
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advances in CO2 activation by frustrated Lewis pairs: from stoichiometric to catalytic reactions
Md. Nasim
Khan
 ab,
Yara
van Ingen
a,
Tribani
Boruah
a,
Adam
McLauchlan
a,
Thomas
Wirth
*b and
Rebecca L.
Melen
*a
ab,
Yara
van Ingen
a,
Tribani
Boruah
a,
Adam
McLauchlan
a,
Thomas
Wirth
*b and
Rebecca L.
Melen
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Translational Research Hub, Maindy Road, Cathays, Cardiff, CF24 4HQ Cymru/Wales, UK. E-mail: MelenR@cardiff.ac.uk
bSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, Cymru/Wales, UK. E-mail: wirthT@cardiff.ac.uk
First published on 9th November 2023
Abstract
The rise of CO2 concentrations in the environment due to anthropogenic activities results in global warming and threatens the future of humanity and biodiversity. To address excessive CO2 emissions and its effects on climate change, efforts towards CO2 capture and conversion into value adduct products such as methane, methanol, acetic acid, and carbonates have grown. Frustrated Lewis pairs (FLPs) can activate small molecules, including CO2 and convert it into value added products. This review covers recent progress and mechanistic insights into intra- and inter-molecular FLPs comprised of varying Lewis acids and bases (from groups 13, 14, 15 of the periodic table as well as transition metals) that activate CO2 in stoichiometric and catalytic fashion towards reduced products.
Introduction
Background
Since the beginning of the industrial revolution, human activity has raised the concentration of carbon dioxide (CO2), amongst other important greenhouse gases, in the environment by over 50%. CO2 gas absorbs and emits radiant energy at infrared wavelengths that causes an increase in atmospheric temperature, thus global warming and has become a main driver of climate change.1 Society has made some progress in finding low-carbon emitting alternatives, for example in sources of energy, and now a scattered dip in CO2 level has been observed. Minimising CO2 emissions by at least 50% to limit the increase in the global average temperature by 2 °C by 2050 has been set as a global target.2 This will require a rapid exploitation of new energy technologies with a low-carbon or zero-carbon energy sources to restore our ecosystem. As one single technology is not expected to solve this problem, global warming alerts have drawn urgent attention to control the expansion of CO2 concentrations in the atmosphere through the framework of carbon capture, storage, and utilisation (CCSU).3 The important challenge remains not only in carbon dioxide capture and storage but also to utilise it for the creation of value-added carbon products.4 CCSU processes add value to the conversion of CO2 into fuels and chemicals, and can compensate the cost of capturing CO2. This approach has generated many new directions in various branches of science and technologies including chemical, biological and material applications.5 Extensive work has been carried out in the past few decades for CO2 capture and utilisation (CO2-CU) using various chemical processes for the reduction of CO2 into products such as formic acid and methanol, or light hydrocarbons such as methane.6 Metal and non-metal derived reagents and catalysts in homogeneous and heterogeneous systems have been explored including promising mediums such as zeolites, metal-organic frameworks (MOFs), covalent organic frameworks (COFs), nanomaterials, as well as electrochemical, photochemical and thermal processes.The activation and chemical conversion of CO2 requires high energy due to its high thermodynamic stability (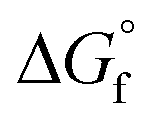 = −396 kJ mol−1).7 Entropy is one important factor that limits CO2 transformations, even for some reactions where ΔH° < 0, ΔG° is positive. Conversion of CO2 can take place at room temperature or at lower temperatures but in such transformations the carbon atom retains its oxidation state of +4 and promotes the formation of carbamates, carbonates, urea and its derivatives, polycarbonates, and polyethers, where OH−, H2O, amines, carbanions, olefins, alkynes, and dienes have been reacted as reagents with CO2. Reactions that produce carboxylates from CO2 are thermodynamically favourable. Carbon product formation such as methanol, carbon monoxide, formaldehyde, methane, and hydrocarbons from CO2 require higher energy, and thus kinetic control to steer away from the thermodynamically favoured products. Kinetically, to obtain CO2 reduced products with a change of oxidation state in the carbon atom requires more energy. To achieve the reduction of CO2, the reagent should bend CO2 to overcome the first energy barrier and to achieve subsequent reduction steps. Preorganised reducing agents kinetically favour the challenging reduction of CO2 beyond carbonate or formate.
= −396 kJ mol−1).7 Entropy is one important factor that limits CO2 transformations, even for some reactions where ΔH° < 0, ΔG° is positive. Conversion of CO2 can take place at room temperature or at lower temperatures but in such transformations the carbon atom retains its oxidation state of +4 and promotes the formation of carbamates, carbonates, urea and its derivatives, polycarbonates, and polyethers, where OH−, H2O, amines, carbanions, olefins, alkynes, and dienes have been reacted as reagents with CO2. Reactions that produce carboxylates from CO2 are thermodynamically favourable. Carbon product formation such as methanol, carbon monoxide, formaldehyde, methane, and hydrocarbons from CO2 require higher energy, and thus kinetic control to steer away from the thermodynamically favoured products. Kinetically, to obtain CO2 reduced products with a change of oxidation state in the carbon atom requires more energy. To achieve the reduction of CO2, the reagent should bend CO2 to overcome the first energy barrier and to achieve subsequent reduction steps. Preorganised reducing agents kinetically favour the challenging reduction of CO2 beyond carbonate or formate.
To overcome the energy barrier, external sources of energy (such as electrochemical, photochemical or thermal energy) are typically required. If value-added products are obtained at higher cost compared to the cost of CO2 capturing and natural fuels, then the process would not be economical and could not be applied industrially.8
Although CO2 has been reduced to obtain chemicals and fuels, the current use of CO2 in chemical synthesis is limited owing to the high thermodynamic stability of CO2 that has to be overcome.9 To control the energy barrier in reduction processes of CO2, metals are being used that are often rare and high cost materials, indeed transition metals are being explored making the process efficient and cost effective. The conversion of CO2 to value-added products is highly desirable, yet the inert and highly energetically stable nature of this small molecule makes this a challenging task.10 CO2 is a linear and an apolar molecule (dipole moment, μ = 0), despite its two polar C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds and is ambiphilic in nature. The length of the CO bond in CO2 is 116.3 pm, shorter than the approximate 140 pm bond length of a typical single C–O bond, and shorter than most other CO double bonds, such as carbonyls.11 It offers two reaction sites, an electrophilic site at the carbon (Lewis acidic centre) due to the low-lying empty antibonding π* orbital (LUMO, lowest unoccupied molecular orbital), and two nucleophilic sites at the oxygen atoms (Lewis basic character) due to an available pair of valence electrons (HOMO, highest occupied molecular orbital) (Fig. 1). Carbon dioxide has an electron affinity (Eea) of about −0.6 eV and a first ionisation potential (IP) of about +13.8 eV which makes it a better electron acceptor than electron donor. Overall, a high energy of about 750 kJ mol−1 is required to break the CO bond. Upon activation, the molecule will distort from its linear sp-hybridised geometry to a sp2-hybridised carbon centre with concurrent elongation of the CO bond and a change in its molecular energy.12
O bonds and is ambiphilic in nature. The length of the CO bond in CO2 is 116.3 pm, shorter than the approximate 140 pm bond length of a typical single C–O bond, and shorter than most other CO double bonds, such as carbonyls.11 It offers two reaction sites, an electrophilic site at the carbon (Lewis acidic centre) due to the low-lying empty antibonding π* orbital (LUMO, lowest unoccupied molecular orbital), and two nucleophilic sites at the oxygen atoms (Lewis basic character) due to an available pair of valence electrons (HOMO, highest occupied molecular orbital) (Fig. 1). Carbon dioxide has an electron affinity (Eea) of about −0.6 eV and a first ionisation potential (IP) of about +13.8 eV which makes it a better electron acceptor than electron donor. Overall, a high energy of about 750 kJ mol−1 is required to break the CO bond. Upon activation, the molecule will distort from its linear sp-hybridised geometry to a sp2-hybridised carbon centre with concurrent elongation of the CO bond and a change in its molecular energy.12
 | ||
| Fig. 1 Molecular properties of carbon dioxide. | ||
Classical activation of CO2 by nucleophilic attack at the carbon atom can be achieved using bases,13 transition metals,14 or by one electron reductions15 to ultimately generate acetates, carbamates, ureas, bicarbonates, oxalates, formates, or carbon monoxide, amongst other products (Fig. 2). Further advances in research have been able to achieve value added carbon products by reducing CO2 to products such as methanol, methane, or higher carbon chains.16
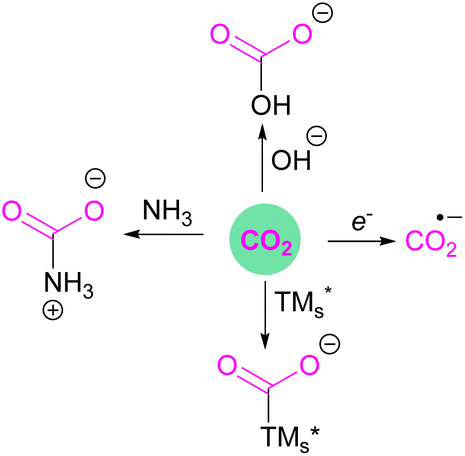 | ||
| Fig. 2 Classical activation reactions of carbon dioxide. | ||
Frustrated Lewis pairs (FLPs)17 are mixtures of a Lewis acid and a Lewis base, that, because of steric hindrance, cannot combine to form a classical adduct. FLPs can perform efficient chemical transformations without losing the individual properties of the FLP system.18 This feature also enables the activation of small molecules including CO2 and is now well-explored in the literature in several reviews.19 Herein, we cover all major developments made using p-block elements and transition metals to activate CO2 with FLP systems with a particular emphasis on more recent reports. In this review we will cover stoichiometric as well as catalytic processes including theoretical efforts to understand the mechanism of CO2 activation and reduction using FLPs.
Frustrated Lewis pairs (FLPs)
In the classic model of Gilbert Lewis (Fig. 3a), a Lewis base with an electron pair in the HOMO donates electron density to the LUMO of the Lewis acid by forming a dative bond.20 This process provides a HOMO of lower energy with a stabilised donor acceptor adduct and quenches the reactivity of both, the Lewis acid and base. A deviation to the classical model was observed after the augmented work reported by Brown,21 in which no adduct formation occurred between BMe3 and 2,6-lutidine, and later Wittig,22 Tochtermann,23 Piers24 and Oestreich.25 Stephan and co-workers coined the chemical term “frustrated Lewis pair” (FLP) that exists with unquenched acidity and basicity in a combination of a sterically hindered Lewis acid and Lewis base (Fig. 3b).26 This inhibition of adduct formation allows for the HOMO of the Lewis base and the LUMO of the Lewis acid to effect non-classical reactivity.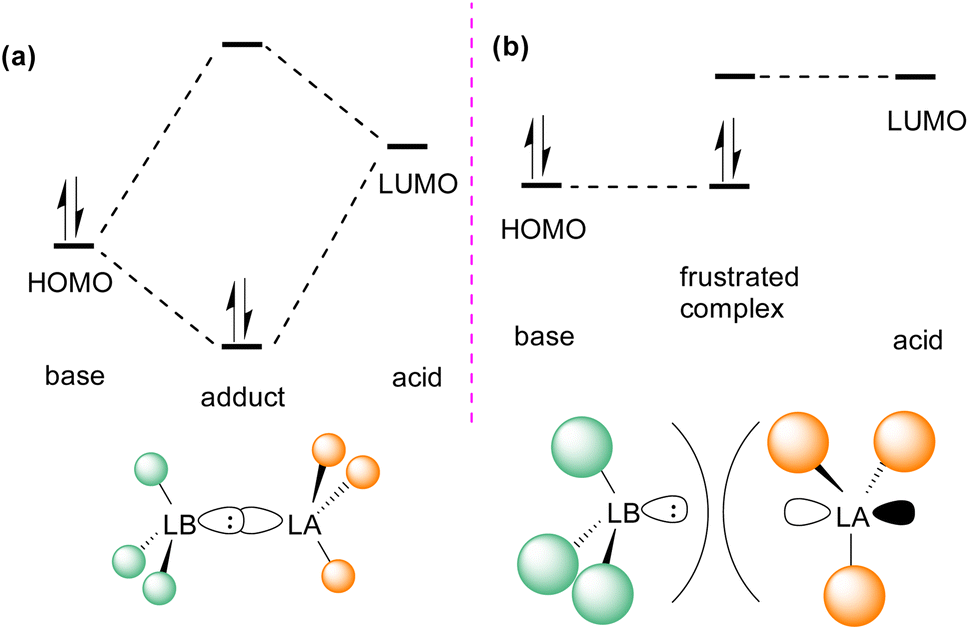 | ||
| Fig. 3 Frontier molecular orbital presentation of (a) a classic Lewis acid–base adduct and (b) a frustrated Lewis pair. LA = Lewis acid; LB = Lewis base. | ||
The pioneering work of splitting dihydrogen heterolytically with the FLP tBu3P/B(C6F5)3 demonstrated that the unquenched reactivity of FLPs could be applied to the activation of small molecules.27 Since then, various FLP systems have been investigated to activate a variety of small molecules. Two types of FLP systems are typically considered: intermolecular or intramolecular (Scheme 1). Intermolecular FLPs are systems where the Lewis acid and Lewis base are two individual molecules that interact through secondary London dispersion interactions to bring the Lewis acid and base together where small molecules insert into the cavity of the FLP such as the combination of tBu3P and B(C6F5)3. In intramolecular FLP systems, the Lewis acid and Lewis base are combined in one molecule by a covalent linker. An example of an intramolecular FLP system is the phoshinoborane 1 shown in Scheme 1, where the Lewis acidic boron centre and the Lewis basic phosphorus centre are separated by an aryl ring. These molecules are also able to heterolytically cleave the small molecule A–B (Scheme 1). When the FLPs heterolytically cleave the bonds in small molecules, the Lewis base donates its electron pair to the electron deficient fragment of the small molecule and the Lewis acid accepts an electron pair from the HOMO of the small molecule. This results in bond formation and an ionic/zwitterionic product. It has also been found that certain combinations of Lewis acids and bases in FLPs can lead to a transfer of one electron from the Lewis basic donor to a Lewis acidic acceptor generating a reactive frustrated radical pair (FRP). This FRP can react in a homolytic way with small molecules (Scheme 2).28
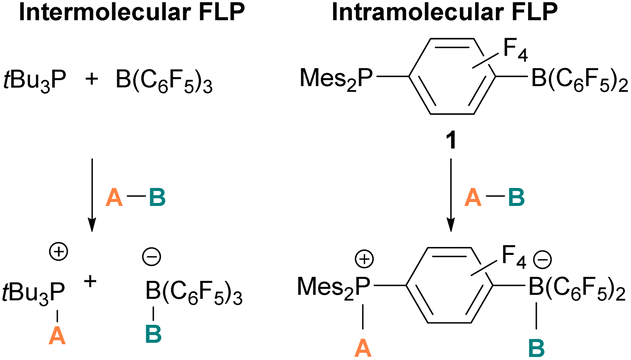 | ||
| Scheme 1 Cleavage of molecule A–B with intermolecular and intramolecular FLPs. Mes = Mesityl. | ||
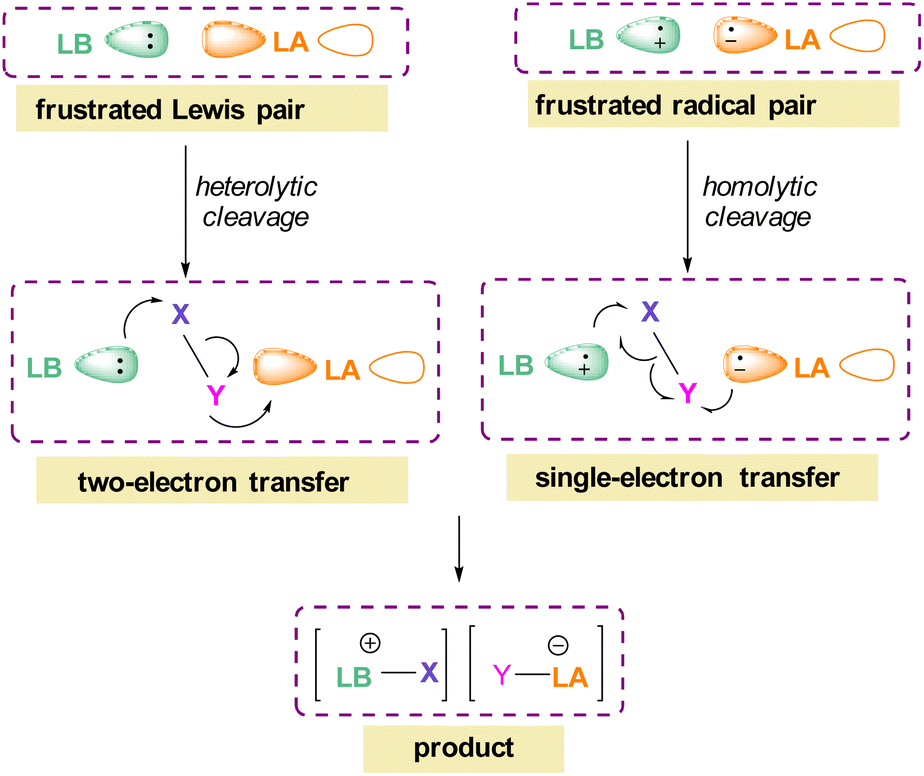 | ||
| Scheme 2 FLP reactivity with small molecules via heterolytic (two e− transfer) and homolytic cleavage (single e− transfer). | ||
Mechanistic aspects of FLP CO2 reduction
Amongst the small molecules activated by FLPs, CO2 has been well-studied owing to the importance of discovering new CCSU processes. Two mechanistic pathways are proposed for the capture and activation of CO2 by FLPs thus far (Scheme 3). One is a concerted mechanism and the other is a two-step process. Two computational models have been explored in the tBu3P/B(C6F5)3 FLP system to study the mode of CO2 activation. In the concerted mechanism, the reactants (FLP and free CO2) and the CO2-FLP adduct is formed by a single transition state (TS) in which the LB–C and LA–O bonds are formed simultaneously.29 Conversely, for the two-step process, when the CO2 moves closer to the FLP system, the P–C bond is formed first, followed by the formation of the B–O bond to give the final CO2-FLP adduct.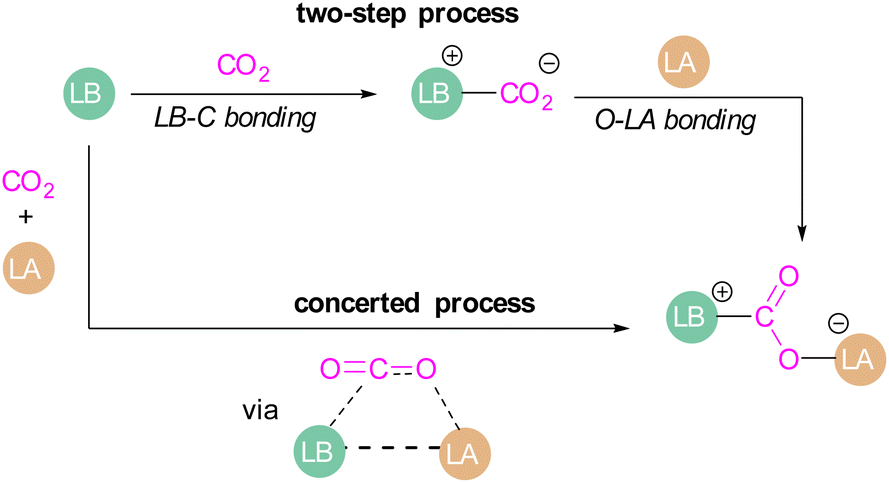 | ||
| Scheme 3 Mechanistic aspects of FLPs in their reaction with CO2. | ||
It is well-documented that the solvent is important to stabilise the final zwitterionic products.30 Liu and co-worker31 led a mechanistic study in the solid-state utilising density functional theory (DFT) simulations, in which they analysed the separate roles of the Lewis acid and base without the presence of a solvent. The authors found that the reaction proceeds in a two-step process where CO2 initially enters the cavity of the tBu3P/B(C6F5)3 FLP. The carbon atom of CO2 then interacts with the phosphorus atom and an oxygen atom interacts with boron leading to a reduction in the O–C–O angle of CO2 to 167.8°. This means that the CO2 species is bent although there are no chemical bonds formed. They believe that this is due to a weak interaction between CO2 and the FLP, where CO2 interacts with crystal fields in the solid state created by the FLP pair. In the solution state, the crystal fields would be replaced by solvent interactions. In studying the separate roles of the Lewis acid and base, the authors suggest that the combination of a strong Lewis acid and a weak Lewis base should be selected to make the CO2 activation thermodynamically feasible. This is due to the formation of the B–O bond being strongly exergonic while the formation of the P–C bond was deduced to be endergonic.
Other sources of energy such as light and/or electric current have been employed in other fields however, the most used source of energy for the activation of CO2 with FLP systems is heat and pressure of CO2.
However, in the FLP adduct of CO2, the CO2 molecule is bent, and a one-electron transfer could facilitate the reduction process. In a homogeneous system, the first electrochemical study was performed on the FLP-CO2 adduct for tBu3P-CO2-B(C6F5)3 (Scheme 4).32 Electrochemically, when an electron is added to tBu3P-CO2-B(C6F5)3, a change in the bond lengths was observed. The carbon oxygen CO bond length increased by 0.05 Å in and the C–O bond length decreased by 0.03 Å. The B–O and P–C bonds both decreased in length by 0.06 Å and 0.01 Å, respectively. Overall, it was observed that addition of electrons to the CO2 adduct tBu3P-CO2-B(C6F5)3 first generated intermediate [tBu3P-CO2-B(C6F5)3]−, then reduced CO2 to CO also generating tBu3P, and [(HO)B(C6F5)3]−. In this system, the intermediate [tBu3P-CO2-B(C6F5)3]− can also react with the solvent THF causing dimerisation.
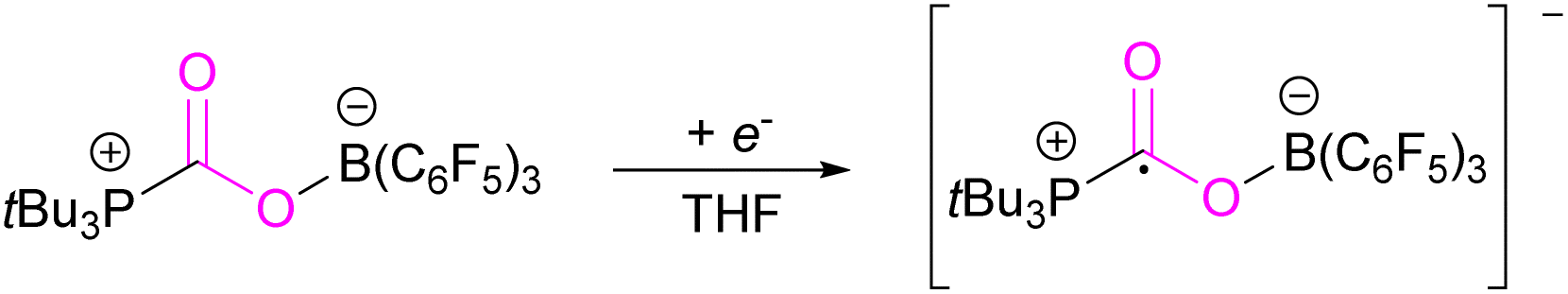 | ||
| Scheme 4 Electrochemical reduction of FLP adduct tBu3P-CO2-B(C6F5)3. | ||
To obtain a controlled reduction of CO2, FLPs in the solid-state have been explored based on the phenomenon of adsorption, activation, and evolution pathways of CO2. For example, Yan and co-workers created a stable FLP system for the activation of carbon dioxide.33 Their system involves a composite material of zinc and tin having different electronegativities. The Lewis pairs first capture and stabilise protons and then selectively activate CO2. Here the zinc oxide surface captures protons and acts as a Lewis base while the tin acts as Lewis acid. The two-electron reduction with two protons start the reaction for CO2 activation and finally resulted in the formation of formic acid.
In this review we will cover FLP mediated activation and reduction of CO2 that has been explored to achieve value-added carbon products. This will include, several inter- and intramolecular FLPs systems which have been designed and developed utilising both p- and d-block elements acting as a Lewis acid in combinations with p- and d-block elements acting as a Lewis base. Each section and sub-sections will detail the different systems developed with stoichiometric and catalytic quantities of FLPs, and will be ordered by the periodic group of the Lewis acid to give structure to this review. The mechanistic and computational insights will be discussed where relevant.
Group 13 Lewis acids
Borane/phosphine FLPs for CO2 activation
Boron is by far the most explored Lewis acidic element in FLPs for CO2 activation. Different Lewis base partners such as phosphrous, nitrogen, carbon as a carbene and metals have been explored in combination with the boron Lewis acid.Many of the first FLP systems for CO2 activation involved phosphorus as the Lewis basic component. Early FLPs utilised in CO2 activation comprised of a phosphine and borane that could reversibly bind and release CO2 including the intermolecular FLP tBu3P/B(C6F5)3 and 2 (Scheme 5).
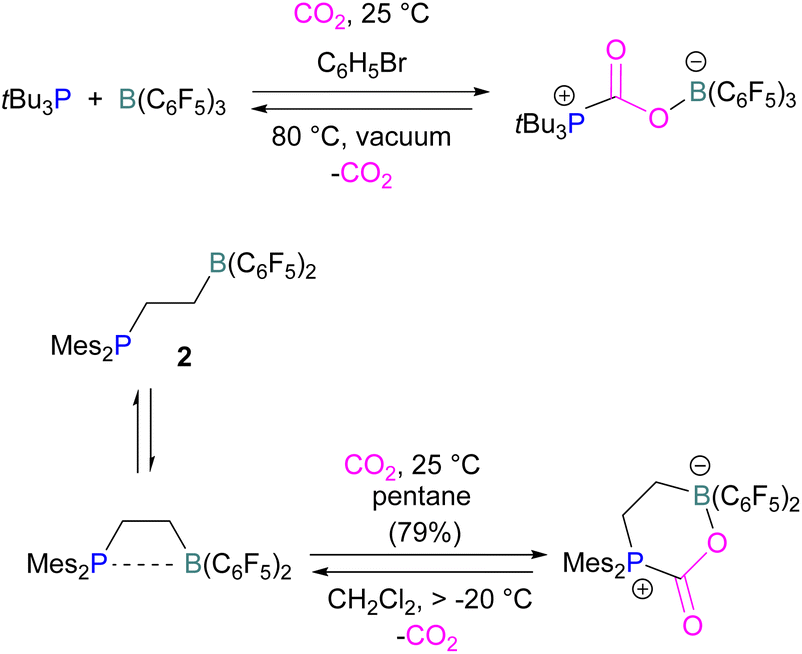 | ||
| Scheme 5 First inter- and intramolecular FLPs utilised in CO2 activation. | ||
At the time, these systems offered rare examples of metal-free CO2 sequestration.34 Theoretical investigations show the mechanism proceeding by simultaneous formation of P–C and O–B bonds from thermochemical computed data (B97-D/TZVPP’, B2PLYP-D/TZVPP’, and B2PLYP-D/QZVP(-g, -f) levels of theory). tBu3P reacts with B(C6F5)3 at room temperature and under 1 bar of CO2 forms the desired stable product tBu3P-CO2-B(C6F5)3, which upon heating at 80 °C under vacuum releases the CO2 molecule and regenerates the starting FLP mixture. Calculations for the formation of tBu3P-CO2-B(C6F5)3 show that the overall reaction is exothermic. Privalov and co-workers calculated several energy pathways for CO2 activation.35 After these first reports of non-metal based inter- and intramolecular FLP-mediated reversible CO2 activation, the scientific community explored a range of new FLPs for CO2 activation, as shown in Fig. 4. Several other phosphine bases and boron acids have been used in the intermolecular system including iPr3P, XPhos and Mes2EtP, and B(p-C6F4H)3, and B(R)(C6F4H)2, (R = hexyl, Cl, cyclohexyl, norbornyl, Ph).36,37 More complex boranes bearing functionalised substituents with cyclic structures were also tested with tBu3P to trap CO2 generating adducts 3 and 4.38
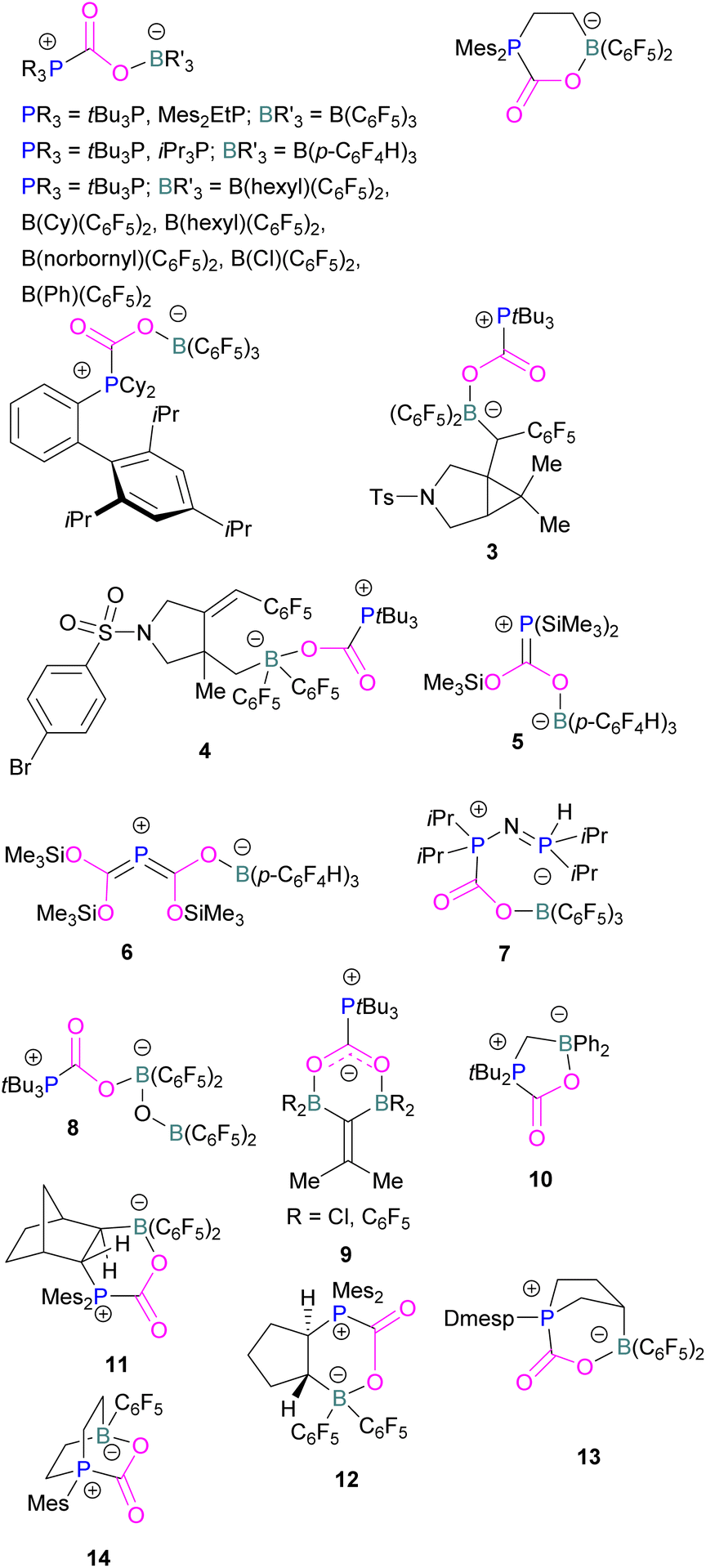 | ||
| Fig. 4 Examples of FLP adducts of boranes with a phosphorus Lewis base. Dmesp = Dimesitylphenyl. | ||
Interestingly, when (Me3Si)3P was utilised as a Lewis base instead of tBu3P, with B(p-C6F4H)3, silyl migration was observed in the final adducts 5 and 6 (Fig. 4 and Scheme 6). tBu3P and (Me3Si)3P with CO2 in pentane at room temperature initially yields the expected adduct (Me3Si)3P-CO2-B(p-C6F4H)3. Subsequently, silyl migration from phosphorus to oxygen forms a stable compound which can be better represented as the zwitterionic compound 5. The same starting Lewis acid and base can also react with two equivalents of CO2 in dichloromethane (CH2Cl2) at room temperature for 24 h, providing silyl migrated product 6. Compound 6 can also be obtained from 5, when 5 is treated with CO2 in CD2Cl2 at room temperature for 24 h (Scheme 6).39 Kemp and co-workers on the other hand, investigated the phosphine base bis(di-i-propylphosphino)amine with B(C6F5)3 which formed the expected 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct 7. The crystal structure of 7 shows that H-isomerisation took place with a migration of the proton from nitrogen to phosphorus (Scheme 7).40
:1 adduct 7. The crystal structure of 7 shows that H-isomerisation took place with a migration of the proton from nitrogen to phosphorus (Scheme 7).40
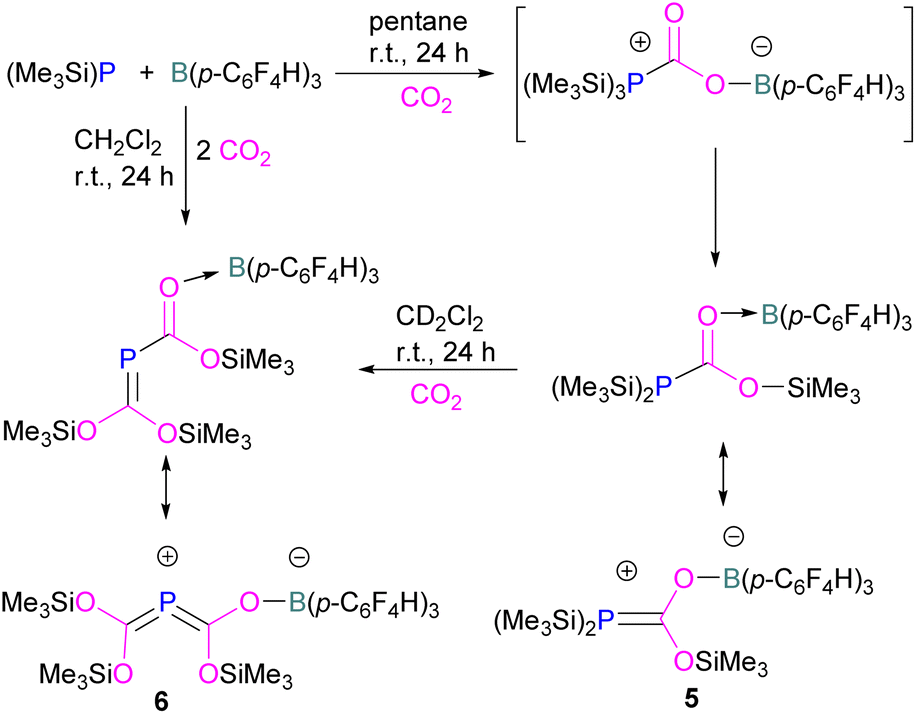 | ||
| Scheme 6 Silyl migration in the (Me3Si)3P and B(p-C6F4H)3 FLP. | ||
 | ||
| Scheme 7 H-isomerisation with a migration of the proton from nitrogen to phosphorus. | ||
Stephan and co-workers have expanded the borane scope to explore the reactivity of bis-boranes to trap CO2 with tBu3P. It was also found that, 1,1-bis-(C6F5)2BOB(C6F5)2 binds with CO2 in a monodentate manner generating 8, whilst bis-boranes of type (R2B)2CCMe2, where R = Cl or C6F5, provide a bidentate chelation of CO2 to obtain a unique type of heterocyclic compounds 9.41 The chelation of CO2 by the two B-centres in 8 was restrained due to steric crowding as well as a significant π-character in the B–O bonds, which was evident from the relatively large B–O–B bond angle of 139.5(2)°. Whereas, in 9 B–C–B angles of 117.3(2)° (R = Cl) and 121.2(2)° (R = C6F5) show a six membered planar structure.
