
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrid bronzes: mixed-valence organic–inorganic metal oxides as a tunable material platform†
W. Lakna N.
Dayaratne‡
 ,
Raúl
Torres-Cadena‡
,
Bennett P.
Schmitt
,
Emma M.
Westrick
and
Adam
Jaffe
*
,
Raúl
Torres-Cadena‡
,
Bennett P.
Schmitt
,
Emma M.
Westrick
and
Adam
Jaffe
*
Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556, USA. E-mail: ajaffe@nd.edu
First published on 13th September 2023
Abstract
We demonstrate that mixed-valence layered organic–inorganic metal oxides of the form (L)zHxMO3 (L = neutral ligand; M = Mo, W; z = 0.5, 1; 0 < x < 2), which we call hybrid bronzes, can be readily synthesized through mild solution-state self-assembly reactions to integrate the stability and electronic utility of inorganic metal oxide bronzes with the chemical diversity and functionality of organic molecules. We use single-crystal and powder X-ray diffraction coupled with X-ray, electronic, and vibrational spectroscopies to show that the products of aqueous pre-, mid-, or post-synthetic reduction are mixed-valence versions of highly crystalline layered hybrid oxides. Pillaring, bilayered, or canted bilayered arrangements of molecular arrays relative to inorganic sheets are dictated by judicious choice of organic ligands that can also incorporate chemical, redox, or photoactive handles. Significantly, bond-valence sum analysis and diffuse reflectance spectroscopy indicate relatively delocalized electronic behavior and four-point variable-temperature electrical transport measurements show that hybrid bronzes have comparable conductivity to their all-inorganic parent compounds. This work establishes a solution-processable, inexpensive, air- and water-stable, and non-toxic material family whose electronic bands can be readily tuned and doped, thereby positioning hybrid bronzes to address myriad material challenges.
1. Introduction
New classes of multifunctional and tunable materials are required to advance a sustainable energy future, enable new information technologies, and expand our fundamental understanding of solid-state electronic phenomena. However, the simultaneous inclusion of versatile electronic properties stemming from an extended electronic structure, mild synthetic conditions, material stability to air and moisture, and controllable chemical functionality within one material platform remains a long-standing challenge.Crystalline metal oxides are ubiquitous and display myriad favorable electronic properties as well as high chemical and thermal stability.1–3 Of great interest are reduced metal oxide bronzes of the form AxMOy (A = cation; M = Mo, W, V, Nb), where the name “bronze” stems from their metallic luster resulting from high quasi-free electron concentrations. These excess electrons are charge-balanced by oxygen defects or intercalated cations such as alkali cations or protons, and x and y therefore depend on the reduction level and metal oxidation state. Metal oxide bronzes have exceptional electronic range from semiconducting to metallic behavior depending on their reduction level.2,4 They have been explored in diverse application contexts including photovoltaics,5 transistors,6 electrochemical cells—including batteries, electrochromics, and fuel cells7,8—and catalysis.9 These mixed-valence metal oxides also exhibit more exotic solid-state physical phenomena such as spin-glass behavior,10 charge-density wave states,11 and superconductivity.12 Inorganic bronzes, however, generally lack molecular-scale tunability,1–3 thus diminishing the ability to control their structure–property relationships, and some can require high-temperature syntheses. In contrast, molecular species offer fine synthetic control but can lack the desirable mechanical or electronic properties of extended solids, including facile electron transport. Hybrid systems striving to meet these challenges13—like metal–organic frameworks,14 hybrid metal chalcogenides,15,16 and halide perovskites17–19—display numerous promising qualities such as tunable light absorption and emission, mild syntheses, and intriguing solid-state physical phenomena, but often suffer from low solvent and temperature stability, localized (not-extended) electronic structures, and an inability to support electronic doping.
Herein, we develop hybrid bronzes (Fig. 1) that combine alternating layers of (1) stable metal-oxide sheets featuring tunable extended electronic structures, variable band gaps, and adjustable electronic charge-carrier concentrations with (2) arrays of molecular species that direct structure and introduce functional handles in addition to their potential for chemical, redox-, or photo-activity. In other words, we place facile charge transport pathways in direct proximity to functional molecules.
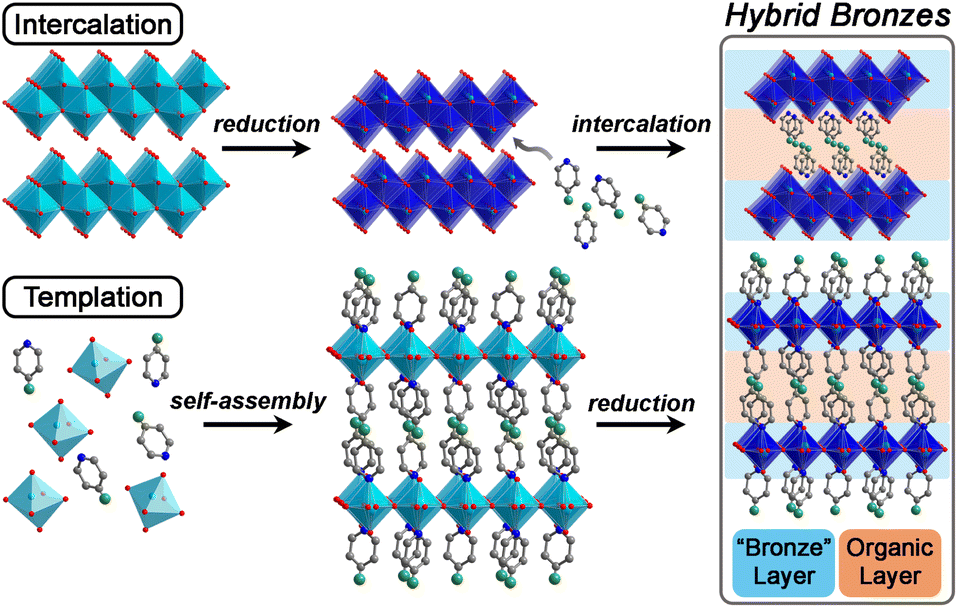 | ||
| Fig. 1 Conceptual strategies for the synthesis of hybrid bronzes that feature alternating layers of reduced metal oxides and molecular arrays. Metals, O, N, and C atoms, and chemically/redox-/photo-active functional groups are represented by light blue, red, blue, gray, and green spheres, respectively. Light octahedra denote oxidized metals and dark octahedra denote reduced metals. Intercalation: layered metal oxides are first reduced, followed by molecular insertion. Templation: molecular and inorganic precursors are self-assembled into a hybrid structure that is subsequently reduced. | ||
Promising examples of layered hybrid metal oxides templated by alkyl and aromatic amines have been reported,20–34 however, much of their (opto)electronic behavior and structure–property relationships remain to be explored. In particular, systematic control over their level of reduction—i.e., their charge carrier concentrations and resulting evolution of their electronic structures—has not been developed. Amine/ammonium intercalation has also been demonstrated for hydrogen bronzes of the form HxMOy and other layered W, Mo, and V oxides,35–37 but without extensive structural characterization or control of molecular functionality. Furthermore, while isolating atomically thin metal-oxide layers yields important electronic phenomena—such as highly correlated electrons, useful model systems for solid-state physics in the context of magnetism and long-range order, and functional systems relevant for next-generation electronics38,39—it remains a long-standing challenge. Hybrid materials represent an opportunity to produce electronically isolated two-dimensional metal-oxide layers in stable, bulk three-dimensional form, yet their exploration in this context also remains underdeveloped.
