
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Elucidating the reaction mechanism of SO2 with Cu-CHA catalysts for NH3-SCR by X-ray absorption spectroscopy†
Anastasia Yu.
Molokova
 ab,
Reza K.
Abasabadi
bc,
Elisa
Borfecchia
b,
Olivier
Mathon
a,
Silvia
Bordiga
b,
Fei
Wen
d,
Gloria
Berlier
b,
Ton V. W.
Janssens
*c and
Kirill A.
Lomachenko
*a
ab,
Reza K.
Abasabadi
bc,
Elisa
Borfecchia
b,
Olivier
Mathon
a,
Silvia
Bordiga
b,
Fei
Wen
d,
Gloria
Berlier
b,
Ton V. W.
Janssens
*c and
Kirill A.
Lomachenko
*a
aEuropean Synchrotron Radiation Facility, 71 avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France. E-mail: lomachenko@esrf.fr
bDepartment of Chemistry and NIS Centre, University of Turin, via Giuria 7, 10125 Turin, Italy
cUmicore Denmark ApS, Kogle Allé 1, 2970 Hørsholm, Denmark. E-mail: TonV.W.Janssens@eu.umicore.com
dUmicore AG & Co, Rodenbacher Chaussee 4, 63457 Hanau, Germany
First published on 10th October 2023
Abstract
The application of Cu-CHA catalysts for the selective catalytic reduction of NOx by ammonia (NH3-SCR) in exhaust systems of diesel vehicles requires the use of fuel with low sulfur content, because the Cu-CHA catalysts are poisoned by higher concentrations of SO2. Understanding the mechanism of the interaction between the Cu-CHA catalyst and SO2 is crucial for elucidating the SO2 poisoning and development of efficient catalysts for SCR reactions. Earlier we have shown that SO2 reacts with the [Cu2II(NH3)4O2]2+ complex that is formed in the pores of Cu-CHA upon activation of O2 in the NH3-SCR cycle. In order to determine the products of this reaction, we use X-ray absorption spectroscopy (XAS) at the Cu K-edge and S K-edge, and X-ray emission spectroscopy (XES) for Cu-CHA catalysts with 0.8 wt% Cu and 3.2 wt% Cu loadings. We find that the mechanism for SO2 uptake is similar for catalysts with low and high Cu content. We show that the SO2 uptake proceeds via an oxidation of SO2 by the [Cu2II(NH3)4O2]2+ complex, resulting in the formation of different CuI species, which do not react with SO2, and a sulfated CuII complex that is accumulated in the pores of the zeolite. The increase of the SO2 uptake upon addition of oxygen to the SO2-containing feed, evidenced by X-ray adsorbate quantification (XAQ) and temperature-programmed desorption of SO2, is explained by the re-oxidation of the CuI species into the [Cu2II(NH3)4O2]2+ complexes, which makes them available for reaction with SO2.
Introduction
The current technology to reduce the harmful NOx emissions from diesel-powered vehicles is based on the selective catalytic reduction of nitrogen oxides (NOx) to N2 and H2O by ammonia (NH3-SCR).1,2 Cu-exchanged chabazite zeolites (Cu-CHA) are preferred catalysts in diesel exhaust systems, due to their high activity in the low-temperature region (150–350 °C) and hydrothermal stability above 500 °C. The low-temperature activity of Cu-CHA-based catalysts, however, is strongly reduced in the presence of SO2, and therefore, application of such catalysts in exhaust systems requires the use of ultra-low sulfur diesel fuel.3,4The mechanism of the NH3-SCR reaction in Cu-CHA based catalysts is a redox cycle,5,6 in which the oxidation state of Cu changes between CuI and CuII. The reaction proceeds via a number of Cu-complexes formed by adsorption and reaction of NO, NH3 and O2 as ligands on the Cu-ions inside the CHA zeolite.7 The oxidation from CuI to CuII occurs by activation of O2, which is a crucial step in the reaction cycle. At temperatures below 250 °C, the CuI species are predominantly represented by [CuI(NH3)2]+ complexes, which are loosely bound to the zeolite, and are therefore mobile under the reaction conditions for NH3-SCR.8–10 The O2 activation then takes place via a reaction of O2 with a pair of [CuI(NH3)2]+ complexes, to form a [Cu2II(NH3)4O2]2+ complex.6,7,11–15 These [Cu2II(NH3)4O2]2+ complexes are then reduced back to the original [CuI(NH3)2]+ complexes by NH3 and NO, under the formation of the reaction products N2 and H2O. The mobility of the [CuI(NH3)2]+ complexes is important, as it facilitates the formation of the required CuI pairs for the O2 activation, enabling the NH3-SCR reaction at low temperatures.
Because the presence of SO2 results in a significantly lower activity of the Cu-CHA catalysts below 300 °C, the SO2 must affect the NH3-SCR reaction cycle. We have recently shown that SO2 reacts with [Cu2II(NH3)4O2]2+ complexes that are formed upon activation of O2.16 That reaction results in the decomposition of the [Cu2II(NH3)4O2]2+ complex, and a partial reduction of CuII to CuI. To determine the effect of this reaction of SO2 with the [Cu2II(NH3)4O2]2+ complex on the rate of the NH3-SCR reaction, we need to know how this reaction proceeds and what reaction products are formed. A recent theoretical study proposes that deactivation by SO2 occurs via the accumulation of ammonium bisulfate (NH4)HSO4 in the zeolite after initial reaction with the [Cu2II(NH3)4O2]2+ complexes, thus limiting the mobility of the [CuI(NH3)2]+ species, and the Cu-pair formation necessary for O2 activation.17 Such a mechanism is consistent with the observations that some SO2 desorption occurs around 400 °C, where ammonium bisulfate decomposes, and that most of the low-temperature activity can be recovered by heating to 550 °C.3 Ammonium bisulfate was also suggested as one of the species forming in the catalyst cages.18–20
Even though the formation of ammonium bisulfate can explain certain aspects of the deactivation by SO2, other observations point towards the formation of species that contain both S and Cu. Indeed, the uptake of SO2 is often saturated at S/Cu ratios below 1,4,19 which suggests that the uptake of SO2 is limited by the amount of Cu in the catalyst. This implies a direct interaction between SO2 and Cu, such that a further reaction with SO2 is not possible. Furthermore, the release of SO2 from a Cu-CHA catalyst exposed to SO2 occurs at a slightly higher temperature as compared to a Cu-CHA catalyst with ammonium bisulfate deposited on it via impregnation.16 This indicates that a sulfate- or sulfite-like compound is formed, that is more stable than ammonium bisulfate, which would be consistent with a direct interaction of SO2 with Cu. Indeed, direct interaction of SO2 with Cu was reported to result in the formation of Cu-sulfate19,20 and Cu-bisulfate,18 or similar species. Finally, Cu-CHA catalysts typically show 10–20 times lower low-temperature activity for NH3-SCR when saturated with SO2,3,4,20,21 but never a complete deactivation. If deactivation were caused by accumulation of ammonium sulfate, it would be expected to be complete upon saturation with SO2, at least for temperatures up to the onset of the decomposition of ammonium bisulfate.