In intramolecular systems, when the Lewis base and acid are sufficiently aligned in a geminal fashion, an increase in reactivity is observed as seen in the formation of adduct 10.42 Another intramolecular FLP with a norbornane structure with a vicinal designed FLP was utilised to trap CO2 to obtain adduct 11.43 Similarly to the vicinal FLP in adduct 11, an FLP based on cyclopentane with trans-1,2-substituents was explored to form 12.44 Other intramolecular B/P FLP systems include an active FLP borylated tetrahydrophosphole which yielded adduct 13,45 and a cyclic six membered FLP with 1,4-phosphane/borane substituents which undergoes an addition reaction with CO2 to form adduct 14. Erker and co-workers observed that heating 14 in n-heptane at 80 °C for 15 min under CO2 converts 14 to its cyclotetrameric macrocyclic oligomer 15. Tetramer 15 is unstable in solution, even in a CO2 atmosphere it slowly converts back to the monomer 14 (Fig. 5).46
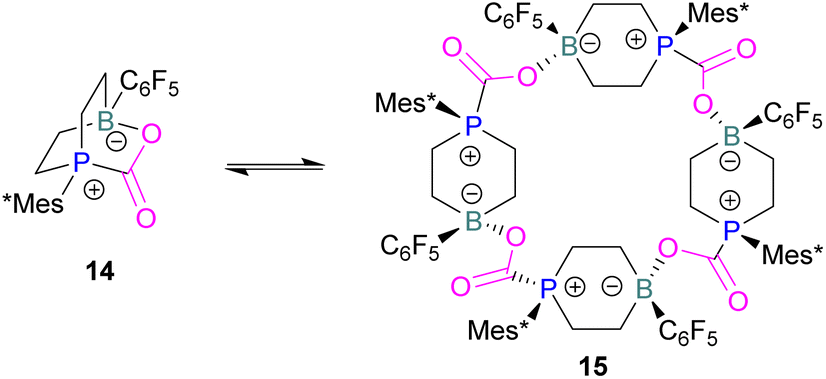 | ||
| Fig. 5 FLP adducts of boranes with P-base. Mes* = 2,4,6-Tri-tert-butylphenyl. | ||
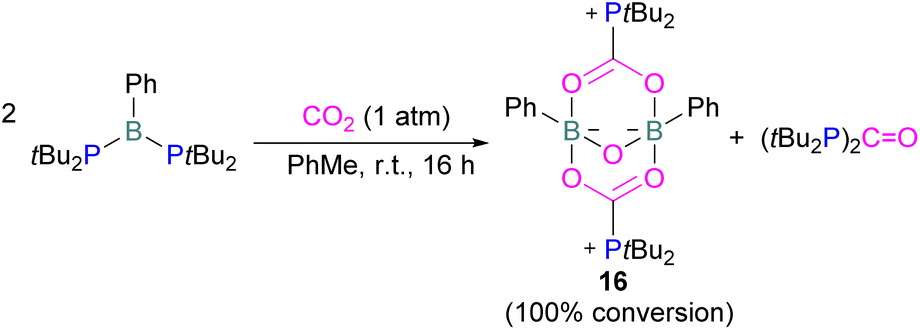
Szynkiewicz and co-workers reported the phosphinoboration and diphosphination of CO2. In 2019, they published the first report of catalytic (with respect to the borane) diphosphination of CO2 with a diphosphane/boron FLP. CO2 was inserted into the relatively weak P–P bond (Scheme 8).47 Furthermore in 2019, they reported the use of diaminophosphinoboranes to phosphinoborate CO2; though not sterically frustrated, this compound still exhibits FLP-like reactivity (Scheme 8, bottom).48 More recently the same group built on this work, reporting the reaction of CO2 (among several other small molecules) with a diphosphinoborane B(PtBu2)2Ph to yield a diphospha-urea and a bicyclic diboroxane 16 (Scheme 9). The reaction proceeds by CO2 insertion into a single B–P bond, elimination of (tBu2P)2CO to give phenyl oxoborane PhBO. Reaction of this species with a further equivalent of the parent diphosphinoborane and 2 equivalents of CO2 gives the product 16. While stable under N2, the product decomposes with loss of CO2 and diphospha-urea to give triphenylboroxine (PhBO)3.49
 | ||
| Scheme 8 Diphosphination (top) and phosphinoboration (bottom) of CO2. | ||
 | ||
| Scheme 9 Synthesis of phenyl oxoborane. | ||
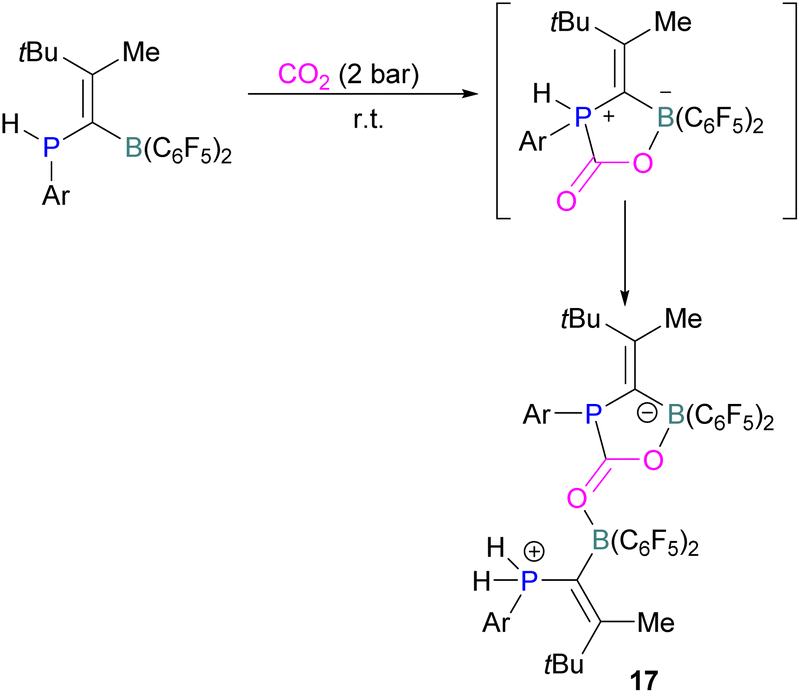
Kessete and co-workers published a computational study evaluating a number of intramolecular phosphine/borane catalysts for CO2 reduction. One finding was that, though electron-withdrawing substituents on borane like fluorine stabilise the CO2-FLP adduct, they also destabilise the transition state (TS), increasing activation energy. Fluorination of the substituents of the phosphorus reduces its basicity and so destabilises both the transition state and the adduct formed. This highlights the importance of tuning the Lewis acidic and basic sites of FLPs to achieve stabilised transition states, but that are also Lewis acidic, or Lewis basic enough centres to bind with CO2.50 Jian and co-workers reported in 2017 that geminal vinylidene-bridged phosphorus/boron Lewis pairs could react with CO2 to give a phosphinodiborated product 17, as shown in Scheme 10. Interestingly, this geminal P/B is supported with an sp2 carbon, which is different from previous reports of geminal FLPs.51
 | ||
| Scheme 10 Reaction of geminal vinylidene-bridged P/B Lewis pair with CO2. | ||
Following activation of CO2, the subsequent transformations have been investigated initially stoichiometrically. Stephan and co-workers reported that the bis-borane, 1,2-C6H4(BCl2)2, forms an adduct with tBu3P and also shows FLP reactivity with CO2 to form the FLP-CO2 zwitterionic compound 18 (Scheme 11). Compound 18 is remarkably more stable, with respect to the loss of CO2, and no decomposition was observed even on heating to 80 °C for 24 h compared to the CO2 adducts obtained from FLPs tBu3P/B(C6F5)3 (loss of CO2 at 80 °C), Mes2PCH2CH2B(C6F5)2 (loss of CO2 at −20 °C), and bis-boranes Me2CC(BR2)2 where RCl, C6F5 with tBu3P (loss of CO2 at 15 °C). The chlorine atom in 18 bridges between the boron centres which enhances the Lewis acidity of the boron bound with the oxygen atom of CO2 and results in a stronger B–O bond making the adduct more thermally stable, than other discussed examples. Hence, the strength of the bond between the Lewis acid and the oxygen atom of CO2 plays a critical role in establishing reversibility, this can be induced by the addition of electron-withdrawing groups, such as Cl in 18. The species 18 was reduced by Me2NHBH3 followed by quenching with deuterated water (D2O) to obtain deuterated methanol (MeOD) as the final product. In another way, 18 was also reduced by [C5H6Me4NH2]/[HB(C6F5)2(C7H11)] (19) and quenched with D2O again yielding H3COD (Scheme 11). Here, two Lewis acidic boron sites are available, and bridging of a chlorine atom between the two stabilises the zwitterionic adduct.52
 | ||
| Scheme 11 Stoichiometric reduction of FLP-CO2 adduct 18. | ||
There remain two major issues with tBu3P/1,2-C6H4(BCl2)2 that pose limitations for a catalytic cycle. The first issue is that H2 cannot be activated, so H2 surrogates such as Me2NHBH3 or [C5H6Me4NH2]/[HB(C6F5)2(C7H11)] were utilised as stoichiometric reductants. Secondly, the boron centre in this FLP is more oxophilic, so the last step required quenching with D2O to cleave the B–O bond.
In another stoichiometric system, O'Hare and co-workers synthesised a series of FLPs based on Lewis acid {C6F4(o-C6F5)}3B and (C6Cl5)3B with trialkylphosphines as Lewis bases (Scheme 12). The idea of synthesising these FLPs was to achieve a weaker B–O bond to facilitate the cleavage of B–O bond upon reduction and potentially generate a catalytic system. The steric congestion factor was applied as steric bulk at the ortho position alone could decrease the B–O bond strength.53 The synthesised FLPs were exposed to H2 to form FLP-H2 as activated salts of the type [R3P–H][H–BR′3]. These salts were then exposed to CO2 (1 atm) to obtain formatoborates of type 20 in the presence of toluene at 140 °C for 24 h using Young's tap NMR tubes. The formatoborates 20 could also be prepared independently from the reaction of the FLP with formic acid in toluene at room temperature for 16 h (Scheme 12).54 The formatoborates 20 were subjected to H2 and heated to 140 °C for 16 h but were not reduced, instead decarboxylation of the formatoborates 20 occurred and hydride salts were formed with no further reductions.
 | ||
| Scheme 12 A series of FLPs consisting of C6F4(o-C6F5)}3B or (C6Cl5)3B with trialkylphosphines, and their reduction reactions. | ||
The higher stability of the formatoborates 20 and their decarboxylation at higher temperatures limited this FLP approach for a catalytic reduction of CO2.
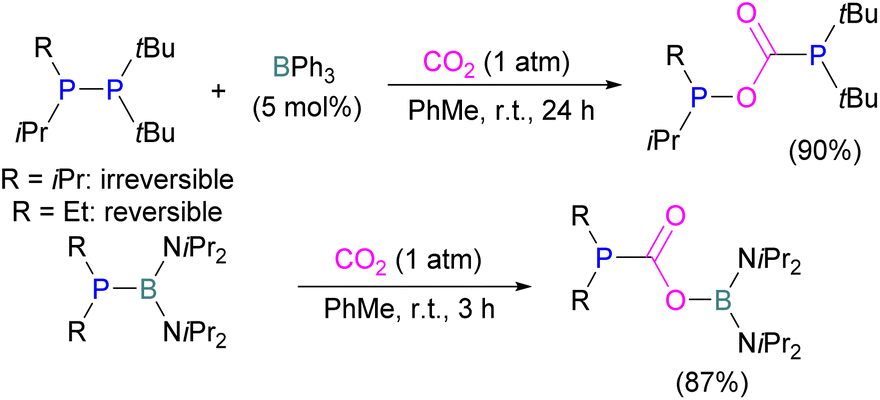
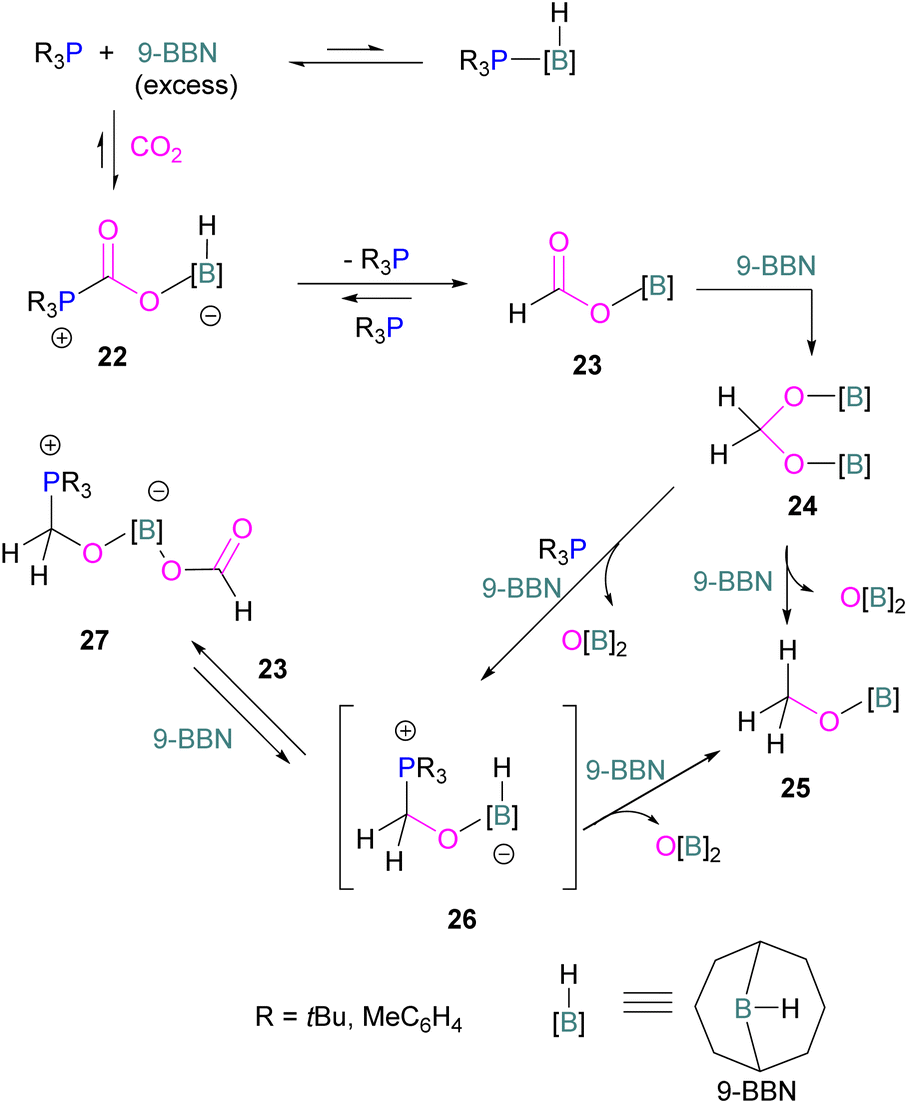
Following reports on stoichiometric reactions, the first catalytic reduction of CO2 with an organocatalyst FLP was explored by Fontaine and co-workers in 2013. They applied hydroboranes HBR2 [HBcat (catecholborane), HBpin (pinacolborane), 9-BBN (9-borabicyclo[3.3.1]nonane), BH3·SMe2 and BH3·THF] to produce CH3OBR2 or (CH3OBO)3 following reduction of CO2. Upon hydrolysis, CH3OBR2 or (CH3OBO)3 yield methanol as the final product in up to 99% yield (Scheme 13) with high turnover numbers (TON > 2950) and turnover frequencies (TOF = 853 h−1). The intramolecular phosphino-borane catalyst 21 was found to be an efficient catalyst for this reaction.55 The same authors studied the mechanism of this hydroboration of CO2 with catalyst 21 using computational and experimental methods. It was found that an intramolecular FLP was involved in every step of the reduction and the simultaneous activation of both, the reducing agent and CO2, were the key to efficient catalysis in every reduction step.56 Furthermore, Fontaine and co-workers synthesised various phosphine–borane derivatives of catalyst 21 with different substituents on boron and phosphorus as shown in Scheme 13 (bottom). These were then tested for hydroboration of CO2 using HBcat or BH3·SMe2 to generate methoxyboranes. The most active species were derivatives with a catechol unit on boron. They also performed isotope labelling experiments and DFT studies and found that once the formaldehyde adduct was generated, the CH2O moiety remained on the catalyst system. The lowest energy barriers were found for concerted activation of catecholborane by the Lewis base and of CO2 by the Lewis acid. The results show higher potency of “O” for the activation of hydroboranes than “P”.57 Overall, FLP 21 acted as an efficient catalyst because of two important features: Firstly, 21 did not form an adduct with CO2, as seen previously with most FLPs that formed a stable CO2 adduct. Exposing 21 to 1 atm of CO2 at room temperature resulted in no spectroscopic change of the solution (by 1H, 31P, and 11B NMR spectroscopy). Also, species 21 remained monomeric in solution without any P–B interaction. Secondly, the CH2O moiety was released upon reduction from the catalyst and made 21 available for another reaction. Hence, the higher high turnover numbers and high turnover frequencies for 21. Later, Stephan and co-workers developed another catalytic method for the reduction of CO2 using 9-BBN as a reducing agent and phosphine as a catalyst (Scheme 14). The reaction proceeds via an FLP-type CO2 activation intermediate 22 and the reduction products include boron-bound formate species, 23, the diolate-linked compound 24, and methoxide product 25. Intermediate 26 could be transferred to 25 in the presence of 9-BBN and to 27 in the presence of the boron-bound formate species 23. Derivatives of 27 were isolated and confirmed with single crystal X-ray diffraction analysis. With 0.02 mol% of tBu3P, product 25 is obtained in 98% yield at reaction temperature 60 °C. In the best scenario, the catalyst tBu3P provides 5556 turnovers of hydride transfers to CO2 and a TOF of 176 h−1.58
 | ||
| Scheme 13 Catalytic performance of 21 in reduction of CO2 for methanol synthesis (top), and different FLP catalysts tested (bottom). | ||
 | ||
| Scheme 14 Phosphine catalysed CO2 reduction with 9-BBN. | ||
Instead of forming a classical adduct of tBu3P and 9-BBN, this system showed an FLP-type CO2 activation and subsequent hydride transfer from boron to the carbonyl carbon in 22, releasing tBu3P for the next cycle and hence this system worked catalytically for the reduction of CO2.
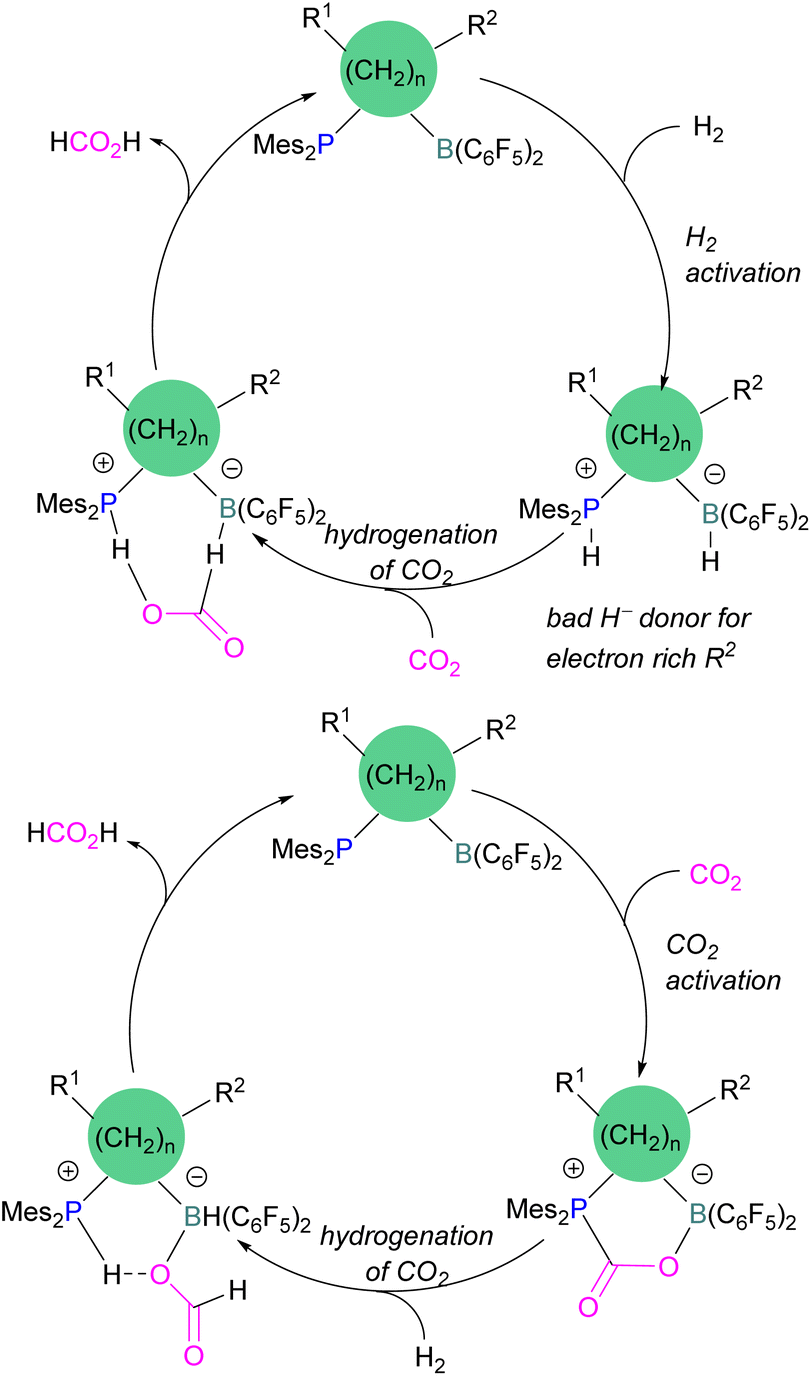
Dang and co-workers reported a theoretical study on a catalytic mechanism for computationally designed bridged P/B FLPs in the activation of H2 and CO2. They found that the reaction follows a one-step concerted mechanism with small reaction barriers (14.8–24.0 kcal mol−1). Among the computationally designed bridged FLPs, some were found to successfully reduce CO2 with molecular hydrogen in two feasible pathways. The first pathway follows immediate hydrogenation of CO2 after H2 activation (Scheme 15, top), the second follows CO2 activation first, then metathesis of H2 followed by reductive elimination (Scheme 15, bottom). Both catalytic cycles provide the product HCO2H from the reduction of CO2. From all computationally designed bridged FLPs, straightforward H2 activation takes place with those that do not have electron donating substitutions on the B's adjacent carbon site, or have a long chain between the B and P.59
 | ||
| Scheme 15 Two different calculated reduction cycles for CO2 in bridged FLPs. | ||
Xanthene FLPs were computationally investigated using DFT methods [level of theory: B3LYP-D3/6-311+G*(*)//M06-2X/6-31G*(*) in bromobenzene], and their reduction of CO2 was modelled as shown in Scheme 16. Differently substituted xanthene backbones were investigated, showing that more rigid backbones have lower activation energies for CO2 hydrogenation.
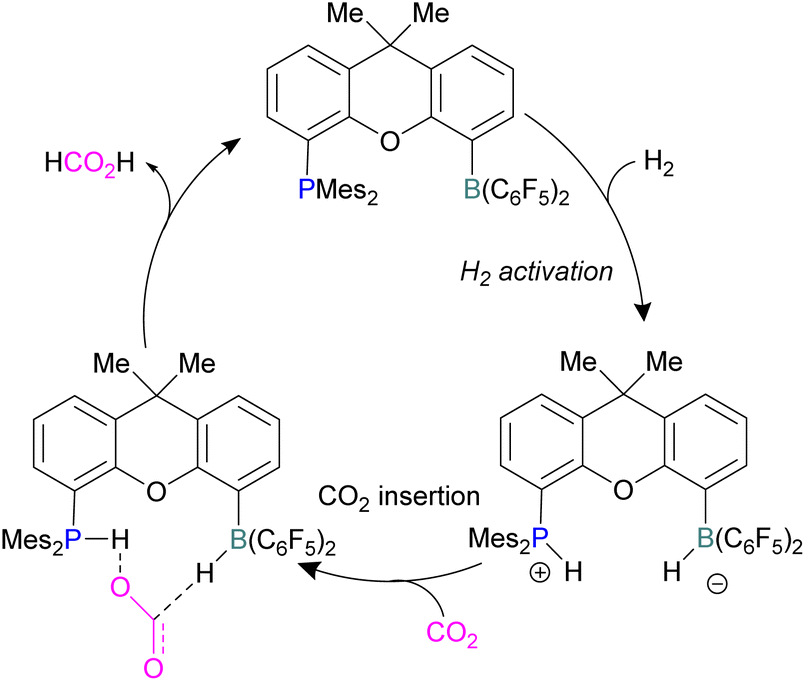 | ||
| Scheme 16 Xanthene FLPs mediated catalytic reduction of CO2 into formic acid. | ||
The formation of P/B–H in the first step was shown to be exergonic and this first intermediate is the catalyst resting stage.60 The hydride transfer from boron to the carbonyl carbon of CO2 produces formate and subsequent protonation resulted in the formation of formic acid bringing the xanthene FLP into the next cycle for the CO2 reduction.
The incorporation of FLPs into polymers has also seen some success in CO2 activation. Shaver and co-workers explored the first use of polymeric FLPs to catalyse the incorporation of CO2 into cyclic ethers for the formation of cyclic carbonates and showed good selectivity (Scheme 17, top). Different phopshines and boranes were explored as the Lewis base and acid in the polymer (Scheme 17, bottom). These poly(FLPs) can easily be recovered and reused after the reaction, however the efficiency of the catalyst gradually decreases due to partial phosphine oxidation and increased crosslinking.61
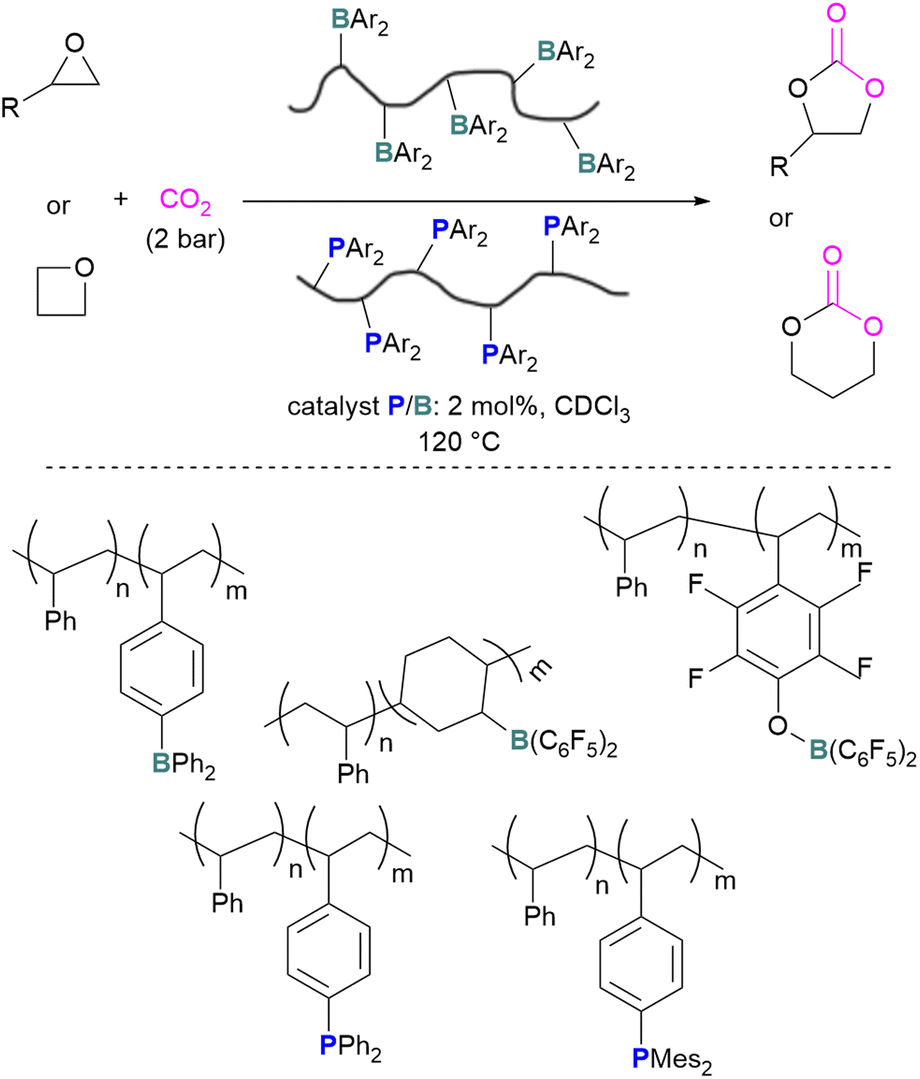 | ||
| Scheme 17 Polymeric FLP-catalysed reaction of ethers and CO2 for cyclic carbonate formation (top), and polymers used (bottom). | ||
Yan and co-workers developed CO2-responsive dynamic gel system based on an FLP for the first time (Scheme 18). Here, CO2 can be regarded as a “gas glue” which crosslinks the Lewis acidic and Lewis basic sites and forms a new type of a FLP network. The trapped CO2 FLP network undergoes reversible release of CO2 upon heating at >60 °C.