In this work, we show that single-crystals and microcrystalline powders of new air- and water-stable hybrid molybdenum and tungsten bronzes can be synthesized through two successful strategies: templation and intercalation. We further demonstrate a simplified and milder synthetic approach for many of these hybrids that does not require cumbersome hydrothermal conditions. Distinct from previous work on amine-templated metal oxides that have mostly been reported in their fully oxidized forms, we focus on exercising fine control over their level of reduction during or after their synthesis, followed by elucidation of their optical, electronic, and charge transport properties. We therefore use the term “hybrid bronze” to refer to hybrid metal oxides of mixed valence. Critically, we show that with minimal optimization, hybrid bronzes display high electronic conductivity, implying their potential for many electronic applications such as in transistors, electrocatalyst supports, energy storage electrodes, and charge transport layers. Additionally, considering the hybrids' high optical contrast between different states of charge and the apparent direct band gaps of the fully oxidized materials that are in the visible region of the spectrum, optical/optoelectronic applications leveraging their electrochromism and light absorption, respectively, are also promising avenues for development. Analysis of the structural elements within these single-phase crystalline systems enables us to begin unveiling structural and electronic property correlations, including how inorganic connectivity motifs and polyhedral tilting—that likely dictate orbital overlap and therefore electronic structure—relate to light absorption and charge transport behavior. We additionally include redox-, photo-, and chemically-active organic moieties to further demonstrate the versatility of this material platform beyond the simple alkyl or aromatic amines that have been reported in early intercalation studies for hydrogen molybdenum bronzes35 or templated metal oxides. In other words, we demonstrate the potential for these organic molecules to provide additional functionality rather than simply acting as inert structural elements.
2. Results and discussion
2.1. Synthetic control of hybrid bronzes
To launch hybrid bronzes as a robust and tunable platform, we sought versatile strategies to combine mixed-valence metal-oxide layers with ordered arrays of potentially functional molecular units. Here, two approaches were pursued to produce hybrid bronzes (Fig. 1). In the first approach that we broadly term “intercalation,” layered metal oxides are pre-reduced followed by suspension in a solvent and insertion of molecular species between the layers—presumably without disrupting their topology. In other words, minimal perturbation is expected for the layers' metal–oxygen bonding and therefore the electronic band structure. In the second approach that we broadly term “templation,” metal and organic precursors are combined in water followed by a self-assembly process involving atomic-level restructuring to yield a hybrid metal oxide featuring inorganic layers exhibiting wholly distinct connectivity and potentially distinct electronic structure. Here, the metal-oxide precursors can be reduced prior to synthesis, or the metal-oxide layers can be reduced either during or after synthesis to produce the hybrid bronze. In this work, each approach proved successful in producing phase-pure, highly crystalline hybrid bronzes under mild conditions—in air, often in the presence of water, and below 100 °C.Suspension of HxMoO3 in a non-polar solvent such as toluene followed by addition of aliphatic or aromatic amines such as piperazine, pyrazine, and pyridine at 80 °C or even room temperature afforded new, highly crystalline materials, as evidenced by sharp powder X-ray diffraction (PXRD) peaks (Fig. 2). Shifting of the low-angle reflection ca. 12.6° that is associated with the interlayer spacing between metal-oxide sheets to lower values of 2θ indicates the intercalative insertion of these organic molecules between the layers and supports the formation of hybrid bronzes. Interestingly, the observed interlayer distances ca. 12.7 Å between inorganic sheets within these intercalated products are consistent with expected values for a bilayer of organic molecules. This behavior alludes to the importance that intermolecular interactions such as π–π stacking, H-bonding, and steric effects will have on ordering of organic species in the intercalated molecular layers. The appearance, disappearance, and shifting of peaks at higher values of 2θ indicate additional changes in bulk symmetry during intercalation. Though previous reports have explored similar molecular intercalation within molybdenum bronzes,35,44,45 we are not aware of reports of such crystalline phases or of such products in which nearly negligible amounts of H0.33MoO3 starting material are observed. However, due to numerous coincidental product diffraction peaks, we cannot definitively rule out its presence in trace quantities. A comparison of Raman spectra (Fig. S3†) for MoO3, a pyrazine-intercalated product, and a pyrazine-templated product (discussed in the next section) suggests that the intercalated phase retains the same connectivity as its parent oxide bronze whereas the templated product is distinct. Here, the intercalated compound exhibits modes at ca. 665 cm−1, 820 cm−1, and 995 cm−1 that are virtually identical to modes in MoO3 assigned to metal bonding to bridging μ3-oxo, bridging μ2-oxo, and terminal μ1-oxo species, respectively.46 These proof-of-concept results demonstrate the intercalation method's promise for the future insertion of redox-, photo-, or chemically active molecules to help build the hybrid bronze platform. The spectroscopic evidence discussed above as well as the optical and electronic behavior discussed below suggest that the metal–oxygen connectivity is retained in the intercalation strategy, preserving a similar degree of orbital overlap and therefore band dispersion, band gap, and charge carrier mobility as the inorganic parent bronze. However, since the isolation of hybrid bronze single crystals synthesized via intercalation that are of sufficient quality for structural solution is difficult, determination of structure–property relationships for materials produced in this manner is more challenging. Hence, the results presented in this work will largely focus on the templation strategy described in the next section.