Since the first step of the SO2 uptake is a reaction with the [Cu2II(NH3)4O2]2+ complexes, the uptake of SO2 may be affected by the Cu content in the catalyst and the gas atmosphere. The propensity toward the formation of the [CuII2(NH3)4O2]2+ complex is determined by the Cu content and partial pressure of oxygen.4,13,22 Furthermore, ammonia is required as well, in order to form the mobile [CuI(NH3)2]+ and the [CuII2(NH3)4O2]2+ complexes. The effect of ammonia is clearly demonstrated by the observation that the CuII species formed upon exposure of Cu-CHA to O2 at 500 °C is much less reactive towards SO2 than the same species exposed to NH3.16 Therefore, the impact of SO2 on the activity will depend on the reaction conditions.4,13,22
In this work, we use in situ X-ray absorption spectroscopy (XAS) to monitor the reaction of SO2 with the [Cu2II(NH3)4O2]2+ complexes in Cu-CHA catalysts with an Si/Al ratio of 6.7, and Cu contents of 3.2 wt% and 0.8 wt%. To maximize the amount of the [Cu2II(NH3)4O2]2+ complexes in the catalysts prior to the reaction with SO2, we expose the catalyst to O2 at 200 °C for 60 minutes after reduction in a mixture of NH3 and NO at the same temperature. This procedure leads to an almost complete conversion of Cu to [Cu2II(NH3)4O2]2+ complexes.11,14,16 A Cu content of 3.2 wt% is typical for technical Cu-CHA based NH3-SCR catalysts. At 0.8 wt%, the low-temperature activity is significantly lower, and therefore this sample represents a situation in which the formation of the [CuII2(NH3)4O2]2+ complex becomes less efficient. The XAS at the Cu K-edge is used to obtain information on the Cu-species formed upon exposure to SO2. Since the reaction of the [Cu2II(NH3)4O2]2+ complex with SO2 leads to a partial reduction of the CuII, the presence of oxygen has an effect on the resulting species after the interaction of the [Cu2II(NH3)4O2]2+ complex with SO2. To determine the effect of oxygen on the interaction with SO2, we compare the changes in the Cu K-edge XAS spectra of the [Cu2II(NH3)4O2]2+ complex during the reaction with SO2 alone and with an SO2 + O2 mixture. The difference in SO2 uptake in the presence and absence of oxygen is determined by X-ray adsorbate quantification (XAQ)23 and temperature programmed desorption of SO2 (SO2-TPD). These results are combined with in situ S K-edge X-ray absorption spectroscopy (XAS) and S Kα X-ray emission spectroscopy (XES), which provide further information about the oxidation state and local environment of the sulfur atoms during and after reaction with the [Cu2II(NH3)4O2]2+ complex.
Experimental
The Cu-CHA catalysts were prepared by impregnation of the parent H-CHA zeolite material (Si/Al = 6.7) with the appropriate amount of an aqueous solution of Cu-nitrate. After impregnation, the samples were dried for 2 h at 90 °C, followed by 1 h of calcination at 500 °C in air to decompose the nitrates. In this study, two Cu-CHA catalysts were used: with a Cu loading of 0.8 wt% (referred to as the low-Cu sample) and with a Cu loading of 3.2 wt% (referred to as the high-Cu sample).The general procedure to investigate how the [Cu2II(NH3)4O2]2+ complex reacts with SO2 is as follows. First, the [Cu2II(NH3)4O2]2+ complex is prepared by heating a fresh catalyst sample to 500 °C in 10% O2, followed by cooling to 200 °C, and a reduction in 500 ppm NO + 600 ppm NH3 at 200 °C. The [CuII2(NH3)4O2]2+ complex is then formed by a reoxidation of the reduced catalyst in 10% O2 at 200 °C. After the [CuII2(NH3)4O2]2+ complexes have been formed, the sample is exposed to either 400 ppm SO2 for 3 hours, or to a mixture of 360 ppm SO2 and 10% O2 at 200 °C, while monitoring the changes in the XAS spectra. All the experiments were performed under flow conditions with He as carrier gas. Total flow was 100 ml min−1 (for stages without SO2) or 50 ml min−1 (for stages with SO2).
The whole procedure was followed by in situ Cu K-edge XAS at the BM23 beamline of the European Synchrotron Radiation Facility (ESRF, Grenoble, France).24 Exposure of the [Cu2II(NH3)4O2]2+ dimer to 400 ppm SO2 was also monitored by in situ S K-edge HERFD XANES spectroscopy during a separate experiment at the ID26 beamline of the ESRF.25 S K-alpha XES spectra were also recorded at ID26 for the stationary points of the treatment protocol.
The evolution of S content in the catalyst during the reaction with SO2 was evaluated by in situ XAQ23 measurements. Total SO2 uptake was also independently determined by temperature programmed desorption of SO2 (SO2-TPD).
Experimental procedures are reported in more details in the ESI.†
Results
Exposure to SO2 without O2
To determine the effect of Cu content on the reaction of SO2 with the Cu-CHA, we compared the Cu K-edge spectra for the low-Cu catalyst after exposure to SO2 using the same pre-treatment protocols to form specific different CuI and CuII species earlier reported for the high-Cu catalyst.16 We observed the same general trend, which means that the [Cu2II(NH3)4O2]2+ complex is the most sensitive to SO2 for the low-Cu catalyst as well. The detailed results of these measurements are reported in Fig. S4 in the ESI.†Fig. 1 shows the Cu K-edge XANES and EXAFS FT data collected during the exposure of the [Cu2II(NH3)4O2]2+ complex to SO2 at 200 °C for the low-Cu/CHA and high-Cu/CHA catalysts. The observed trends in the XANES and EXAFS spectra in these measurements are quite similar, indicating that the reaction of SO2 proceeds in a similar way for both catalysts. There is a clear increase of the XANES peak at 8983 eV, indicating the partial reduction of CuII to CuI, and a decrease in the intensity of the first shell in the EXAFS FT, indicating a reduction of the coordination number, due to decomposition of the [Cu2II(NH3)4O2]2+ complex.
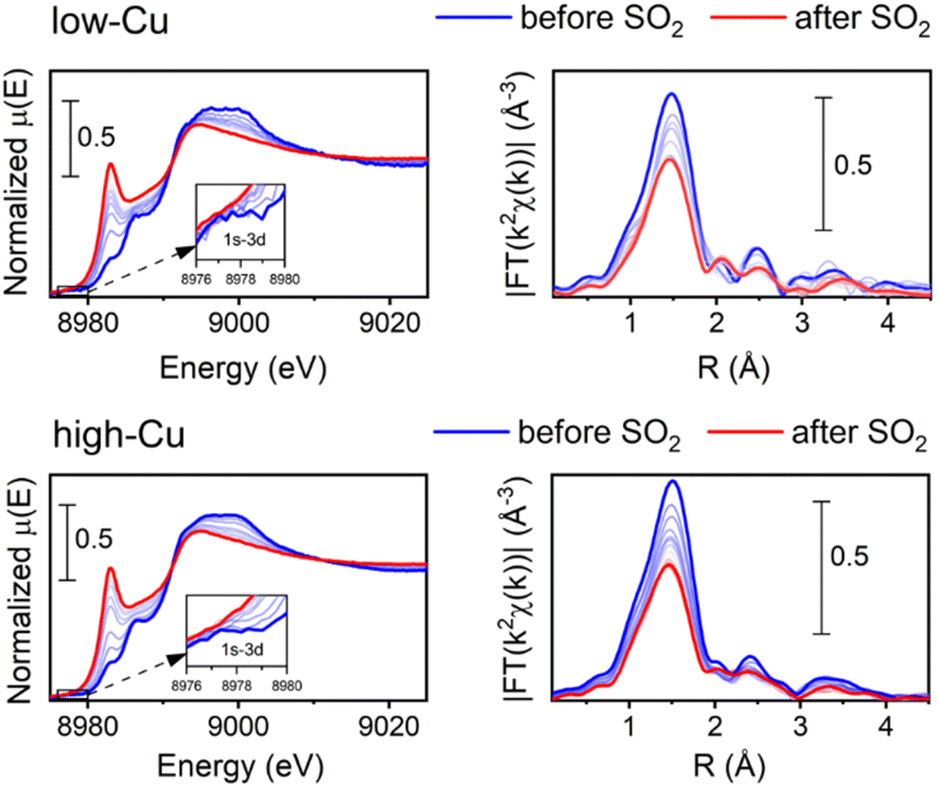 | ||
| Fig. 1 Cu K-edge XANES (a) and FT-EXAFS spectra (b) collected in situ during the exposure of the [Cu2II(NH3)4O2]2+ species to 400 ppm SO2 at 200 °C for low-Cu (0.8 wt% Cu/CHA) and high-Cu (3.2 wt% Cu/CHA) catalysts. | ||
To develop a better understanding of the reaction of SO2 with the [Cu2II(NH3)4O2]2+ complex, we identify the reaction intermediates and reaction products by applying a combination of multivariate curve resolution alternating least squares method (MCR-ALS)26,27 and linear combination fitting (LCF). This hybrid approach consists of three stages. First, we apply MCR-ALS to the ensemble of the experimental spectra to deduce the shape of principal components. Then, the MCR components that can be readily associated with the known Cu species whose experimental spectra are available are substituted by the experimental Cu K-edge spectra of these species. Finally, LCF is performed over the same experimental dataset, using the experimental spectra selected at the previous step and the remaining MCR components. It allows at the same time to minimize the spectral artefacts induced by the MCR algorithm, and get a reasonable estimate of the spectra for the species that cannot be readily identified.