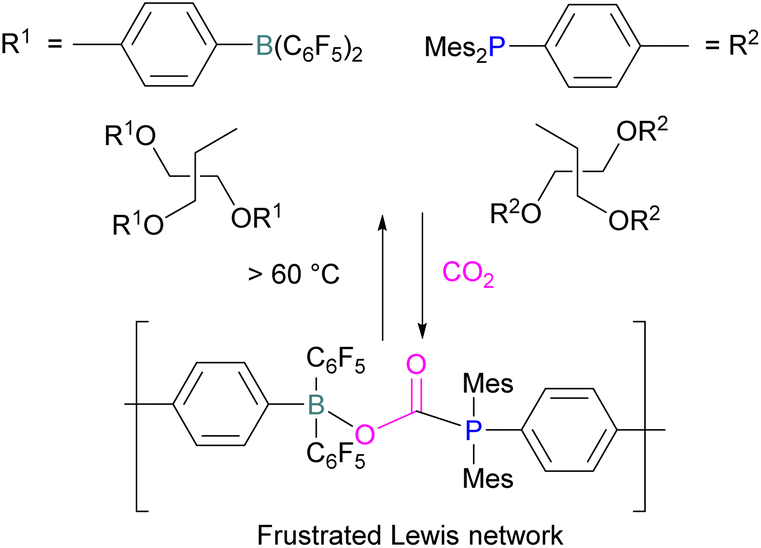 | ||
| Scheme 18 CO2-responsive dynamic gel system based on a FLPs. | ||
The authors found that CO2-bridging crosslinks in the network are dynamic covalent linkages, which provides the gel with unique gas-tuneable viscoelastic, mechanical, and self-healing characteristics.62 The authors showed that the same (–B–CO2–P–) poly-FLPs are efficient catalysts in transforming amine substrates to formamide derivatives using the CO2 poly-FLP as the starting point. Various amines were screened and the yields for the formamide products were in the range of 41–99% with TON = 420–14800. The highest TON of 14800 was observed for diethylamine giving 99% yield of the corresponding diethylformamide product (Scheme 19).63 CO2 bridges the polymer chains and a CO2 -triggered micellisation was obtained. Addition of PhSiH3 and R1R2NH resulted in the desired formamide products and regenerated the polymer. After separation of the products re-micellisation of the polymers was performed with CO2 and a reusable catalytic system was established with a high turnover number.
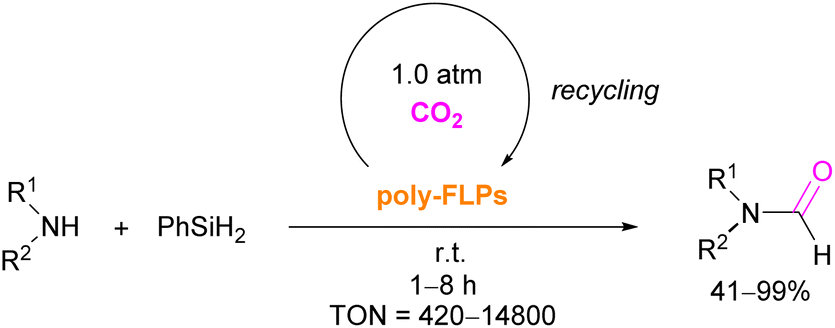 | ||
| Scheme 19 –B–CO2–P– poly-FLPs in formamide synthesis. | ||
Erasmus and co-workers developed efficient FLPs supported on silica nano-powder for CO2 capture.64 A series of CO2 adducts 28 were synthesised by reacting silica nanopowder supported Lewis acids and dissolved Lewis bases in pentane with CO2 (2 bar) which was passed through the pentane mixture at −65 °C. At room temperature these adducts were observed to be reversible in nature (Scheme 20, top).
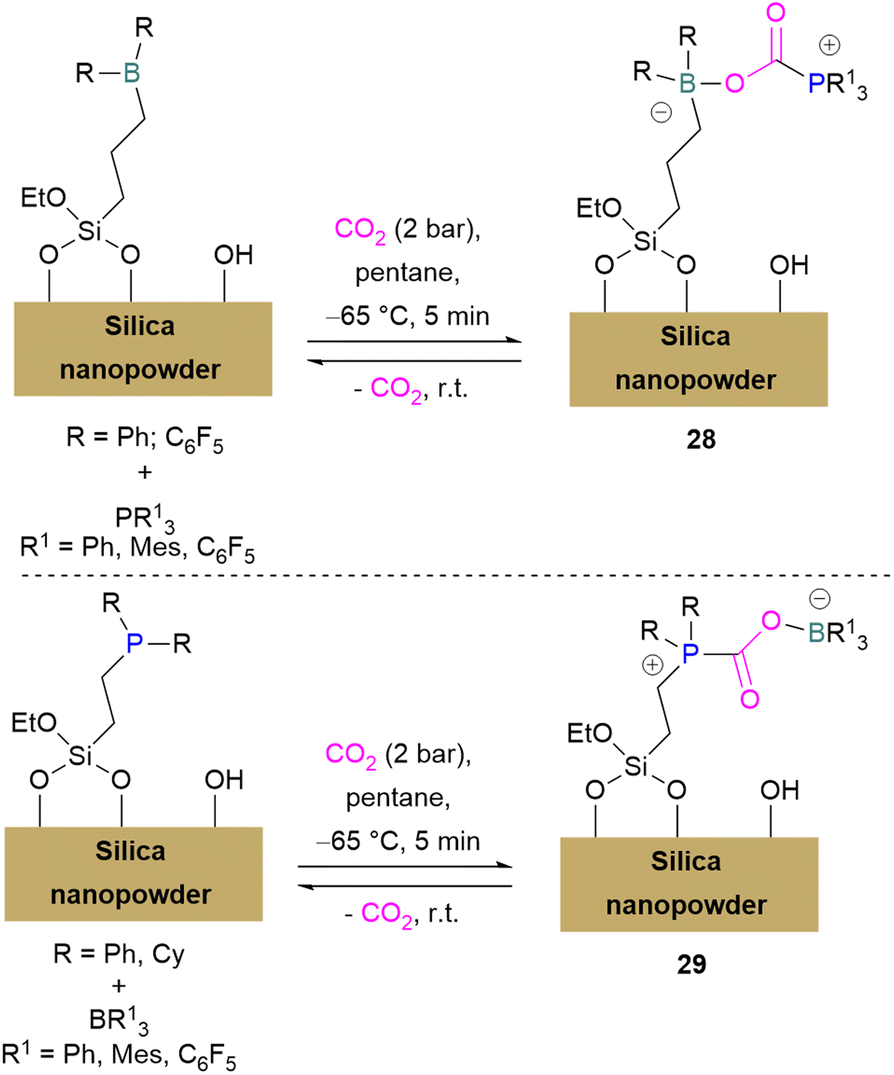 | ||
| Scheme 20 FLPs supported on silica nano-powder for CO2 capture. | ||
In a similar manner a series of silica nano-powder supported FLP-CO2 adducts 29 were synthesised from silica nano-powder supported Lewis bases and dissolved Lewis acids. The silica nanopowder supported FLPs were also explored for the conversion of CO2 to formic acid using hydrogen gas. Initially, the activation of H2 was done by the supported Lewis acid/bases with FLP partners to obtain [–BH]− [HP–]+ salts. Furthermore, introducing CO2 to these salts resulted in HCO2H and regenerated the FLPs. HCO2H is a protic polar molecule and has tendency to form O⋯H bonds with the free –OH functionalities on the silica. The main reason for the release of HCO2H from the system after reduction was the immobility of silica nanopowder bound Lewis acids (or Lewis bases) and so did not inhibit the activity of the FLPs.
In FLP systems having a P-basic centre, activation of CO2 proceeds via the formation of a P–C bond, and depending on the type of reactive acidic site a B–O, Al–O or Ga–O bonds are generated, often in a reversible manner. Alkyl phosphines iPr3P or tBu3P alone could not activate the CO2 molecule. It is known that the presence of a Lewis acidic component is not necessary for capturing CO2 when very electron rich P-nucleophiles are used.65
Borane/nitrogen FLPs for CO2 activation
In addition to phosphorus as a Lewis base in FLP-CO2 activation and reduction, there has been a wealth of FLPs described in the literature that use a nitrogen Lewis base in combination with a boron Lewis acid. A selection of the corresponding FLP-CO2 adducts are displayed in Fig. 6.66,67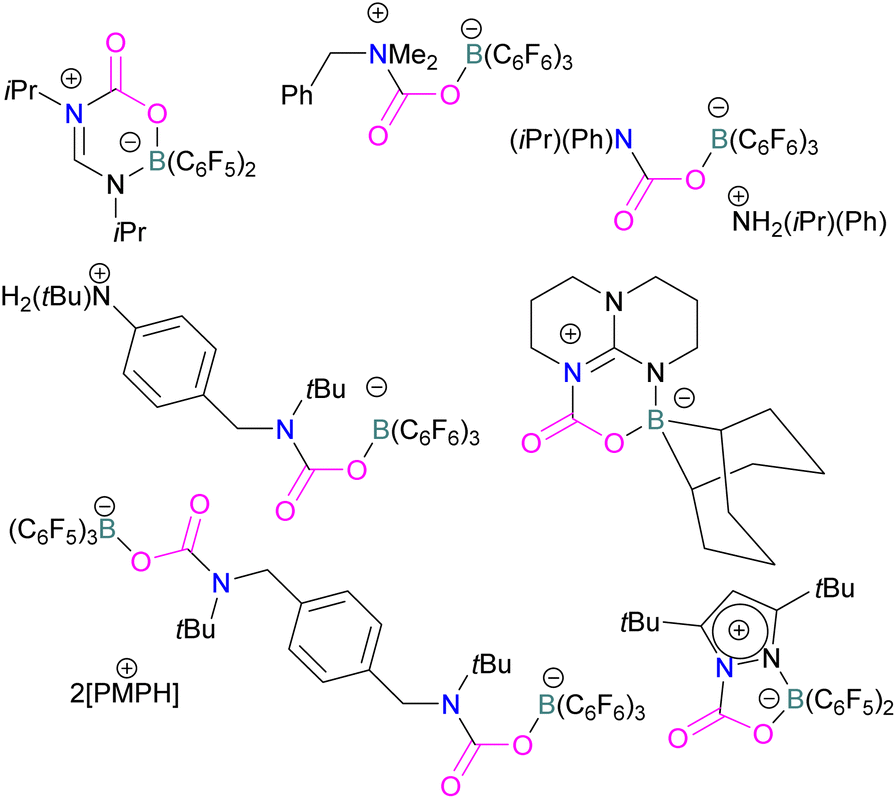 | ||
| Fig. 6 Structures of N–CO2–B adducts. PMP = 1,2,2,6,6-Pentamethylpiperidine. | ||
Stephan and co-workers reported a new synthetic method for making boron amidinates. The strained ring boron amidinate derivative 30 was prepared by reacting Piers' borane, HB(C6F5)2, with isopropyl carbodiimide. 30 was then successfully employed to trap CO2 incorporated into a new heterocycle 31 (Scheme 21). Compound 31 was fully characterised along with a single crystal X-ray diffraction structure.68 A theoretical study on the reaction of 30 with CO2 found a concerted addition mechanism.
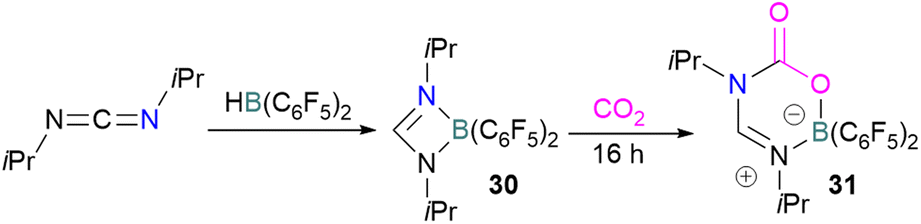 | ||
| Scheme 21 Synthesis of boron amidinates and reaction with CO2. | ||
In this reaction, the C-atom and O-atom of CO2 inserts into the B–N bond of 30 and forms the C–N and B–O bonds simultaneously. The frontier orbitals involved in the reaction mechanism were investigated as well as electric charge analysis and showed that results were consistent with charge transfer from HOMO of 30 to the LUMO of CO2.69 In another findings, Chattaraj and co-workers have studied this boron amidinate 30 as a bridged B/N FLP.70 They compared 30 with a P/B bridged system shown in Scheme 15 which describes two types of cycles. In this work, a similar process shows that CO2 hydrogenation with amidinate 30 leads to formic acid (HCO2H) as the final product. In the proposed mechanisms, either H2 is activated by the Lewis basic centre of the FLP, and CO2 is activated by the Lewis acidic centre of the FLP, or alternatively, CO2 can be activated by Lewis basic centre of the FLP and H2 by Lewis acidic centre of the FLP. In both cases, simultaneous activation of CO2 and H2 by a single TS was confirmed by Natural Bond Orbital (NBO) analysis and this TS is the rate determining step. From energy decomposition analysis (EDA), in the TS geometry it was found that electron density was donated from the HOMO of FLP to the LUMO of H2 and electron density from HOMO of H2 molecule to the LUMO of CO2.70 Stephan and co-workers have also utilised phosphinimines and B(C6F5)3 to explore FLP reactivity, Ph3PNR with B(C6F5)3 and CO2 produced the adducts 32 (R = Ph, C6F5) (Scheme 22).71 Figueroa and co-workers observed that (boryl)iminomethane 33 reacts intramolecularly with CO2 and forms a five-membered ring 34 in a 1,2-cyclohexyl shift (Scheme 23). Due to the 1,2-cyclohexyl shift, product 34 is stable and prevents the release of CO2, exhibiting irreversibility. Heating of the solution of 34 to 80 °C showed no release of CO2. Likewise, heating of the solid sample of 34 to 150 °C under vacuum did not display any CO2 release either.72
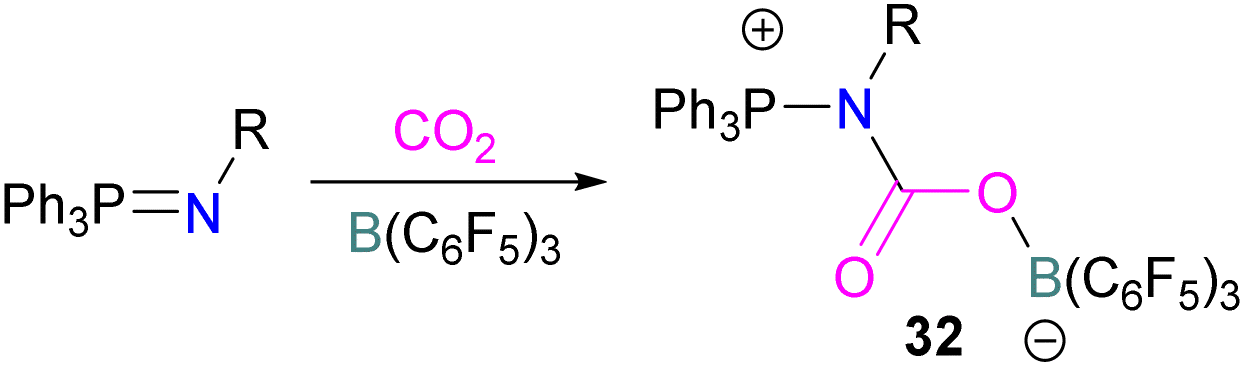 | ||
| Scheme 22 Activation of CO2 using phosphinimines and B(C6F5)3. | ||
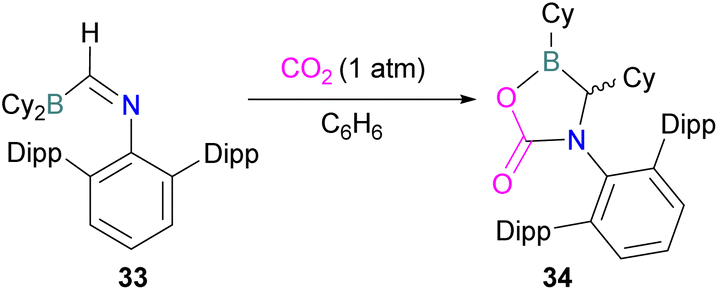 | ||
| Scheme 23 CO2 adduct of (boryl)iminomethane. Dipp = 2,6-Diisopropylphenyl. | ||
N/B CO2 adducts have also been used in subsequent stoichiometric transformations. It has been previously reported that TMP [2,2,6,6-(tetramethylpiperidine)] along with B(C6F5)3 splits H2 heterolytically and forms the ion pair [TMPH][HB(C6F5)3].73 O'Hare and co-workers utilised this ion pair [TMPH][HB(C6F5)3] to insert CO2 into the B–H bond forming a formatoborate complex 35 at elevated temperatures. Compound 35 can also be obtained from the reaction of TMP, B(C6F5)3 and HCO2H (Scheme 24). The structure of 35 was confirmed by single crystal X-ray diffraction analysis.74 The formatoborate complex 35 could be transformed to produce MeOH by applying more equivalent of ion pair [TMPH][HB(C6F5)3]. The formation of [(C6F5)3B–OH]− is an obstacle for this method to be developed into a catalytic transformation. A similar FLP system consisting of 2,6-lutidine/B(C6F5)3 has also been shown to split H2 heterolytically to form borohydride salt [(CH3)2C5H3NH][HB(C6F5)3].75 Mayer and co-workers applied this salt for the activation of CO2 at 4 atm pressure and at room temperature. The air-stable formatoborate complex 36 (Scheme 25) resulted and its structure was confirmed by X-ray diffraction analysis.76 Compared to 35, Mayer and co-workers observed that 36 on heating to 80 °C resulted only in decomposition instead of transforming to other CO2 reduced products. This restricts the method to obtain only formatoborate complex 36 in a stoichiometric way. Fontaine and co-workers explored the hydrogenation of carbon dioxide using intramolecular o-phenylene bridged B/N FLPs 37 (Scheme 26). When R = 2,4,6-Me3C6H2, the FLP species forms the formyl, acetal and methoxy derivatives 38, but when R = 2,4,5-Me3C6H2, the boron-linked product 39 formed instead.77
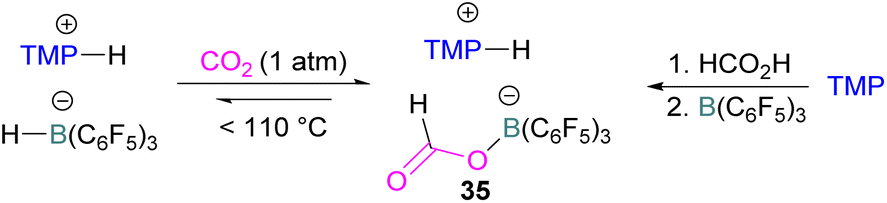 | ||
| Scheme 24 Formation of a formatoborate complex with the TMP/B(C6F5)3 FLP. | ||
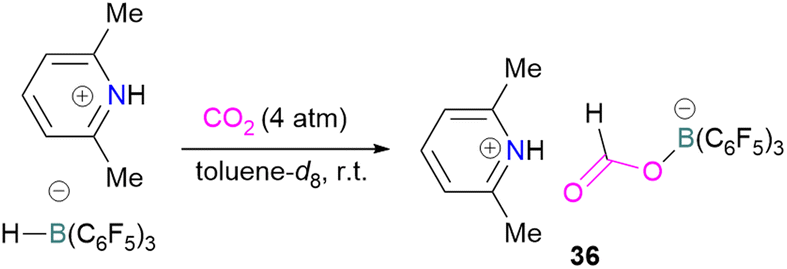 | ||
| Scheme 25 Formation of a formatoborate complex with the 2,6-lutidine/B(C6F5)3 FLP. | ||
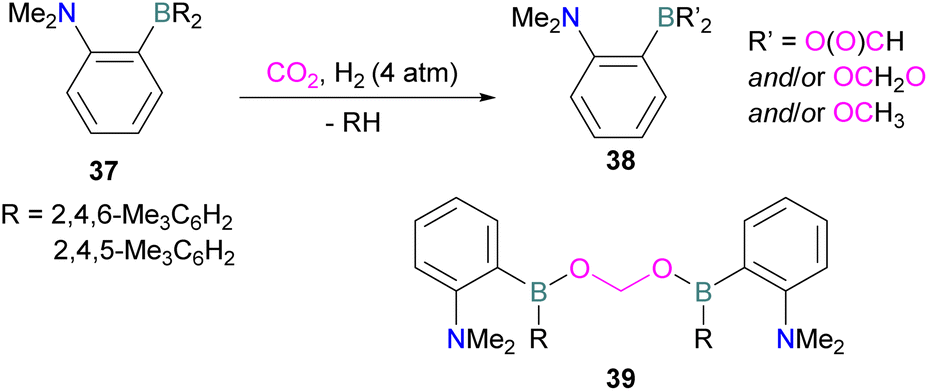 | ||
| Scheme 26 Hydrogenation of CO2 using intramolecular B/N FLPs. | ||
Catalytic transformations of CO2 have also been successful using B/N FLP systems. To address the catalytic shortcomings of the reaction developed by O'Hare and his group using the FLP TMP/B(C6F5)3 for the reduction of CO2 with H2 to form methanol, Piers and co-workers developed a catalytic method by adding silane to the reaction mixture with excess B(C6F5)3 to form methane (Scheme 27).78 They also reported that when Et3SiH was not added to the reaction, then the CO2 adduct as the salt [TMP-CO2-B(C6F5)3][TMPH] was formed. As seen in other systems, the formation of the CO2-adduct is reversible, however, when Et3SiH is added then it provided the [TMPH][HB(C6F5)3] salt along with a triethylsilyl carbamate 40.78
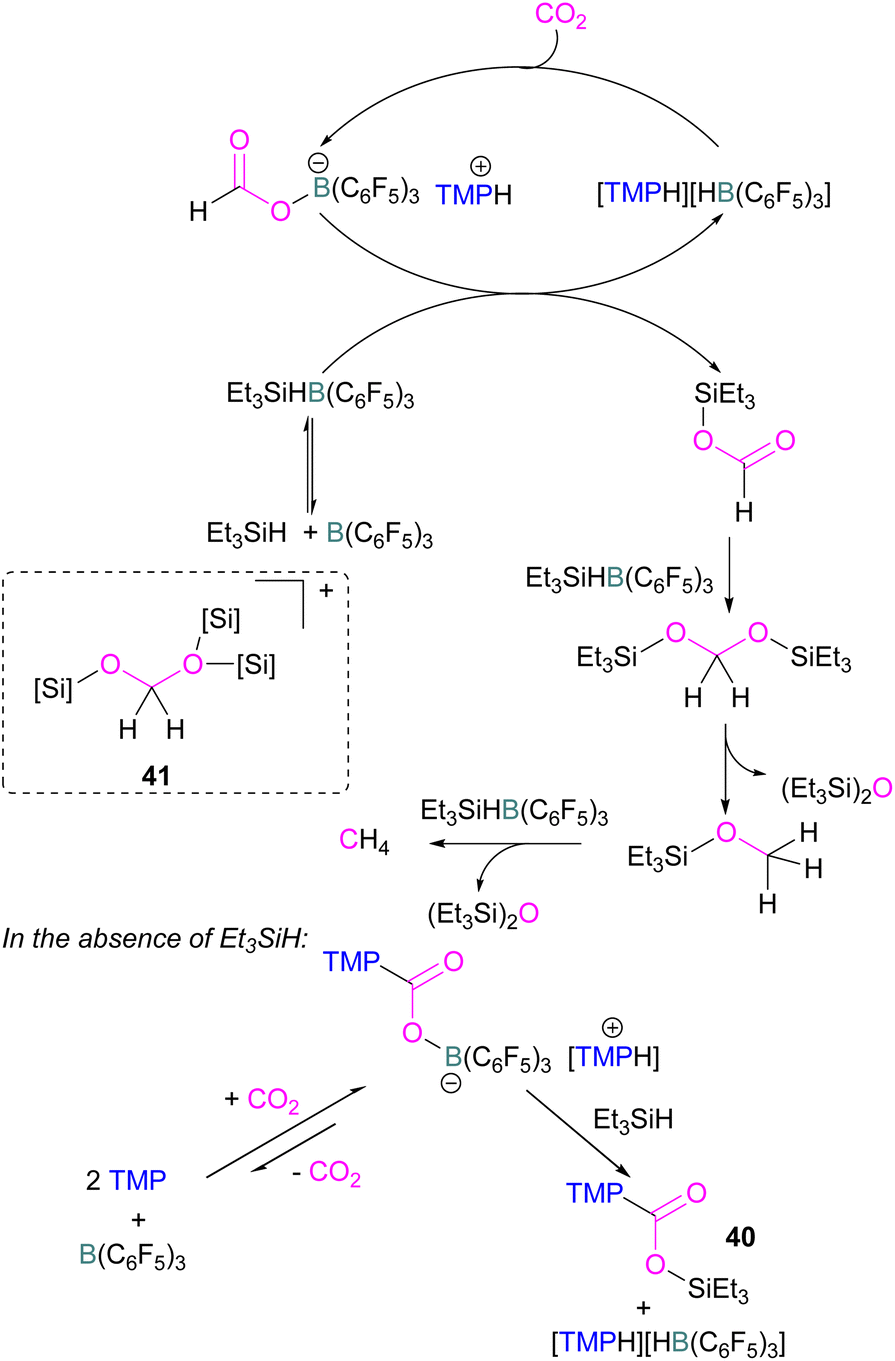 | ||
| Scheme 27 Converting stoichiometric CO2 reduction into a catalytic process by adding excess B(C6F5)3 and Et3SiH to the TMP/B(C6F5)3 FLP system. | ||
For this reaction, Wang and co-workers carried out computational studies to look at the mechanism of CO2 reduction to methane with Et3SiH catalysed by the ion pair [TMPH][HB(C6F5)3] in combination with B(C6F5)3 in detail. The mechanism proposed by Piers was confirmed to be energetically feasible in this study. The reduction proceeds via CO2 insertion into [TMPH][HB(C6F5)3], followed by three successive hydride transfers from Et3SiH to the CO2 centre. It was confirmed that the insertion of CO2 into the H–B bond of [TMPH][HB(C6F5)3] proceeds in a stepwise manner with Hδ+ and Hδ− in the salt first transferring to CO2 to form 41 (Scheme 27, insert).
The role of B(C6F5)3 was also found to be important since it promotes hydride transfer and acts as a shuttle to bring Hδ− from Et3SiH to CO2.79 Overall, additional B(C6F5)3 activates the silane reducing agent, Et3SiH, producing Et3Si+ as a good oxygen acceptor and thus promotes the catalytic deoxygenation of CO2 to CH4.
Cantat and co-workers explored nitrogen bases such as TBD (triazabicyclodecene), Me-TBD (MTBD), DBU (1,8-diazabicyclo[5.4.0]undec-7-ene), and others for the reduction of CO2 in the presence of 9-BBN or CatBH. The reactions were performed at room temperature and a TON of up to 648 was achieved (Scheme 28). In this process, CO2 is initially reduced to a borylformate which then undergoes reduction firstly to an acetal and then a methoxyborane. The stoichiometric reaction of TBD–CO2 and 9-BBN in THF forms product 42 along with other reduced products (Scheme 28). Compound 42 was analysed by single crystal X-ray diffraction and it was found that the acidic NH proton in the TBD–CO2 adduct was replaced with a 9-BBN unit. Compound 42 can be considered as a nitrogen/boron FLP system trapped with CO2. A mechanism was proposed based on rigorous control experiments. In 42, CO2 behaves as a Lewis base and coordinates to the hydroborane R2BH to form adduct 43, which enables hydride transfer from the borane to carbon and forms 44. Compound 42 is regenerated when CO2 is applied, releasing the boron formate and thus catalysed the system for CO2 hydroboration. Finally, the boron formate is reduced to the methoxyborane. It is important to note that for MTBD it was found that the reaction proceeds with the activation of borane followed by the capture of CO2.80
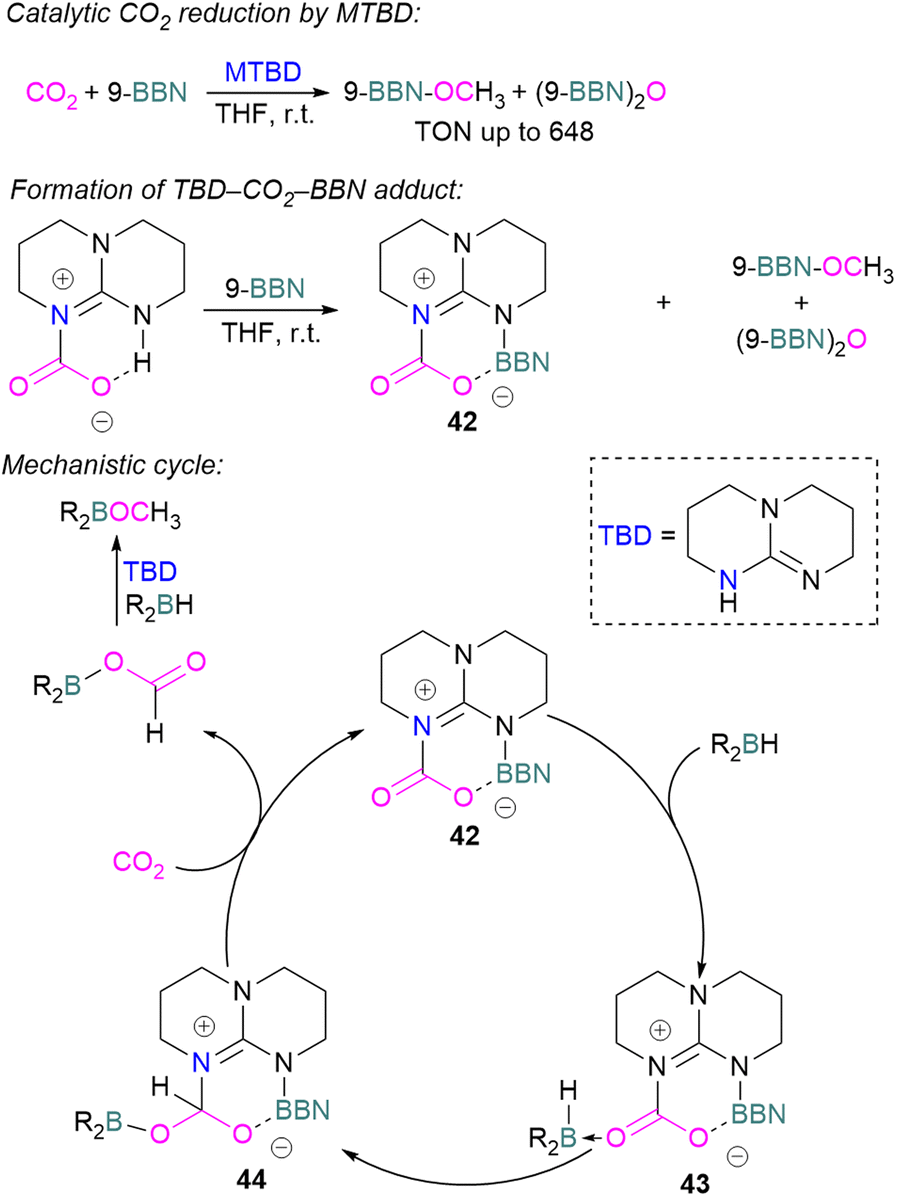 | ||
| Scheme 28 Catalytic reduction of CO2 using N-bases and B–H reducing agents. | ||
Stephan and co-workers inadvertently discovered a new class of N/B molecule 45, that consists of a strong Lewis basic phosphorus centre and weak Lewis acidic boron centre which makes it a suitable FLP system. They utilised FLP 45 in the reduction of CO2 (5 atm) at 60 °C with BH3·SMe2 as a reducing agent and obtained a boroxine product (Scheme 29).81 The reduction of CO2 was observed catalytically in this case due to the presence of a strong basic centre and a weak Lewis acid that facilitates lability of the reduced CO2 fragments. This shows a difference to FLPs composed of a strong Lewis acid in which only stoichiometric reduction was observed, as in the case of 18 where 1,2-C6H4(BCl2)2 is the Lewis acid (Scheme 11).
 | ||
| Scheme 29 Catalytic reduction of CO2 using FLP 45. | ||
Zhang et al. found that 4 equivalents of BH3·NMe3 and catalytic 6-amino-2-picoline could be used to formylate secondary amines. The proposed mechanism proceeds though dehydrocoupling of the amineborane and catalyst to form an intramolecular FLP 46, which reacts with CO2. The activated CO2 is then inserted into the N–B bond which is subsequently reduced by borane with loss of H2BOBH2 to give the methylated amine (Scheme 30).82
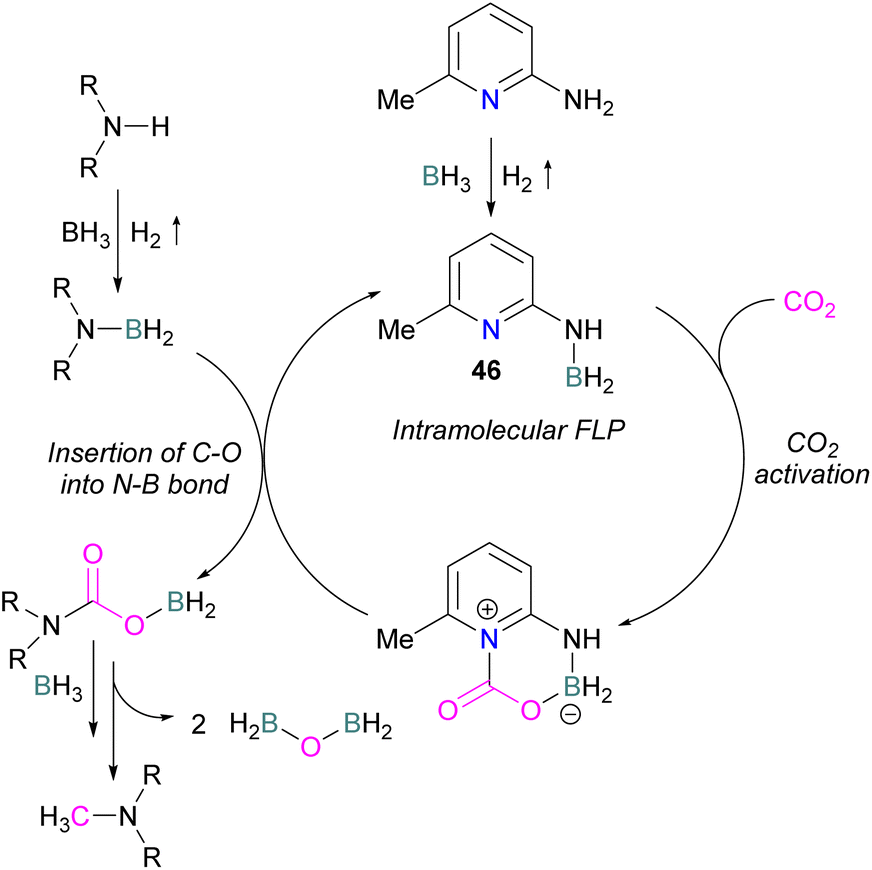 | ||
| Scheme 30 Catalytic transformation of CO2 by N-methyl formylation of secondary amines. | ||
For the activation of CO2 using FLPs, many arrangements of plausible Lewis pairs are possible. Hence, it is a challenge to find a particular combination that is superior for catalysing CO2 reduction.