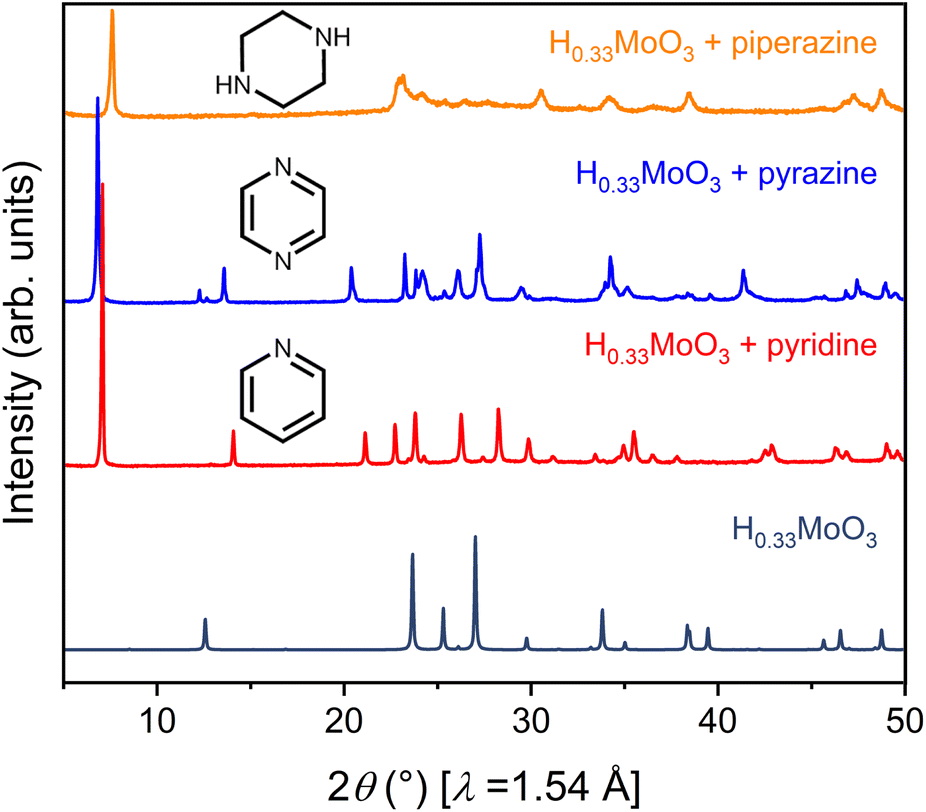 | ||
| Fig. 2 Powder X-ray diffraction patterns for H0.33MoO3 (navy) intercalated with piperazine (orange), pyrazine (blue), and pyridine (red). | ||
| Compound | Organic molecule | Structure type | Metal (M) | Synthesis method(s)a | Successful reduction method(s)b | Reduction level(s) (x)c |
|---|---|---|---|---|---|---|
| a Synthesis methods are abbreviated as follows: hyd = hydrothermal, stir = stirring. b Reduction methods are abbreviated as follows: pre = with HxMO3 precursor, in situ = with in situ Mo or W metal, post = post-synthetically with Na2S2O4. c Determined by XPS. Error on x values for Mo is ca. 0.05. Error on x values for W is ca. 0.1. | ||||||
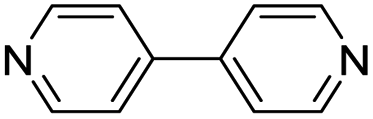
| (4,4′-bipy)0.5HxMO3 |
|
Pillared | Mo | hyd, stir | pre, in situ, post | 0,250.17, 0.26, 0.38, 0.45, 0.47,250.69 |
| W | hyd, stir | in situ, post | 0,260.3, 0.4, 0.7 | |||
| (pyz)0.5HxMO3 |
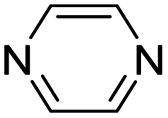
|
Pillared | Mo | hyd, stir | in situ | 0.3, 0.34,*270.37, 0.44 |
| W | hyd, stir | in situ | 0,260.2 | |||
| (azp)0.5HxMO3 |
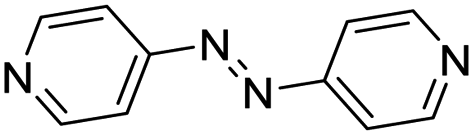
|
Pillared | Mo | hyd, stir | in situ | 0, 0.64 |
| W | hyd, stir | — | 0 | |||
| (4,4′-bipy-ethane)0.5HxMO3 |
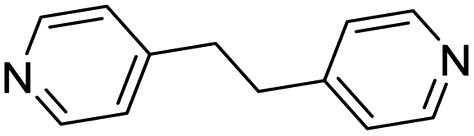
|
Pillared | Mo | — | — | — |
| W | hyd, stir | in situ | 0,320.5 | |||
| (4,4′-bipy-ethene)0.5HxMO3 |
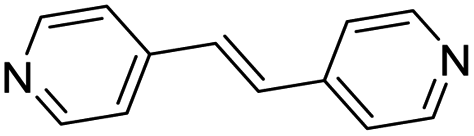
|
Pillared | Mo | stir | in situ | 0.43 |
| W | hyd, stir | in situ | 0, 0.3 | |||
| (dps)0.5HxMO3 |
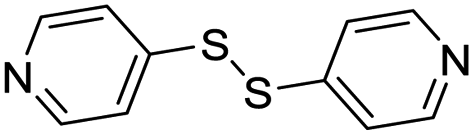
|
Pillared | Mo | — | — | — |
| W | stir | — | 0 | |||
| (4H-trz)0.5HxMO3 |
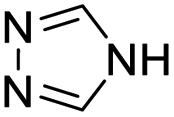
|
Bilayer (bridged adj. octahedra) | Mo | hyd, stir | pre, in situ, post | 0,250.09, 0.33, 0.40, 0.48 |
| W | hyd, stir | in situ, post | 0,330.3 | |||
| (py)HxMO3 |
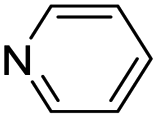
|
Bilayer | Mo | — | — | — |
| W | hyd, stir | post | 0,200.4 | |||
| (py-4-vinyl)HxMO3 |
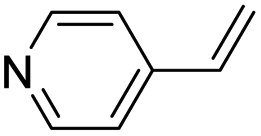
|
Bilayer | Mo | stir | pre | unknown |
| W | — | — | — | |||
| (py-4-OH)HxMO3 |
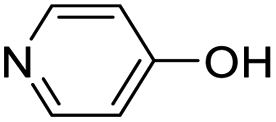
|
Bilayer | Mo | stir | — | 0 |
| W | — | — | — | |||
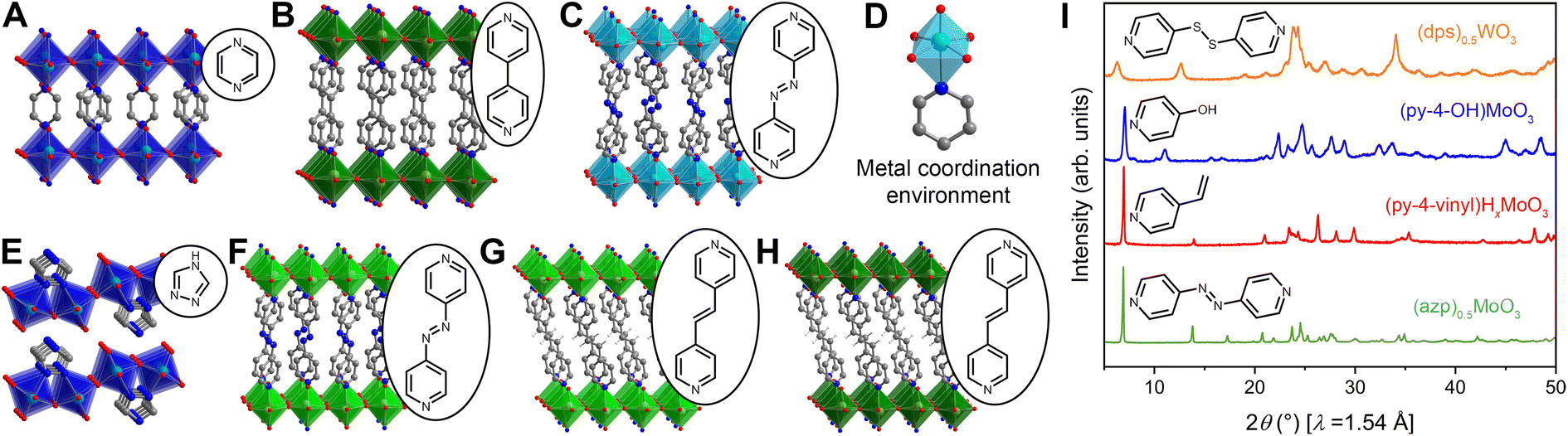 | ||
| Fig. 3 Single-crystal X-ray structures of (A) (pyz)0.5H0.3MoO3, (B) (4,4′-bipy)0.5H0.3WO3, and (C) (azp)0.5MoO3. Light blue, green, gray, blue, and red spheres represent Mo, W, C, N, and O atoms, respectively. H atoms omitted for clarity. Polyhedra are drawn to illustrate connectivity. Light-shade polyhedra denote an oxidized structure while dark-shade polyhedra denote a reduced structure. (D) Characteristic metal coordination environment featuring an MO5N octahedral configuration. Crystal structures derived from Le Bail refinements of modified single-crystal crystallographic information files for (E) (4H-trz)0.5H0.09MoO3, (F) (azp)0.5WO3, (G) (4,4′-bipy-ethene)0.5WO3, and (H) (4,4′-bipy-ethene)0.5H0.3WO3. Vinyl H atoms for the 4,4′-bipy-ethene molecules are shown as white spheres. (I) Powder X-ray diffraction patterns for new hybrid metal oxides containing redox-, photo-, and/or chemically active functional groups. The patterns corroborate a high degree of structural similarity between materials for which single crystals have not yet been isolated and those that have. | ||
Hybrid metal oxides and their hybrid bronzes display almost identical structural behavior when comparing the Mo and W analogs. One distinguishing structural feature observed throughout this family is that each metal center is octahedrally coordinated by five oxygen atoms and one datively bound ligand nitrogen atom (Fig. 3D). This direct metal–ligand bond further emphasizes the disruption and reformation of inorganic connectivity during the self-assembly process. These octahedra then form layers via corner-sharing in the equatorial plane. Intriguingly, though three-dimensionally corner-sharing metal–oxygen octahedra are common in the perovskite and rhenium trioxide structure types, including in the WO3 parent compound, connectivity exhibiting only corner-sharing and not edge-sharing is less common in two dimensions.38 Three major bonding motifs between the organic molecules and inorganic layers are observed within the structures discussed in this work. In the first, monotopic ligands such as pyridine produce hybrid materials of the general formula (L)MO3 (in the oxidized form; L = ligand, M = metal) in which the inorganic metal-oxide layers alternate with bilayers of organic molecules. In the second, some ditopic ligands such as 4,4′-bipy yield materials with the formula (L)0.5MO3 (when in the oxidized form) in which the organic ligands order in a monolayer with each ligand acting as a pillar bridging between two metal centers in opposing inorganic layers (Fig. 3A–C and F–H). In the third, other ditopic ligands such as 4H-trz bridge adjacent metal centers within the same inorganic layer, leading to MO5N octahedral tilting and a puckering of the inorganic layer that produces a corrugated motif (Fig. 3E). In this latter case, the general formula is also (L)0.5MO3, but the organic species can technically be described as existing in a bilayer. Several groups have provided in-depth reviews on these and other structural motifs for hybrid metal oxides.23,47