In the reported procedures, it was possible to select as references the experimental spectra for framework-bound CuII (fw-CuII), for the [CuI(NH3)2]+ complex, and for the [Cu2II(NH3)4O2]2+ complex. Conversely, CuI directly bound to the zeolite framework (fw-CuI) was represented by calculated MCR component. The second MCR component that was used in the LCF was designated “sulfated component”, since it appeared only after the samples were exposed to SO2-containing mixtures.
Fig. 2 shows all the reference components used in the linear combination fits (three experimental spectra and two MCR components). More details on the choice of the reference spectra for the linear combination fitting procedure are given in the ESI.†
 | ||
| Fig. 2 Components used as references in the linear combination fit. The fw-CuII, [CuI(NH3)2]+ and [Cu2II(NH3)4O2]2+ components are experimental spectra. The spectra of fw-CuI and the sulfated component are calculated by MCR-ALS. | ||
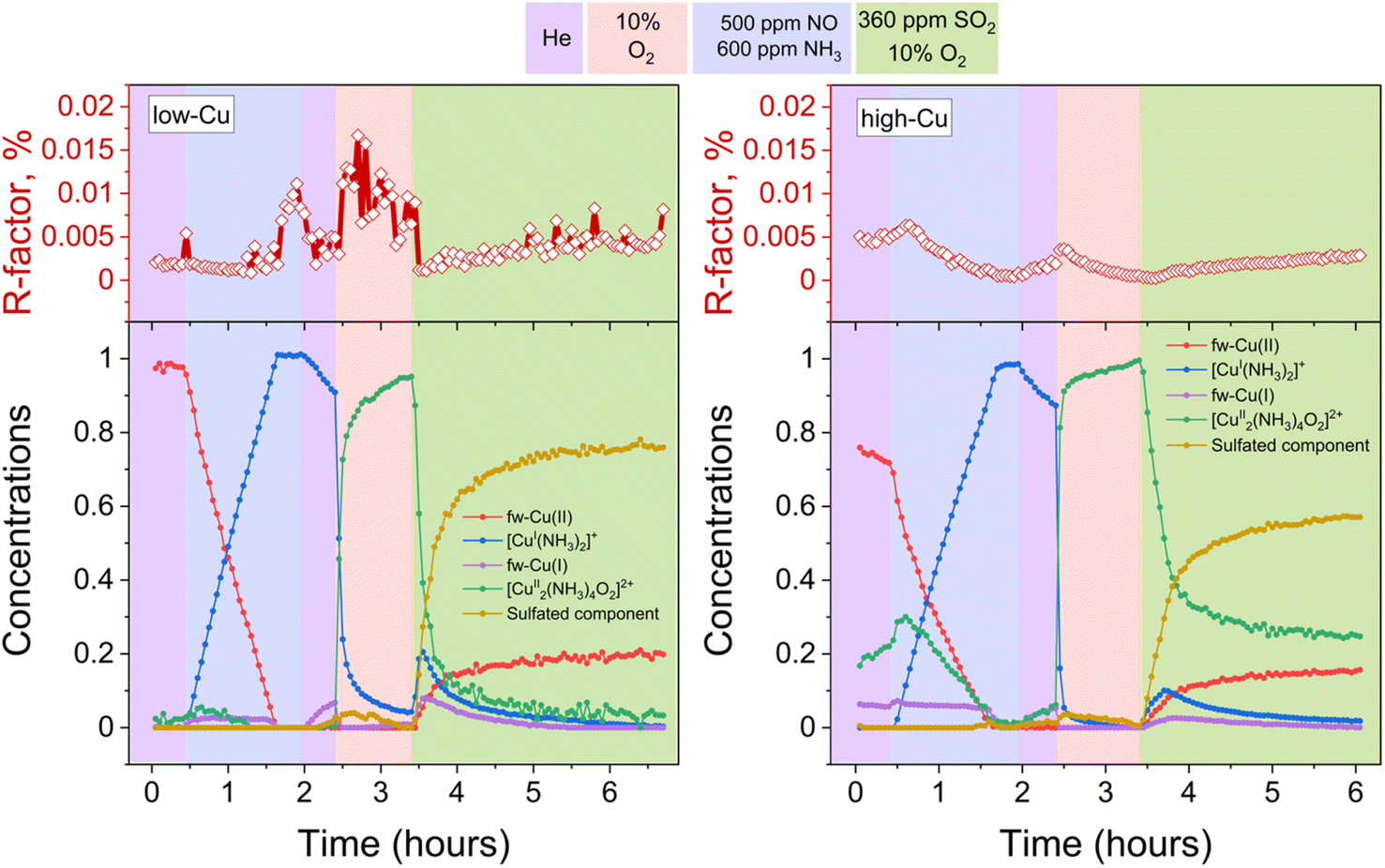
Fig. 3 presents the concentration profiles of the reference components for both the low-Cu and the high-Cu catalysts during the pre-treatment and exposure to SO2. The concentration of the sulfated component increases when the [Cu2II(NH3)4O2]2+-complex is exposed to SO2. The final fraction of the sulfated component is 17% in the high-Cu sample, and 22% in the low Cu/CHA sample. After the exposure to SO2, we find around 50% of the Cu present as the linear [CuI(NH3)2]+ complex, and 25% as fw-CuI, indicating that 75% of the Cu in the catalysts has undergone reduction to CuI. We note that the [Cu2II(NH3)4O2]2+-complex is no longer present after the exposure to SO2, consistent with the high reactivity of this complex with SO2. These results indicate that the reaction of the [Cu2II(NH3)4O2]2+-complex with SO2 is not significantly affected by the variation of Cu content.
 | ||
| Fig. 3 Quantification of Cu speciation during the in situ Cu K-edge XANES measurements from LCF, using the reference spectra shown in Fig. 2. Upper panels: R-factors of the linear combination fits. Lower panels: concentration profiles for the different Cu species during reduction in NH3 + NO, the formation of the [Cu2II(NH3)4O2]2+-complex, and exposure of the [Cu2II(NH3)4O2]2+-complex to SO2, for the low-Cu (left) and high-Cu (right) catalysts. | ||
Exposure to SO2 in the presence of O2
Following the exposure of [Cu2II(NH3)4O2]2+ dimers to SO2, approximately half of the Cu species undergoes a transformation into [CuI(NH3)2]+. It is well known that the exposure of [CuI(NH3)2]+ to O2 leads to the formation of [Cu2II(NH3)4O2]2+ complexes,11,13,14 which, in turn, can react with SO2. Consequently, in order to increase the SO2 uptake, the samples were exposed to a mixture of SO2 + O2, as it makes possible the regeneration of [Cu2II(NH3)4O2]2+ complexes from [CuI(NH3)2]+.Fig. 4 shows the evolution of XANES and EXAFS FT when exposing the [Cu2II(NH3)4O2]2+ complex to a mixture of 360 ppm SO2 and 10% O2 at 200 °C. XANES spectra reveal a slight initial increase and subsequent decrease of the peak at 8983 eV. This indicates that transient CuI species are formed in the process, which react further with the oxygen to form CuII, resulting in the final oxidation state of Cu being +2. This is further corroborated by the 1s–3d transition at 8978 eV indicating the presence of CuII, as this transition is not present in CuI.
 | ||
| Fig. 4 Cu K-edge XANES (a) and FT-EXAFS spectra (b) collected in situ during the exposure of the [Cu2II(NH3)4O2]2+ species to 360 ppm SO2 and 10% O2 at 200 °C for low-Cu (0.8 wt% Cu/CHA) and high-Cu (3.2 wt% Cu/CHA) catalysts. | ||
The intensity of the first peak of the EXAFS FT for the final spectrum after exposure to SO2 + O2 is close to the initial intensity, indicating that the average coordination number of the first shell of Cu remains close to four. That shows that 4-coordinated Cu species are dominant after the exposure to SO2 + O2. Moreover, the second peak becomes more pronounced, evidencing the presence of a relatively heavy neighbor at a well-defined distance in the second shell of Cu. The possible candidates are Si or Al from the zeolitic framework or S in case of formation of sulfated species.