With this in mind, Corminboeuf and co-workers proposed a map of chemical composition of FLPs for their activity towards formate product by catalytic hydrogenation of CO2. They built the map upon linear scaling relationships, pinpointing specific FLP combinations with complementary acidity and basicity to optimally balance the energetics of the catalytic cycle. Amongst such combinations, they created a library of 60 P/N Lewis bases and 64 triaryl boranes as Lewis acids resulting in a library of 3840 FLPs. Out of these, they experimentally demonstrated the catalytic transformation of CO2 to formate by using an inverse FLP system obtained from tris(p-bromo)tridurylborane (tbtb) as Lewis acid and DBU as the Lewis base. A turnover number of 24 ± 3 was found for this catalytic reaction (Scheme 31). This is the first example of a metal-free CO2 hydrogenation in which stoichiometric addition of a silylhalide was not required. This was achieved through the fine-tuning of the Lewis acid and base based on their energies of hydride and proton attachment, respectively. Here, the authors conclude that inverse FLPs, with a weaker Lewis acid and strong Lewis base or strong Lewis acid with weaker Lewis base, yet with cumulative high acid–base strength, is the ideal combination to achieve CO2 hydrogenation. The authors highlight the importance of overcoming both activation barriers to CO2 activation as well as H2 activation when targeting catalytic CO2 hydrogenation.83
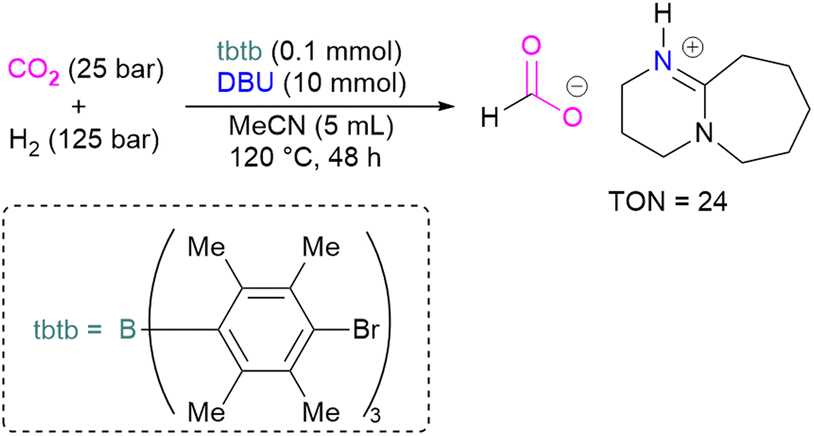 | ||
| Scheme 31 Catalytic transformation of CO2 to formate using the inverse FLP consisting of tbtb and DBU. | ||
In 2022, Palomero and Jones reported the preparation of bis(boramidinate)ferrocenes 47 and 48 by hydroboration of 1,1′-dicarbodiimidoferrocenes. The resulting compounds reacted with CO2. The reaction of the BBN derivative 47 with CO2 (10 atm) to form the mono-CO2-bound product as a yellow precipitate (90% conversion) in a process that was highly reversible (Scheme 32). Whilst this precluded isolation of the CO2-bound products, such reversibility may be preferable for applications in catalytic hydrogenation, facilitating release of the reduced product and catalytic turnover. Lower pressures of CO2 were shown to reduce conversion.84 To allow the use of lower pressures, the more electron-poor bis(pentafluorophenyl)borane analogue 48 was employed. At 5 atm CO2, it activated 2 equivalents of CO2, although the reaction required two weeks to go to completion.
 | ||
| Scheme 32 CO2 adducts of bis(boramidinate)ferrocenes. | ||
Borane/carbon FLPs for CO2 activation
Stable N-heterocyclic carbenes (NHCs) upon reaction with CO2 form a mesomeric betaine 49 having a C–C bond between the carbene and CO2. On the other hand, very reactive carbenes can form oxiranones 50 (Scheme 33). The product remains in equilibrium with the starting substrates. Most attention has been focused on stable sterically hindered NHCs amongst all carbenes for the activation of small molecules,85 and several carbenes in FLP systems have been explored and found to be efficient in the activation of CO2.86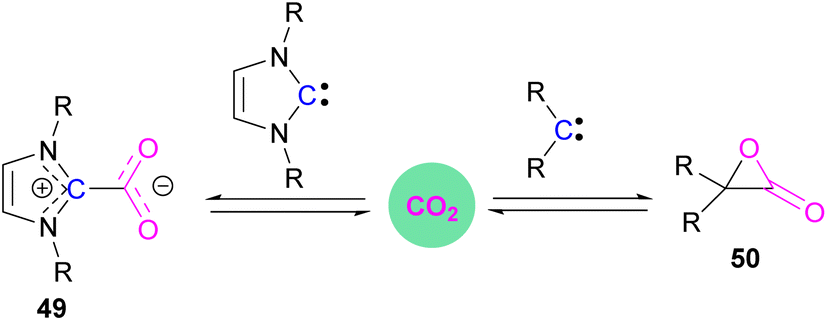 | ||
| Scheme 33 Reaction of carbenes with CO2. | ||
The first carbene based FLP system to activate CO2 was reported by Tamm and co-workers in 2012 (Scheme 34).87 They showed that exposure of CO2 to a solution of a bulky carbene (1,3-di-tert-butylimidazolin-2-ylidene) and tris[3,5-bis(trifluoromethyl)phenyl]borane, B(3,5-(CF3)2C6H3)3, in benzene at 60–70 °C, a white precipitate, identified as the FLP-CO2 adduct was formed. The adduct was isolated in 66% yield. At room temperature this adduct was also obtained on exposure of CO2 to the solution of the FLP in benzene with a 24 h reaction time and a higher yield of 86% was isolated.
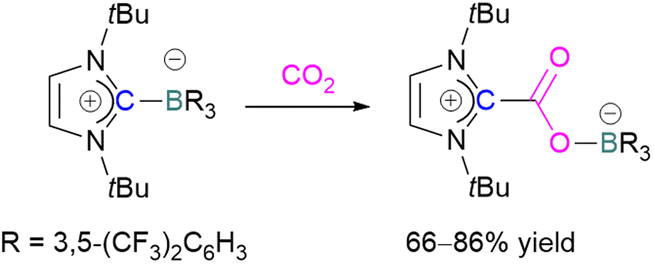 | ||
| Scheme 34 First carbene based FLP system for CO2 activation. | ||
Later, Tamm and his group synthesised a library of carbene based FLP CO2-adducts and studied their reaction profile computationally (level of theory: M05-2X/6-311G**). A 1:1 mixture of a bulky carbene and B(C6F5)3 with CO2 provided NHC-CO2-B(C6F5)3 products 51 in 89% and 68% yield depending on the starting carbene (Scheme 35). From DFT calculations a low energy barrier was observed for the NHC-CO2-B(C6F5)3 adduct formation (10.4 (R = H) and 12.2 (R = Me) kcal mol−1), and it was concluded that steric changes on the NHC were more pronounced than electronic impacts.88
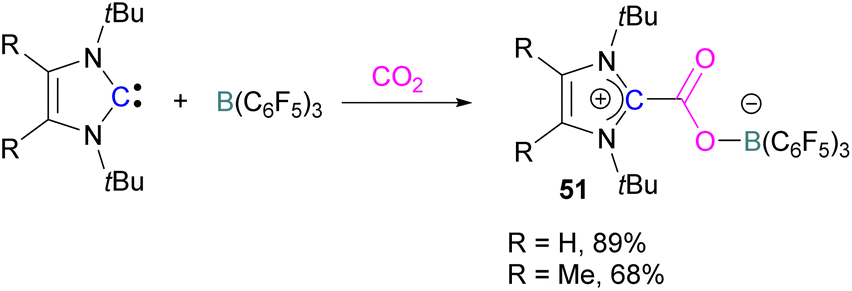 | ||
| Scheme 35 Capture of CO2 with NHC/B(C6F5)3 FLPs. | ||
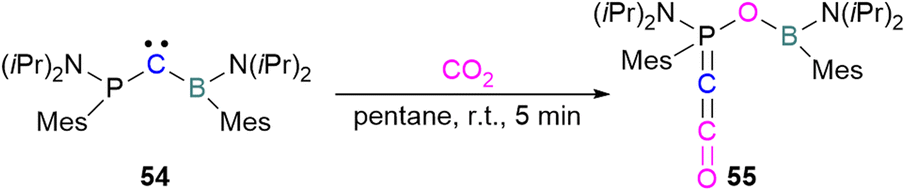
Zhu and co-workers computationally designed a boron-based carbene intramolecular FLP 52 and calculated its reactivity with various small molecules, including CO2. This FLP with CO2 forms a zwitterionic species 53 and the authors discuss the important driving force of aromaticity in the final adduct (Scheme 36).89 Baceiredo and co-workers exposed boryl(phosphine)carbene 54 to CO2 (1 atm) and an unusual product 55 was observed. After analysis of the product's structure, it was found that the carbene inserted into the CO bond of the CO2. Thus, incorporating carbon dioxide into the corresponding phosphoryl ketenylidene derivative (Scheme 37).90
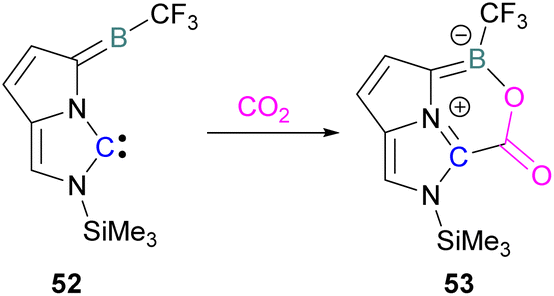 | ||
| Scheme 36 Computationally designed carbene/borane derived FLP. | ||
 | ||
| Scheme 37 CO2 adduct of a boryl(phosphine)carbene. | ||
Stoichiometric reduction reactions with carbene/borane FLPs have been reported.91 In 2019, Mandal and co-workers prepared an N-heterocyclic carbene-boron adduct 56 by reacting an abnormal heterocyclic carbene (aNHC) with 9-BBN. The synthesised NHC-boron adduct 56 was utilised to capture CO2 from the atmosphere under ambient conditions in benzene overnight. Product 57 was obtained, due to moisture in the air leading to hydrolysis of 9-BBN and boric acid formation with the release of a cyclooctane molecule. The CO2 was incorporated as a formate ion. Further treatment of 57 with excess 9-BBN leads to the formation of compound CH2(OBBN)2 with the release of H2, and finally converts this to CH3OBBN (Scheme 38).91 This work was presented as a first metal-free system to reduce CO2 by capturing it from the atmosphere under ambient conditions where CO2 remains in a concentration of ∼400 ppm. Mandal and co-workers later reported the use of the same FLP system for a catalytic reduction of CO2 in the presence of a range of hydroboranes leading to methoxyborane (Scheme 39). Reaction of the carbene with CO2 firstly gave the adduct whilst reaction of the carbene with 3 equivalents of 9-BBN in the presence of CO2, provided boron diformate 58. Zwitterionic boron diformate 58 was utilised catalytically with a loading of 0.005 mol% for the conversion of 9-BBN to the methoxide derivative CH3O-BBN under a CO2 atmosphere. Catalyst 58 leads to a TON of 6000, which is the highest TON observed among all the metal-free catalysts investigated at ambient conditions. The key feature of this catalytic process is the formation two equivalents of 9-BBN formate, BBN(OCHO), from the reaction of catalyst 58 with an equivalent of 9-BBN resulting in the release of dihydrogen.
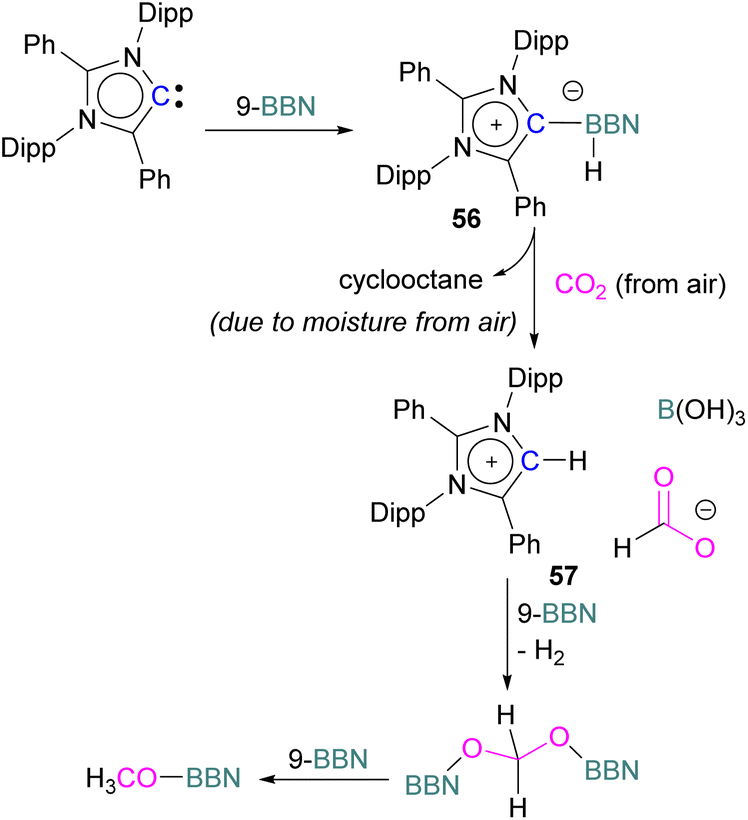 | ||
| Scheme 38 NHC-BBN adduct captures of CO2 from the air under ambient conditions and reduction to formate and methoxide. | ||
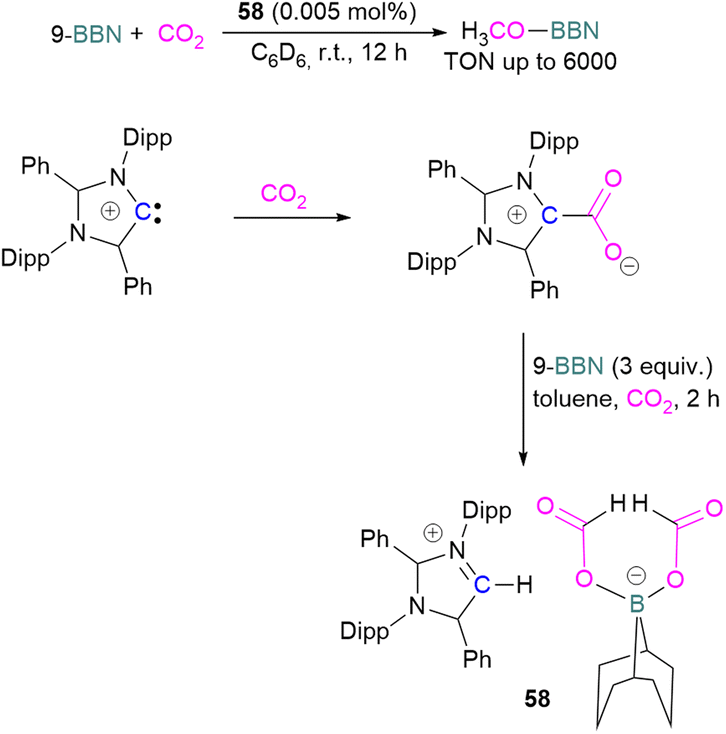 | ||
| Scheme 39 Preparation of zwitterionic boron diformate 58 and its use as a catalyst for the reduction of CO2. | ||
This generates the carbene which further captures a CO2 molecule regenerating 58 with 9-BBN and thus providing a catalytic process. The 9-BBN formate is finally reduced and hydroborated to CH3O–B in a series of steps in the presence of an excess of 9-BBN.92
Borane/silicon FLPs for CO2 activation
To date there is just one example of a boron/silicon FLP for CO2 activation. Very recently Mo and co-workers reported the synthesis of a geometrically constrained bis(silylene)-stabilised borylene 59.93 Spectroscopic and X-ray analyses reveal that structure 59 has a tricoordinate boron centre with a distorted T-shaped geometry. Computational analysis shows that the HOMO comprises a lone pair of electrons on the boron centre and is delocalised over the Si–B–Si unit. Compound 59 shows single electron transfer reactivity towards B(C6F5)3 forming a frustrated radical pair [(SiNSi)B]˙+[B(C6F5)3]˙−. The reaction of 59 with CO2 (1 atm) in C6D6 at room temperature forms a new product 60 by cleaving CO2 which, upon hydroboration with two equivalent of 9-BBN, forms compound 61 in quantitative yields. The structure of 61 showed that boron and silicon atoms are bridged by boryloxymethylene (CHOBR2) formed by the hydroboration of the CO group. The Si–O–B bridge in 60 was cleaved along with the formation of BH and SiOBR2 units. Compound 61 can also be obtained directly by treating 59 with CO2 and 2 equivalents of 9-BBN in C6D6 at room temperature with an isolated yield of 45% (Scheme 40). The catalytic performance of 59 (5 mol%) and 60 (5 mol%) shows an efficient transformation of N-methylaniline into the corresponding formamide (92% yields in each case) by capturing and hydroborating CO2 with HBpin (Scheme 40).93
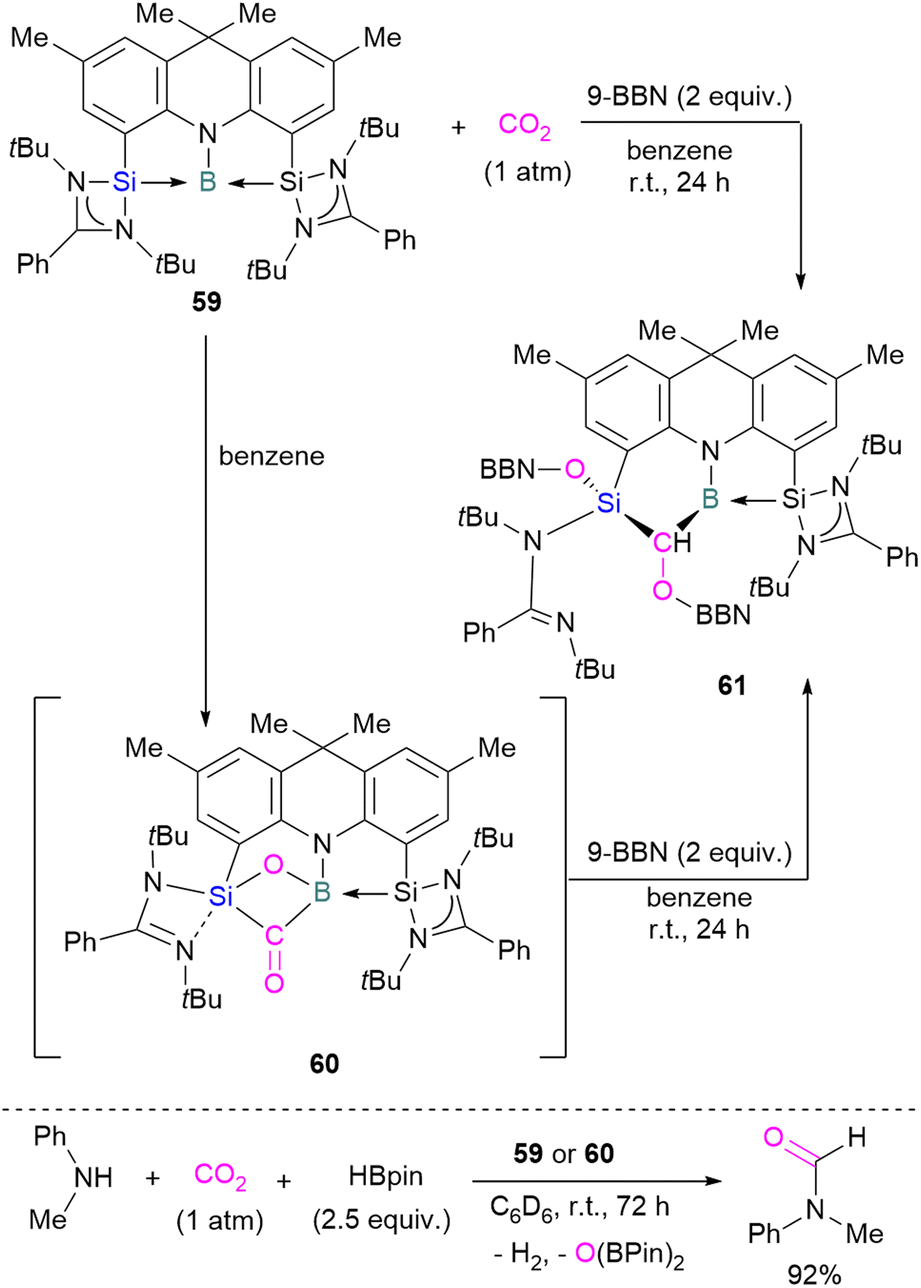 | ||
| Scheme 40 Cooperative bond activation of CO2 with a borylene/silylene compound and catalytic application. | ||
Borane/germanium FLPs for CO2 activation
Similar to the silylene described above, germylenes are also reported as the Lewis base component of an FLP for CO2 reduction. Kato and co-workers reported an interesting N,P-heterocyclic germylene 62 in 2016, that bears several reactive sites (including a germylene centre) and can activate two CO2 molecules simultaneously.94 Compared to classical FLPs, 62 showed unusual behaviour of multi-reactive sites and has been utilised as a donor component in the Lewis acid–base pair. In fluorobenzene, the N,P-heterocyclic germylene 62 reacts with B(C6F5)3 at room temperature to give the corresponding adduct 62 B(C6F5)3 as colourless crystals in 67% yield (Scheme 41). The authors explored the catalytic reduction of CO2 with 5 mol% of the FLP adduct 62 B(C6F5)3 and the reducing agent Et3SiH. The proposed mechanism for CO2 activation showed that 62 B(C6F5)3 reacts with silane Et3SiH and forms a cationic germylene 63 which promotes CO2 hydrosilylation catalytically via two possible activation modes A and B to obtain product H2C(OSiEt3)2 selectively.94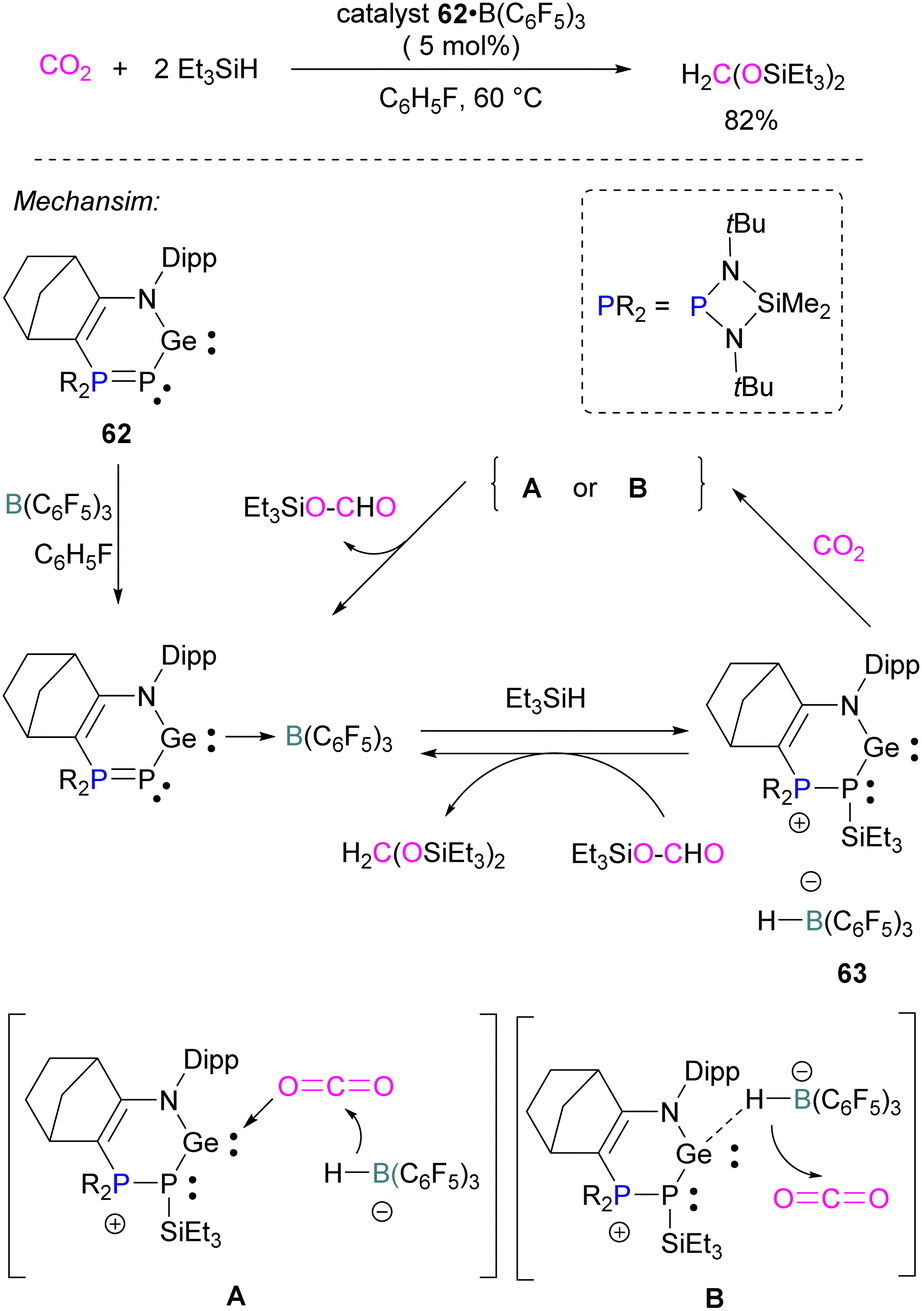 | ||
| Scheme 41 N,P-heterocyclic germylene in catalytic hydrosilylation of CO2. | ||
Borane/boron FLPs for CO2 activation
FLP systems where the same element is used both as the Lewis acidic and Lewis basic reactive centres are rarely observed. One such example was reported by Kinjo and co-workers in 2015 with 1,3,2,5-diazadiborinine 64 featuring nucleophilic and electrophilic boron centres within the same molecule.95This first reported intramolecular boron–boron FLP showed high regioselectivity in the reaction with CO2, yielding a bicyclic product 65 (Scheme 42). Interestingly, CO2 activation by 64 was reversible at 90 °C. To obtain insight into electronic features of 1,3,2,5-diazadiborinine 64, the authors performed DFT calculations [level of theory: B3LYP/6-311G+(d,p)]. NBO analysis showed that the compound possess both nucleophilic and electrophilic boron centres with a formal B(+I)/B(+III) mixed valence system.95 Later, Zhao and co-workers performed more detailed computational analyses of 64.96 They reported π delocalisation over the central ring which extends from the lone pair on (O) → π*(N–C), and favourable orbital overlap with CO2 is generated from the electrophilic interaction with the Lewis acidic boron centre and nucleophilic donation to the LUMO+3 of the other boron centre.
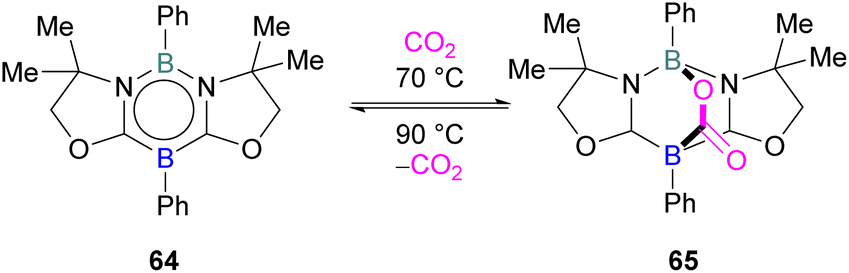 | ||
| Scheme 42 Reaction of a 1,3,2,5-diazadiborinine with CO2. | ||
Kinjo and co-workers synthesised another class of boron compounds in which the boron acts as a Lewis basic centre.