Of the five oxygen ligands, four are formally μ2 bridging O2− species while the fifth is a terminal (μ1) oxo featuring a shorter bond length ca. 1.6 Å consistent with a M–O bond order of approximately two. Fourier-transform infrared (FT-IR) absorption and Raman spectra collected across the entire series of metal and ligand combinations corroborate this metal–oxygen bonding behavior with strong vibrational spectra peaks associated with the μ2- and μ1-oxos in the 600–1000 cm−1 range (Fig. S3 and S8–S12†). In many cases, the MO5N octahedra are severely distorted and M–O bond lengths to the μ2 oxos vary between 1.8 and 2.0 Å, demonstrating the degree of structural variation and complexity in these materials. This detailed bond-length information allows for bond-valence sum analysis on single-crystal structures to support elucidation of electronic behavior in hybrid bronzes and will be discussed in the next section.
The templation approach generally consists of combining either molybdenum precursors such as MoO3 or (NH4)6Mo7O24 (i.e., ammonium heptamolybdate) or tungsten precursors such as Na6[H2W12O40] (i.e., sodium metatungstate) or WO3·H2O (i.e., tungstic acid) under neutral aqueous conditions with templating organic molecules to initiate self-assembly reactions, yielding layered hybrid metal oxides. In typical reports of hybrid metal oxides,20–24,26,28–32,34 these syntheses have been almost exclusively performed under hydrothermal conditions in PTFE-lined pressure vessels above the boiling point of water ca. 180 °C. The typically colorless or yellow/orange crystalline powders or single crystals that result indicate that Mo and W are generally in their hexavalent oxidation states.
In our work, hydrothermal syntheses were utilized to produce single crystals for structural determination. These conditions are often still cumbersome, however, requiring temperatures between 150 and 180 °C, long reaction times of over a week, and relatively low yields. Here, we have also developed a far more rapid and reproducible synthetic protocol in which the same organic and inorganic precursors described above can be stirred at 80 °C in water overnight to produce highly crystalline powders of hybrid bronzes and their parent hybrid oxides in their oxidized state (Fig. S13†). This robust methodology has allowed for the incorporation of a greater number of more functional molecules within this work and will enable the rapid expansion of the hybrid bronze phase space in the future.
One of the most important aspects of the templation-based synthetic approach is the establishment of control over the level of reduction in hybrid bronzes. To our knowledge, scant development of this mixed-valence control or exploration of the resultant changes in electronic behavior have been reported. As mentioned previously, reductants can be incorporated either during or after synthesis to yield the desired hybrid bronze product. The resultant powders or crystals are significantly darker—typically appearing dark blue or black—and usually feature a more metallic luster relative to their oxidized congeners that tend to be light orange, yellow, or white. We were able to incorporate Mo or W metal as the reductant during either our newly developed low-temperature stirring method or hydrothermal syntheses. No evidence of zero-valent metal is observed in X-ray diffraction or X-ray photoelectron spectroscopy (XPS) (Fig. S4–S7 and S14–S27†), implying these metal atoms are incorporated within the newly generated hybrid material or remain in the solution phase. We also considered the possibility of using reduced inorganic metal oxide bronzes with precisely known stoichiometry as precursors to hybrid bronzes. Indeed, using the hydrogen molybdenum bronzes (HxMoO3) described in Section 2.1.1 in either hydrothermal or low-temperature stirring reactions also afforded the desired hybrid bronzes with high crystallinity and purity. Further, we found that post-synthetic reduction of many hybrid metal oxides with aqueous solutions of Na2S2O4 produced the desired reduced hybrid bronzes in high purity. Though hybrid bronzes display subtle structural differences to their oxidized analogs, when comparing across reduced materials, the method or level of reduction do not lead to discernible differences in symmetry, lattice constants, and material crystallinity (Fig. S4–S7†). These methods are summarized in Table 1 and discussed in detail in the ESI.† We note that in addition to their aqueous syntheses in the presence of air, hybrid bronzes are stable in air and even submerged in water, as evidenced by retention of crystallinity in powder diffraction patterns for representative hybrid bronzes kept in water for over a month (Fig. S28†).