Fig. 5 displays the results of the combined MCR-ALS and LCF fitting of the XANES spectra collected during the formation of the [Cu2II(NH3)4O2]2+ followed by the exposure to the SO2 + O2 mixture. Compared to the results obtained after exposure to SO2 in the absence of O2 using the same set of reference spectral components, the fraction of the sulfated component has become significantly larger, especially for the low-Cu sample. Furthermore, both samples show a transient increase of fw-CuI and [CuI(NH3)2]+ components in the beginning of the exposure to SO2 + O2, but very rapidly their concentration goes to zero. Interestingly, for the high-Cu sample, the significant fraction of component assigned to the [Cu2II(NH3)4O2]2+ complex remains even after the exposure to SO2 + O2 (see the Discussion). Finally, for both the low-Cu and the high-Cu catalysts, the fw-CuII component appears at the final stage of the process.
 | ||
| Fig. 5 Quantification of Cu compounds during the in situ Cu K-edge XANES measurements from LCF, using the reference spectra shown in Fig. 2. Upper panels: R-factors of the linear combination fits. Lower panels: concentration profiles for the different Cu species during reduction in NH3 + NO, the formation of the [Cu2II(NH3)4O2]2+-complex, and exposure of the [Cu2II(NH3)4O2]2+-complex to SO2 + O2, for the low-Cu (left) and high-Cu (right) catalysts. | ||
Alternating exposure to SO2 and O2
To discern the individual effects of SO2 and O2, a third experiment was conducted for both the low-Cu and high-Cu samples, wherein the dimers were exposed to alternating switches between SO2 and O2; the results of the LCF are shown in Fig. 6. Upon exposure to SO2 in the first two cycles, the [Cu2II(NH3)4O2]2+ complexes underwent decomposition, resulting in the formation of fw-CuI, [CuI(NH3)2]+ and the species corresponding to the sulfated component. However, in the subsequent 3rd and 4th cycles, the sulfated component did not exhibit significant growth during the exposure to SO2.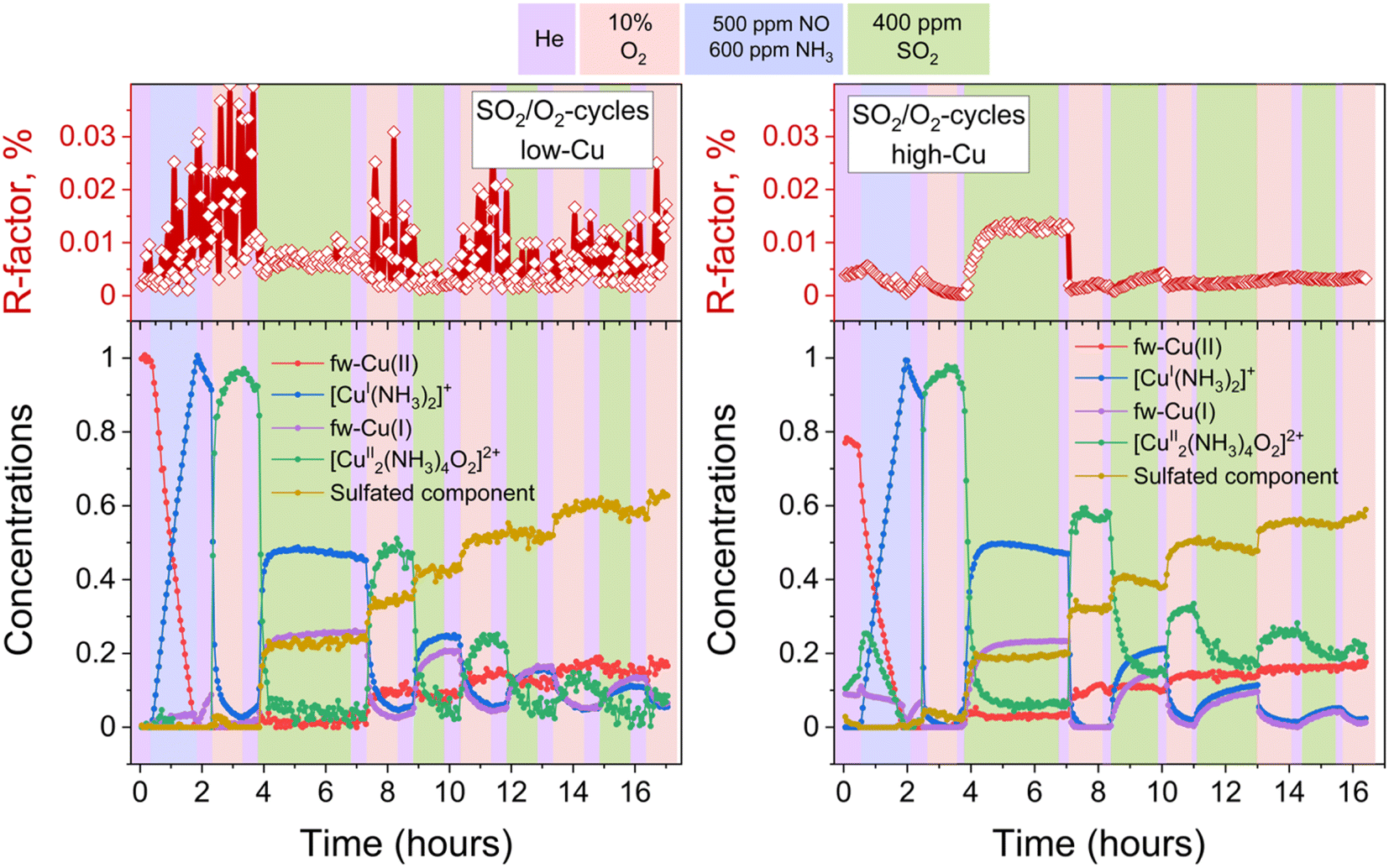 | ||
| Fig. 6 Quantification of Cu compounds during the in situ Cu K-edge XANES measurements from linear combination fits, using the reference spectra shown in Fig. 2. Upper panels: R-factors of the linear combination fits. Lower panels: concentration profiles for the different Cu species during reduction in NH3 + NO, the formation of the [Cu2II(NH3)4O2]2+ complex, and consequent exposures of the [Cu2II(NH3)4O2]2+ complex to SO2 and O2, for the low-Cu (left) and high-Cu (right) catalysts. | ||
In the case of the low-Cu sample, the conversion of [Cu2II(NH3)4O2]2+ complexes at each step was nearly complete. In contrast, for the high-Cu sample, almost complete conversion was observed only after the initial exposure to SO2. Conversely, during each consequent exposure to SO2, a significant amount of the component previously assigned to the [Cu2II(NH3)4O2]2+ complex remained. This “unreactive” [Cu2II(NH3)4O2]2+ component forms after the first exposure of the sulfated sample to oxygen and, after subsequent exposure to SO2, stabilizes at the same level as after exposure of the high-Cu catalyst to SO2 + O2 (Fig. 5). At each subsequent exposure to SO2, less and less CuI is formed, concomitantly with a decreasing amount of the “reactive” [Cu2II(NH3)4O2]2+ component.
Uptake of SO2 monitored by XAQ and SO2-TPD
The presence of oxygen during the SO2 exposure not only affects the final oxidation state of the Cu, but also leads to an increased uptake of SO2. This increased amount of SO2 in the catalyst can be quantified by X-ray adsorbate quantification (XAQ)23 and SO2-TPD.The XAQ technique relies on the phenomenon that the total absorption of X-rays depends on the composition of the sample. Therefore, the increase in total X-ray absorption during SO2 exposure reflects the increase in the amount of SO2 in the sample. This quantitative information was used to calculate the S/Cu ratios in situ during the exposure of the samples to the gas mixtures containing SO2. Fig. 7 shows the measured XAQ signals for the low Cu and high Cu catalysts during exposure to SO2 and SO2 + O2. All S/Cu curves have similar shape, exhibiting rather fast changes in the first 30–40 minutes of the measurement, followed by a much slower growth at a later stage. The final S/Cu levels reached in the presented experiments are 0.32 for the low-Cu sample and 0.22 for the high-Cu sample. In the presence of O2, the final levels increase to S/Cu = 1.04 and 0.63 for the low-Cu and high-Cu catalysts, respectively.
