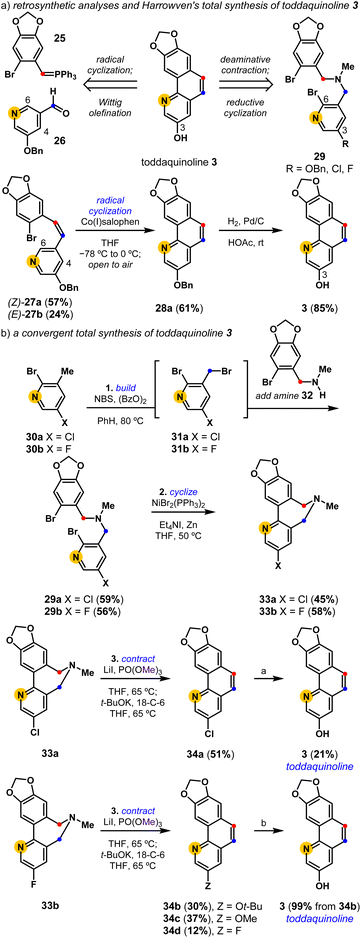 |
| | Scheme 1 A build-cyclize-contract total synthesis of toddaquinoline 3. (a) 3 equiv. KOH, 10 mol% Pd2dba3, 40 mol% Me4t-BuXPhos, 1,4-dioxane/H2O, 100 °C; (b) CF3CO2H, CH2Cl2, rt. dba = dibenzylideneacetone. | |
An iterative strategy to access aminated benzo[h]quinolines and aza[n]helicenes
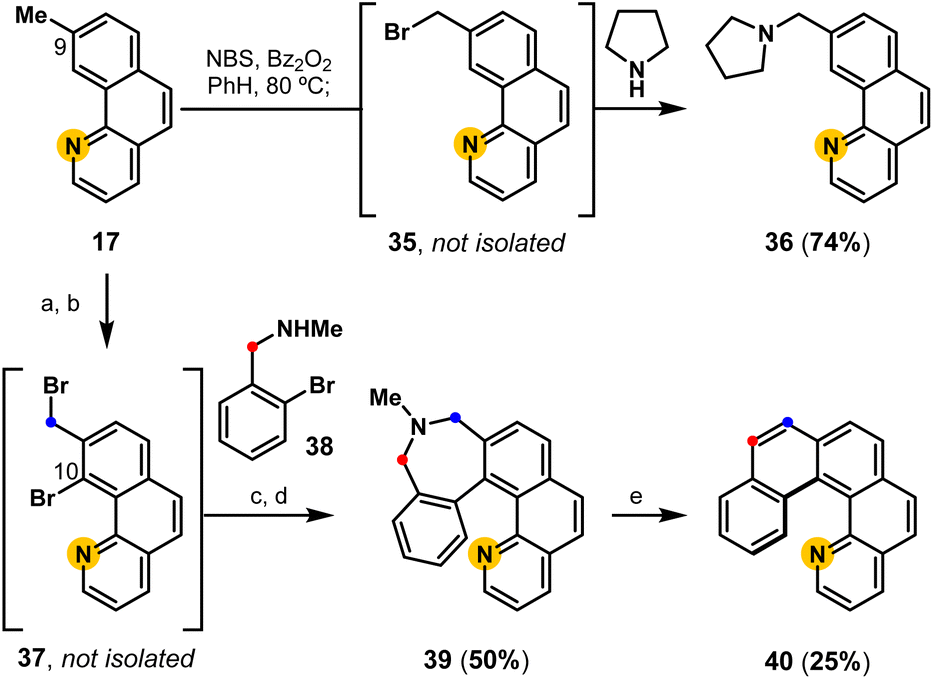
The C9-methyl substituent in benzo[h]quinoline 17 provides an additional opportunity to controllably extend the aromatic system to access aminated benzo[h]quinolines and aza[5]helicenes (Fig. 5).47 A proof-of-concept experiment demonstrates that aminated derivatives are directly available from 17 following the developed one-pot radical halogenation/alkylation procedure to access pyrrolidine derivative 36via benzyl bromide intermediate 35. Using Sanford's C–H functionalization protocol,50 the same 9-methyl benzo[h]quinoline (17) can be regioselectively brominated (C10) and subsequently converted into a pyridyl-containing dihydroazepine derivative 39 following radical halogenation/alkylation, and reductive cyclization. Finally, biaryl-linked dihydroazepine 39 undergoes deaminative contraction to access aza-pentahelicene 40 (phenanthro[3,4-h]quinoline), demonstrating a modular strategy to access pentahelicenes35,50 that may enable the precision-controlled introduction of substituents by the strategic addition of aromatic rings.