2.2. Optical and electronic correlations to structure
We initially probed reduction level in templated hybrid bronzes through bond-valence sum (BVS) analysis48,49 of single-crystal X-ray structures (Table S4, details in ESI†). As a representative example, the bond lengths within the MoO5N octahedra in (4,4′-bipy)0.5MoO3 are consistent with fully oxidized Mo6+. Despite minimal overall perturbations of the lattice parameters or topology, small changes in Mo–O and Mo–N bond lengths in the case of the reduced hybrid bronze (4,4′-bipy)0.5HxMoO3 indicate the valence of Mo centers is +5.63(1) which is consistent with the range of 0.17 < x < 0.69 measured in this work by XPS (see below). Furthermore, the intermediate value between 5 and 6, rather than crystallographically distinct Mo centers with near-integer values of either 5 or 6, is suggestive of highly delocalized electrons similar to type III Robin–Day mixed valence compounds.50X-ray photoelectron spectroscopy was used to interrogate the oxidation state of metal centers within hybrid bronzes more directly. Clear evidence is observed for a combination of M6+, M5+, and M4+ (M = Mo, W) centers within the series of hybrid bronzes presented in Fig. 4 and S14–S27,† in contrast with predominantly M6+ centers in the fully oxidized hybrid metal oxides. In the case of molybdenum-based materials, paired peaks at binding energy ranges of 232.2–233.0 eV and 235.4–236.2 eV are attributable to the 3d5/2 and 3d3/2 spin–orbit doublet states of Mo(VI), while paired peaks at binding energy ranges of 230.7–231.5 eV (3d5/2) and 234.0–234.7 eV (3d3/2) support the presence of Mo(V).51,52 This assignment corroborates chemical reduction through the methods described above, accompanied by intercalation of protons for charge balance. The fitting and deconvolution of molybdenum oxide XPS spectra has historically presented significant challenges owing to the complex interplay of localized and delocalized core and valence electrons.52,53 However, reasonable fits are achieved without overparameterization using a single pseudo-Voigt function for each spin–orbit peak, allowing us to determine the level of reduction and therefore the value of x in (L)zHxMoO3 (Table 1). A complete description of XPS fitting methods is included in the ESI.† Tungsten-based hybrid materials were fit in a similar manner, wherein doublets at 36.0 ± 0.5 eV (4f7/2) and 38.1 ± 0.5 eV (4f5/2), 34.7 ± 0.5 eV (4f7/2) and 36.9 ± 0.5 eV (4f5/2), and 33.3 ± 0.3 eV (4f7/2) and 35.4 ± 0.3 eV (4f5/2) correspond to W(VI), W(V), and W(IV), respectively.54 To confirm that our hybrid bronzes were homogeneously reduced throughout each crystal/crystallite, we also collected XPS spectra for single crystals of representative samples, followed by spectra on the same crystals that we ground to expose fresh surfaces of the crystal interiors (Fig. S27†). From these measurements, we determined x values for (pyz)0.5HxMoO3 of 0.30 and 0.29 for the crystals and ground crystals, respectively, and x values of 0.28 and 0.3 for (4,4′-bipy)0.5HxWO3, i.e., within experimental error, thus supporting uniform reduction throughout the hybrid bronzes. We note that in the case of nominally unreduced W-based samples, some W5+ is observed, consistent with previous reports of tungsten reduction induced by the combination of high vacuum and charge neutralization in the XPS chamber,55 likely from creation of oxygen defects. Since the degree to which this effect occurs for already reduced tungsten-based oxide materials is not well-known, we incorporate the magnitude of this discrepancy as error in our calculated values of x in (L)zHxWO3 (Table 1). It should also be noted that XPS is a relatively surface-sensitive technique, and it is possible that minor surface oxidation upon exposure to air—especially in the cases of more reduced hybrid bronzes—could render the observed ratio of M6+ to M5+/4+ to be higher than the predicted value. Nevertheless, across the series of hybrid molybdenum and tungsten bronzes, the measured ratios are well-aligned with expected nominal levels of reduction determined by the ratio of reductant to M6+ included in syntheses or the initial value of x in inorganic hydrogen molybdenum bronze precursors, HxMoO3.
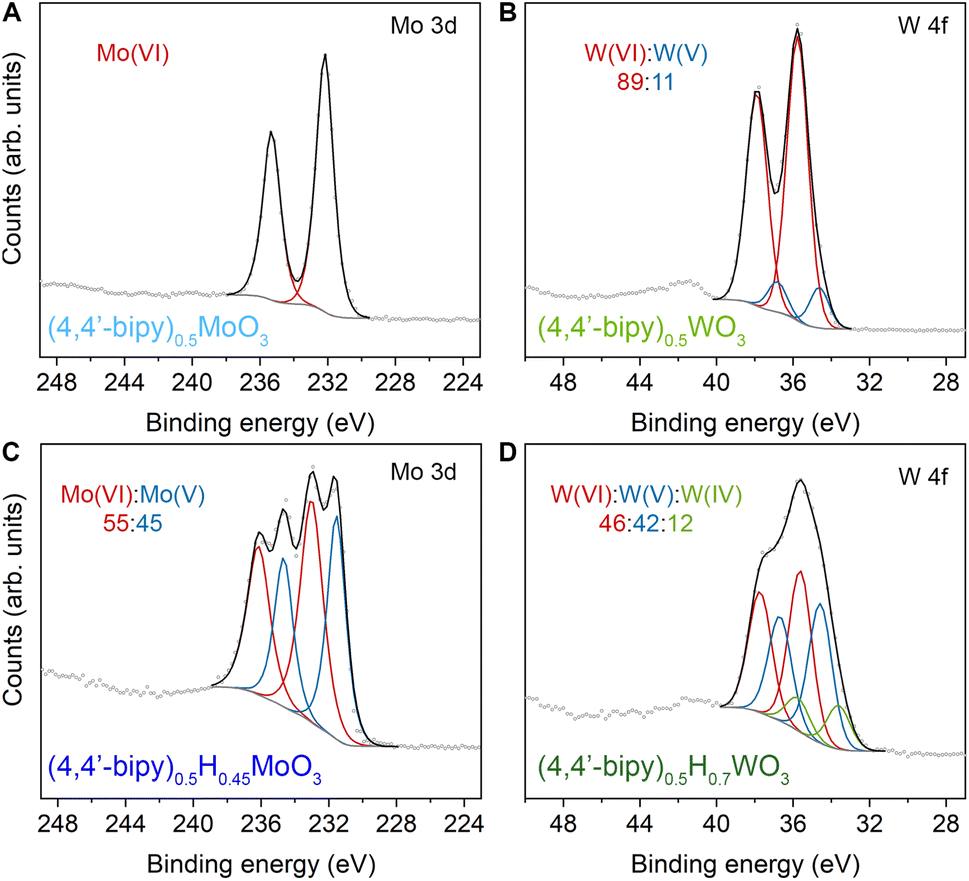 | ||