 | ||
| Fig. 7 S/Cu ratio calculated from XAQ during exposure to SO2 and SO2 + O2 for low-Cu (0.8 wt% Cu/CHA) and high-Cu (3.2 wt% Cu/CHA) catalysts. | ||
The second method that was employed to measure SO2 uptake is SO2-TPD, where the desorption of SO2 is recorded as a function of temperature during heating of the catalyst. In contrast to XAQ, where the amount of SO2 adsorbed on the catalyst is monitored in situ, SO2-TPD measures the amount of SO2 released from a saturated sample. Fig. 8 shows the SO2-TPD data for the two catalysts after exposure to SO2 and SO2 + O2; the SO2-TPD of a Cu-free CHA impregnated with (NH4)2SO4 is included for comparison. All desorption curves for the Cu-CHA catalysts show a desorption feature in the range 400–600 °C, and one in the range 750–1000 °C. Because these profiles are clearly different from that of the adsorbed (NH4)2SO4 on the Cu-free zeolite reference, we conclude that the SO2 in the Cu-CHA catalysts predominantly interacts with the Cu, without a significant amount of free (NH4)2SO4. This is in good agreement with the conclusion that SO2 mainly reacts with the [Cu2II(NH3)4O2]2+ complex. The features in the range 750–1000 °C remain largely unaffected by the presence of O2, while the peak around 420 °C becomes larger, and shows a slight shift towards higher temperatures. Overall, SO2-TPD results indicate that the presence of O2 leads to a larger amount of SO2 in the Cu-CHA catalysts, and that SO2 binds predominantly to the Cu ions in the zeolite, in agreement with our earlier results.16
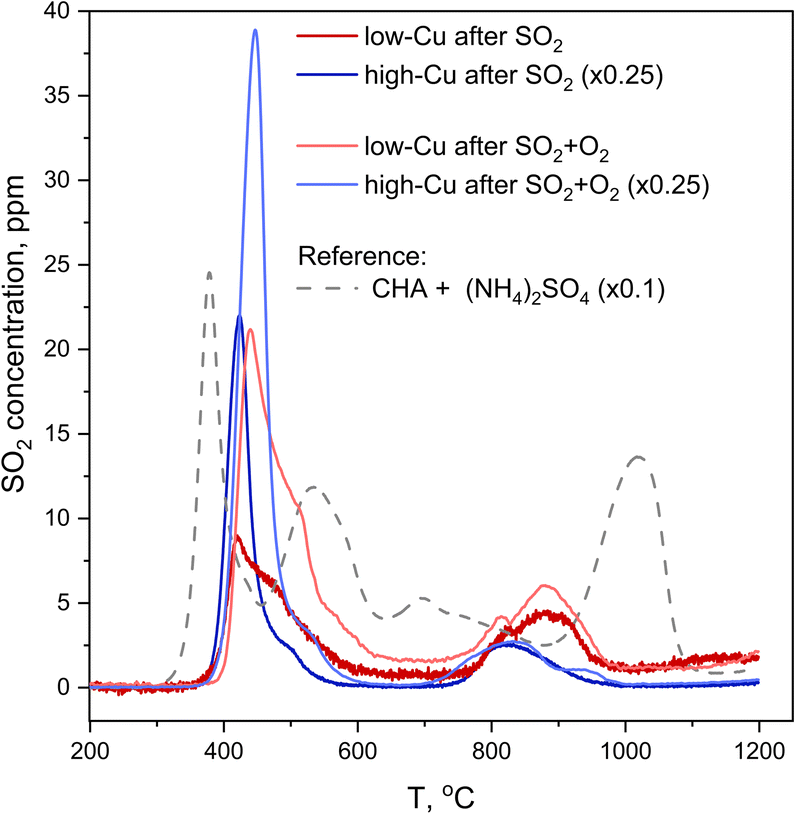 | ||
| Fig. 8 SO2-TPD profiles collected after exposure of the catalysts to SO2 (red and blue lines) and SO2 + O2 (orange and light blue lines) in comparison to a reference SO2-TPD curve of a Cu-free CHA zeolite impregnated with 20 wt% (NH4)2SO4, downscaled ×10. Pre-treatment is the same as for the procedures followed by in situ XAS. The curves for the high Cu catalysts (3.2 wt% Cu/CHA) (blue and light blue lines) are downscaled ×4. | ||
XAQ and TPD show that exposure to SO2 + O2 leads to a greater sulfur uptake compared to the exposure to only SO2 (Fig. 9). Importantly, the S/Cu molar ratio is in good correspondence with the concentration of the sulfated component in the XANES LCF, which confirms the assignment of the latter mainly to sulfated species.
 | ||
| Fig. 9 S/Cu ratios in the low-Cu (0.8 wt% Cu/CHA) and high-Cu (3.2 wt% Cu/CHA) samples after exposure to SO2 and SO2 + O2 obtained from XAQ and SO2-TPD compared to the concentration of the sulfated component obtained from the XANES LCF. | ||
Sulfur K-edge XANES and Kα XES
To resolve the oxidation state of sulfur and the configuration of sulfur species in the sulfated Cu-CHA catalyst we measured S K-edge XANES during the exposure of the [Cu2II(NH3)4O2]2+ complex to SO2 and Kα-XES spectra at the end of the exposure. Fig. 10 shows the evolution of S K-edge XANES spectra during exposure of the [Cu2II(NH3)4O2]2+ complex to SO2. The spectra were collected in HERFD mode. The increase of the edge jump corresponds to the increasing concentration of sulfur, which means that S is accumulated in the sample. From the position of the edge and the shape of the spectrum we can identify the oxidation state of S and possible local environment by comparing with references. Fig. 11a shows that the sulfur in the sample predominately exists in the S6+ oxidation state, forming an SO42− group. Fig. 11b presents Kα XES spectra of the [Cu2II(NH3)4O2]2+ complex after exposure to SO2 in comparison with reference compounds. The positions of the two features at 2.3066 and 2.3078 keV in the Kα XES of the complex agree well with the references containing S6+ in the SO42− group, which is in line with the findings from S K-edge XANES.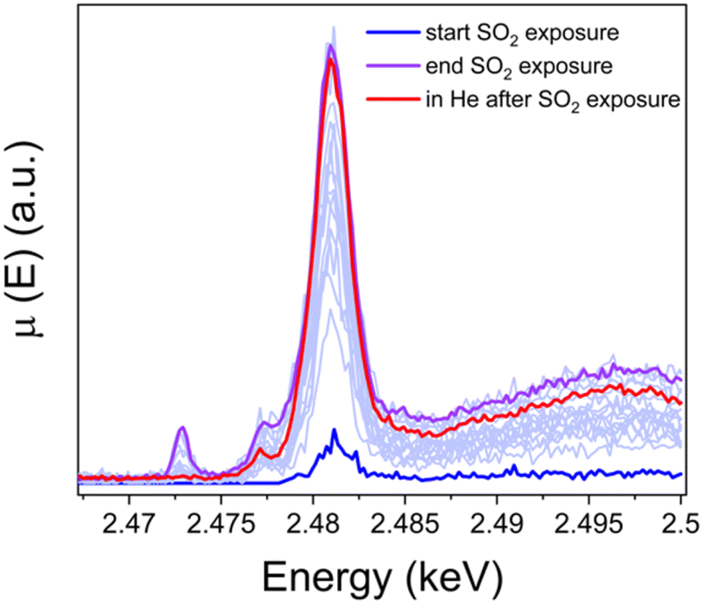 | ||
| Fig. 10 S K-edge XANES spectra of the high-Cu catalyst (3.2 wt% Cu/CHA) collected in situ during exposure of the [Cu2II(NH3)4O2]2+ complex to SO2 at 200 °C and in He after exposure to SO2. | ||
 | ||
| Fig. 11 S K-edge XANES spectra of the [Cu2II(NH3)4O2]2+ complex exposed to SO2 and of the reference compounds. K-alpha XES spectra of the [Cu2II(NH3)4O2]2+ complex exposed to SO2 and references. High-Cu catalyst (3.2 wt% Cu/CHA). | ||
The sulfated component
From our measurements, we can identify the [CuI(NH3)2]+, fw-CuII, fw-CuI and reactive [Cu2II(NH3)4O2]2+ complexes, even though the exact structure of the framework-bound complexes is still under debate. Following the exposure of the Cu-CHA catalysts to SO2, both in the presence and in the absence of O2, a new species, whose spectrum we designated a “sulfated component”, appears.In order to elucidate the structure of the species associated with the sulfated component, we compared the spectrum generated by MCR-ALS with the experimental spectra of three references: [CuII(NH3)4]SO4·H2O, the [Cu2II(NH3)4O2]2+ complex formed in Cu-CHA and [Cu2II(NH3)4]2+ complex in solution (Fig. 12). The sulfated component has striking similarity to the spectrum of Cu(NH3)4SO4·H2O. Therefore, we propose that the species that give rise to the sulfated component contain 4-coordinated CuII in the square-planar environment similar to the one of Cu(NH3)4SO4, where a square-planar CuII ion is coordinated to four NH3 ligands. It is also possible that a geometrically similar CuII configuration with mixed NH3/O ligands is realized in the zeolite, since the XANES spectrum is expected to be very similar. Because the S atoms are not directly coordinated to the Cu in the structure of the [CuII(NH3)4]SO4, and the Cu–S distance is about 4.6 Å, it is not possible to determine the precise location of the S atom in the sulfated species in the zeolite based on XANES data alone.