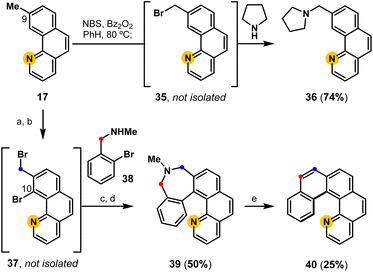 |
| | Fig. 5 Functionalization of methyl-substituted benzo[h]quinolines to access aminated benzo[h]quinolines and aza-helicenes: (a) Pd(OAc)2, NBS, CH3CN; (b) NBS, Bz2O2, PhH, 80 °C; (c) add 38 in THF; (d) NiBr2(PPh3)2, Et4NI, Zn, THF, 50 °C; (e) LiI, PO(OMe)3, THF, 65 °C; t-BuOK, 18-C-6, THF, 65 °C. | |
Conclusions
We developed a build-cyclize-contract strategy to access polycyclic (hetero)aromatics. The key developed deaminative ring contraction involves a proposed in situ methylation followed by base induced [1,2]-Stevens rearrangement/dehydroamination cascade reaction. We report 25 examples of reductive cyclization and deaminative contraction (28–97% yields) processes to yield polycyclic (hetero)aromatics, culminating in a four-step total synthesis of toddaquinoline. Given the simple starting materials, this strategy can be readily applied to prepare substituted polycyclic (hetero)aromatics with utility as the diversifiable cores of many bioactive polycyclic natural products,3,28 ligands for catalysis,25 and the organic components for the discovery of novel materials.26,51
Data availability
The data supporting this article has been uploaded as part of the ESI.†
Author contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for financial support of this research from the University of Utah and the Donors of the American Chemical Society Petroleum Research Fund (ACS PRF 62220-DNI1). This research is based upon work supported by the National Science Foundation under Grant No. 2247651. We thank Prof. Ryan E. Looper, Prof. Aaron W. Puri, Prof. Jon D. Rainier, and Prof. Matthew S. Sigman for thoughtful discussions and their review of this manuscript. We thank Dr Chideraa I. Nwachukwu, Timothy P. McFadden, and Mason Lunceford for experimental assistance. The NMR spectroscopy results included in this report were recorded at the David M. Grant NMR Center, a University of Utah Core Facility. Funds for construction of the Center and the helium recovery system were obtained from the University of Utah and the National Institutes of Health awards 1C06RR017539-01A1 and 3R01GM063540-17W1, respectively. NMR spectroscopy instruments were purchased with support of the University of Utah and the National Institutes of Health award 1S10OD25241-01. There are no conflicts to declare.
Notes and references
- For selected reviews, see:
(a) A. Kovács, A. Vasas and J. Hohmann, Phytochemistry, 2008, 69, 1084–1110 CrossRef PubMed;
(b) Z.-X. Qing, P. Yang, Q. Tang, P. Cheng, X.-B. Liu, Y. Zheng, Y.-S. Liu and J.-G. Zeng, Curr. Org. Chem., 2017, 21, 1920–1934 CrossRef CAS;
(c) M. D. Douka and K. E. Litinas, Molecules, 2022, 27, 7256 CrossRef CAS PubMed.
- For selected recent examples, see:
(a) M.-E. Sahuc, R. Sahli, C. Rivière, V. Pène, M. Lavie, A. Vandeputte, P. Brodin, A. R. Rosenberg, J. Dubuisson, R. Ksouri, Y. Rouillé, S. Sahpaz and K. Séron, J. Virol., 2019, 93, e02009–e02018 CrossRef CAS PubMed;
(b) P. Patra and S. Patra, Heterocycles, 2023, 106, 241 CrossRef CAS.