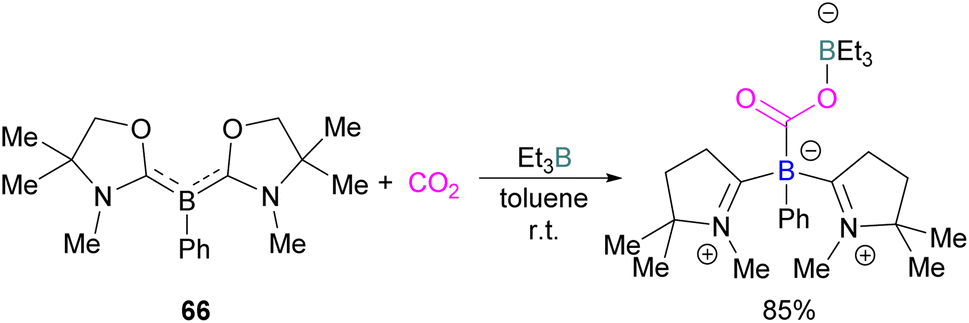
It was found previously that tricoordinate organoboron L2PhB: (L = oxazol-2-ylidene) compound 66 does not react with BEt3 This is perhaps due to a mismatch of the softness/hardness of the respective boron centres in 66 and BEt3 based on HSAB (Hard Soft Acid Base) theory in addition to steric hindrance. As compound 66 and BEt3 do not react, they act like an FLP. Thus 66 and BEt3 were reacted with CO2 in toluene at room temperature and the FLP-CO2 adduct was isolated in 85% yield (Scheme 43).97
 | ||
| Scheme 43 Reaction of tricoordinate organoboron compound with Et3B and CO2. | ||
Borane/metal FLPs for CO2 activation
In addition to p-block Lewis bases, transition metal complexes can also act as the Lewis base component of an FLP with boron as the Lewis acid. This is demonstrated using a ruthenium acetylide which is an electron rich species. This can create a Lewis basic β-carbon centre and form an FLP when combined with a Lewis acid (Scheme 44). Stephan and co-workers in 2013 showed that when [(η5-indenyl)Ru(PPh3)2(CCPh)] was reacted with B(p-C6F4H)3 no reactivity was observed, indicating that their combination is an FLP in nature. A solution of this FLP, when exposed to CO2 for 12 h, provided an orange solid in 70% yield and was fully characterised using NMR and single crystal X-ray crystallography as the FLP-CO2 adduct where the β-carbon centre had attacked the electrophilic carbon centre of CO2 (Scheme 44).98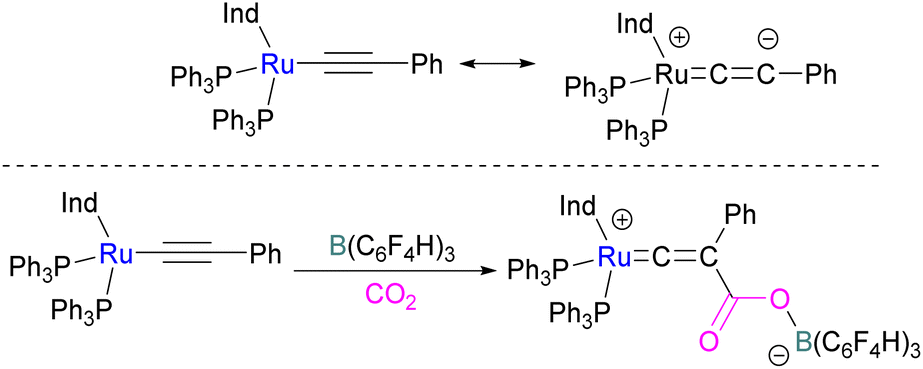 | ||
| Scheme 44 CO2 adduct of [(η5-indenyl)Ru(PPh3)2(CCPh)] and B(C6F4H)3. Ind = Indenyl. | ||
Wass and co-workers explored reactions with a platinum(0) complex as a Lewis base in the activation of small molecules using a sterically congested boron-based Lewis acid. They found that pairing a Lewis basic platinum(0)–CO complex supported by a diphosphine ligand with B(C6F5)3 acts as a frustrated Lewis pair, to activate CO2 (Scheme 45). The presence of B(C6F5)3 is important as no activity was observed between the platinum complex and CO2 in the absence of the borane. In this scenario, Pt(0) acts as a donor of electron and the boron atom acts as the acceptor forming a coordinated Pt–CO2–B system. A substitution of CO by CO2 on platinum was observed after the loss of the CO molecule.
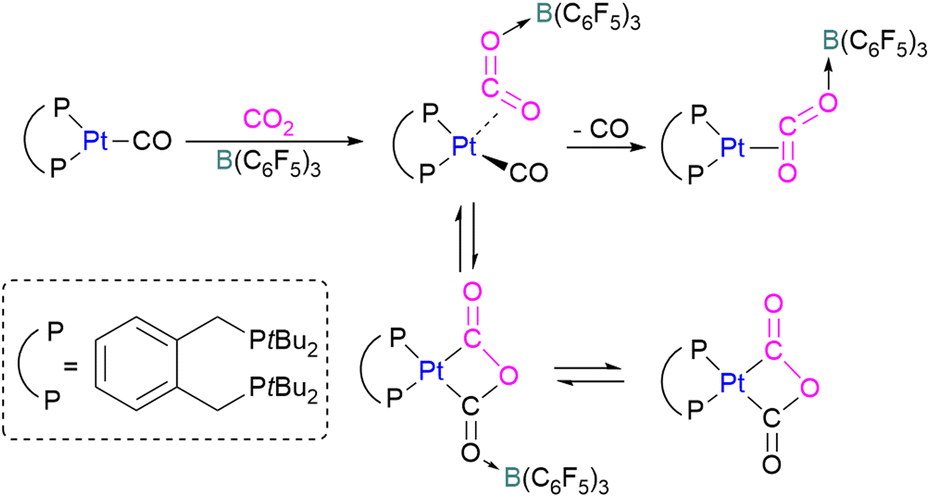 | ||
| Scheme 45 Activation of CO2 using Lewis basic platinum(0)–CO complex/B(C6F5)3. | ||
In this process 95% isotopically pure 13CO2 was used but the 31P NMR analysis of the product showed a mixture of 13C labelled and unlabelled product in a ratio of 4:1. The source of unlabelled product must be from the 12CO in ligand of the starting material. This suggests that a symmetrical [C2O3]2− complex forms during the reaction pathway. The proposed mechanism in Scheme 45 suggests that the reaction of the platinum(0)–CO complex and B(C6F5)3 with CO2 is a metal-mediated oxygen transfer between CO2 and CO rather than a simple ligand substitution.99
In 2013, Berke and co-workers showed an FLP-type activation of CO2 using a [Re]–H/B(C6F5)3 system where the Re–H bond acts as a Lewis base. Catalysts 67 and 68 were prepared stepwise from a rhenium hydride precursor [ReH(PR3)2(NO)Br] (Scheme 46). Initially, the precursor was reacted with B(C6F5)3 and CO2 in benzene to form the FLP-CO2 adduct. With a PiPr3 ligand on the rhenium precursor, insertion of the Re–H into the FLP-bound CO2 molecule was observed generating 67. Compound 67 could be hydrogenated with H2 (1 bar) in toluene at 60 °C for 1 h to give 68 in 99% yield. Both 67 and 68 were screened for the hydrosilylation of CO2 using Et3SiH as a reducing agent (Scheme 46). Catalyst 67 with a loading of 1 mol% provided the (Et3SiO)2CH2 product in 87% yield (TON = 89, TOF = 5.9 h−1), while 68 provided the reduced product in 89% yield (TON = 95, TOF = 7.3 h−1). Similarly, catalysts 67 and 68 were utilised for CO2 hydrogenation (PH2/CO2 = 40/20 bar) in the presence of TMP as a base. Catalyst 68 provided the formate salt of TMP in 46% yield (TON = 92, TOF = 6.1 h−1).100
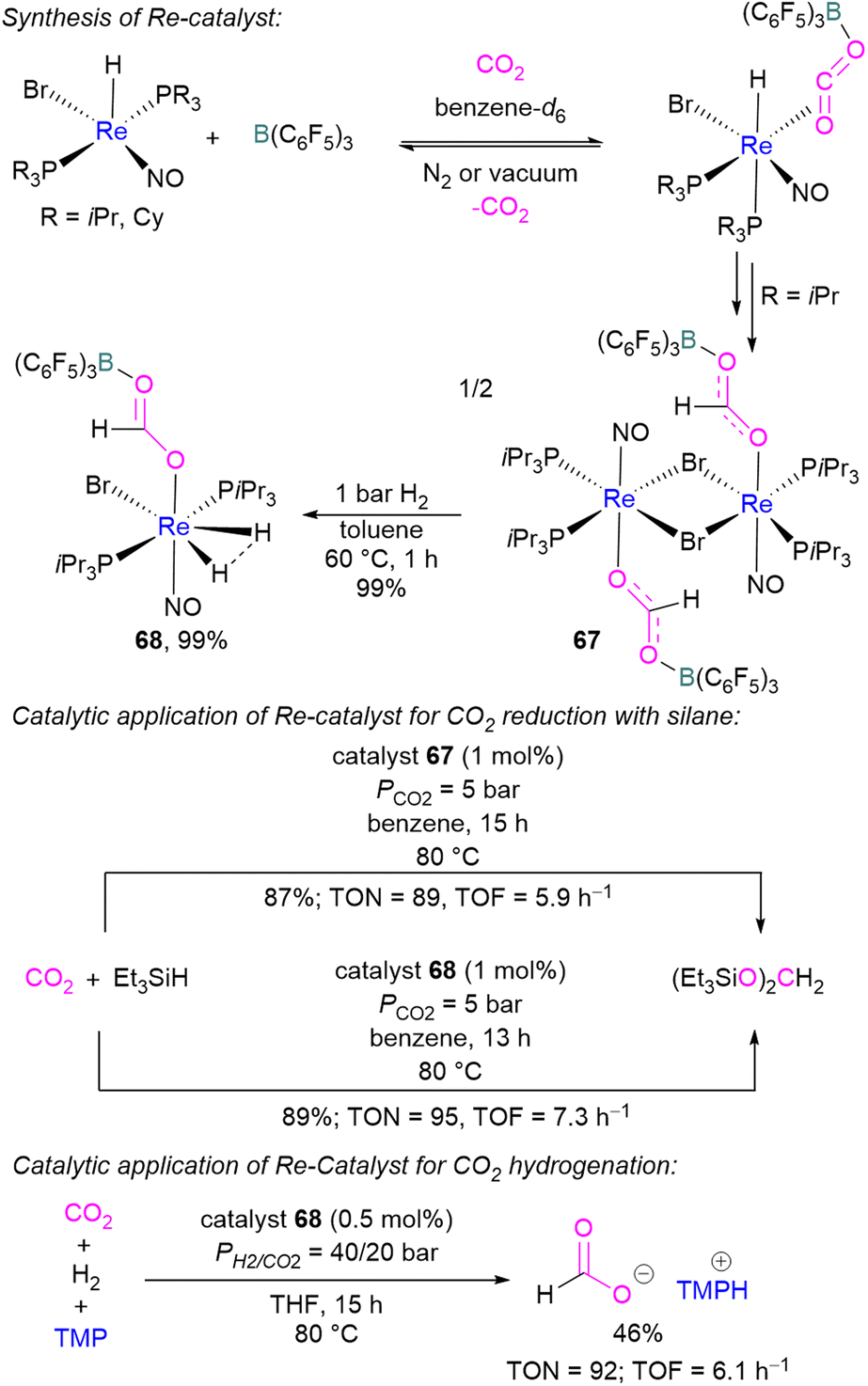 | ||
| Scheme 46 Synthesis of [Re]-CO2-B(C6F5)3] adduct and utilisation as a catalyst in CO2 reduction. | ||
In another study, Agapie and co-workers investigated the effects of the Lewis acid B(C6F5)3 towards the conversion of CO2 to CO and water using a molybdenum complex (Scheme 47).101 The activation of CO2 was found to be linearly related to the strength of the Lewis acid. When a labile Mo(0)-CO2 adduct interacts, it will increase both the degree of activation and the kinetic stability of bound CO2 as shown in Scheme 47. In contrast to the CO2 displacement by a solvent that is predominantly observed in the absence of a Lewis acid, in the presence of B(C6F5)3 and [H(Et2O)2][BArF24] (BArF24 = tetrakis[3,5-bis-(trifluoromethyl)phenyl]borate) CO2 cleavage occurs. This demonstrates the significance of kinetic and thermodynamic aspects in the effective CO2 reduction chemistry primarily relying upon bond activation and the residence period of the associated small molecule.
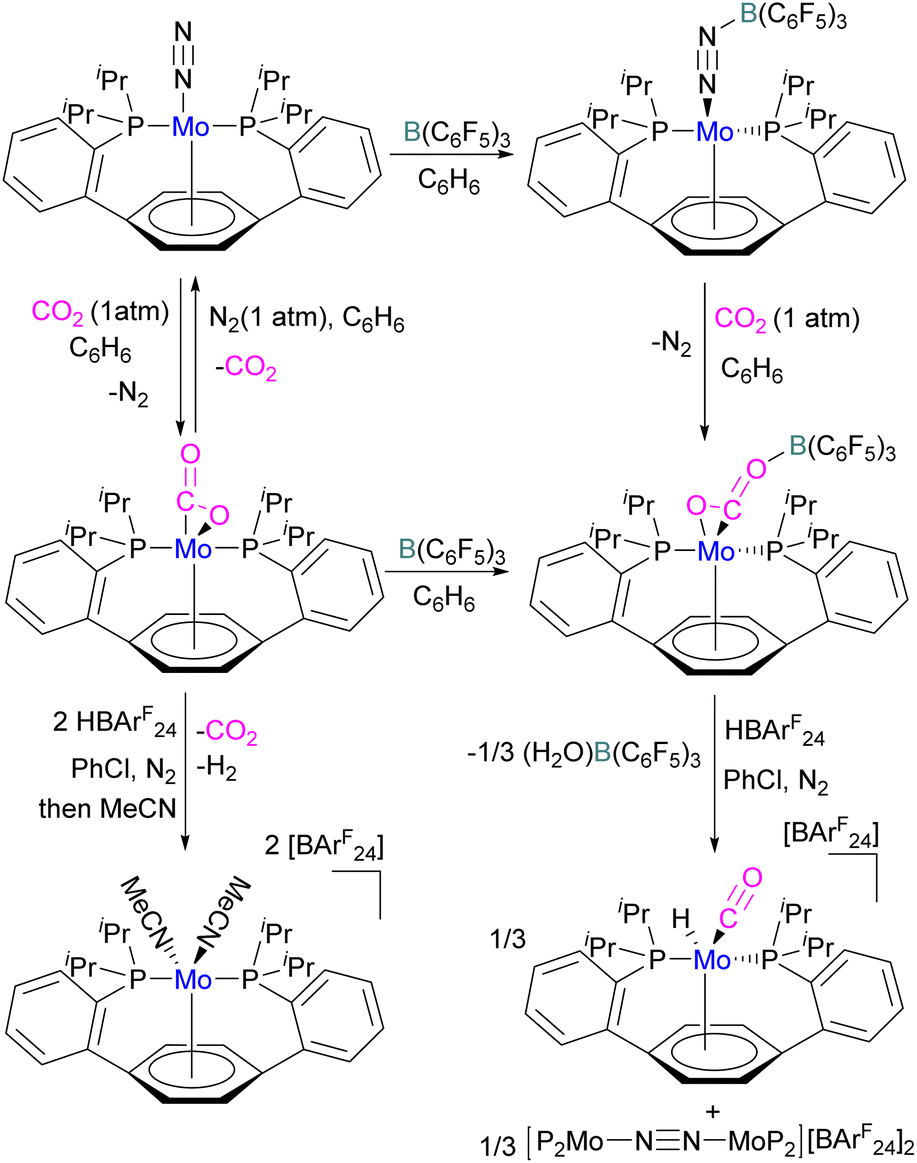 | ||
| Scheme 47 Mo based FLP for CO2 activation. BArF24 = tetrakis(3,5-bis(trifluoromethyl)phenyl)borate. | ||
The authors demonstrated that the chemistry of the labile substrate is greatly influenced by the time the substrate resides in the metal's coordination sphere. It is shown that Lewis acid additives promote CO2 cleavage via kinetic stabilisation rather than merely by thermodynamic activation.
One final system to note here uses the boron Lewis acid B(C6F5)3 with s-block metal carbonates M2CO3 (M = Na, K, and Cs) for the highly efficient reduction of CO2 to formate. Amongst the screened metal carbonates, Cs2CO3 showed the highest TON of 3941.102
Aluminium FLPs for CO2 activation
A number of FLPs based on aluminium Lewis acids have also been reported for CO2 capture and reduction, although the greater oxophilicity of aluminium (potentially inhibiting product release) means that catalytic hydrogenation has yet to be reported. This oxophilicity also means that, whereas FLPs containing boron Lewis acids typically bind CO2 in a 1:1:1 Lewis acid:Lewis base:CO2 ratio, Al-containing FLPs often bind it in a 2:1:1 ratio, with both oxygen atoms binding an aluminium centre.103 Studying FLPs comprised of phosphines and aluminium esters, Smythe et al., showed that the ratio of mono-to bis-bound adduct varies with Lewis acidity.104
After exposure to 1 atm of CO2, Al(O–C6H2Cl3)3 bound CO2 in a predominantly mono fashion (ca. 95% mono), whereas the greater Lewis acidity of Al(OC6Cl5)3 (ca. 1:1 mono:bis) and Al(OC6F5)3 (ca. 75% bis) favoured the bis-bound adduct.
Like boron CO2 adducts, a range of aluminium FLP-CO2 adducts are reported (Scheme 48). Uhl and co-workers reported the synthesis of geminal ambiphilic phosphine–aluminium FLPs that can activate CO2 to a form a cyclic adduct 69.105 Later, the same authors synthesised AlPC2O type heterocycle having cis/trans isomeric compounds.106 Uhl also reported a P–H functionalised Al/P FLP in 2019. The FLP reacts with CO2 to give a five-membered zwitterionic cycle similar to that in 69, as typical for vicinal intramolecular FLPs. However, the enhanced acidity of the phosphine means that it can be deprotonated by addition of a base (such as DABCO or nBuLi) to give a more stable precipitate.107 Similarly, Fontaine and co-workers studied the reactivity of the stable Lewis adducts [R2PCH2AlMe2 (R = Me, Ph)] and found adducts 70 and 71.108 Harder and co-workers reported a geminal Al/P FLP, with a nitrogen rather than the more common carbon linker. Like the carbon-linked Al/P FLP reported earlier by Fontaine, this reacts with CO2 to give a 2:2 eight-membered ring product 72 by insertion of CO2 into the Al-linker bond with cis- and trans-isomers.109 Limberg et al. utilised a biphenylene backbone to prepare a strained intramolecular P/Al-based FLP which was reacted with CO2 (2 bar) at room temperature for 5 minutes in deuterated dichloromethane to obtain adduct 73.110 They also reported xanthene-linked intramolecular Al/P FLPs containing two Al centres which are able to activate CO2.111
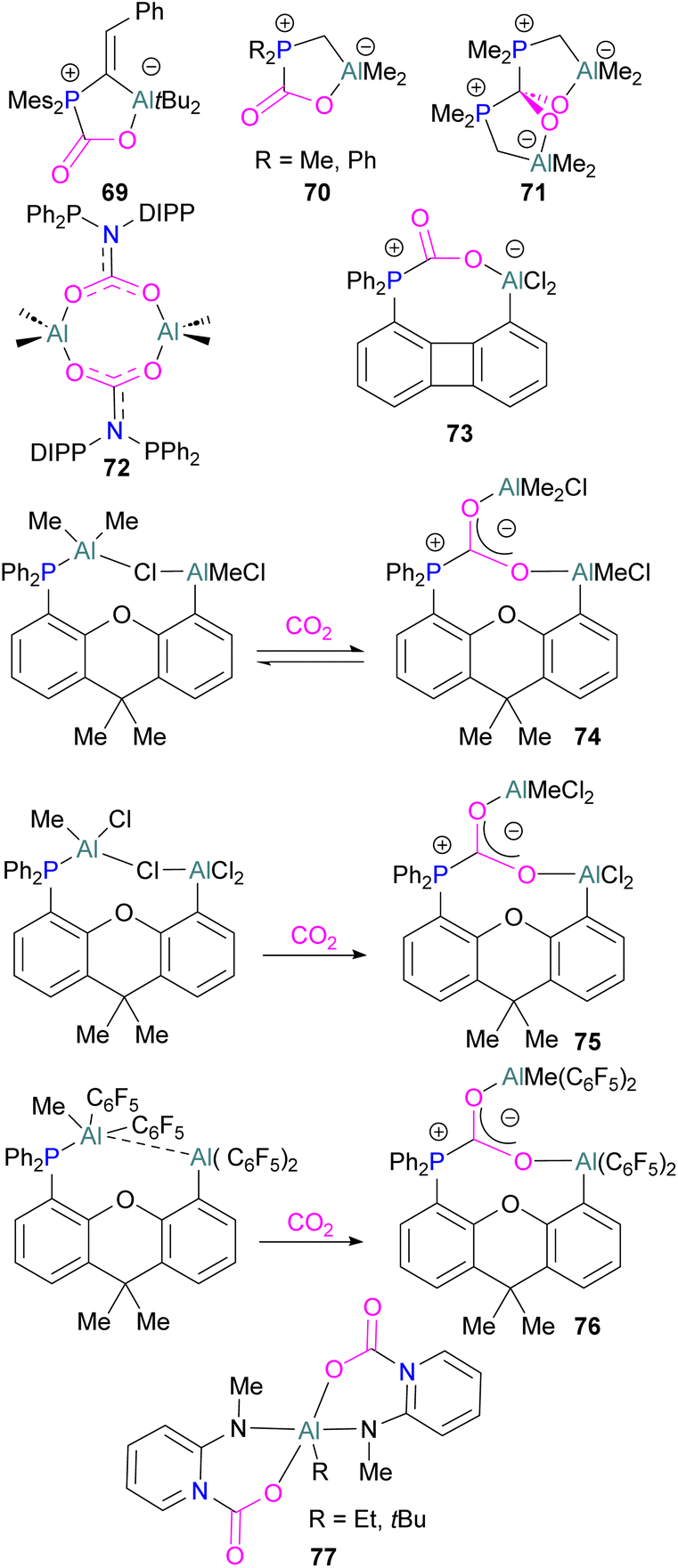 | ||
| Scheme 48 Aluminium based CO2 adducts. | ||
In the products 74–76, the two aluminium centres each bind to one of the CO2's oxygen atoms. The binding strength could be tuned by varying the substituents on aluminium. The more Lewis acidic xanthene-AlCl2 and xanthene-Al(C6F5)2 fragments bind CO2 irreversibly giving 75 and 76 (Scheme 48), while xanthene-MeClAl binds CO2 reversibly giving 74 under 2 bar CO2, liberating CO2 when this excess pressure was released. For catalytic applications, this reversible binding is necessary to enable release of the product. Although most CO2-binding Al FLPs are Al/P rather than Al/N, one example of an Al/N FLP was described by Brewster in 2020 using the readily available 2-(methylamino)pyridine as ligand yielding 77 upon reaction with 2 equivalents of CO2.112 An example of an FLP-CO2 adduct with a metal as a Lewis base was provided by Bourissou and co-workers who utilised geminal P–Al ligand [Mes2PC(CHPh)AltBu2/Pt(PPh3)] in the activation of CO2 molecule to obtain an adduct.
In this bimetallic system, platinum acts as the Lewis base and activates the CO2 molecule by reacting at the carbon centre of the CO2 molecule and the formed negative charge on one of the oxygen atoms is stabilised by the Lewis acidic aluminium centre (Scheme 49).113
 | ||
| Scheme 49 Pt/Al bimetallic FLP system in CO2 activation. | ||
Several of these adducts have been used in stoichiometric and catalytic transformations of CO2. Stephan and co-workers, reported the synthesis of CO2 adducts 78 between AlX3 (X = Cl, Br) and PR3 (R = Mes) (Fig. 7). Upon treatment of these adducts with excess ammonia borane (NH3·BH3), an Al-methoxy species was generated which after hydrolysis resulted in the formation of MeOH at room temperature.103 To study the steps involved in the reaction, Me3N·BH3 was utilised to reduce the FLP-CO2 adduct 78 (X = C6F5, R = o-Tol). Along with the methoxy derivatives of alane, compound 79 was isolated and fully characterised.114 Later, other groups have studied the mechanism for this reaction computationally and explained the reduction of CO2 trapped FLPs.115 In other reactions, Stephan and co-workers explored the adducts of 78 (X = Cl, Br, I, C6F5, OC(CF3)3 and R = Mes, o-tolyl) for the stoichiometric transformation of CO2 to CO.116
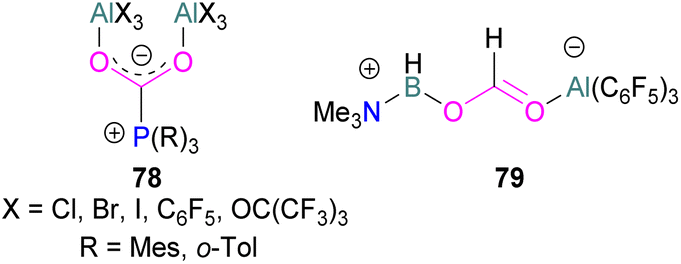 | ||
| Fig. 7 Aluminium based FLPs-CO2 adducts used in CO2 reduction. | ||
While fluorination of substituents is a common way to increase Lewis acidity in FLP design, an alternative is the use of a cationic Lewis acid. Harder reported an FLP comprised of Lewis basic PPh3 and a cationic [Dipp-NacNacAlMe]+ (NacNac = β-diketiminate ligand) Lewis acid 80.117 Exposure of this FLP to CO2 results in the rapid formation of a stable adduct which upon stoichiometric hydrosilylation with triethylsilane forms compound 81 (Scheme 50). Hydride transfer from Et3SiH to the carbon atom of CO2 generates Et3Si+ which is trapped by the base, PPh3, and forms an ion pair [Et3SiPPh3][B(C6F5)4]. The insertion of Et3Si+ into an ion pair restricts the system to stoichiometric CO2 reduction. Otherwise, cleavage of the Al–O bond and transfer of formate ion HCO2− to Et3Si+ would have made the system catalytic.
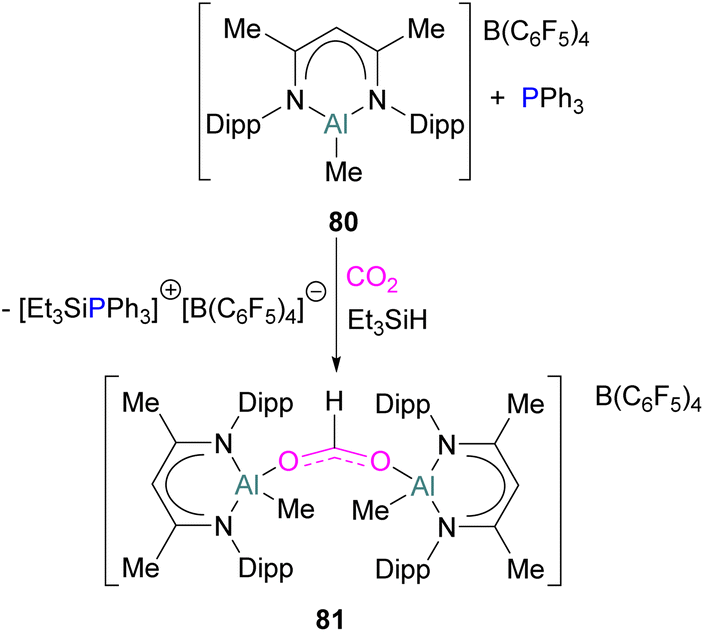 | ||
| Scheme 50 Aluminium based FLP for the stoichiometric reduction of CO2. | ||
An unusual report of an oxygen-bridged geminal Al/P FLP 82 was made by Wickemeyer et al. (Scheme 51). It was prepared by reaction of the parent alane and phosphine oxide, giving an initial zwitterionic compound 83 which slowly eliminates H2 to give the FLP 82. 82 was found to bind CO2 to give the heterocyclic CO2 adduct 84. The hydrogenated zwitterion 83 can also be generated in small quantities by exposure of the FLP to H2. This species bound CO2 irreversibly, and exposure of the hydrogen adduct to CO2 gave stoichiometric CO2 reduction to the aluminium bound formate 85.
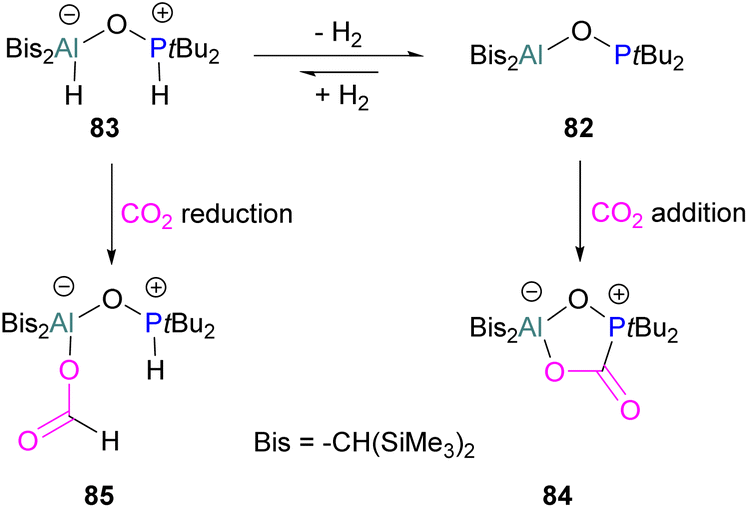 | ||
| Scheme 51 Oxygen-bridged geminal Al/P FLP for CO2 activation and reduction. | ||
However, the instability of the hydrogen adduct and strong Al–O bond make the system not well suited for catalytic applications (Scheme 51).118
Huang et al. reported a variety of group 12 and group 13 formamidinate FLPs. While formamidinates are able to coordinate as bidentate ligands, the incorporation of strongly electron-withdrawing C6F5 substituents on nitrogen increases the preference of the monodentate species with a vacant coordination site on the metal. The free “N” and unsaturated metal in proximity are able to act as an FLP,119 and the compounds' potential for catalytic CO2 hydrosilylation. While the formamidinates investigated (B, Al, Ga, In and Zn) showed poor activity for this reaction on their own, significantly improved performance was seen when combined with B(C6F5)3 or Al(C6F5)3. The highest activity for complete conversion of triethylsilane under 1 bar CO2 after 10 h at 80 °C was observed with the aluminium formamidinate/B(C6F5)3, yielding almost exclusively CH4. Replacing Et3SiH with Ph2SiH2 gave selective formation of the bis(silyl ether).
However, mechanistic studies involving the aluminium formamidinate suggest that the catalyst decomposes under the reaction conditions to generate other aluminium species, which were the catalytically active species, and were not identified.