 | ||
| Fig. 12 Comparison of the sulfated component obtained with MCR-ALS with the Cu K-edge XANES spectra of the [Cu(NH3)4]SO4·H2O, [Cu2II(NH3)4O2]2+ complex inside the CHA and CuII2(NH3)4 in solution (left); the fitting results for the reconstructed EXAFS of the sulfated component (right). | ||
To locate the S atoms in the sulfated species, we extracted the EXAFS part of the sulfated component by subtracting the weighted reference spectrum for fw-CuII from the spectrum after exposure of the low-Cu catalyst to SO2 and O2 (see Fig. 4 and 5), using the weight coefficients derived from the LCF. We then fitted the EXAFS spectrum of the sulfated component with two N and two O in the first shell and S in the second shell. The fitting results are reported in Table 1. The resulting fit in comparison with the EXAFS of the sulfated component with a highlighted contribution of the Cu–S path is presented in Fig. 12. From this analysis, we find a Cu–S distance of 2.58 Å, which is much shorter than in the Cu(NH3)4SO4·H2O reference compound (4.65 Å). This means that the structure of the sulfated species is different from Cu(NH3)4SO4·H2O.
| Path | N | S 0 2 | σ 2, Å2 | R, Å | E 0, eV | R-Factor |
|---|---|---|---|---|---|---|
| Cu–O | 2 | 0.9(1) | 0.002(2) | 1.94(2) | 2.4 | 0.02 |
| Cu–N | 2 | 0.003(3) | 2.09(2) | |||
| Cu–S | 1 | 0.006(2) | 2.58(2) |
Discussion
The unreactive part of the [Cu2II(NH3)4O2]2+ component
For the high-Cu catalyst, the concentration of the component assigned to [Cu2II(NH3)4O2]2+ complexes does not go to zero during repeated exposures to SO2 followed by exposure to O2, remaining above 18% after each exposure to SO2 (see Fig. 6).The first explanation for such behavior may be that a fraction of the [Cu2II(NH3)4O2]2+ complexes become inaccessible for SO2 and therefore do not react. This interpretation resembles the mechanism for deactivation of Cu-CHA catalysts as proposed by Bjerregaard et al.17 A further consequence of this interpretation is that it limits the uptake of SO2 in a Cu-CHA catalyst with a sufficiently high Cu content (ca. 3 wt%), while, at the same time, a part of the Cu does not react with SO2. Such a conclusion is in good agreement with earlier measurements of a limited uptake of SO2 in Cu-CHA catalysts,3,4 and the observation that Cu-CHA catalysts show a residual activity after saturation with SO2.3 With such an interpretation, the structure of the [Cu2II(NH3)4O2]2+ complex remains unchanged, which is directly reflected in the XANES spectra.
The second explanation for the persistence of the component assigned to the [Cu2II(NH3)4O2]2+ complex involves a transformation of the [Cu2II(NH3)4O2]2+ complex to a different structure, having similar coordination of the Cu-ions. A possibility is the formation of peroxo–dicopper complexes CuxOy attached to the framework,28 which can be formed by exposure of the Cu-CHA to O2 at 400–500 °C. Such species are expected to have a very similar XANES spectrum to the [Cu2II(NH3)4O2]2+ complexes, because coordination of the Cu-ions is similar, and oxidation state is the same. Therefore, XANES spectra of such framework-bound complexes may be difficult to distinguish from those of the [Cu2II(NH3)4O2]2+ complex by MCR-ALS analysis, which merges them into a single component. Such an interpretation is supported by the observation that the component assigned to [Cu2II(NH3)4O2]2+ complexes appears also during the initial oxidation of the high-Cu catalyst at 500 °C (green curve in Fig. 6, right panel, at t = 0) with the concentration of around 20%, which is very close to the concentration observed after exposure to SO2 + O2. Clearly, it is not possible to form [Cu2II(NH3)4O2]2+ complexes at the activation stage, since the required NH3 is not yet available and the temperature is too high. As such moieties are essentially fw-CuII species, we expect that their reactivity towards SO2 is very low, as we have shown in our previous work,16 thus resulting in an accumulation of these species in the sample.
Unraveling the mechanism of the sulfation reaction
The results presented above have some implications for a mechanism describing the reaction of SO2 with Cu-CHA catalysts. Sulfur XES and XANES show that S is stored in the sample as S6+ within (SO4)2− tetrahedra. This indicates an oxidation of SO2 by the [Cu2II(NH3)4O2]2+ complex. A formation of an (SO4)2− group in the absence of O2 in the gas implies oxygen transfer from the [Cu2II(NH3)4O2]2+ complexes to the SO2 molecule.In the reaction of SO2 with the [Cu2II(NH3)4O2]2+ complexes in the absence of O2, the S/Cu ratio reaches only around 25% (Fig. 9). Nevertheless, all Cu in the catalyst changes its coordination environment, since all [Cu2II(NH3)4O2]2+ complexes are converted (Fig. 3). The fact that all Cu in the catalyst is affected at a S/Cu uptake of 0.25 suggests that a single SO2 molecule reacts with at least two [Cu2II(NH3)4O2]2+ complexes. This requires either sufficient mobility of the [Cu2II(NH3)4O2]2+ complexes or that first a mobile intermediate product is formed in the reaction between SO2 and the first complex, which then reacts with a second [Cu2II(NH3)4O2]2+. Since the mobility of the bulky [Cu2II(NH3)4O2]2+ complex is expected to be limited, the formation of a smaller mobile intermediate product seems more likely.
Bringing these considerations together, we arrive at a reaction pathway consisting of two steps. In the first step (eqn (1)), SO2 reacts with a [Cu2II(NH3)4O2]2+ complex, the peroxo bond breaks, a mobile SOaX intermediate forms, and CuII reduces to CuI. The CuI appears in the form of [CuI(NH3)2]+ and fw-CuI, as indicated by the LCF (Fig. 3). A possible candidate for the mobile intermediate SOaX is SO3, formed by oxidation of SO2, upon the formation of CuI-species. It is known that SO3 has a similar effect on the NH3-SCR activity of Cu-CHA as SO2,3,20,29,30 which would be consistent with the proposed reaction scheme. However, our data do not allow the exact structure of the mobile SOaX complex to be determined, so the formation of SO3 remains to be proven (or ruled out) experimentally.
| Cu2II(NH3)4O2 + SO2 → CuI(NH3)2 + fw-CuI + SOaX + … | (1) |
| SOaX + Cu2II(NH3)4O2 → CuI(NH3)2 + CuIISO4Z + … | (2) |
In the second step (eqn (2)), the mobile SOaX intermediate reacts with another [Cu2II(NH3)4O2]2+ complex, breaking the peroxo-bond to form the (SO4)2− group within the sulfated Cu-species associated with sulfated XAS component (CuIISO4Z) and another linear diamino complex [CuI(NH3)2]+. Z in the sulfated species CuIISO4Z may comprise O, framework O or NH3 to result in a square-planar coordination of Cu with 4-ligands proven by our EXAFS and XANES results (Fig. 12). This two-step reaction scheme is in line with the Cu concentration profiles obtained by the LCF analysis in the case of exposure to SO2 in the absence of O2. Note that eqn (1) and (2) aim to summarize the experimental findings in this article, and therefore do not represent a complete mechanism of the reaction of SO2 with the Cu-CHA catalyst.