- For representative total syntheses of substituted polycyclic aromatics including natural products, see:
(a) A. S. Kende and D. P. Curran, J. Am. Chem. Soc., 1979, 101, 1857–1864 CrossRef CAS;
(b) X. Wang and V. Snieckus, Tetrahedron Lett., 1991, 32, 4879–4882 CrossRef CAS;
(c) X. Wang and V. Snieckus, Tetrahedron Lett., 1991, 32, 4883–4884 CrossRef CAS;
(d) C. Hoarau, A. Couture, H. Cornet, E. Deniau and P. Grandclaudon, J. Org. Chem., 2001, 66, 8064–8069 CrossRef CAS PubMed;
(e) S. B. Jones, L. He and S. L. Castle, Org. Lett., 2006, 8, 3757–3760 CrossRef CAS PubMed;
(f) M. D. Markey, Y. Fu and T. R. Kelly, Org. Lett., 2007, 9, 3255–3257 CrossRef CAS PubMed;
(g) F. Gosselin, S. Lau, C. Nadeau, T. Trinh, P. D. O'Shea and I. W. Davies, J. Org. Chem., 2009, 74, 7790–7797 CrossRef CAS PubMed.
- H. Sarkar, M. Zerezghi and J. Bhattacharyya, Phytochemistry, 1988, 27, 3006–3008 CrossRef CAS.
- Y.-C. Wu, J.-Y. Liou, C.-Y. Duh, S.-S. Lee and S.-T. Lu, Tetrahedron Lett., 1991, 32, 4169–4170 CrossRef CAS.
- I.-S. Chen, I.-L. Tsai, S.-J. Wu, W.-S. Sheen, T. Ishikawa and H. Ishii, Phytochemistry, 1993, 34, 1449–1451 CrossRef CAS.
- E. Valencia, A. Patra, A. J. Freyer, M. Shamma and V. Fajardo, Tetrahedron Lett., 1984, 25, 3163–3166 CrossRef CAS.
-
(a) J. B. Bremner and K. N. Winzenberg, Aust. J. Chem., 1978, 31, 313–320 CrossRef CAS;
(b) S.-T. Lu and I.-L. Tsai, Heterocycles, 1988, 27, 751 CrossRef CAS;
(c) S.-S. Lee, Y.-J. Lin, M.-Z. Chen, Y.-C. Wu and C.-H. Chen, Tetrahedron Lett., 1992, 33, 6309–6310 CrossRef CAS;
(d) S.-S. Lee, C.-M. Chiou, H.-Y. Lin and C.-H. Chen, Tetrahedron Lett., 1995, 36, 1531–1532 CrossRef CAS;
(e) Y. Masahide, H. Takamitsu, A. Yasuhiro and S. Kensuke, Bull. Chem. Soc. Jpn., 1993, 66, 1451–1455 CrossRef.
- S. V. Kini and M. M. V. Ramana, Tetrahedron Lett., 2004, 45, 4171–4173 CrossRef CAS.
- J. A. Vanecko, H. Wan and F. G. West, Tetrahedron, 2006, 62, 1043–1062 CrossRef CAS.
- Z. Schwartz, C. Valiton, M. Lovasz and A. G. Roberts, Synthesis, 2023, 55 DOI:10.1055/s-0042-1751446.
- A. J. Floyd, S. F. Dyke and S. E. Ward, Chem. Rev., 1976, 76, 509–562 CrossRef CAS.
-
D. F. DeTar, in Org. React., John Wiley & Sons, Ltd, 2011, pp. 409–462 Search PubMed.
-
(a)
F. B. Mallory and C. W. Mallory, in Org. React., John Wiley & Sons, Ltd, 2005, pp. 1–456 Search PubMed;
(b) K. B. Jørgensen, Molecules, 2010, 15, 4334–4358 CrossRef PubMed;
(c) A. G. Lvov, J. Org. Chem., 2020, 85, 8749–8759 CrossRef CAS PubMed.
- For a pertinent review on photoelectrocyclization reactions and their applications in total synthesis, see: X. Zhao and J. D. Rainier, Synthesis, 2021, 53, 1200–1212 CrossRef CAS PubMed.
- L. Liu, B. Yang, T. J. Katz and M. K. Poindexter, J. Org. Chem., 1991, 56, 3769–3775 CrossRef CAS.
- Y. Li, D. E. Wise, J. K. Mitchell and M. Parasram, J. Org. Chem., 2023, 88, 717–721 CrossRef CAS PubMed.
- For pertinent discussions, see:
(a)
N. Miyaura, in Adv. Met.-Org. Chem., ed. L.S. Liebeskind, JAI, 1998, pp. 187–243 Search PubMed;
(b) X. Cai, S. Brown, P. Hodson and V. Snieckus, Can. J. Chem., 2004, 82, 195–205 CrossRef CAS.