Surawatanawong and co-workers compared the reactivity of geminal P/Al and B/P FLPs with CO2 and H2 based on systems previously published by Lammertsma et al.42 The compounds investigated consisted of an sp2-carbon bridged FLP (Mes2P–C(CHPh)–EtBu2) and an sp3-carbon bridged FLP (tBu2P-CH2-EPh2) (E = B, Al). In their comparative study between the geminal B/P and Al/P FLP activation of CO2 and H2 (Scheme 52), the main conclusions the authors drew are that the FLPs are more reactive towards CO2 than H2, and that the geminal B/P FLPs involve stronger orbital interactions with CO2 than their Al/P counterparts. Distortion–interaction decomposition showed that the distortion energy in the H2 fragment is higher than that in the CO2 transition state leading to a higher energy barrier for H2 activation than CO2 activation. This again highlights the importance of considering energy barriers to the activation of both CO2 and H2, similarly highlighted by the work of Corminboeuf above (Scheme 31). The type of geminal linker, sp2 or sp3, was found not to affect the reactivity.120
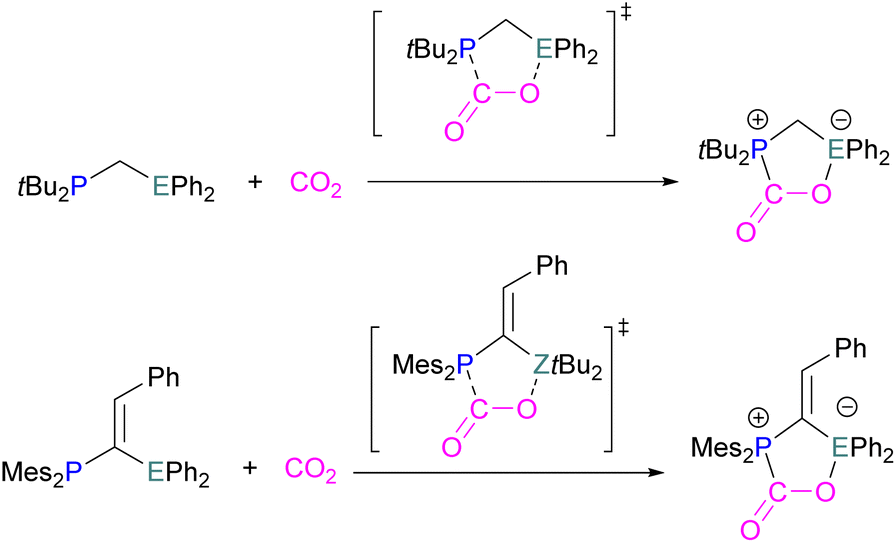 | ||
| Scheme 52 Geminal P/B and P/Al FLPs with CO2. | ||
Base-free CO2 reduction with group 13 Lewis acids
In the final example using only Group 13 Lewis acids, without a base, Chen and co-workers reported the first example of a mixed Lewis acid system consisting of Al(C6F5)3 and B(C6F5)3 for the highly selective reduction of CO2 into CH4via a tandem hydrosilylation (Scheme 53). The reaction proceeds in a catalytic manner. In the first step, Al(C6F5)3 effectively mediates the overall hydrosilylation cycle fixing CO2 into HCO2SiEt3 by activating the carbonyl group. For this initial transformation B(C6F5)3 was found to be inefficient but for the subsequent reduction steps to CH4 (Scheme 53, steps 2–4) B(C6F5)3 was found to be crucial to give CH4 in up to 94% yield through a frustrated Lewis pair (FLP)-type Si–H activation. The higher Lewis acidity of Al(C6F5)3 relative to the corresponding borane led to the formation of stable intermediates ([Al]-substrate adducts and [Al]-intermediates). In this reaction for the overall reduction of CO2 to CH4, the role observed for both Lewis acids are not only complementary but also synergic where the first reduction step is initiated by the aluminium catalyst and later by the boron catalyst.121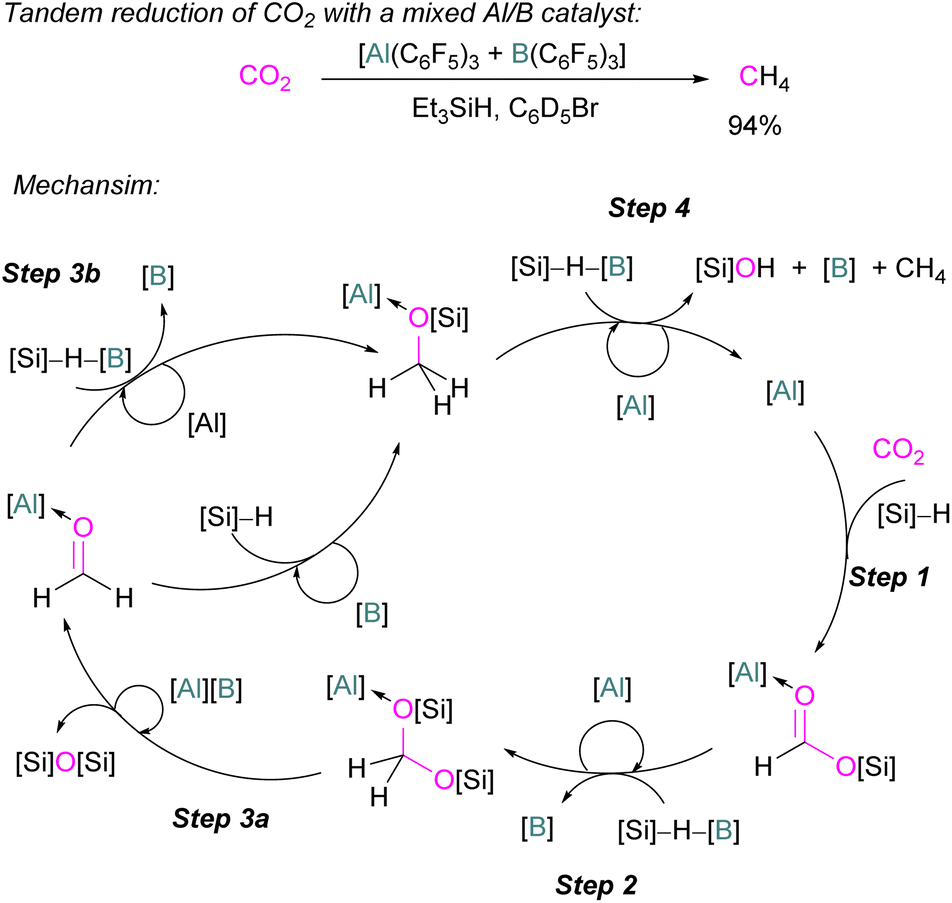 | ||
| Scheme 53 Catalytic reduction of CO2 by mixed Lewis acids [Al(C6F5)3 and B(C6F5)3]. [B] = B(C6F5)3; [Al] = Al(C6F5)3; [Si] = SiEt3. | ||
Gallium and indium FLPs for CO2 activation
Examples of homogenous gallium and indium containing FLPs in CO2 activation are rare although several heterogenous systems are known for indium (see later). Uhl and co-workers reported a dimeric gallium hydrazide displaying FLP-like reactivity able to insert CO2 into the Ga–N bond, yielding a seven-membered C2O4Ga2 cycle 86 (Scheme 54, top).122 An atypical example of FLP-like reactivity with CO2 was also reported by Goicoechea using a phosphanyl phosphagallene.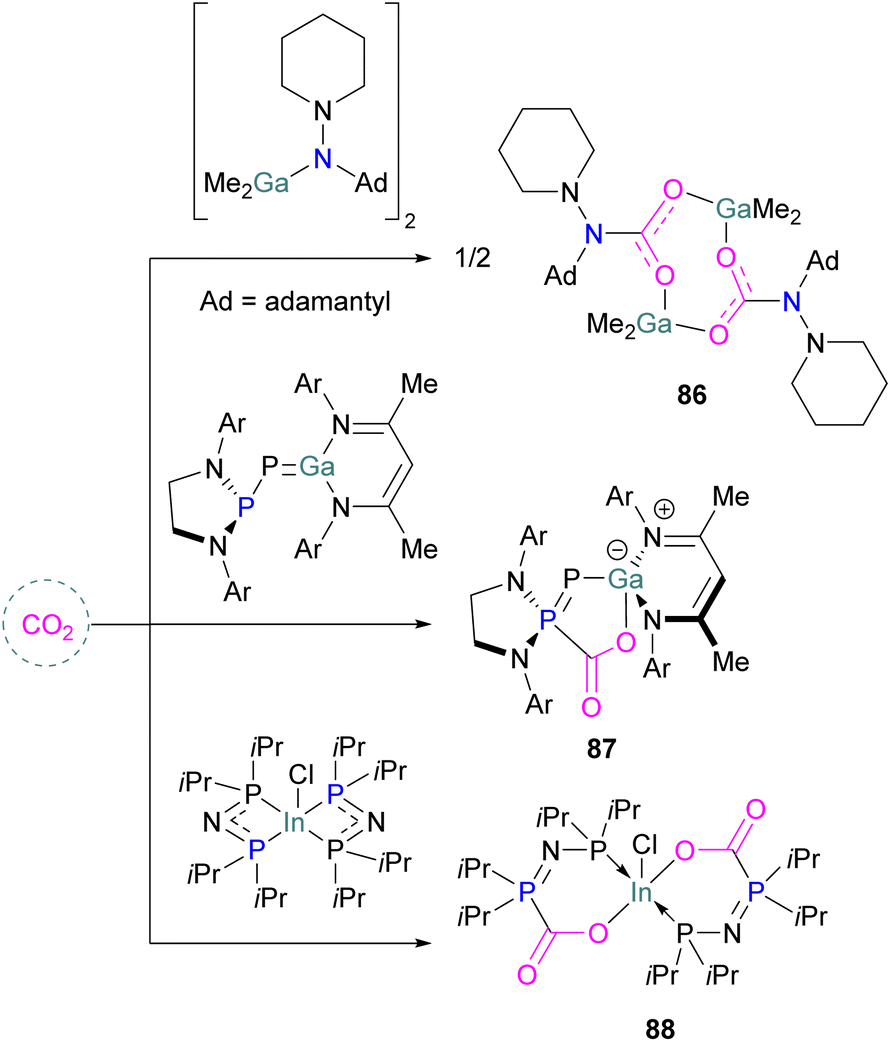 | ||
| Scheme 54 FLP type reactivity of homogenous indium and gallium systems with CO2. | ||
The compound adds to CO2 with oxidation of the phosphanyl phosphorus, with gallium bound to the phosphanyl phosphorus and one of the oxygen atoms bound to gallium 87 (Scheme 54, middle).123 Kemp and co-workers prepared a P,P-chelated heteroleptic complex bis[bis-(diisopropylphosphino)amido]indium chloride [(iPr2P)2N]2InCl. In both the solid-state and solution, it was found that CO2 inserted into two of the four M–P bonds to produce [O2CP(iPr2)NP(iPr2)]2InCl 88 (Scheme 54, bottom). Experimental analysis showed that the time taken for the insertion of CO2 at room temperature in solution condition was less than 1 minute and less than 2 h in the solid–gas reaction. The complex was stable up to 60 °C under vacuum but released CO2 when heated above 75 °C.124
Group 14 Lewis acids
Compared to group 13, group 14 elements have been less studied as Lewis acid components of FLPs for CO2 activation and conversion. Although carbenium ions such as trityl are isoelectronic with boron, the activation of CO2 with carbon Lewis acids within an FLP are not known to the best of our knowledge. Examples with heavier Group 14 Lewis acids are known, however, and are described below.Silicon FLPs for CO2 activation
Silicon cations are highly electrophilic and are therefore good candidates as the Lewis acid component of FLPs for small molecules activation. In addition, CO2 transformation into products such as benzoic acid, formic acid, and methanol using silicon cations formed from a [Ph3C][B(C6F5)4]/R3SiH system in different solvents has already been shown to be effective.125With sterically hindered phosphines, triarylsilylium borates [Ar3Si+][B(C6F5)4] form FLPs. Müller and co-workers studied a series of silylium ion/phosphane Lewis pairs [Ar3Si+/PR3]. When Ar = Me5C6 and R = tBu or Cy these FLPs were able to activate CO2 (1 atm, 30 min) in benzene at room temperature to obtain the FLP-CO2 adducts [R3P–CO2–SiAr3] (Fig. 8).126 In 2015, Mitzel and co-workers reported the first synthesis of a neutral Si/P FLP (C2F5)3SiCH2PtBu2 and this was utilised in trapping CO2 at room temperature as a cyclic adduct 89 in quantitative yields.127
 | ||
| Fig. 8 Si-based CO2 adducts. | ||
The stability of the adduct of a trimethylsilylium and a congested N-heterocyclic carbene (90) was found to be strongly dependent on the nature of the counterion used in the reaction. The stability was found to increase with decreasing nucleophilicity of the ion X, or increasing Lewis acidity of the silylating agent Me3SiX (X = I, OTf, NTf2; Tf = SO2CF3).128 Tamm and co-workers explored the N-heterocyclic carbene-silylium ion frustrated FLP for the synthesis of adduct 90 (Fig. 8).129
Like the N/B intramolecular FLP described earlier, Cantat and co-workers synthesised a series of TBDR2SiX [R = Me, iPr, Ph; X = Cl, B(C6F5)4, I; TBD = triazabicyclodecene] compounds and utilised them for CO2 capture to obtain N/Si+ FLP-CO2 adducts 91 (Fig. 8). The formation and stability of the adducts are dependent on the steric and electronic environment at the silicon centre.
Among the synthesised series, R = Me and X = Cl was found to be a good FLP adduct in reducing CO2 to methoxyboranes (R2BOMe) using 9-BBN as a reducing agent both in a stoichiometric and catalytic way. The authors carried out DFT calculations in support of their experimental results to explore the role of N/Si+ FLP-CO2 adducts in the catalytic reduction of CO2 with different boranes.130 They synthesised a series of o-phenylene-bridged phosphorus–silicon Lewis pairs and investigated their reactivity towards CO2 but no reaction was observed.131 Stephan and co-workers applied silyl triflates of the form R4−nSi(OTf)n (R = C6F5, Ph, Me; n = 1, 2; OTf = OSO2CF3) to activate CO2 for adduct formation with bulky amines and phosphines (Scheme 55). Silyl triflates Ph2SiOTf2 and R3SiOTf (R = C6F5, Ph, Me) with TMP formed the silyl carbamates 92. Trialkylphosphines also activate CO2 in combination with the silyl triflate generating FLP-CO2 adducts 93, with the silyl triflates R3SiOTf showing reversible CO2 binding. The bis-CO2 adduct 94 was obtained at lower temperature (–40 °C) and using excess phosphine.132
 | ||
| Scheme 55 Si-based CO2 adducts using silyl triflates as Lewis acids. | ||
Germanium and tin FLPs for CO2 activation
It has been proven that the cleavage of FLP-CO2 adducts are quite difficult to great extent due to the strong hard–hard interaction of oxygen and the typical hard Lewis acids used in the FLP system according to the HSAB principle.133 Ge and Sn are softer elements and are less oxophilic, thus their use could provide beneficial to enable catalytic CO2 reduction. Although FLPs with a germanium Lewis acidic centre are known and have been shown to activate small molecules, their use in CO2 activation and conversion is not yet reported.134Mitzel and co-workers reported in 2019 the synthesis of a geminal Sn/P FLP (F5C2)3SnCH2PtBu2 by reacting LiCH2PtBu2 with (F5C2)3SnCl. When the FLP (F5C2)3SnCH2PtBu2 was exposed to CO2 at −70 °C, it formed an adduct that was found to be reversible at 25 °C.135 Fernandez performed a theoretical analysis of the FLP systems (F5C2)3E–CH2–PtBu2 (E = Si, Ge, Sn) to understand the effect of the nature of these group 14 elements on their reactivity. Moving down the group, the reactivity of these species is kinetically enhanced (Si < Ge < Sn). Quantitatively, this trend of reactivity was analysed by the “activation strain model” of reactivity in combination with the energy decomposition analysis method. A five-membered TS with CO2 lead to the experimentally observed zwitterionic products. The model identifies the interaction energy between the deformed reactants as the main factor controlling the reactivity of these geminal FLPs containing Si/Ge/Sn, where the lone pair of phosphorus donates into the π* orbital of CO and a stronger electrostatic and orbital interaction is observed for Sn over Si.136 Similarly, Pati and co-workers computationally explored the ability of the FLP system (F5C2)3E–CH2–D(tBu)2 where E = Si, Ge, Sn and D = P, N to act as hydrogenation catalysts using CO2 as a substrate.137 For the FLPs where D = N, simultaneous proton and hydride migration take place, whereas for D = P FLPs, proton transfer is followed by hydride transfer. NBO analysis shows that LP(O) → σ* (D–H) and σ(E–H) → π* (CO) dominate along the energy profile. From their studies, they predict that FLP (C2F5)3Sn–CH2–N(tBu)2 would be able to perform CO2 hydrogenation particularly well.
Hulla reported an application of tin-based FLPs in the form R3SnX/N-base (R = alkyl and X = OTf− or NTf2−] which can catalyse the formation of azoles from ortho-substituted anilines via complete deoxygenation of CO2 in the presence of H2.138
Computational insights into group 13 and 14 Lewis acids in FLP catalysed CO2 activation
Grimme reported mechanistic insights, based on extensive DFT calculations, on all steps of the FLP catalysed reduction of CO2 to boryl formate, H2CO, bis(boryl) acetal, and methoxyl borane products in 2020.139 The work addressed three FLP catalysts that had been previously reported; (i) Fontaine's B/P intramolecular FLP reported in 2013,55,56 (ii) Stephan's intermolecular FLP consisting of tBu3P and 9-BBN from 2014,58 and (iii) Cantat's 2016 Si/N FLP with 9-BBN (Fig. 9, top).130 The report unveils the importance of the Lewis-basic CH2O “oxide” site in promoting a hydride transfer, from calculations (PW6B95-D3+COSMO-RS//TPSS–D3+COSMO level of theory in THF). Initial formation of a zwitterionic FLP-H2CO adduct had been proposed previously and was verified in this report, in the intramolecular FLP reported by Fontaine, this is generated through the Lewis-basic Bcat oxygen atoms 95 (Fig. 9).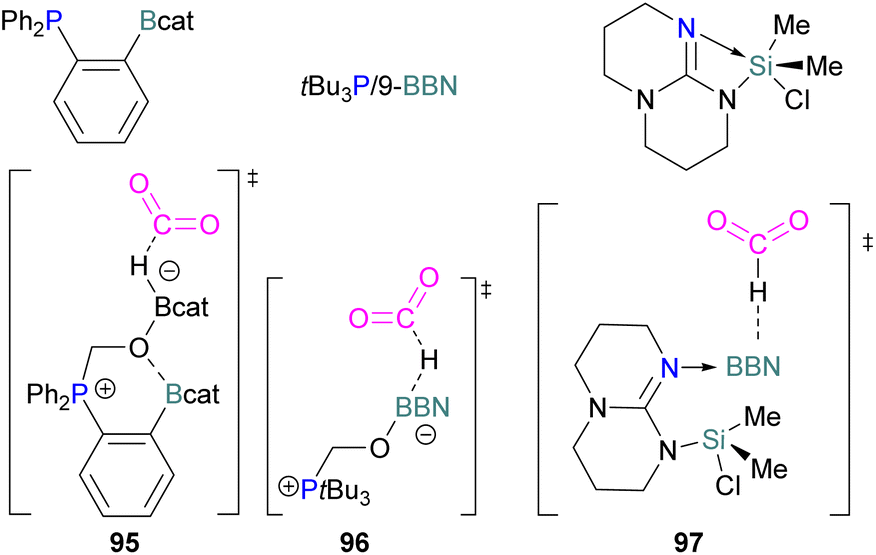 | ||
| Fig. 9 Substrates and intermediates involved in the FLP activation of CO2. | ||
Subsequent hydride transfer from the FLP-H2CO adduct to CO2 then forms boryl formate HCOOBcat through a series of steps, identifying the Lewis acidic Bcat group as the ‘base shuttle’. For Stephan's intermolecular tBu3P/9-BBN FLP, a hydride transfer from tBu3P–CH2O–9-BBN to CO2via96 (Fig. 9) is exergonic by −16.1 kcal mol−1 with a barrier of 7.2 kcal mol−1, which is feasible at room temperature. The final reduction step from H2C(O-9-BBN)2 into H3CO-9-BBN is the slowest reduction step, with a barrier of 23.3 kcal mol−1. Lastly, when investigating the mechanism for Cantat's Si/N FLP, Grimme and co-workers found the neutral adduct between the Lewis-basic N and 9-BBN to be the most energetically favourable starting point. Here, the B–H is partially activated by the Si/N centres. Hydride transfer to CO2 is then exergonic by −7.5 kcal mol−1via97 (Fig. 9). In summary, zwitterionic FLP-H2CO adducts were found to be the active catalysts, strong oxygen and nitrogen Lewis bases were found to stabilise the hydride transfer steps to CO2, and finally, Lewis-acidic groups such as Bcat were found to act as a base shuttle.
Group 15 Lewis acids
Generally, in FLP chemistry, group 15 elements are employed as the Lewis base component due to the presence of a lone pair when in the +3 oxidation state. Although nitrogen Lewis acids are known in FLPs, and have been used for small molecule activation, their application for CO2 activation has not been explored.140 On the other hand, there are a few examples using phosphorus as the Lewis acid. An example was reported by Stephan who prepared a CO2-adduct 98 based on intramolecular amidophosphoranes where the phosphorus acts as a Lewis acidic centre and a nitrogen centre in the parent FLP acts as a nucleophilic centre to capture CO2 (1 atm) at ambient temperatures. Similarly, the bis-CO2-adduct 99 (Fig. 10) was also prepared under the same reaction conditions.141 A detailed computational mechanism was studied for the adduct 98 by Zhu and co-workers. They investigated that ring strain, and the trans-influence are the key factors in amidophosphoranes to capture CO2.142 | ||
| Fig. 10 Phosphorus as a Lewis acid in CO2 capture. | ||
Transition metal Lewis acids
Earlier we have discussed examples of how low valent transition metals can act as the Lewis base of an FLP when combined with boron Lewis acids. In this section we will discuss selected reports where the transition metal behaves as the Lewis acid to activate CO2 in an FLP fashion. Several early examples by Piers reported the use of Lewis acidic scandium complexes in combination with B(C6F5)3 and a silane to be operative under an FLP type mechanism to reduce CO2.143,144 Examples by Wass and co-workers in 2011, however, were the first to extend the concept of FLPs to transition metals through the use of cationic zirconocene–phosphinoaryloxide complexes.145 Wass reported the synthesis of zirconocene–phosphinoaryloxide complexes 100 and their applications in the FLP activation of H2 to generate 101 and activation of CO2 to give the FLP-CO2 adduct 102. 102 showed no further reaction with H2, however 101 could insert into CO2 under mild conditions to generate 103 (Scheme 56, top). A similar system also reported by Wass focuses on intermolecular zirconium/phosphorus FLPs where a zirconium(IV) cation 104 is combined with a tertiary phosphine. Activation of CO2 occurred under mild conditions to yield the adduct 105 (Scheme 56, bottom).146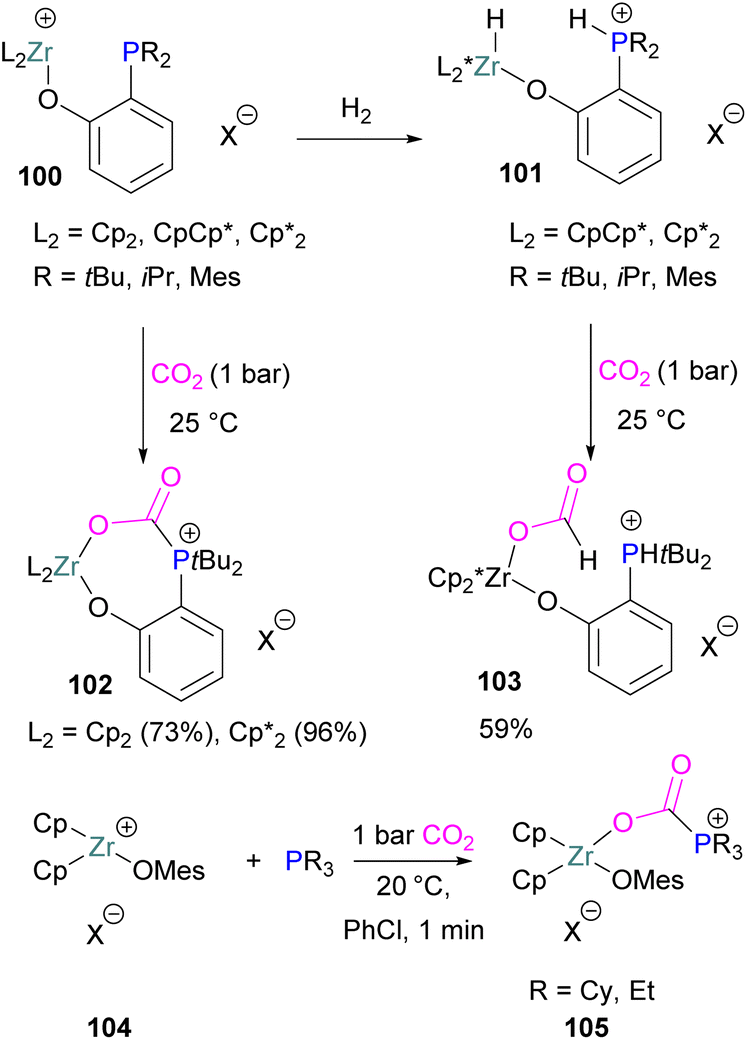 | ||
| Scheme 56 Reaction of zirconium FLPs with H2 and with CO2. X− = [B(C6F5)3]−, Cp = cyclopentadienyl, C6H5; Cp* = pentamethylcyclopentadienyl, C5Me5. | ||
Systematic modification of the phosphine Lewis base showed that FLPs with modest Tolman steric parameters are highly reactive and have the maximum selectivity for the intended product. The base was found to affect the selectivity, and PEt3 gave the cleanest results. These later findings demonstrate that transition metal FLPs do not require intramolecular systems and allow for the construction of intermolecular transition metal frustrated or cooperative Lewis pairs. Another zirconium based FLP has been reported by Erker in the form of an intramolecular cationic geminal Zr+/P pair 106 which could react with CO2 to form a five-membered metallaheterocyclic adduct 107 (Scheme 57).147 Systems based on 106 have been the subject of theoretical studies for the reactivity of the Zr+/P pair system in the activation of CO2.148 Whereas, computational investigations reveal that the activation reaction between CO2 and Zr+/P-based FLP-associated compounds is exothermic and coherent, generating a cyclic ring. The Zr+–P bond length contributes to the reactivity of these compounds. The donor–acceptor relationship was also found to determine the bonding nature of the activation reactions between CO2 and Zr+/P-based FLP-related compounds. Accordingly, the calculated OCO bond stretching distance and OCO bending angle relate to the activation energy for CO2 activation reactions with Zr+/P-based FLP-related compounds, in line with Hammond's postulate.
 | ||
| Scheme 57 Zr+/P Pair system in activation of CO2. | ||
The heavier group 4 metal hafnium has also been shown to undergo FLP-type CO2 activation between the metal centre and a pendant Lewis basic centre on the ligand.
The hafninum complex 108 was found to react with one or two equivalents of CO2 to give a series of monometallic and bimetallic CO2 activated products (109–111) depending upon the substituents on the phosphine ligand (Scheme 58).149 In these complexes, the phosphinoamines binds to hafnium via the nitrogen atom, and binds weakly through the softer phosphorus atom. Reaction of metallocene cation complexes [Cp*2HfMe][B(C6F5)4] with trimethylsilyl-(diarylphosphino)acetylenes yielded internal phosphane stabilised hafnium cations [Cp*2Hf–C(Me)–C(SiMe3)PPh2][B(C6F5)4]. As with other vicinal compounds, Hf+/P 112 exhibits FLP-like reactivity and generates the adduct 113 when reacted with CO2 (Scheme 59).150
 | ||
| Scheme 58 CO2 adduct formation using an intramolecular hafnium FLP. | ||
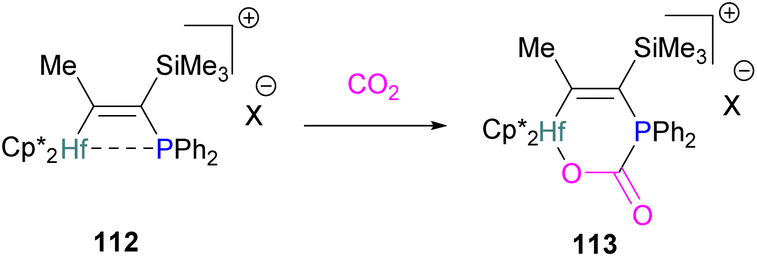 | ||
| Scheme 59 Vicinal hafnium FLP for CO2 activation. X− = [B(C6F5)4]−. | ||
An alternative approach is to use a transition metal to assist CO2 activation as demonstrated by Streubel and co-workers. The 3-imino-azaphosphiridine complex 114 was prepared and reacted with CO2 to obtain a heterocyclic compound 115 (Scheme 60).151
 | ||
| Scheme 60 Tungsten-accelerated CO2 activation. | ||
While numerous creative strategies are being developed for CO2 capture, there is an increasing interest in using carbon dioxide as a C1 carbon source. The hydrogenation of CO2 to formic acid and its derivatives is one such well-developed strategy based on Ru.152 In 2012, Stephan and co-workers developed an elegant catalytic system based on ruthenium hydride 116 (Scheme 61). This salt was explored in the reduction of CO2 catalytically using HBpin as a reducing agent. One equivalent of 116 and 18 equivalents of HBpin under an atmosphere of CO2 gave the MeOBPin product catalytically after 96 h at 50 °C. Increasing the ratio of 116:HBpin to 1:100 resulted in a small increase in the TON.153 In this system the RuNP ring in the catalyst is similar to FLP systems, and the binding of CO2 depends upon the cooperative action of the Lewis acidic metal centre with one of the Lewis basic phosphine centres in the ligand. Cleavage of the C–P bond upon reduction with HBpin and a transfer of oxygen from Ru to Bpin allows the system for a catalytic hydroboration of CO2. A similar cooperativity between metal and ligand was observed by Crispin and co-workers154 who reported the synthesis of iridium–pyridylidene complexes [TpMe2Ir(C6H5)2(C(CH)3C(R)NH] (TpMe2 = hydrotris(3,5-dimethylpyrazolyl)borate; R = H, Me, Ph) 117.
 | ||
| Scheme 61 Catalytic reduction of CO2 using a Ru–H salt and pinBH. | ||
These species can activate a range of small molecules including CO2 (RH) in an FLP fashion between a Lewis acidic iridium centre and a Lewis basic nitrogen atom on the pyridyl ligand (Scheme 62).
 | ||
| Scheme 62 Cooperative activation of CO2 using an iridium catalyst. | ||
Similar to the tungsten system described earlier, other metals have been employed to assist classical FLPs in CO2 reduction. A copper-hydride system has been developed by Bertrand and co-workers for the activation and reduction of CO2 in synergy with a N/B FLP. Amongst several screened reactions, both stoichiometric as well as catalytic, the authors found that a catalytic system consisting of a (CAAC)CuH (CAAC = cyclic (alkyl)(amino)carbene) 118 with B(C6F5)3 in a 1:2 ratio and DBU (10 mmol) forms the formate salts of DBU from CO2 (15 bar) and H2 (45 bar) when heating the reaction mixture at 100 °C for 24 h in THF (Scheme 63). The key step in this reaction is the insertion of CO2 into the copper hydride and regeneration of copper hydride with H2. While the Cu–H bond readily inserts CO2, it is difficult for copper to activate H2. Thus, the FLP assists by activating H2 to allow regeneration of the copper hydride. The TON for this reaction is observed as 1881.155
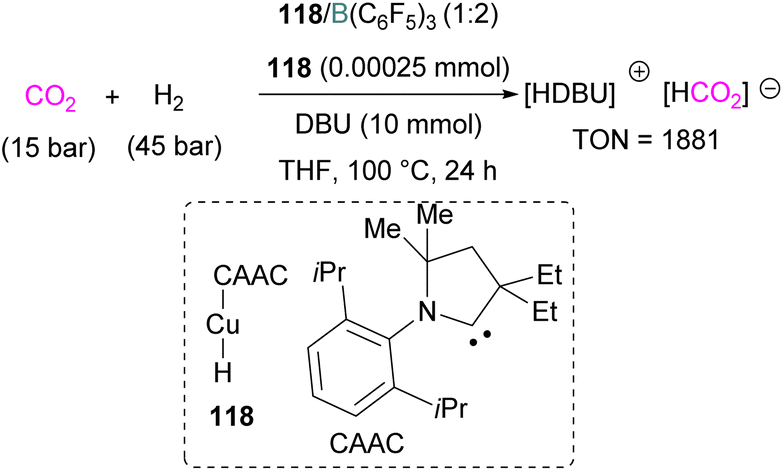 | ||
| Scheme 63 Catalytic reduction of CO2 with Cu–H/Lewis pair system. | ||
Zinc metal has been explored for the reduction of CO2 to CO. Stephan reported the in situ formation of the catalytically active species Et3PCPEt3 through the reduction of CO2 to CO employing CH2I2.
This (bis)ylide was found to interact with CO2, eliminating the phosphine oxide Et3PO as a by-product and forming an interim phosphaketene. The addition of catalytic ZnBr2 was found to facilitate the process through an FLP-type activation mode and was important for the regeneration of the (bis)ylide with simultaneous removal of CO (Scheme 64).156 The same authors described Zn-based FLP chemistry for functionalising CO2, using tBu3P/ZnEt2 FLPs.157
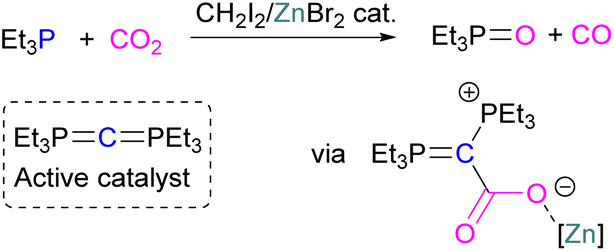 | ||
| Scheme 64 Reduction of CO2 using phosphaketene/Zn FLPs. | ||
The transition metal based Lewis acids in the activation of CO2 benefit from a higher coordination number when compared to the lighter main-group Lewis acids that were discussed previously. The ability of the TM species, such as in 110 and 116, to bind CO2 as well as ligand systems in more intricate intermediary species offers a higher degree of fine-tuning of the stability of adducts formed. Hence, higher turnover numbers may be observed for transition metal systems, as regeneration of the active catalyst species is kinetically more favourable. This may also explain for the more accessible use of H2 as the reducing source compared to silanes or hydrogen surrogates often used for the main group systems. This is seen for 118 and 100, whereas main group based Lewis acids frequently require pre-organised reducing sources, limiting the reaction to hydroboration or hydrosilylation of CO2 rather than direct hydrogenation. Nonetheless, concerns of toxicity and environmental impact drive an increased interest in transition metal free reagents. The work on main-group CO2 activation has focused on mimicking transition metals in synergistically accepting and donating electrons in the activation of CO2 with species that combine filled and vacant orbitals.