When the sample is exposed to SO2 in the presence of O2, we observe an initial transient formation of the same CuI intermediates, as in case of the exposure to SO2 alone (Fig. 5). This indicates that O2 reoxidizes the [CuI(NH3)2]+ complexes into new [Cu2II(NH3)4O2]2+ species. In this way, the CuI-species formed in the reaction with SO2 become available again for further reaction with SO2, which explains the increased SO2 uptake compared to exposure to SO2 without oxygen. This is corroborated by the results obtained with alternating exposure of the Cu-CHA catalysts to SO2 and O2 (Fig. 6), showing alternating conversion of the [Cu2II(NH3)4O2]2+ complexes in the SO2 phases and their reconstruction from the CuI during the exposures to O2.
However, the exposure of the sample to alternating SO2/O2 cycles (Fig. 6) also reveals that the growth of the sulfated component occurs not only in SO2 phases but also when the sample is exposed to O2. Moreover, during the later stages of the cycles (3rd and 4th cycles), the growth of the sulfated component is observed only in the presence of O2, while the exposure to SO2 at this stage does not result in the formation of the sulfated component at all. Nonetheless, even at this stage the reactive [Cu2II(NH3)4O2]2+ complexes do undergo decomposition in the presence of SO2, resulting in sulfur uptake, as evidenced by the evolution of the S/Cu ratio obtained from the in situ XAQ signal (Fig. 13).
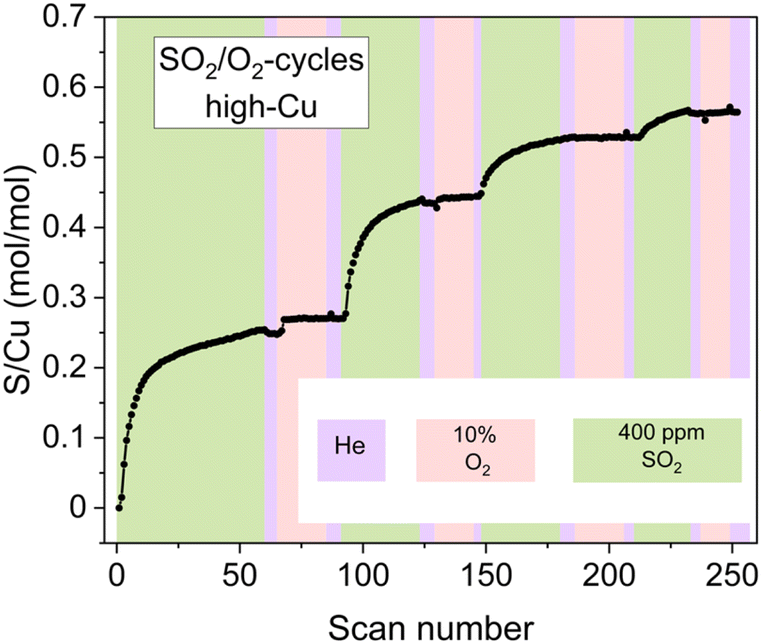 | ||
| Fig. 13 The evolution of the S/Cu ratio during the SO2/O2-cycles for the high-Cu sample (3.2 wt% Cu/CHA) deduced from the XAQ signal. | ||
A possible explanation for this effect is that at the late stages of the cycles, when the reactive Cu species are few, it is less likely to have two [Cu2II(NH3)4O2]2+ complexes close enough to perform the second step of the sulfation reaction (eqn (2)). Therefore, the mobile sulfur species SOaX formed in the first step (eqn (1)) can be converted into the sulfated Cu species only after the stock of [Cu2II(NH3)4O2]2+ complexes is replenished upon the exposure to O2. It is possible that in such regime SOaX undergo further transformations (e.g. reacting with NH3 or NH4+) before reacting with newly formed [Cu2II(NH3)4O2]2+ and yielding the sulfated Cu species, but the obtained data do not allow unambiguous identification of the corresponding reaction pathways. Nonetheless, the observed effect serves as indirect confirmation of the multi-step nature of the sulfation process involving at least two [Cu2II(NH3)4O2]2+ complexes per SO2.
Conclusions
In this study, we applied in situ XAS at Cu and S K-edges, S Kα XES, XAQ and SO2-TPD to investigate the interaction mechanism between the [Cu2II(NH3)4O2]2+ complex in the Cu-CHA catalyst and SO2.Upon reacting the [Cu2II(NH3)4O2]2+ complex with SO2, a mixture of fw-CuI (approximately 1/4 of total Cu), [CuI(NH3)2]+ complexes (approximately 1/2) and a new sulfated CuII compound (approximately 1/4) are formed. The presence of oxygen in the gas mixture with SO2 enhances the reaction, leading to higher concentrations of the sulfated species and an increased S/Cu ratio in the sample. This effect is explained by reoxidation of the [CuI(NH3)2]+ species to the reactive [Cu2II(NH3)4O2]2+ complexes. The catalysts with 0.8 wt% Cu/CHA and 3.2 wt% Cu/CHA demonstrated similar results.
Following a multi-technique experimental approach, the structure of the Cu and S local environment of sulfated species accumulated in the sample was uncovered. Copper in the sulfated species exists as Cu2+ and adopts a square-planar coordination with four light ligands in the first coordination shell, which, most probably, are NH3 and O. Sulfur in the sulfated species is in the S6+ oxidation state, forming an SO4 group. The sulfur atom is located in the second shell of Cu at an approximate distance of 2.6 Å, suggesting that Cu and S are connected through two oxygen ligands.
Data availability
The datasets presented in this article are available at the ESRF repository: https://cloud.esrf.fr/s/2HQDGDp3w7DX4zX.Author contributions
The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreements no. 847439 (InnovaXN) and no. 955839 (CHASS). We thank V. A. Saveleva and B. Detlefs for the help during the XAS and XES experiments at ID26. The assistance of C. Atzori at the BM23 beamline is kindly acknowledged. We are grateful to Peter N. R. Vennestrøm and A. Martini for fruitful discussions. Authors acknowledge support from the Project CH4.0 under the MUR program “Dipartimenti di Eccellenza 2023-2027” (CUP: D13C22003520001).References
- C. K. Lambert, Perspective on SCR NOx control for diesel vehicles, React. Chem. Eng., 2019, 4, 969–974 RSC
.