-
(a) R. C. Larock, M. J. Doty, Q. Tian and J. M. Zenner, J. Org. Chem., 1997, 62, 7536–7537 CrossRef CAS;
(b) A.-E. Gies and M. Pfeffer, J. Org. Chem., 1999, 64, 3650–3654 CrossRef CAS PubMed;
(c) V. Mamane, P. Hannen and A. Fürstner, Chem. - Eur. J., 2004, 10, 4556–4575 CrossRef CAS PubMed;
(d) A. Iuliano, P. Piccioli and D. Fabbri, Org. Lett., 2004, 6, 3711–3714 CrossRef CAS PubMed;
(e) T. J. Donohoe, A. J. Orr and M. Bingham, Angew. Chem., Int. Ed., 2006, 45, 2664–2670 CrossRef CAS PubMed;
(f) W. A. L. van Otterlo and C. B. de Koning, Chem. Rev., 2009, 109, 3743–3782 CrossRef CAS PubMed;
(g) C. Wang, S. Rakshit and F. Glorius, J. Am. Chem. Soc., 2010, 132, 14006–14008 CrossRef CAS PubMed;
(h) Y. Xia, Z. Liu, Q. Xiao, P. Qu, R. Ge, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 5714–5717 CrossRef CAS PubMed;
(i) W. C. Fu, Z. Wang, W. T. K. Chan, Z. Lin and F. Y. Kwong, Angew. Chem., Int. Ed., 2017, 56, 7166–7170 CrossRef CAS PubMed.
- Z. Zhao, L. H. Britt and G. K. Murphy, Chem. - Eur. J., 2018, 24, 17002–17005 CrossRef CAS PubMed.
-
(a) M. Murai, N. Hosokawa, D. Roy and K. Takai, Org. Lett., 2014, 16, 4134–4137 CrossRef CAS PubMed;
(b) C. C. McAtee, P. S. Riehl and C. S. Schindler, J. Am. Chem. Soc., 2017, 139, 2960–2963 CrossRef CAS PubMed.
- H. Albright, A. J. Davis, J. L. Gomez-Lopez, H. L. Vonesh, P. K. Quach, T. H. Lambert and C. S. Schindler, Chem. Rev., 2021, 121, 9359–9406 CrossRef CAS PubMed.
- V. Mamane, F. Louërat, J. Iehl, M. Abboud and Y. Fort, Tetrahedron, 2008, 64, 10699–10705 CrossRef CAS.
- Nitrogen-containing Heterocycles are present in 59% of U.S. FDA approved small-molecule drugs:
(a) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed ; For a pertinent example of a benzo[h]quinoline in drug discovery, see:;
(b) N. Atatreh, C. Stojkoski, P. Smith, G. W. Booker, C. Dive, A. D. Frenkel, S. Freeman and R. A. Bryce, Bioorg. Med. Chem. Lett., 2008, 18, 1217–1222 CrossRef CAS PubMed.
- E. C. Riesgo, X. Jin and R. P. Thummel, J. Org. Chem., 1996, 61, 3017–3022 CrossRef CAS PubMed.
- E. K. Cho, P. K. Quach, Y. Zhang, J. H. Sim and T. H. Lambert, Chem. Sci., 2022, 13, 2418–2422 RSC.
-
(a) D. C. Harrowven, I. L. Guy and L. Nanson, Angew. Chem., Int. Ed., 2006, 45, 2242–2245 CrossRef CAS PubMed;
(b) G. Chelucci, D. Addis and S. Baldino, Tetrahedron Lett., 2007, 48, 3359–3362 CrossRef CAS.
- T. P. McFadden, C. I. Nwachukwu and A. G. Roberts, Org. Biomol. Chem., 2022, 20, 1379–1385 RSC.
- T. Ross Kelly, Q. Li and V. Bhushan, Tetrahedron Lett., 1990, 31, 161–164 CrossRef.
- M. Iyoda, H. Otsuka, K. Sato, N. Nisato and M. Oda, Bull. Chem. Soc. Jpn., 1990, 63, 80–87 CrossRef CAS.
- S. Zhang, D. Zhang and L. S. Liebeskind, J. Org. Chem., 1997, 62, 2312–2313 CrossRef CAS PubMed.
- S. Leleu, C. Papamicaël, F. Marsais, G. Dupas and V. Levacher, Tetrahedron: Asymmetry, 2004, 15, 3919–3928 CrossRef CAS.
- W. Xue, H. Xu, Z. Liang, Q. Qian and H. Gong, Org. Lett., 2014, 16, 4984–4987 CrossRef CAS PubMed.