| Fig. 4 XPS spectra for hybrid molybdenum and tungsten oxides and their corresponding reduced hybrid bronze forms. Individual peak fits are shown in red, blue, and green. Peak fit backgrounds and envelopes are shown with gray and black lines, respectively. (A) Mo 3d spectrum for (4,4′-bipy)0.5MoO3. (B) W 4f spectrum for (4,4′-bipy)0.5WO3. (C) Mo 3d spectrum for (4,4′-bipy)0.5H0.45MoO3. (D) W 4f spectrum for (4,4′-bipy)0.5H0.7WO3. | ||
Diffuse reflectance spectra measured across ultraviolet and visible wavelengths for the fully oxidized hybrid metal oxides reveal steep absorption edges ca. 2.3–2.9 eV (Fig. 5 and S29–S31†). These spectral features are consistent with electronic excitation from valence bands primarily derived from O 2p states to conduction bands consisting of predominantly metal d states with contributions from O 2p states, as described in literature for MoO3, WO3, and (4,4′-bipy)MO3 (M = Mo, W).32,56–58 The latter 4,4′-bipyridine-templated hybrid structures are the only hybrid metal oxides within this material family for which we are aware of reported electronic structure calculations,32,58 but the observed steep and strong electronic absorption features as well as these electronic band structures suggest the hybrids are direct or nearly direct band gap semiconductors in contrast to the parent α-MoO3 and cubic WO3 metal oxides that exhibit indirect gaps. Due to the two-dimensional inorganic connectivity in these layered materials that gives rise to a two-dimensional density of states for their extended electronic structures, the Tauc method59 that was originally developed for band gap determination of three dimensional semiconductors is not appropriate. Therefore, linear extrapolation of α/S vs. energy plots (where transformation of the diffuse reflectance data according to the Kubelka–Munk equation gives α as the absorbance and S as the scattering coefficient) were utilized and yielded band gap values similar to those described for reported hybrid metal oxides (Table S5, Fig. S29–S31†). Interestingly, in comparing (4H-trz)0.5MoO3 to (4,4′-bipy)0.5MoO3, the former displays a higher band gap value of ca. 2.9 eV while the latter shows a band gap of ca. 2.4 eV. Likewise (4H-trz)0.5WO3 features a band gap of ca. 2.9 eV, and (4,4′-bipy)0.5WO3 has a band gap of ca. 2.5 eV. All structures contain MO5N octahedra, but the inorganic layers within the triazole structures are corrugated to allow the organic ligand to bridge adjacent metal centers. One potential explanation for the larger band gap is that octahedral tilting diminishes orbital overlap and therefore band dispersion, thus increasing the separation of the valence band maximum and conduction band minimum.
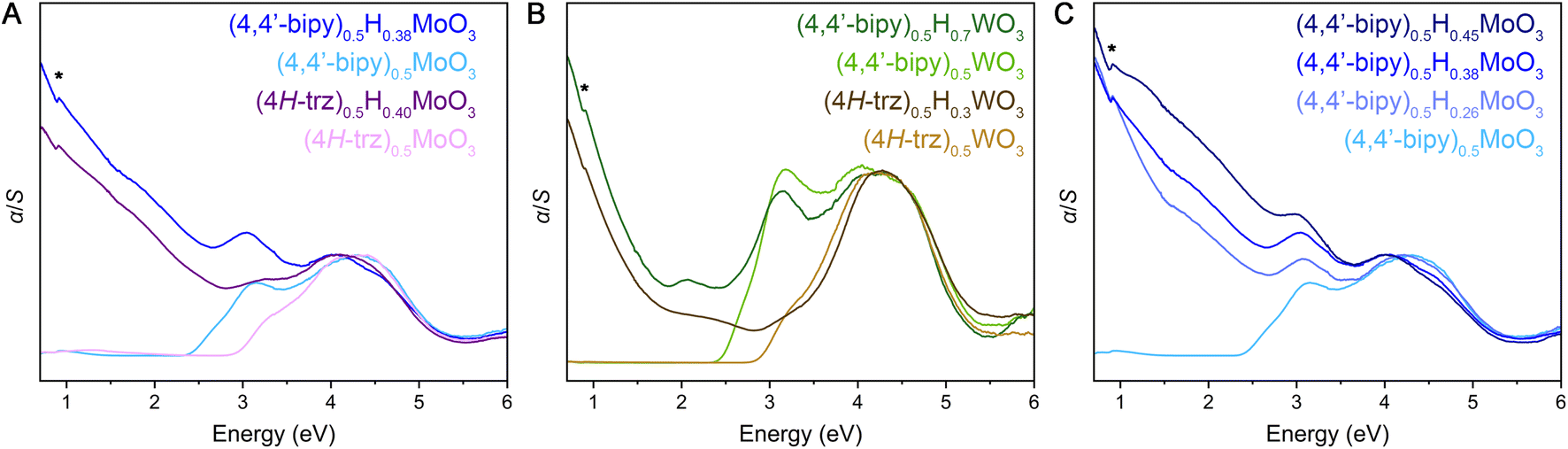 | ||
| Fig. 5 Diffuse reflectance spectra for hybrid bronzes and their oxidized analogs. (A) (4,4′-bipy)0.5HxMoO3 (x = 0, 0.38) and (4H-trz)0.5HxMoO3 (x = 0, 0.40). (B) (4,4′-bipy)0.5HxWO3 (x = 0, 0.7) and (4H-trz)0.5HxWO3 (x = 0, 0.3). (C) Comparison of spectra for (4,4′-bipy)0.5HxMoO3 (x = 0, 0.26, 0.38, and 0.45), i.e., at different levels of reduction. Spectra are normalized to the high-energy absorption feature ca. 4.5 eV that is attributed to a band-to-band transition. BaSO4 background peaks are indicated with an asterisk. | ||
Evidence for an appreciable concentration of fully or partially delocalized electrons in the reduced (mixed-valence) metal-oxide layers within hybrid bronzes is observed in their diffuse reflectance spectra (Fig. 5 and S29–S32†). New, strong absorption is observed spanning visible and near-IR energies—from above 3 eV and extending below 0.7 eV—where the degree of absorption rapidly increases with decreasing energy. The intensity of this new spectral feature scales with increasing level of reduction (Fig. 5C and S32†), achieved either by starting with a more reduced inorganic HxMO3 bronze or through larger stoichiometric equivalents of reductant incorporation during or after synthesis. This scaling behavior corroborates assignment of the low-energy feature to electronic excitations from newly introduced charge carriers. Similar spectral features are also observed for HxMoO3 (x = 0.33, 0.95, 1.68; Fig. S33†) and a representative intercalated material, H0.15MoO3(py)y (Fig. S34†), despite their distinct metal-oxide connectivity relative to the templated materials. Thus, though some differences in band structure are likely between the intercalated and templated hybrids, their optical signatures point to similar electronic behavior.