Rare earth metals
Rare earth metals are often employed as Lewis acids for a variety of reactions, and have also been employed as Lewis acids in FLPs. Dihydrogen was readily activated by combination of homoleptic rare-earth metal aryloxides, RE(OAr)3 (RE = La, Sm, and Y) with N-heterocyclic carbenes (NHCs) under mild conditions. In addition, the La/NHC pair exhibited FLP-like reactivity towards carbon dioxide, affording 1,2-addition products 119, as shown in Scheme 65.158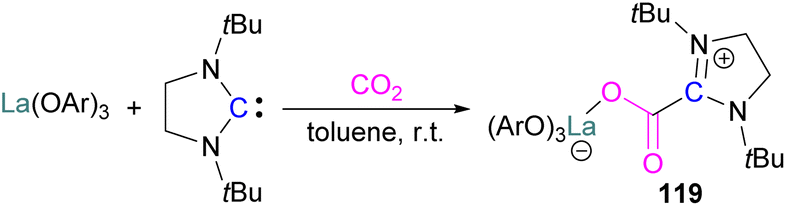 | ||
| Scheme 65 La-in CO2 activation. Ar = 2,6-tBu2C6H3. | ||
Heterogenous FLPs
Recent discoveries of FLPs as homogeneous catalysts has more recently turned to heterogeneous catalysts which operate by an FLP-type mechanism in which the Lewis acidic and Lewis basic centres in the solid structure activate CO2. Here, the understanding the chemistry of reactants, intermediates, and products on surfaces is crucial for designing catalytic nanostructures that transform carbon dioxide into carbon-based fuels. Several systems have been reported using indium as a Lewis acid. For example, indium oxide nanocrystals, In2O3x(OH)y, can catalyse the reverse water gas shift reaction, reducing carbon dioxide to carbon monoxide and water.159 Surface hydroxide groups and oxygen vacancies facilitate this reaction as shown in Scheme 66.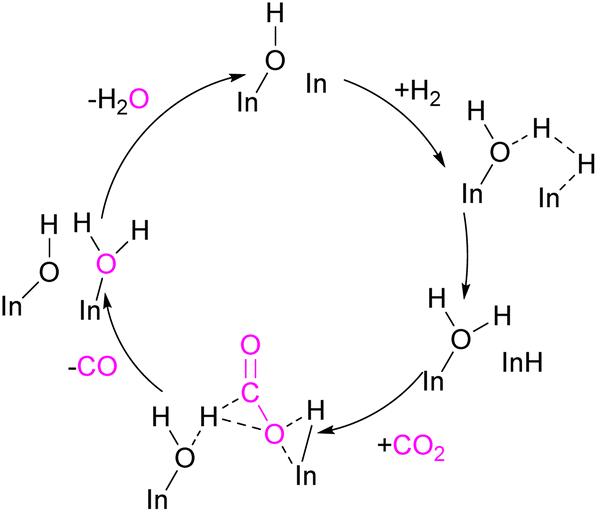 | ||
| Scheme 66 In-FLP mediated CO2 reduction. | ||
The enhancement of activity in the gas-phase reverse water gas shift process has also been investigated, as well as the distinct photoactive behaviour of pristine and defective indium oxide surfaces.160 Based on TD-DFT calculations, this study discovered that surface FLP in In2O3x(OH)y has Lewis acidic indium sites close to a Lewis basic surface hydroxide. These acquired more acidity/basicity making them more active in the excited state relative to the ground state. In the photochemical reaction this reduces the activation energy relative to the thermal reaction, and could provide a mechanism to design improved photocatalytic systems for solar fuel production. In 2018, Ozin and co-workers, reported a similar system based on a rod-like nanocrystal superstructure of In2O3−x(OH)y, that could effectively catalyse the hydrogenation of CO2 to methanol under light at atmospheric pressure. The rate of conversion was found to be 0.06 mmol g−1 h−1 with a 50% selectivity and a long-term working stability.161
Another heterogenous system based on indium has been developed by Wang and co-workers (Fig. 11). The authors developed a photocatalytic material based on a ZnIn2S4/In(OH)3 − x heterojunction that works in a cooperative fashion to reduce CO2 into CO driven by light. The ZnIn2S4 functions to harvest the light and transport an electron to the FLP-activated CO2 on the In(OH)3−x surface. In In(OH)3−x, the hydroxyl-deficient vacancies (OHVs) acts as a Lewis acid, and the adjacent hydroxyl groups act as a Lewis base generating the FLP which activates CO2. This composite showed a CO formation rate of 1945.5 μmol g−1 h−1.162
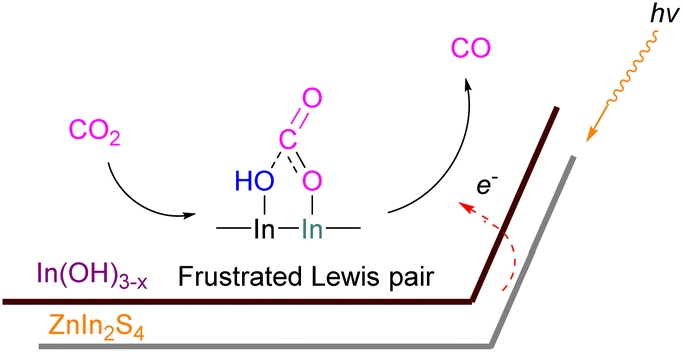 | ||
| Fig. 11 An FLP ZnIn2S4/In(OH)3 − x (ZIOS) heterojunction system for the reduction of CO2 to CO. | ||
Above we have seen that heterogenous FLPs can capture and react with H2 and CO2, boosting photocatalytic CO2 reduction. Isomorphous substitution of In3+ with Bi3+ has been found to increase catalytically active surface FLPs. Isomorphous substitution optimises surface catalytic active sites and affects optoelectronic characteristics, improving our understanding of photocatalytic CO2 reduction. Such isomorphous substitution will help to develop CO2 reduction materials with higher catalytic performance by tuning surface FLP site strength.163 Another bismuth containing heterogenous system has been reported by Wang who synthesised Sn-doped BiOBr with oxygen vacancies. The synthesised material possesses surface frustrated Lewis acidic (bismuth) and Lewis basic (lattice oxygen) pairs in BiOBr through the substitution of Bi3+ with Sn4+. 4Sn–BiOBr showed the best performance for photocatalytic CO2 reduction into CO with a yield of 165.6 μmol g−1 h−1.164 Another main group heterogenous system also reported is B3P3 doped hexa-cata-hexabenzocoronene, a model of nanographene (B3P3·NG) which reacted with carbon dioxide. This multi FLP device binds three CO2 molecules sequentially or simultaneously on the B3P3·NG surface.165 For the CO2 reduction via dissociative chemisorption of H2, nanocarbon-based FLP bifunctional catalysts are becoming promising due to their unquenched electron transport capability. One study proposes a nanocarbon-based FLP catalyst for the CO2 reduction via the dissociative chemisorption of H2.166
The catalyst consists of nitrogen/phosphorus doped graphene and M(C6F5)3 (M = B, Al, Ga, In) as Lewis acids. The study demonstrates the potential of doped carbon-based FLPs as innovative nanostructure catalysts for CO2 reduction via molecular hydrogen. N-doped FLP catalysts with activation barriers between 0.01 and 0.11 eV are promising for CO2 reduction, potentially enabling CO2 reduction catalytic material design.
Converting and storing solar energy through light-driven CO2 reduction is a promising area of research. A particular approach for converting CO2 to methane gas uses hydroxyls inherent on an oxyhydroxide photocatalyst, such as in CoGeO2(OH)2, as a proton source. Irradiation of CoGeO2(OH)2 causes the lattice hydroxyls to be oxidised by photogenerated holes, leading to the formation of oxygen vacancies (OVs) and protons. These OVs and Lewis acid-base pairs bind CO2 and protons to activate it before reducing it to CH4. In the presence of water molecules, the surface lattice hydroxyls regenerate, allowing for continuous CO2 conversion as shown in Scheme 67. This strategy has the potential to pave the way for a novel use of photocatalysis in the field of energy conversion.167
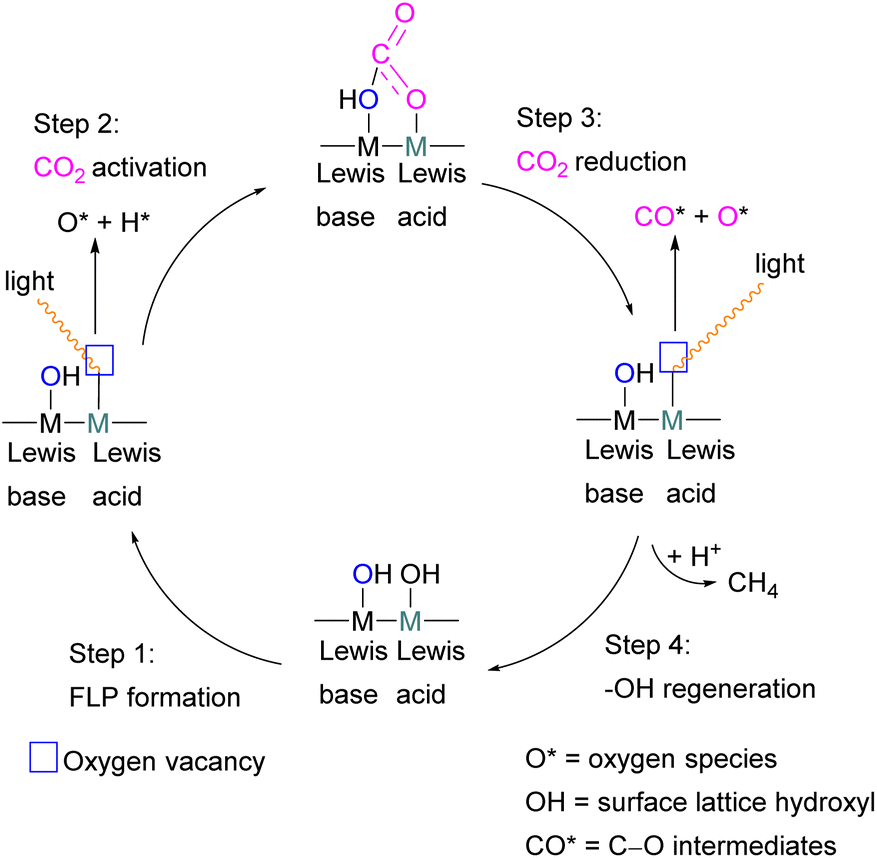 | ||
| Scheme 67 Heterogenous Ge FLP in the reduction of CO2 to CH4. M = Metallic element. | ||
Zinc based heterogenous systems have also been reported. Neumann and co-workers studied the coordination of CO2 to a Zn(II) Lewis acid site in Wells–Dawson type polyoxometalates α2-(P2W17O61Zn)8− which bound CO2 in an FLP fashion (Fig. 12). This system reveals two distinct binding modes: stronger “side-on” binding at higher temperatures and weaker “end-on” architectures at lower temperatures.168 This interaction with 2,4,6-collidine is possible through the development of a frustrated Lewis pair at lower temperatures.
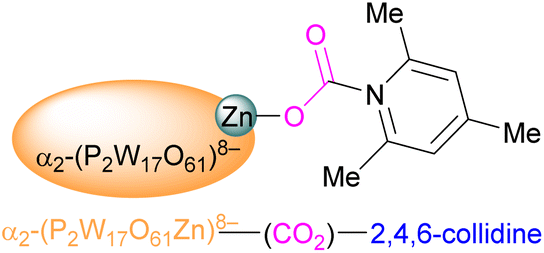 | ||
| Fig. 12 CO2 coordinated with α2-(P2W17O61Zn)8− and 2,4,6-collidine. | ||
Like the indium oxide systems described above, efficient photocatalysts for CO2 reduction have also been reported using titanium. Anatase TiO2−x hierarchical hollow boxes with FLPs can be synthesised through in situ topological modification of perovskite as shown in Fig. 13. These structures possess strong adsorption and activation properties, converting CO2 to CO without auxiliary substances. This innovative approach converts solar energy into chemical energy.169
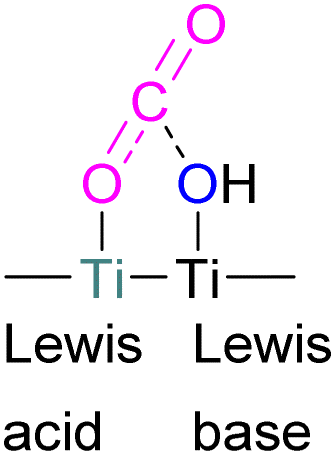 | ||
| Fig. 13 Photocatalytic reduction of CO2 with FLP-TiO2−x. | ||
The above examples of heterogeneous FLPs are important for the activation of CO2 compared to synthesising FLP-CO2 adducts.
There are some challenges to select suitable methods for achieving light absorption, electron–hole separation, energy gap matching for the reduction of CO2 to different products (product selectivity) in a photochemical way. The examples In2O3x(OH)y, ZnIn2S4/In(OH)3−x, 4Sn–BiOBr, and CoGeO2(OH)2 are promising systems for the photochemical reduction of CO2. The use of clean sources of the reducing agent H2 in these systems will suppress the chemical waste which is generated when activated reducing agents for the reduction of CO2 adducts are used.
Several reports of cerium as a Lewis acid in heterogenous FLPs are reported. Qu synthesised a defect-enriched cerium oxides (CeO2) with constructed interfacial FLPs (Ce3+···O2−) that activate CO2 efficiently via the interactions between the carbon atom of the CO2 molecule with the Lewis basic lattice O2− in CeO2, and the two oxygen atoms of CO2 with two adjacent Lewis acidic Ce3+ centres in CeO2. This CeO2 solid material showed FLP-inspired tandem activation of CO2 and reactions with alkenes to catalytically form selective cyclic carbonates.170 Davide et al. reported that CO2 activation is shown to occur via a bidentate carbonate bridging the FLP through a Ce3+-to-CO2 charge transfer (Scheme 68).171 The authors performed a detailed study of the system in which an FLP was formed over a highly defective sample of CeO2.
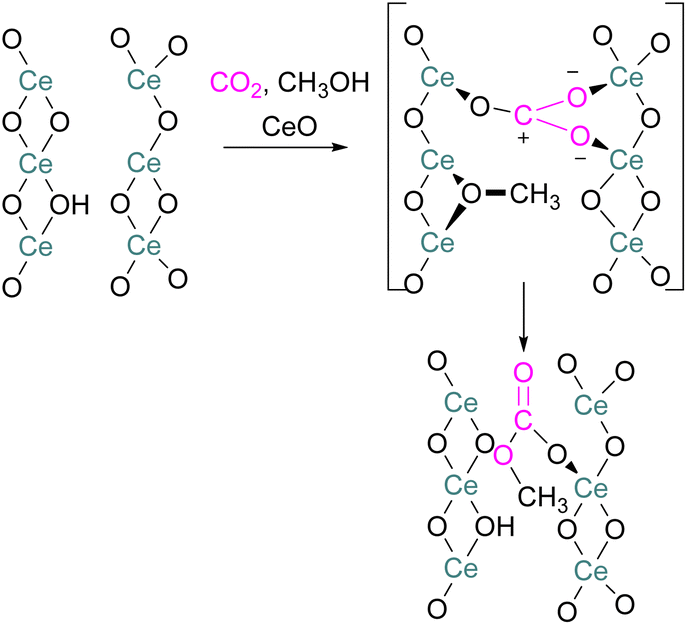 | ||
| Scheme 68 CO2 reduction using a CeO2 surface FLP to monomethylcarbonate. | ||
The reaction of CO2 with MeOH formed monomethylcarbonate through an FLP mechanism involving Ce3+ and oxygen vacancies.
Recently, other cerium based FLP systems have also been explored for the reduction of CO2 into products such as CH4 and carbonates.172
Finally, it should be noted that there are several metal organic framework (MOF) systems that have Lewis acidic metal centres and Lewis basic ligands that can also act in an FLP manner to activate CO2.173–175
Stoichiometric and catalytic reduction of CO2: scope and limitations
So far, we have witnessed a wealth of FLP systems for CO2 activation and reduction to different products. In this final section we summarise the key findings of the different FLP systems described in terms of CO2 activation and the stability of the CO2 adducts, as well as the CO2 reduction strategies using hydrogen, silane or borane reducing agents.An insight into the stability of CO2 adducts
In this review many different FLP adducts with CO2 are discussed. Stability of the FLP-CO2 adducts is dependent on different factors, such as the state of the system (solid or solution phase), temperature and conditions (e.g. such as applying vacuum), the strength of the Lewis base-C (CO2) and Lewis acid-O (CO2) bonds, steric effects around the ligand attached to the acidic or basic reactive centre, and the geometry of the FLP system either as intra- or intermolecular systems. Several systems undergo reversible CO2 activation which is highly dependent upon the geometry of the FLP (intra- or intermolecular) as well as the electronic and steric effects at the Lewis acid and basic sites. Among the described examples of the FLP systems for CO2 activation, the first was described for the B/P intra- and intermolecular systems. The FLP tBu3P/B(C6F5)3 with CO2 was observed to form a stable adduct tBu3P-CO2-B(C6F5)3 at room temperature but, upon heating under vacuum releases CO2 and regenerates the FLP. The same outcome was observed for intramolecular system 2, which produces a cyclic adduct with CO2 with lower stability which decomposes even at −20 °C.34 When the Lewis base and acid are aligned in a geminal fashion, an increase in reactivity is observed as seen in the formation of adduct 10 with a non-fluorinated FLP.42 A unique binding mode of CO2 in the FLP system bis-borane was observed that resulted in compound 9 as a six-membered stable adduct in which two boron Lewis acidic centres in the FLP bind to the two oxygen atoms in the CO2 molecule. This, however, was not the case for the FLP tBu3P/O(B(C6F5)2)2, where chelation of CO2 by two B-centres was not observed due to steric effects and as well as a significant π-character in the B–O bonds supported by crystal structure information.41 With the more Lewis acidic and oxophilic aluminium Lewis acids, more stable CO2 adducts were typically observed and in several cases both oxygen atoms of CO2 bound to a Lewis acidic site, through coordination to two aluminium centres for example in compounds 74–76 and 78.103,111 The binding strength could be tuned by varying the substituents on aluminium. The more Lewis acidic centres –AlCl2 and –Al(C6F5)2 bind CO2 irreversibly, while less Lewis acidic –AlMeCl binds CO2 reversibly under 2 bar CO2, liberating CO2 when the excess pressure is released. For catalytic applications, this reversible binding is necessary to enable release of the product. In several cases, not only CO2 activation is observed but also further reactivity of the adduct with the FLP to generate more stable CO2 activated products. These reactions, however, would be irreversible and therefore stoichiometric. Examples of this have been observed in the B/N FLP 34 in which a cyclohexyl group migrates from boron to an adjacent carbon centre.72 Overall, in the activation of CO2 molecule specially in the homogeneous FLP-CO2 adduct systems, a species that can reversibly form weak adducts of CO2 with almost no energy barrier in either direction would be an incredible valuable tool to enable catalytic transformations. Alternatively, combinations of LA and LB are promising that show reversible CO2 binding. Homogeneous and heterogenous metal based FLP systems have been shown to be very promising in the activation of CO2 through an FLP mechanism and offer much promise for catalytic turnover (see later).CO2 reductions in FLP systems
As we have seen in this review, the binding and activation modes of small molecules (in this case CO2 and H2) are different in homogeneous (metals and non-metal in inter- and intramolecular) and heterogeneous FLP systems. In homogeneous systems, the combination of Lewis base and acid in frustrated pairs typically cleave the H–H bond heterolytically and form ion pairs [LB–H]+[LA–H]−. [LA–H]− acts as a hydride source and can reduce CO2 by transferring H− to the carbon atom. The oxygen anion generated is then trapped by the Lewis acid generating formates (Fig. 14A). The formation of formate salts of CO2 with H2 surrogates [LB–H]+[LA–H]− are shown as examples in Schemes 12, 24 and 25. To utilise this strategy for CO2 reduction in a catalytic way has been studied computationally showing a possible reduction of CO2 to HCO2H in Schemes 15 and 16 but practically has not been demonstrated. For a practical feasibility, the ion pair [LB–H]+ should be able to supply H+ to the formed formate ion [HCOO–LA]−. If this occurs, then the FLP would catalyse hydrogenation of CO2, but the limiting factor is the release of the formate from [HCOO–LA]− due to the strength of the O–LA bond. In other words, for a catalytic hydrogenation of CO2 using FLPs, the ion pair [LB–H]+[LA–H]− should regenerate the free Lewis acid and base following CO2 reduction. This remains a key challenge in main group FLP-CO2 reduction and the use of a strong Lewis acid often precludes product release. One strategy to overcome this could be to use inverse frustrated Lewis pair systems. This was observed for the use of excess Lewis base DBU in combination with the Lewis acid tbtb (tris(p-bromo)tridurylborane), an inverse FLP as shown in Scheme 31.83 Another example of catalytic hydrogenation of CO2 to formate was explored by using K2CO3/B(C6F5)3 with H2.102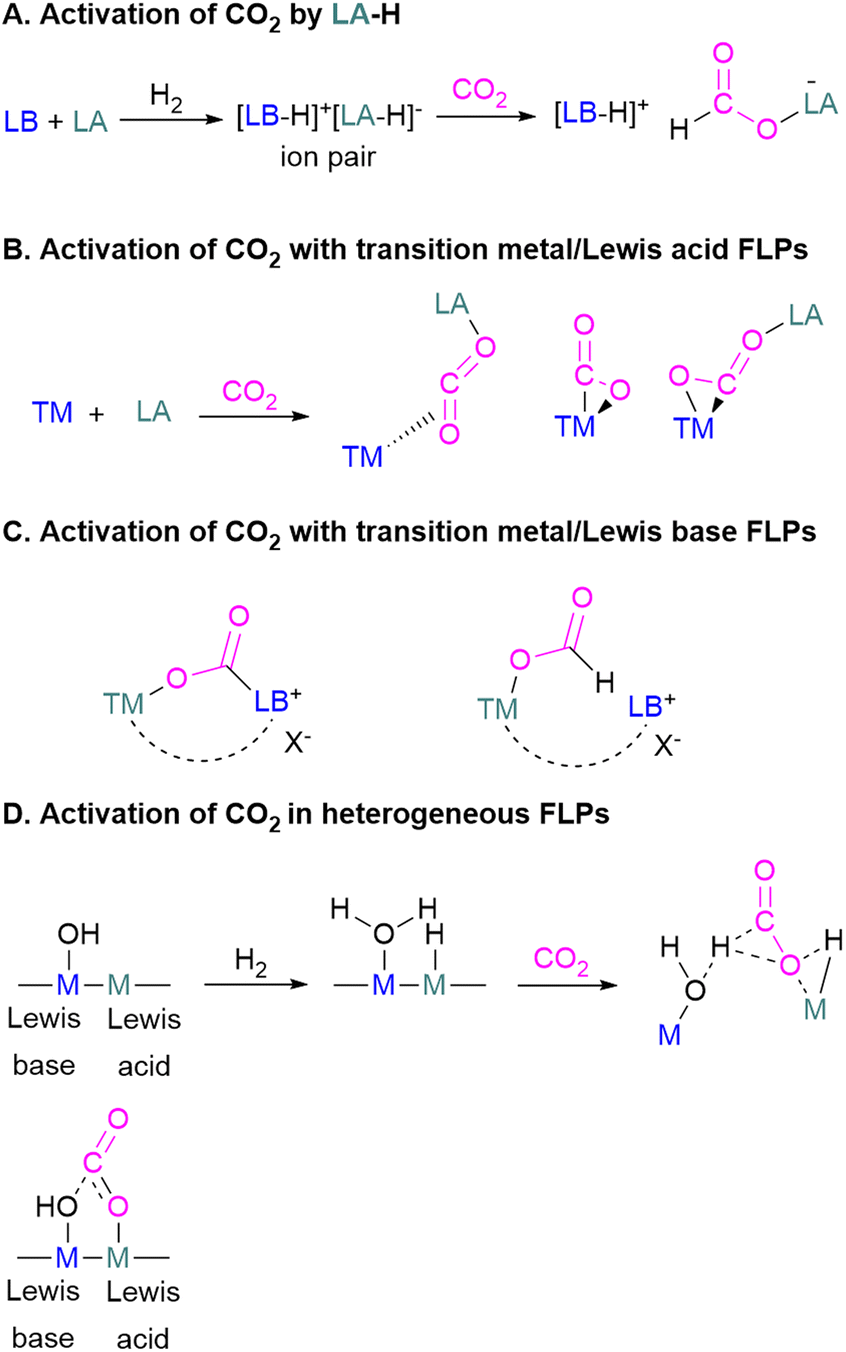 | ||
| Fig. 14 CO2 activation mode in different FLP systems. | ||
Metal systems as a Lewis basic centre in combination with a Lewis acid show a different way of activating CO2 and coordinate to the CO2 molecule through the CO bond (Fig. 14B) as seen with Lewis basic Pt, Re and Mo systems. The Lewis acidic component of the FLP then activates the oxygen atom. CO2 can be found in a reduced state with a Re system where reduction to formate is observed through CO2 hydrogenation using H2. When the transition metal is incorporated as the Lewis acid component of an FLP, then the mode of CO2 activation is similar to that observed for the main group Lewis acids with TM–O bond formation (Fig. 14C). Here, it is interesting to note that the system before reaction with CO2 can cleave H2 heterolytically, similar to other FLPs and the TM–H bond then inserts into the CO2 molecule. An example for this type of reaction is shown for cationic zirconocene–phosphinoaryloxide complexes in Scheme 56.
Heterogeneous FLPs systems activate CO2 using a similar concept but via a different mechanism (Fig. 14D) and are generally not limited by some of the challenges that main group systems face. In these systems the surface has Lewis acidic metal sites such as indium or titanium. However, the Lewis basic sites are typically an oxygen atom (often as a hydroxy group). This tolerance to hydroxy functional groups is a significant advantage in the heterogenous systems and provides easier routes for CO2 reduction. In many of the main group FLP systems described herein the strong binding to oxygen centres, while beneficial for CO2 activation, is detrimental to catalytic turnover.
CO2 reduction by FLPs: using H2, activated B–H and Si–H
Several catalytic reduction methods have been developed utilising different FLP systems using different reducing agents. For CO2 reduction, direct hydrogenation provides the best approach as it is clean and generates no waste. However, many FLP systems, especially those using the main group elements, have used other reducing agents such as silanes or boranes due to the inability of the system to activate H2 in conjunction with CO2. Thus, although some FLPs showed promising results for the CO2 reduction with a direct use of H2 gas as a reducing agent, heterogeneous systems have shown to be more promising with the direct use of H2. As described by several computational studies considering hydrogenation of CO2 with only H2 as the reducing agent, the energy barrier to the activation of H2 by the FLP system is generally higher than the activation barrier for that of CO2. Fine-tuning of both the Lewis acid's ability to accept a hydride from H2 and the Lewis base's ability to accept a proton from H2 is necessary to consequently reduce activated CO2. The computational work herein have highlighted the importance for a cumulative high Lewis basicity and acidity, whilst avoiding the combination of a very strong Lewis acid with a very strong Lewis base as this negatively impacts both the activation barriers to H2 and CO2.For many systems seen in this review, boron reducing agents have commonly been employed in homogeneous systems, for example R2BH/HBpin. Here either the Lewis base or acid activates the reducing agent towards reduction, as shown in Fig. 15. The first mode of CO2 reduction with R2BH and the Lewis base is the nucleophilic activation of R2BH. Strong nucleophiles favor hydride transfer to CO2 by increasing the hydridicity of the B–H bond (Fig. 15A). Alternatively, the Lewis acids may abstract the hydride of the B–H bond to yield a boron electrophile and convert the Lewis acid catalyst to a strong hydride donor (Fig. 15B).176 In these two mechanisms, the Lewis acid component is then trapped by the generated oxygen anion. In these cases, the formation of stable CO2 adducts often hampers catalysis by stabilising the catalyst's resting state. As we have seen above, the FLP catalyst can also activate the CO2 molecule directly. As CO2 is a weak Lewis base, this mode of action usually involves a bifunctional activation of CO2 with the cooperative effect of a Lewis base and a Lewis acid (Fig. 15C). The borohydride reducing agent then can directly hydrogenate this species. In many cases, “activation” of CO2 in the form of an adduct is both deleterious and necessary to the catalytic activity, as it stabilises the lowest intermediate in the potential energy surface yet prepares CO2 for the subsequent reduction steps by removing electron density from the carbon by coordination to the Lewis acid. It is noteworthy that future catalytic systems based on this approach should be target compounds that show a lower affinity for CO2, based on thermodynamics, yet increase the electrophilicity of the carbon centre. Instead of borane reducing agents, silanes such as Et3SiH have also been used for catalytic CO2 reduction. For example, using R3SiH with a Lewis acid, an electrophilic activation is observed similar to the way shown Fig. 15B. Although several examples use H2 as the reducing agent, this remains a challenge with main group systems and more success has been observed in this regard when using either homogenous or heterogenous transition metal systems.