- R. Gounder and A. Moini, Automotive NOx abatement using zeolite-based technologies, React. Chem. Eng., 2019, 4, 966–968 RSC
- P. S. Hammershoi, Y. Jangjou, W. S. Epling, A. D. Jensen and T. V. W. Janssens, Reversible and irreversible deactivation of Cu-CHA NH3-SCR catalysts by SO2 and SO3, Appl. Catal., B, 2018, 226, 38–45 CrossRef
- P. S. Hammershoi, A. D. Jensen and T. V. W. Janssens, Impact of SO2-poisoning over the lifetime of a Cu-CHA catalyst for NH3-SCR, Appl. Catal., B, 2018, 238, 104–110 CrossRef
- L. Chen, T. V. W. Janssens, P. N. R. Vennestrom, J. Jansson, M. Skoglundh and H. Gronbeck, A Complete Multisite Reaction Mechanism for Low-Temperature NH3-SCR over Cu-CHA, ACS Catal., 2020, 10, 5646–5656 CrossRef CAS
- Y. Feng, X. Wang, T. V. W. Janssens, P. N. R. Vennestrøm, J. Jansson, M. Skoglundh and H. Grönbeck, First-Principles Microkinetic Model for Low-Temperature NH3-Assisted Selective Catalytic Reduction of NO over Cu-CHA, ACS Catal., 2021, 11, 14395–14407 CrossRef CAS
- T. V. W. Janssens, H. Falsig, L. F. Lundegaard, P. N. R. Vennestrom, S. B. Rasmussen, P. G. Moses, F. Giordanino, E. Borfecchia, K. A. Lomachenko, C. Lamberti, S. Bordiga, A. Godiksen, S. Mossin and P. Beato, A Consistent Reaction Scheme for the Selective Catalytic Reduction of Nitrogen Oxides with Ammonia, ACS Catal., 2015, 5, 2832–2845 CrossRef CAS
- K. A. Lomachenko, E. Borfecchia, C. Negri, G. Berlier, C. Lamberti, P. Beato, H. Falsig and S. Bordiga, The Cu-CHA deNOx catalyst in action: temperature-dependent NH3-assisted selective catalytic reduction monitored by operando XAS and XES, J. Am. Chem. Soc., 2016, 138, 12025–12028 CrossRef CAS PubMed
- R. Millan, P. Cnudde, V. van Speybroeck and M. Boronat, Mobility and Reactivity of Cu+ Species in Cu-CHA Catalysts under NH3-SCR-NOx Reaction Conditions: Insights from AIMD Simulations, JACS Au, 2021, 1, 1778–1787 CrossRef CAS PubMed
- I. A. Pankin, H. Issa Hamoud, K. A. Lomachenko, S. B. Rasmussen, A. Martini, P. Bazin, V. Valtchev, M. Daturi, C. Lamberti and S. Bordiga, Cu- and Fe-speciation in a composite zeolite catalyst for selective catalytic reduction of NOx: insights from operando XAS, Catal. Sci. Technol., 2021, 11, 846–860 RSC
- C. Negri, T. Selleri, E. Borfecchia, A. Martini, K. A. Lomachenko, T. V. W. Janssens, M. Cutini, S. Bordiga and G. Berlier, Structure and Reactivity of Oxygen-Bridged Diamino Dicopper(II) Complexes in Cu-Ion-Exchanged Chabazite Catalyst for NH3-Mediated Selective Catalytic Reduction, J. Am. Chem. Soc., 2020, 142, 15884–15896 CrossRef CAS PubMed
- F. Gao, D. Mei, Y. Wang, J. Szanyi and C. H. Peden, Selective Catalytic Reduction over Cu/SSZ-13: Linking Homo- and Heterogeneous Catalysis, J. Am. Chem. Soc., 2017, 139, 4935–4942 CrossRef CAS PubMed
- C. Paolucci, I. Khurana, A. A. Parekh, S. C. Li, A. J. Shih, H. Li, J. R. Di Iorio, J. D. Albarracin-Caballero, A. Yezerets, J. T. Miller, W. N. Delgass, F. H. Ribeiro, W. F. Schneider and R. Gounder, Dynamic multinuclear sites formed by mobilized copper ions in NOx selective catalytic reduction, Science, 2017, 357, 898–903 CrossRef CAS PubMed
- A. Martini, C. Negri, L. Bugarin, G. Deplano, R. K. Abasabadi, K. A. Lomachenko, T. V. W. Janssens, S. Bordiga, G. Berlier and E. Borfecchia, Assessing the Influence of Zeolite Composition on Oxygen-Bridged Diamino Dicopper(II) Complexes in Cu-CHA DeNOx Catalysts by Machine Learning-Assisted X-ray Absorption Spectroscopy, J. Phys. Chem. Lett., 2022, 13, 6164–6170 CrossRef CAS PubMed
- C. Negri, A. Martini, G. Deplano, K. A. Lomachenko, T. V. W. Janssens, E. Borfecchia, G. Berlier and S. Bordiga, Investigating the role of Cu-oxo species in Cu-nitrate formation over Cu-CHA catalysts, Phys. Chem. Chem. Phys., 2021, 23, 18322–18337 RSC
- A. Y. Molokova, E. Borfecchia, A. Martini, I. A. Pankin, C. Atzori, O. Mathon, S. Bordiga, F. Wen, P. N. R. Vennestrøm, G. Berlier, T. V. W. Janssens and K. A. Lomachenko, SO2 Poisoning of Cu-CHA deNOx Catalyst: The Most Vulnerable Cu Species Identified by X-ray Absorption Spectroscopy, JACS Au, 2022, 2, 787–792 CrossRef CAS PubMed
- J. D. Bjerregaard, M. Votsmeier and H. Grönbeck, Mechanism for SO2 poisoning of Cu-CHA during low temperature NH3-SCR, J. Catal., 2023, 417, 497–506 CrossRef CAS
- Y. Jangjou, Q. Do, Y. T. Gu, L. G. Lim, H. Sun, D. Wang, A. Kumar, J. H. Li, L. C. Grabow and W. S. Epling, Nature of Cu Active Centers in Cu-SSZ-13 and Their Responses to SO2 Exposure, ACS Catal., 2018, 8, 1325–1337 CrossRef CAS
- K. Wijayanti, K. Xie, A. Kumar, K. Kamasamudram and L. Olsson, Effect of gas compositions on SO2 poisoning over Cu/SSZ-13 used for NH3-SCR, Appl. Catal., B, 2017, 219, 142–154 CrossRef CAS
- Y. Jangjou, D. Wang, A. Kumar, J. Li and W. S. Epling, SO2 Poisoning of the NH3-SCR Reaction over Cu-SAPO-34: Effect of Ammonium Sulfate versus Other S-Containing Species, ACS Catal., 2016, 6, 6612–6622 CrossRef CAS
- L. Zhang, D. Wang, Y. Liu, K. Kamasamudram, J. Li and W. Epling, SO2 poisoning impact on the NH3-SCR reaction over a commercial Cu-SAPO-34 SCR catalyst, Appl. Catal., B, 2014, 156–157, 371–377 CrossRef CAS
- R. Villamaina, S. Liu, I. Nova, E. Tronconi, M. P. Ruggeri, J. Collier, A. York and D. Thompsett, Speciation of Cu Cations in Cu-CHA Catalysts for NH3-SCR: Effects of SiO2/AlO3 Ratio and Cu-Loading Investigated by Transient Response Methods, ACS Catal., 2019, 9, 8916–8927 CrossRef CAS
- K. A. Lomachenko, A. Y. Molokova, C. Atzori and O. Mathon, Quantification of Adsorbates by X-ray Absorption Spectroscopy: Getting TGA-like Information for Free, J. Phys. Chem. C, 2022, 126, 5175–5179 CrossRef CAS PubMed
- O. Mathon, A. Beteva, J. Borrel, D. Bugnazet, S. Gatla, R. Hino, I. Kantor, T. Mairs, M. Munoz, S. Pasternak, F. Perrin and S. Pascarelli, The time-resolved and extreme conditions XAS (TEXAS) facility at the European Synchrotron Radiation Facility: the general-purpose EXAFS bending-magnet beamline BM23, J. Synchrotron Radiat., 2015, 22, 1548–1554 CrossRef CAS PubMed
- M. Rovezzi, A. Harris, B. Detlefs, T. Bohdan, A. Svyazhin, A. Santambrogio, D. Degler, R. Baran, B. Reynier, P. N. Crespo, C. Heyman, H. P. Van Der Kleij, P. Van Vaerenbergh, P. Marion, H. Vitoux, C. Lapras, R. Verbeni, M. M. Kocsis, A. Manceau and P. Glatzel, TEXS: in-vacuum tender X-ray emission spectrometer with 11 Johansson crystal analyzers, J. Synchrotron Radiat., 2020, 27, 813–826 CrossRef CAS PubMed
- A. Martini and E. Borfecchia, Spectral Decomposition of X-ray Absorption Spectroscopy Datasets: Methods and Applications, Crystals, 2020, 10, 46 CrossRef
- A. Martini, E. Borfecchia, K. A. Lomachenko, I. A. Pankin, C. Negri, G. Berlier, P. Beato, H. Falsig, S. Bordiga and C. Lamberti, Composition-driven Cu-speciation and reducibility in Cu-CHA zeolite catalysts: a multivariate XAS/FTIR approach to complexity, Chem. Sci., 2017, 8, 6836–6851 RSC
- B. Ipek, M. J. Wulfers, H. Kim, F. Göltl, I. Hermans, J. P. Smith, K. S. Booksh, C. M. Brown and R. F. Lobo, Formation of [Cu2O2]2+ and [Cu2O]2+ toward C–H Bond Activation in Cu-SSZ-13 and Cu-SSZ-39, ACS Catal., 2017, 7, 4291–4303 CrossRef CAS
- Y. S. Cheng, C. Lambert, D. H. Kim, J. H. Kwak, S. J. Cho and C. H. F. Peden, The different impacts of SO2 and SO3 on Cu/zeolite SCR catalysts, Catal. Today, 2010, 151, 266–270 CrossRef CAS
- X. Auvray, M. Arvanitidou, Å. Högström, J. Jansson, S. Fouladvand and L. Olsson, Comparative Study of SO2 and SO2/SO3 Poisoning and Regeneration of Cu/BEA and Cu/SSZ-13 for NH3 SCR, Emiss. Control Sci. Technol., 2021, 7, 232–246 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03924b |
| This journal is © The Royal Society of Chemistry 2023 |