Several regimes of electronic behavior can be considered to explain the observed increased low-energy absorption. If this were a case of plasmonic resonance behavior due to a high concentration of collectively interacting free carriers, a new peak would be expected in the measured spectral range. Indeed, highly reduced HxMoO3 (x > 0.9) has been reported to display a plasmonic resonance feature in the visible region of the spectrum42,60 and a peak at approximately 2 eV on top of the broad sloping feature is observed in our diffuse reflectance spectrum for H1.68MoO3. On the other hand, a peak would also be observed in the case of a light-induced intervalence charge transfer (IVCT) process of relatively localized electrons between neighboring redox sites61,62 or in the case of polaronic absorption—optical transfer of conduction electrons, that are self-localized through electron–lattice coupling, out of their self-induced potential wells to neighboring undistorted sites.63–67 Theory developed by Hush61,62 would predict an IVCT-derived absorption band to appear with an energy approximately equal to four times the activation energy, Ea, for a symmetrical one-electron transfer. Likewise, polaron theory for relatively strongly bound electrons would predict a peak in the near-IR. Since no discernible peak is observed with decreasing energy down to 0.5 eV in the diffuse reflectance spectra, nor is one readily discernible in FT-IR spectra (Fig. S8–S10†), these data are most consistent with delocalized Drude-like electrons or relatively delocalized electron movement that has a low activation energy associated with either IVCT or polaron-like electron hopping behavior. This assignment is supported by extensive optical and electronic study of reduced molybdenum and tungsten oxides, whose behavior is typically described in a polaronic context.63–67 Further, the electronic confinement and structural flexibility arising from the two-dimensional and hybrid nature of these structures is likely conducive to polaronic electron–lattice coupling. For example, 2D hybrid metal halide perovskites that feature qualitatively similar organic–inorganic layer topology to hybrid bronzes along with the 3D halide perovskite congeners have well-documented electron or exciton coupling to the softer metal halide lattice.68–72 Organic–inorganic titanium oxides have also been discussed in the context of polaronic behavior.73 Low-barrier IVCT and polaronic descriptions are ultimately quite similar in that, in either case, charge carriers are potentially quite mobile. Variable temperature and mid-IR absorption measurements will likely provide additional insight, aided by electronic structure calculations. Further mechanistic discussion of charge transport is reserved for the next section.
2.3. Exploration of charge transport and transfer
Hybrid bronzes synthesized through both intercalation and templation strategies exhibit comparable electronic conductivity to their inorganic HxMO3 congeners (Table 2; Fig. 6 and S35–S37†). These measurements corroborate the observed spectroscopic signatures of relatively delocalized electrons in the previous section and demonstrate successful retention of facile charge transport despite dimensional reduction of the inorganic connectivity in the case of the templated materials. For example, the inorganic bronze H0.49MoO3 displays an electronic conductivity of 2.6(1) × 10−3 S cm−1 as determined through four-point dc measurements on pressed powder pellets. In the case of hybrid bronzes, templated structures such as (4,4′-bipy)0.5HxMoO3 (x = 0.17, 0.38, 0.45, 0.47) display conductivity ranging from 4.21(1) × 10−4 to 1.01(1) × 10−2 S cm−1 where conductivity increases with increasing level of reduction and therefore increasing charge carrier concentration. Similarly, (4H-trz)0.5HxMoO3 (x = 0.09, 0.33, 0.40, 0.48) range in conductivity from 1.7(1) × 10−4 to 8.4(1) × 10−2 S cm−1. For the tungsten analogs, comparable conductivities were also observed, with the highest being 1(1) × 10−2 S cm−1 for (4H-trz)0.5H0.3WO3. Intercalated bronzes such as H0.15MoO3(py)y also exhibited similar values ca. 1.8(1) × 10−4 S cm−1. Much like in our optical characterization, this implies that despite differences in metal-oxide connectivity between the intercalated and templated structure types, their electron transport behavior is similar. It is important to note that the conductivities of these samples are likely underestimated owing to the grain-boundary resistance within pressed pellets.14 Furthermore, in the case of the hybrid bronzes, charge transport is expected to be anisotropic. Within the 2D inorganic layers, electronic bands are likely to be dispersed due to significant metal–oxygen orbital overlap, but this connectivity does not extend in the third dimension. Intrinsic conductivity within these inorganic sheets is therefore likely higher than in the perpendicular direction, meaning tortuosity of charge transport is increased in a pressed pellet of randomly oriented crystallites, further decreasing the measured conductivity values.| Material | Reduction method | Conductivity (S cm−1) |
|---|---|---|
| (4,4′-bipy)0.5H0.47MoO3 | In situ (Mo metal) | 1.01(1) × 10−2 |
| (4,4′-bipy)0.5H0.45MoO3 | In situ (Mo metal, inert conditions) | 9.21(9) × 10−3 |
| (4,4′-bipy)0.5H0.38MoO3 | In situ (Mo metal) | 4.62(1) × 10−3 |
| (4,4′-bipy)0.5H0.17MoO3 | MoO3 pre-reduction (H0.33MoO3), then templation | 4.21(1) × 10−4 |
| (4H-trz)0.5H0.48MoO3 | MoO3 pre-reduction (H1.68MoO3), then templation | 8.4(1) × 10−2 |
| (4H-trz)0.5H0.33MoO3 | In situ (Mo metal) | 9.1(1) × 10−3 |
| (4H-trz)0.5H0.40MoO3 | In situ (Mo metal) | 5.7(3) × 10−4 |
| (4H-trz)0.5H0.09MoO3 | MoO3 pre-reduction (H0.33MoO3), then templation | 1.7(1) × 10−4 |
| (pyz)0.5H0.44MoO3 | In situ (Mo metal) | 5.06(2) × 10−2 |
| H0.15MoO3(py)y | MoO3 pre-reduction (H0.33MoO3), then intercalation | 1.8(1) × 10−4 |
| H0.49MoO3 | SnCl2 in 4 M HCl | 2.6(1) × 10−3 |
| H1.3MoO3 | Zn metal in 12 M HCl | 5.37(1) × 10−1 |
| (4,4′-bipy-ethene)0.5H0.3WO3 | In situ (W metal) | 2.2(4) × 10−5 |
| (4,4′-bipy)0.5H0.7WO3 | In situ (W metal) | 3.3(2) × 10−4 |
| (4,4′-bipy)0.5H0.4WO3 | In situ (W metal) | 2.52(1) × 10−4 |
| (4H-trz)0.5H0.3WO3 | In situ (W metal) | 1(1) × 10−2 |
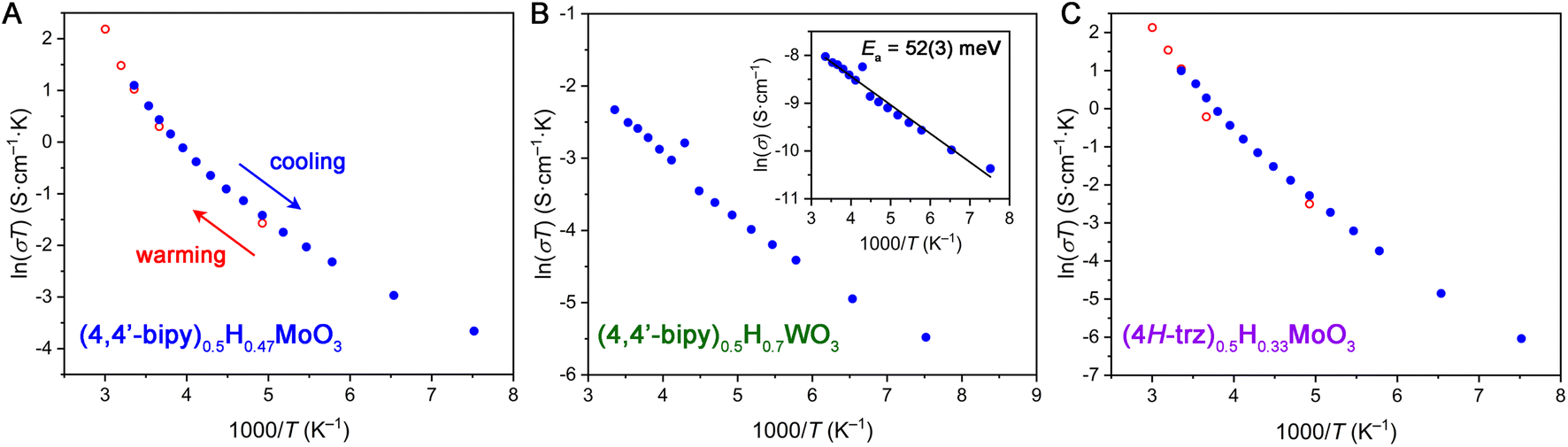 | ||