- C. I. Nwachukwu, T. P. McFadden and A. G. Roberts, J. Org. Chem., 2020, 85, 9979–9992 CrossRef CAS PubMed.
- I. G. Stara, I. Stary, M. Tichy, J. Zavada and V. Hanus, J. Am. Chem. Soc., 1994, 116, 5084–5088 CrossRef CAS.
- H. Joshua, R. Gans and K. Mislow, J. Am. Chem. Soc., 1968, 90, 4884–4892 CrossRef CAS.
- Z.-T. He, H. Li, A. M. Haydl, G. T. Whiteker and J. F. Hartwig, J. Am. Chem. Soc., 2018, 140, 17197–17202 CrossRef CAS PubMed.
- S. Hanessian and M. Mauduit, Angew. Chem., Int. Ed., 2001, 40, 3810–3813 CrossRef CAS.
- A. S. Abel, A. Yu Mitrofanov, A. A. Yakushev, I. S. Zenkov, G. V. Morozkov, A. D. Averin, I. P. Beletskaya, J. Michalak, S. Brandès and A. Bessmertnykh-Lemeune, Asian J. Org. Chem., 2019, 8, 2128–2142 CrossRef CAS.
- For the oxidative deaminative ring contraction of secondary amines, see:
(a) S. H. Kennedy, B. D. Dherange, K. J. Berger and M. D. Levin, Nature, 2021, 593, 223–227 CrossRef CAS PubMed;
(b) H. Qin, W. Cai, S. Wang, T. Guo, G. Li and H. Lu, Angew. Chem., Int. Ed., 2021, 60, 20678–20683 CrossRef CAS PubMed;
(c) C. Hui, L. Brieger, C. Strohmann and A. P. Antonchick, J. Am. Chem. Soc., 2021, 143, 18864–18870 CrossRef CAS PubMed;
(d) H. Na and L. M. Mirica, Nat. Commun., 2022, 13, 1313 CrossRef CAS PubMed;
(e) B. A. Wright, A. Matviitsuk, M. J. Black, P. García-Reynaga, L. E. Hanna, A. T. Herrmann, M. K. Ameriks, R. Sarpong and T. P. Lebold, J. Am. Chem. Soc., 2023, 145, 10960–10966 CrossRef CAS PubMed.
- J. Jurczyk, J. Woo, S. F. Kim, B. D. Dherange, R. Sarpong and M. D. Levin, Nat. Synth., 2022, 1, 352–364 CrossRef PubMed.
- M. W. Danneman, K. B. Hong and J. N. Johnston, Org. Lett., 2015, 17, 3806–3809 CrossRef CAS PubMed.
- R. Ruzziconi, S. Spizzichino, L. Lunazzi, A. Mazzanti and M. Schlosser, Chem. - Eur. J., 2009, 15, 2645–2652 CrossRef CAS PubMed.
- H. Abe, G. Serban, Y. Shigeta, H. Nishioka, Y. Takeuchi and T. Harayama, Heterocycles, 2007, 71, 1623 CrossRef.
- D. C. Harrowven, M. I. T. Nunn, N. J. Blumire and D. R. Fenwick, Tetrahedron Lett., 2000, 41, 6681–6683 CrossRef CAS.
- D. C. Harrowven, M. I. T. Nunn, N. J. Blumire and D. R. Fenwick, Tetrahedron, 2001, 57, 4447–4454 CrossRef CAS.
- N. Takenaka, R. S. Sarangthem and B. Captain, Angew. Chem., Int. Ed., 2008, 47, 9708–9710 CrossRef CAS PubMed.
- K. W. Anderson, T. Ikawa, R. E. Tundel and S. L. Buchwald, J. Am. Chem. Soc., 2006, 128, 10694–10695 CrossRef CAS PubMed.
- H. S. Tae, J. Hines, A. R. Schneekloth and C. M. Crews, Org. Lett., 2010, 12, 4308–4311 CrossRef CAS PubMed.
- A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 2300–2301 CrossRef CAS PubMed.
-
(a) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed;
(b) X. Feng, Y. Bai, M. Liu, Y. Li, H. Yang, X. Wang and C. Wu, Energy Environ. Sci., 2021, 14, 2036–2089 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data. See DOI: https://doi.org/10.1039/d3sc03936f |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Zachary T.
Schwartz‡
,
Zachary T.
Schwartz‡