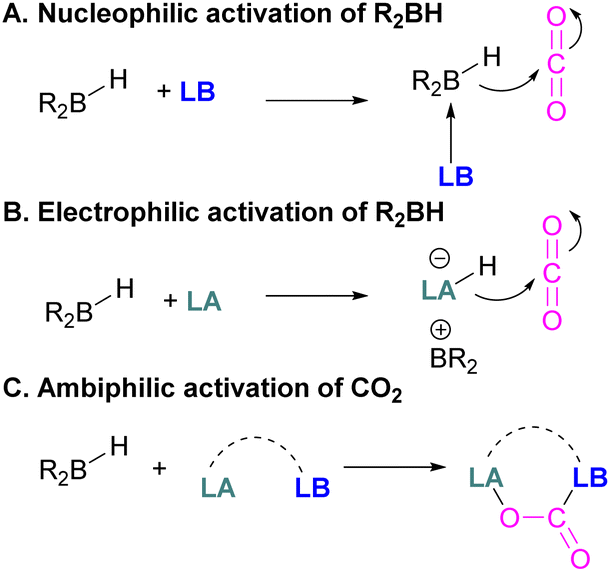 | ||
| Fig. 15 Activation of boranes in CO2 reduction. | ||
Stoichiometric and catalytic reduction of CO2 in FLPs
Various homogeneous and heterogeneous FLPs systems have been investigated for the reduction of CO2. It depends on certain properties of the FLP system to guide the CO2 reduction either for a stoichiometric or a catalytic reaction pathway. For CO2 adducts, a strong Lewis base and a weak Lewis acid adduct of CO2 appear suitable for catalytic CO2 reduction whereas stronger Lewis acids form stable CO2 adducts. To carry out a catalytic reduction of CO2 in a zwitterionic system, a first condition is that the bond between O atom of CO2 and the Lewis acid should be weak, i.e. the Lewis acid should not be too acidic or oxophilic (see Fig. 16). Secondly, the cation of the activated reducing agents (R2B–H or R3Si–H) after supplying hydride ion should be able to trap the oxygen atom, cleaving the O–LA bond. Here, both the Lewis acid and base are not coordinated, and the system will be able to reversibly activate and reduce CO2 leading to a catalytic pathway. Several CO2 adduct systems have been discussed for catalytic CO2 reductions. Intramolecular FLP 21 acts as an efficient catalyst because it does not form an adduct with CO2 (as most of the FLPs form stable CO2 adducts), and the CH2O moiety is released upon reduction from the catalyst and thus makes 21 active for another turnover.55–57 In the tBu3P/9-BBN FLP system, hydride transfer from boron to the carbonyl carbon releases tBu3P for the next cycle, and hence this system also works catalytically.58 Another interesting catalytic example is for the TMP/B(C6F5)3 FLP system, where Et3SiH is employed as a silane reducing agent producing Et3Si+ following B(C6F5)3 Si–H activation.78 Et3Si+ is a good oxygen acceptor and thus promotes the catalytic deoxygenation of CO2 to CH4 and (Et3Si)2O. Although catalytic, the stoichiometric use of silane is not desirable in the longer term and routes that employ H2 as a hydrogen source should be sought. Making use of FLPs in direct catalytic hydrogenations of CO2 remain difficult. Although hydride transfer from [LA–H] to CO2 have been described in this review, the proton transfer from [LB–H] does not occur readily due to the formation of a strong O–LA bond (Fig. 16). Hence, most FLP systems are seen to terminate at adduct formation as O–LA, and although the H2 activation barrier may have been overcome, full hydrogenation of CO2 is prohibited. Some success has been achieved with inverse FLP systems (e.g. tris(p-bromo)tridurylborane (tbtb)/DBU).83 To obtain a suitable FLP for the direct catalytic hydrogenation of CO2, the intrinsic reactivity of each of the components is very important i.e., free energy of proton attachment to the Lewis base and free energy of hydride attachment to the Lewis acid. For high turnover numbers and turnover frequencies, a catalyst should be stable enough and should regenerate in the catalytic cycle. In some of the examples, the highest in carbene system 58 using 9-BBN as the reducing agent, FLPs have shown a good TON and TOF for the reduction of CO2. As shown in the latter part of this review, metals and heterogeneous FLPs are generally efficient to activate and use H2 as a direct reducing source for CO2. For example, indium oxide has been found to be a particularly good example of a heterogeneous FLP system where H2 is directly utilised for the reduction of CO2.159 The potential of transition metals is that they provide reactive sites for the activation of H2 as well as CO2, but in several cases, they are not cost efficient. | ||
| Fig. 16 Mode for facile reduction of CO2. | ||
Conclusions
Tremendous progress in CO2 activation and reduction to value added products in both homogeneous and heterogeneous systems on bench scale has been seen. Both homogenous and heterogenous transition metal systems, as Lewis acids and bases have been efficient for the reduction of CO2. However, main group elements have emerged as alternatives and remarkable progress has been made here. Group 13 elements, boron and aluminium as Lewis acids have been heavily explored combined with Lewis bases such as phosphines, amines, or carbenes. In many cases, activation of CO2 has been achieved up to full conversion under mild conditions, and in several examples reduced products can be obtained in the form of methanol, formates, acetates and CH4 upon addition of a silane or borane, or in some cases H2. Most commonly, the formation of a zwitterionic product is the initial key activation step in the reduction of CO2. However, subsequent product liberation has been seen to be the most limiting step in these reactions, thus limiting the scope of catalytically viable reactions. Currently H2 is utilised as a reducing agent on industrial scale as it is cheap and widely available, but its use in CO2 reduction with FLPs is limited and other reducing agents such as hydrosilanes, hydroboranes or ammonia boranes are often used. However, a drawback of these reducing agents in CO2 reduction is that they form strong Si–O and B–O bonds and form oxidised products such as siloxanes or boroxanes, making the process less atom economic. Thus, there still need to be significant development of FLP CO2 reduction using H2 as the reducing agent. Compared to main group FLP-CO2 adduct systems, transition metals possessing empty orbitals that offer site selective coordination have been applied as more suitable systems to activate the non-polar covalent bond in H2 and utilise this as a direct reducing source. Conversely, heterogeneous systems provide the opportunity of recycling and have also been seen to achieve higher TONs and TOFs. Present chemical methods of recycling siloxanes or boroxanes to the corresponding hydrosilanes or hydroboranes are energy intense. Electrochemical methods are efficient and have made material recycling possible. Therefore, electrochemical methods may be an attractive and efficient approach over chemical routes for recycling of the oxides to their corresponding hydrides. The key to obtain high TON for hydrosilylation, hydroboration, or hydrogenation relies on the stability of the catalyst, the release of the reduced products and then catalyst regeneration. Thermodynamic control is the main limitation in accessing CO2 reduced products beyond carboxylates, whilst kinetic control is the main limitation in achieving high output catalytic cycles that regenerate the catalyst. Although some progress has been made to achieve CO2 reduction in a catalytic manner, the challenge is still to achieve a robust and effective FLP system that can catalyse CO2 reduction at ambient temperature and pressure utilising H2 as a reducing agent. In addition, there must be more focus on selective reduction reactions to give valuable products that are of interest to industry. Further investigations should focus on developing highly active and selective FLP systems for the catalytic conversion to C1 or C2 products, and could include various methods for conversion such as thermal, photochemical, or electrochemical methods.Author contributions
All authors contributed to the writing and revisions of the review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank BP (Alejandro G. Barrado and Sheetal Handa) for funding, and for their guidance in preparing this review.References
-
(a) K. Abbass, M. Z. Qasim, H. Song, M. Murshed, H. Mahmood and I. Younis, Environ. Sci. Pollut. Res., 2022, 29, 42539 CrossRef
; (b) P. C. Jain, Renewable Energy, 1993, 3, 403 CrossRef CAS
- C. Figueres, C. Le Quere, A. Mahindra, O. Bate, G. Whiteman, G. Peters and D. Guan, Nature, 2018, 564, 27 CrossRef CAS
-
(a) A. Bhavsar, D. Hingar, S. Ostwal, I. Thakkar, S. Jadeja and M. Shah, Case Stud. Chem. Environ. Eng., 2023, 8, 100368 CrossRef CAS
-
(a) For examples see: W. Gao, S. Liang, R. Wang, Q. Jiang, Y. Zhang, Q. Zheng, B. Xie, C. Y. Toe, X. Zhu, J. Wang, L. Huang, Y. Gao, Z. Wang, C. Jo, Q. Wang, L. Wang, Y. Liu, B. Louis, J. Scott, A.-C. Roger, R. Amal, H. Heh and S.-E. Park, Chem. Soc. Rev., 2020, 49, 8584 RSC
-
(a) For examples see: H. Onyeaka and O. C. Ekwebelem, Int. J. Environ. Sci. Technol., 2023, 20, 4635 CrossRef CAS
-
(a) For recent examples see: A. Jana, S. W. Snyder, E. J. Crumlin and J. Qian, Front. Chem., 2023, 11, 1135829 CrossRef CAS
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709 CrossRef CAS
-
I. Tebbiche, J. Mocellin, L. T. Huong and L.-C. Pasquier, 27 - Circular Economy and Carbon Capture, Utilization, and Storage, in Circular Bioeconomy - Current Status and Future Outlook, Biomass, Biofuels, Biochemicals, 2021, pp. 813–851 Search PubMed
-
(a) E. A. Quadrelli, G. Centi, J.-L. Duplan and S. Perathoner, ChemSusChem, 2011, 4, 1194 CrossRef CAS
- J. Schneider, H. Jia, J. T. Muckerman and E. Fujita, Chem. Soc. Rev., 2012, 41, 2036 RSC
-
N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Butterworth-Heinemann, 2nd edn, 1997, vol. 305. ISBN 978-0-08-037941-8 Search PubMed
- U. J. Etim, C. Zhang and Z. Zhong, Nanomaterials, 2021, 11, 3265 CrossRef CAS
-
(a) For examples see: A. Takahashi, K. Minami, K. Noda, K. Sakurai and T. Kawamoto, ACS Sustainable Chem. Eng., 2021, 9, 16865 CrossRef CAS
- S. Chakraborty, O. Blacque and H. Berke, Dalton Trans., 2015, 44, 6560 RSC
- N. W. Kinzel, C. Werlé and W. Leitner, Angew. Chem., Int. Ed., 2021, 60, 11628 CrossRef CAS
-
(a) For examples see: Q.-J. Wu, J. Liang, Y.-B. Huang and R. Cao, Acc. Chem. Res., 2022, 55, 2978 CrossRef CAS
- G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124 CrossRef CAS
- F.-G. Fontaine and D. W. Stephan, Philos. Trans. R. Soc., A, 2017, 375, 20170004 CrossRef
-
(a) For CO2 activation see: F.-G. Fontaine and D. W. Stephan, Curr. Opin. Green Sustainable Chem., 2017, 3, 28 CrossRef
-
G. N. Lewis, Valence and the Structure of Atoms and Molecules, Chemical Catalogue Company, Inc., New York, 1923 Search PubMed
- H. C. Brown, H. I. Schlesinger and S. Z. Cardon, J. Am. Chem. Soc., 1942, 64, 325 CrossRef CAS
- G. Wittig and A. Rückert, Adv. Cycloaddit., 1950, 566, 101 CAS
- W. Tochtermann, Angew. Chem., Int. Ed., 1966, 5, 351 CrossRef CAS
- D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440 CrossRef CAS
- S. Rendler and M. Oestreich, Angew. Chem., Int. Ed., 2008, 47, 5997 CrossRef CAS
- J. S. J. McCahill, G. C. Welch and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 4968 CrossRef CAS
- G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880 CrossRef CAS
-
(a) L. J. C. van der Zee, S. Pahar, E. Richards, R. L. Melen and J. C. Slootweg, Chem. Rev., 2023, 123, 9653 CrossRef CAS
- J. Zhu and K. An, Chem.–Asian J., 2013, 8, 3147 CrossRef CAS
-
(a) T. Özgün, K. Bergander, L. Liu, C. G. Daniliuc, S. Grimme, G. Kehr and G. Erker, Chem.–Euro. J., 2016, 22, 11958 CrossRef
- L. Liu, B. Lukose and B. Ensing, ACS Catal., 2018, 8, 3376 CrossRef CAS
- B. L. Thompson and Z. M. Heiden, Tetrahedron, 2019, 75, 2099 CrossRef CAS
- Y. Yan, J. Yu, Y. Du, S. Yan, M. Gu, W. Zhou and Z. Zou, Cell Rep. Phys. Sci., 2023, 4, 101406 CrossRef CAS
- C. M. Mömming, E. Otten, G. Kehr, R. Fröhlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643 CrossRef
-
(a) M. Puand and T. Privalov, Chem.–Euro. J., 2015, 21, 17708 CrossRef
- I. Peuser, R. C. Neu, X. Zhao, M. Ulrich, B. Schirmer, J. A. Tannert, G. Kehr, R. Fröhlich, S. Grimme, G. Erker and D. W. Stephan, Chem.–Euro. J., 2011, 17, 9640 CrossRef CAS
- M. Harhausen, R. Fröhlich, G. Kehr and G. Erker, Organometallics, 2012, 31, 2801 CrossRef CAS
- M. M. Hansmann, R. L. Melen, M. Rudolph, F. Rominger, H. Wadepohl, D. W. Stephan and A. S. K. Hashmi, J. Am. Chem. Soc., 2015, 137, 15469 CrossRef CAS
- K. Takeuchia and D. W. Stephan, Chem. Commun., 2012, 48, 11304 RSC
- B. M. Barry, D. A. Dickie, L. J. Murphy, J. A. C. Clyburne and R. A. Kemp, Inorg. Chem., 2013, 52, 8312 CrossRef CAS
- X. Zhao and D. W. Stephan, Chem. Commun., 2011, 47, 1833 RSC
- F. Bertini, V. Lyaskovskyy, B. J. J. Timmer, F. J. J. de Kanter, M. Lutz, A. W. Ehlers, J. C. Slootweg and K. Lammertsma, J. Am. Chem. Soc., 2012, 134, 201 CrossRef CAS
- M. Sajid, G. Kehr, T. Wiegand, H. Eckert, C. Schwickert, R. Pöttgen, A. J. P. Cardenas, T. H. Warren, R. Fröhlich, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2013, 135, 8882 CrossRef CAS
- L.-M. Elmer, G. Kehr, C. G. Daniliuc, M. Siedow, H. Eckert, M. Tesch, A. Studer, K. Williams, T. H. Warren and G. Erker, Chem.–Euro. J., 2017, 23, 6056 CrossRef CAS
- C. Chen, C. G. Daniliuc, C. Mück-Lichtenfeld, G. Kehr and G. Erker, Chem. Commun., 2020, 56, 8806 RSC
- X. Jie, Q. Sun, C. G. Daniliuc, R. Knitsch, M. R. Hansen, H. Eckert, G. Kehr and G. Erker, Chem.–Euro. J., 2020, 26, 1269 CrossRef CAS
- N. Szynkiewicz, Ł. Ponikiewski and R. Grubba, Chem. Commun., 2019, 55, 2928 RSC
- N. Szynkiewicz, A. Ordyszewska, J. Chojnacki and R. Grubba, RSC Adv., 2019, 9, 27749 RSC
- N. Szynkiewicz, A. Ordyszewska, J. Chojnacki and R. Grubba, Inorg. Chem., 2021, 60, 3794 CrossRef CAS
- J. M. Kessete, T. B. Demissie, M. Chilume, A. M. Mohammed and V. Andrushchenko, Mol. Phys., 2022, 120, e2087566 CrossRef CAS
- Z. Jian, G. Kehr, C. G. Daniliuc, B. Wibbeling and G. Erker, Dalton Trans., 2017, 46, 11715 RSC
- M. J. Sgro, J. Dömera and D. W. Stephan, Chem. Commun., 2012, 48, 7253 RSC
- S. C. Binding, H. Zaher, F. M. Chadwick and D. O'Hare, Dalton Trans., 2012, 41, 9061 RSC
- A. L. Travis, S. C. Binding, H. Zaher, T. A. Q. Arnold, J.-C. Buffeta and D. O'Hare, Dalton Trans., 2013, 42, 2431 RSC
- M.-A. Courtemanche, M.-A. Légaré, L. Maron and F.-G. Fontaine, J. Am. Chem. Soc., 2013, 135, 9326 CrossRef CAS
- M.-A. Courtemanche, M.-A. Légaré, L. Maron and F.-G. Fontaine, J. Am. Chem. Soc., 2014, 136, 10708 CrossRef CAS
- R. Declercq, G. Bouhadir, D. Bourissou, M.-A. Légaré, M.-A. Courtemanche, K. Sy. Nahi, N. Bouchard, F.-G. Fontaine and L. Maron, ACS Catal., 2015, 5, 2513 CrossRef CAS
- T. Wang and D. W. Stephan, Chem. Commun., 2014, 50, 7007 RSC
- B. Jiang, Q. Zhang and L. Dang, Org. Chem. Front., 2018, 5, 1905 RSC
- M. Delarmelina, J. W. de M. Carneiro, C. R. A. Catlow and M. Bühl, Catal. Commun., 2022, 162, 106385 CrossRef CAS
- T. A. R. Horton, M. Wang and M. P. Shaver, Chem. Sci., 2022, 13, 3845 RSC
- L. Chen, R. Liu, X. Hao and Q. Yan, Angew. Chem., Int. Ed., 2019, 58, 264 CrossRef CAS
- L. Chen, R. Liu and Q. Yan, Angew. Chem., Int. Ed., 2018, 57, 9336 CrossRef CAS
- K. Mentoor, L. Twigge, J. W. H. Niemantsverdriet, J. C. Swarts and E. Erasmus, Inorg. Chem., 2021, 60, 55 CrossRef CAS
- F. Buß, P. Mehlmann, C. Mück-Lichtenfeld, K. Bergander and F. Dielmann, J. Am. Chem. Soc., 2016, 138, 1840 CrossRef
- T. Voss, T. Mahdi, E. Otten, R. Fröhlich, G. Kehr, D. W. Stephan and G. Erker, Organometallics, 2012, 31, 2367 CrossRef CAS
- E. Theuergarten, J. Schlösser, D. Schlüns, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2012, 41, 9101 RSC
- M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2010, 132, 13559 CrossRef CAS
- L. Yang, X. Ren, H. Wang, N. Zhang and S. Hong, Res. Chem. Intermed., 2012, 38, 113 CrossRef CAS
- M. Ghara and P. K. Chattaraj, Struct. Chem., 2019, 30, 1067 CrossRef CAS
- C. Jianga and D. W. Stephan, Dalton Trans., 2013, 42, 630 RSC
- B. R. Barnett, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Chem. Commun., 2015, 51, 541 RSC
- V. Sumerin, F. Schulz, M. Nieger, M. Leskelä, T. Repo and B. Rieger, Angew. Chem., Int. Ed., 2008, 47, 6001 CrossRef CAS
- A. E. Ashley, A. L. Thompson and D. O'Hare, Angew. Chem., Int. Ed., 2009, 48, 9839 CrossRef CAS
- S. J. Geier and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 3476 CrossRef CAS
- S. D. Tran, T. A. Tronic, W. Kaminsky, D. M. Heinekey and J. M. Mayer, Inorg. Chim. Acta, 2011, 369, 126 CrossRef CAS
- M.-A. Courtemanche, A. P. Pulis, É. Rochette, M.-A. Légaré, D. W. Stephan and F.-G. Fontaine, Chem. Commun., 2015, 51, 9797 RSC
- A. Berkefeld, W. E. Piers and M. Parvez, J. Am. Chem. Soc., 2010, 132, 10660 CrossRef CAS
- M. Wen, F. Huang, G. Lu and Z.-X. Wang, Inorg. Chem., 2013, 52, 12098 CrossRef CAS
- C. D. N. Gomes, E. Blondiaux, P. Thuéry and T. Cantat, Chem.–Euro. J., 2014, 20, 7098 CrossRef
- T. Wang and D. W. Stephan, Chem.–Euro. J., 2014, 20, 3036 CrossRef CAS
- Y. Zhang, H. Zhang and K. Gao, Org. Lett., 2021, 23, 8282 CrossRef CAS
- S. Das, R. C. Turnell-Ritson, P. J. Dyson and C. Corminboeuf, Angew. Chem., Int. Ed., 2022, 61, e202208987 CrossRef CAS
- O. E. Palomero and R. A. Jones, Dalton Trans., 2022, 51, 6275 RSC
- L. Yang and H. Wang, ChemSusChem, 2014, 7, 962 CrossRef CAS
- S. Y.-F. Ho, C.-W. So, N. Saffon-Merceronc and N. Mézailles, Chem. Commun., 2015, 51, 2107 RSC
- E. L. Kolychev, T. Bannenberg, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Chem.–Euro. J., 2012, 18, 16938 CrossRef CAS
- E. Theuergarten, T. Bannenberg, M. D. Walter, D. Holschumacher, M. Freytag, C. G. Daniliuc, P. G. Jonesa and M. Tamm, Dalton Trans., 2014, 43, 1651 RSC
- J. Zeng, R. Qiu and J. Zhu, Chem.–Asian. J., 2023, 18, e20220123 Search PubMed
- F. Lavigne, E. Maerten, G. Alcaraz, V. Branchadell, N. Saffon-Merceron and A. Baceiredo, Angew. Chem., Int. Ed., 2012, 51, 2489 CrossRef CAS
- S. C. Sau, R. Bhattacharjee, P. K. Hota, P. K. Vardhanapu, G. Vijaykumar, R. Govindarajan, A. Datta and S. K. Mandal, Chem. Sci., 2019, 10, 1879 RSC
- S. C. Sau, Ra. Bhattacharjee, P. K. Vardhanapu, G. Vijaykumar, A. Datta and S. K. Mandal, Angew. Chem., Int. Ed., 2016, 55, 15147 CrossRef CAS
- X. Chen, Y. Yang, H. Wang and Z. Mo, J. Am. Chem. Soc., 2023, 145, 7011 CrossRef CAS
- N. D. Rio, M. Lopez-Reyes, A. Baceiredo, N. Saffon-Merceron, D. Lutters, T. Müller and T. Kato, Angew. Chem., Int. Ed., 2017, 56, 1365 CrossRef
- D. Wu, L. Kong, Y. Li, R. Ganguly and R. Kinjo, Nat. Commun., 2015, 6, 7340 CrossRef CAS
- L. L. Liu, C. Chan, J. Zhu, C.-H. Cheng and Y. Zhao, J. Org. Chem., 2015, 80, 8790 CrossRef CAS
- L. Kong, W. Lu, L. Yongxin, R. Ganguly and R. Kinjo, Inorg. Chem., 2017, 56, 5586 CrossRef CAS
- M. P. Boone and D. W. Stephan, Organometallics, 2014, 33, 387 CrossRef CAS
- S. J. K. Forrest, J. Clifton, N. Fey, P. G. Pringle, H. A. Sparkes and D. F. Wass, Angew. Chem., Int. Ed., 2015, 54, 2223 CrossRef CAS
- Y. Jiang, O. Blacque, T. Fox and H. Berke, J. Am. Chem. Soc., 2013, 135, 7751 CrossRef CAS
- J. A. Buss, D. G. V. Velde and T. Agapie, J. Am. Chem. Soc., 2018, 140, 10121 CrossRef CAS
- T. Zhao, X. Hu, Y. Wu and Z. Zhang, Angew. Chem., Int. Ed., 2019, 58, 722 CrossRef CAS
- G. Menard and D. W. Stephan, J. Am. Chem. Soc., 2010, 132, 1796 CrossRef CAS
- N. C. Smythe, D. A. Dixon, E. B. Garner, M. M. Rickard, M. Mendéz, B. L. Scott, B. Zelenay and A. D. Sutton, Inorg. Chem. Commun., 2015, 61, 207 CrossRef CAS
- C. Appelt, H. Westenberg, F. Bertini, A. W. Ehlers, J. C. Slootweg, K. Lammertsma and W. Uhl, Angew. Chem., Int. Ed., 2011, 50, 3925 CrossRef CAS
- S. Roters, C. Appelt, H. Westenberg, A. Hepp, J. C. Slootweg, K. Lammertsm and W. Uhl, Dalton Trans., 2012, 41, 9033 RSC
- N. Aders, L. Keweloh, D. Pleschka, A. Hepp, M. Layh, F. Rogel and W. Uhl, Organometallics, 2019, 38, 2839 CrossRef CAS
- J. Boudreau, M. A. Courtemanche and F. G. Fontaine, Chem. Commun., 2011, 47, 11131 RSC
- H. S. Zijlstra, J. Pahl, J. Penafiel and S. Harder, Dalton Trans., 2017, 46, 3601 RSC
- P. Federmann, T. Bosse, S. Wolff, B. Cula, C. Herwig and C. Limberg, Chem. Commun., 2022, 58, 13451 RSC
- P. Federmann, R. Müller, F. Beckmann, C. Lau, B. Cula, M. Kaupp and C. Limberg, Chem.–Euro. J., 2022, 28, e2022004 CrossRef
- T. W. Yokley, H. Tupkar, N. D. Schley, N. J. DeYonker and T. P. Brewster, Eur. J. Inorg. Chem., 2020, 2958 CrossRef CAS
- M. Devillard, R. Declercq, E. Nicolas, A. W. Ehlers, J. Backs, N. Saffon-Merceron, G. Bouhadir, J. C. Slootweg, W. Uhl and D. Bourissou, J. Am. Chem. Soc., 2016, 138, 4917 CrossRef CAS
- G. Menard and D. W. Stephan, Dalton Trans., 2013, 42, 5447 RSC
-
(a) C. H. Lim, A. M. Holder, J. T. Hynes and C. B. Musgrave, Inorg. Chem., 2013, 52, 10062 CrossRef CAS
-
(a) G. Menard, T. M. Gilbert, J. A. Hatnean, A. Kraft, I. Krossing and D. W. Stephan, Organometallics, 2013, 32, 4416 CrossRef CAS
- T. E. Stennett, J. Pahl, H. S. Zijlstra, F. W. Seidel and S. Harder, Organometallics, 2016, 35, 207 CrossRef CAS
- L. Wickemeyer, N. Aders, A. Mix, B. Neumann, H. G. Stammler, J. J. C. Trujillo, I. Fernandez and N. W. Mitze, Chem. Sci., 2022, 13, 8088 RSC
- W. Huang, T. Roisnel, V. Dorcet, C. Orione and E. Kirillov, Organometallics, 2020, 39, 698 CrossRef CAS
- M. D. Ramadhan and P. Surawatanawong, Dalton Trans., 2021, 50, 11307 RSC
- J. Chen, L. Falivene, L. Caporaso, L. Cavallo and E. Y.-X. Chen, J. Am. Chem. Soc., 2016, 138, 5321 CrossRef CAS
- W. Uhl, M. Willeke, A. Hepp, D. Pleschka and M. Layh, Z. Anorg. Allg. Chem., 2017, 643, 387 CrossRef CAS
- D. W. N. Wilson, J. Feld and J. M. Goicoechea, Angew. Chem., Int. Ed., 2020, 59, 20914 CrossRef CAS
- D. A. Dickie, M. T. Barker, M. A. Land, K. E. Hughes, J. A. C. Clyburne and A. Kemp, Inorg. Chem., 2015, 54, 11121 CrossRef CAS
- A. Schäfer, W. Saak, D. Haase and T. Müller, Angew. Chem., Int. Ed., 2012, 51, 2981 CrossRef
- M. Reißmann, A. Schäfer, S. Jung and T. Müller, Organometallics, 2013, 32, 6736 CrossRef
- B. Waerder, M. Pieper, L. A. Körte, T. A. Kinder, A. Mix, B. Neumann, H. G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2015, 54, 13416 CrossRef CAS
-
(a) S. Antoniotti, V. Dalla and E. Dunach, Angew. Chem., Int.
Ed., 2010, 49, 7860 CrossRef CAS
- M. F. S. Valverde, E. Theuergarten, T. Bannenberg, M. Freytag, P. G. Jones and M. Tamm, Dalton Trans., 2015, 44, 9400 RSC
- N. von Wolff, G. Lefèvre, J.-C. Berthet, P. Thuéry and T. Cantat, ACS Catal., 2016, 6, 4526 CrossRef CAS
- L. Wickemeyer, J. Schwabedissen, P. C. Trapp, B. Neumann, H.-G. Stammler and N. W. Mitzel, Dalton Trans., 2023, 52, 2611 RSC
- S. A. Weicker and D. W. Stephan, Chem.–Euro. J., 2015, 21, 13027 CrossRef CAS
-
(a) D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef
-
(a) Q. Yang, L. Wang, Y. Li, L. Zhou and Z. Li, J. Organomet. Chem., 2021, 954, 122071 CrossRef
- P. Holtkamp, F. Friedrich, E. Stratmann, A. Mix, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2019, 58, 5114 CrossRef CAS
- J. J. Cabrera-Trujillo and I. Fernandez, J. Phys. Chem. A, 2019, 123, 10095 CrossRef CAS
- P. Sarkar, S. Das and S. K. Pati, J. Phys. Chem. C, 2021, 125, 22522 CrossRef CAS
- A. Paparakis and M. Hulla, ChemCatChem, 2023, 15, e202300510 CrossRef CAS
- Z. W. Qu, H. Zhu and S. Grimme, ChemCatChem, 2020, 12, 3656 CrossRef CAS
-
(a) I. Avigdori, K. Singh, A. Pogoreltsev, A. Kaushansky, N. Fridman and M. Gandelman, Z. Anorg. Allg. Chem., 2023, 649, e202200326 CrossRef CAS
- L. J. Hounjet, C. B. Caputo and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 51, 4714 CrossRef CAS
- J. Zhu and K. An, Chem.–Asian J., 2013, 8, 3147 CrossRef CAS
- A. Berkefeld, W. E. Piers, M. Parvez, L. Castro, L. Maron and O. Eisenstein, Chem. Sci., 2013, 4, 2152 RSC
- F. A. Le Blanc, W. E. Piers and M. Parvez, Angew. Chem., Int. Ed., 2014, 53, 789 CrossRef CAS
- A. M. Chapman, M. F. Haddow and D. F. Wass, J. Am. Chem. Soc., 2011, 133, 18463 CrossRef CAS
- O. J. Metters, S. J. K. Forrest, H. A. Sparkes, I. Manners and D. F. Wass, J. Am. Chem. Soc., 2016, 138, 1994 CrossRef CAS
- X. Xu, G. Kehr, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2013, 135, 6465 CrossRef CAS
-
(a) C.-S. Wu and M.-D. Su, Phys. Chem. Chem. Phys., 2023, 25, 20618 RSC
- M. J. Sgro and D. W. Stephan, Chem. Commun., 2013, 49, 2610 RSC
- X. Xu, G. Kehr, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2014, 136, 12431 CrossRef CAS
- J. M. V. Franco, G. Schnakenburg, T. Sasamori, A. E. Ferao and R. Streubel, Chem.–Euro. J., 2015, 21, 9650 CrossRef
-
(a) S. Wesselbaum, T. vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2012, 51, 7499 CrossRef CAS
- M. J. Sgro and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 51, 11343 CrossRef CAS
- C. Cristóbal, Y. A. Hernández, J. López-Serrano, M. Paneque, A. Petronilho, M. L. Poveda, V. Salazar, F. Vattier, E. Álvarez, C. Maya and E. Carmona, Chem.–Euro. J., 2013, 19, 4003 CrossRef
- E. A. Romero, T. Zhao, R. Nakano, X. Hu, Y. Wu, R. Jazzar and G. Bertrand, Nat. Catal., 2018, 1, 743 CrossRef CAS
- R. Dobrovetsky and D. W. Stephan, Angew. Chem., Int. Ed., 2013, 52, 2516 CrossRef CAS
- R. Dobrovetsky and W. S. Douglas, Isr. J. Chem., 2015, 54, 206 CrossRef
- K. Chang, Y. Dong and X. Xu, Chem. Commun., 2019, 55, 12777 RSC
- K. K. Ghuman, T. E. Wood, L. B. Hoch, C. A. Mims, G. A. Ozin and C. V. Singh, Phys. Chem. Chem. Phys., 2015, 17, 14623 RSC
- K. K. Ghuman, L. B. Hoch, P. Szymanski, J. Y. Y. Loh, N. P. Kherani, M. A. El-Sayed, G. A. Ozin and C. V. Singh, J. Am. Chem. Soc., 2016, 138, 1206 CrossRef CAS
- L. Wang, M. Ghoussoub, H. Wang, Y. Shao, W. Sun, A. A. Tountas, T. E. Wood, H. Li, J. Y. Y. Loh, Y. Dong, M. Xia, Y. Li, S. Wang, J. Jia, C. Qiu, C. Qian, N. P. Kherani, L. He, X. Zhang and G. A. Ozin, Joule, 2018, 2, 1369 CrossRef CAS
- X. Liang, X. Wang, X. Zhang, S. Lin, M. Ji and M. Wang, ACS Catal., 2023, 13, 6214 CrossRef CAS
- Y. Dong, K. K. Ghuman, R. Popescu, P. N. Duchesne, W. Zhou, J. Y. Y. Loh, F. M. Ali, J. Jia, D. Wang, X. Mu, C. Kübel, L. Wang, L. He, M. Ghoussoub, Q. Wang, T. E. Wood, L. M. Reyes, P. Zhang, N. P. Kherani, C. V. Singh and G. A. Ozin, Adv. Sci., 2018, 5, 1700732 CrossRef
- J. Hao, Y. Zhang, L. Zhang, J. Shen, L. Meng and X. Wang, Chem. Eng. J., 2023, 464, 142536 CrossRef CAS
- M. Ferrer, I. Alkorta, J. Elguero and J. M. Oliva-Enrich, Sci. Rep., 2023, 13, 2407 CrossRef CAS
- T. G. Senthamaraikannan, S. Krishnamurty and S. Kaliaperumal, New J. Chem., 2021, 45, 9959 RSC
- X. Wang, L. Lu, B. Wang, Z. Xu, Z. Xin, S. Yan, Z. Geng and Z. Zou, Adv. Funct. Mater., 2018, 28, 1804191 CrossRef
- B. Chen and R. Neumann, Eur. J. Inorg. Chem., 2018, 791 CrossRef CAS
- C. Jia, X. Kan, X. Zhang, G. Lin, W. Liu, Z. Wang, S. Zhu, D. Ju and J. Liu, Chem. Eng. J., 2022, 427, 131554 CrossRef CAS
- S. Zhang, Z. Xia, Y. Zou, F. Cao, Y. Liu, Y. Ma and Y. Qu, J. Am. Chem. Soc., 2019, 141, 11353 CrossRef CAS
- D. Salusso, G. Grillo, M. Manzoli, M. Signorile, S. Zafeiratos, M. Barreau, A. Damin, V. Crocellà, G. Cravotto and S. Bordiga, ACS Appl. Mater. Interfaces, 2023, 15, 15396 CrossRef CAS
-
(a) S. Zhang, Z. Tian, Y. Ma and Y. Qu, ACS Catal., 2023, 13, 4629 CrossRef CAS
- T. M. Rayder, F. Formalik, S. M. Vornholt, H. Frank, S. Lee, M. Alzayer, Z. Chen, D. Sengupta, T. Islamoglu, F. Paesani, K. W. Chapman, R. Q. Snurr and O. K. Farha, J. Am. Chem. Soc., 2023, 145, 11195 CrossRef CAS
- Y. Jiang, X. Zhang and H. Fei, Dalton Trans., 2020, 49, 6548 RSC
- Y. Zhang, S. Chen, A. M. Al-Enizi, A. Nafady, Z. Tang and S. Ma, Angew. Chem., Int. Ed., 2023, 62, e202213399 CrossRef CAS
- R. Declercq, G. Bouhadir, D. Bourissou, M.-A. Légaré, M.-A. Courtemanche, K. S. Nahi, N. Bouchard, F.-G. Fontaine and L. Maron, ACS Catal., 2015, 5, 2513 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2023 |