| Fig. 6 Variable-temperature four-point conductivity measurements on pressed powder pellets of hybrid bronzes, plotted as ln(σT) vs. 1000/T according to the polaronic model of charge transport. (A) (4,4′-bipy)0.5H0.47MoO3. (B) (4,4′-bipy)0.5H0.7WO3. Inset: variable temperature conductivity plotted according to the linearized Arrhenius model with a fitted activation energy of conduction of 52(3) meV. (C) (4H-trz)0.5H0.33MoO3. | ||
To further elucidate the charge transport mechanism within hybrid bronzes, variable-temperature four-point transport measurements were carried out on representative examples: (4,4′-bipy)0.5H0.47MoO3, (4H-trz)0.5H0.33MoO3, (pyz)0.5H0.44MoO3, (4,4′-bipy)0.5H0.7WO3, and (4H-trz)0.5H0.3WO3 (Fig. 6 and S37†). For all hybrid bronzes, conductivity decreased with decreasing temperature, ruling out Drude-like (i.e., metallic) charge carrier behavior. In the case of the hybrid molybdenum bronzes, the conductivity data do not show fully linear behavior in either a plot of ln(σ) vs. 1/T relevant for Arrhenius (σ = σ0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp[−Ea/kT]) behavior or in a plot of ln(σT) vs. 1/T, as is used to fit the polaronic charge transport equation (σT = σ0exp[−Ea/kT]) in its linearized form. Here, σ is conductivity, Ea is the activation energy of conduction, k is Boltzmann's constant, and T is temperature. In the polaronic model, charge carriers couple to lattice vibrations that are temperature dependent, hence the additional temperature term. The observed non-linear behavior is in agreement with reported non-linear temperature dependence of charge transport in hydrogen molybdenum bronzes that has been attributed to charge-density wave contributions.11 The transport mechanism within hybrid molybdenum bronzes may therefore be similarly as complex as in the inorganic bronzes, warranting deeper investigation in the future.
exp[−Ea/kT]) behavior or in a plot of ln(σT) vs. 1/T, as is used to fit the polaronic charge transport equation (σT = σ0exp[−Ea/kT]) in its linearized form. Here, σ is conductivity, Ea is the activation energy of conduction, k is Boltzmann's constant, and T is temperature. In the polaronic model, charge carriers couple to lattice vibrations that are temperature dependent, hence the additional temperature term. The observed non-linear behavior is in agreement with reported non-linear temperature dependence of charge transport in hydrogen molybdenum bronzes that has been attributed to charge-density wave contributions.11 The transport mechanism within hybrid molybdenum bronzes may therefore be similarly as complex as in the inorganic bronzes, warranting deeper investigation in the future.
The hybrid tungsten bronzes such as (4,4′-bipy)0.5H0.7WO3 show more linear behavior in both Arrhenius-type and polaronic-type treatments (Fig. 6B), implying subtle differences in their charge transport mechanisms from the molybdenum analogs. A linear fit of ln(σ) vs. 1/T for (4,4′-bipy)0.5H0.7WO3 yields a small Ea value of 52(3) meV, while (4H-trz)0.5H0.3WO3 gives a somewhat higher value of 179(2) meV. Despite their non-linearity, Arrhenius fits to the approximately linear higher-temperature region above 210 K for (4,4′-bipy)0.5H0.47MoO3, (4H-trz)0.5H0.33MoO3, and (pyz)0.5H0.44MoO3 give ca. 139 meV, 178 meV, and 92 meV, respectively. Notably, the triazole-templated structures show higher activation energies than the bipyridine-templated materials for both Mo and W. This behavior may relate to orbital overlap effects stemming from the octahedral tilting discussed above, though a wider series of materials with equivalent reduction levels and systematically varying levels of tilting in conjunction with electronic structure calculations would be required to further evaluate this hypothesis. The relatively low calculated activation energy values are consistent with the lack of observed absorption peaks in the UV-visible spectral range discussed above. Furthermore, they support either an IVCT or polaronic charge transport mechanism featuring a low activation barrier. The presence of defects leading to shallow trap states that localize charge carriers also cannot be ruled out, and some contribution to the variable-temperature transport behavior likely comes from the inherently thermally activated grain boundary resistance in pressed pellets.14 Single-crystal transport measurements and electronic structure calculations will help parse these possibilities further. Overall, the appreciable electrical conductivities achieved within hybrid molybdenum and tungsten bronzes illustrate the impact of the hybrids' extended inorganic connectivity.
Finally, to test the electronic communication between the inorganic metal-oxide layers and the organic molecular arrays in hybrid metal oxides/bronzes, we measured the electrochemical behavior of (azp)0.5MoO3 in comparison to 4,4′-azopyridine itself. The molecular species alone shows a reversible redox couple with reductive and oxidative peaks at 0.57 and 0.65 V vs. Ag/AgCl in 1 M H2SO4 (Fig. S38†). Upon templation of the redox-active 4,4′-azopyridine within the inorganic layers, this redox couple still appears to be relatively reversible but with shifted reduction and oxidation peak potentials of 0.54 and 0.69 V, respectively. These small perturbations of the electrochemical behavior are consistent with retention access to the molecular redox couple and electronic communication between the organic and inorganic layers. This suggests that electrochemical reduction and ionic intercalation is a promising strategy for tuning the behavior of hybrid bronzes.
3. Conclusions
Hybrid materials offer the potential to simultaneously overcome challenges posed both by solid-state materials' relative inertness to synthetic tuning and by the isolated electronic structures of molecules that preclude charge transport. However, the synchronous combination of facile syntheses, air- and water-stability, low cost, low toxicity, and perhaps most importantly, relatively dispersed electronic bands that can support appreciable charge carrier concentrations into one material system remains an outstanding challenge. Here, we have presented hybrid bronzes as a promising family of materials that features extended inorganic metal-oxide connectivity within two-dimensional layers between which potentially functional organic molecules order in precise arrays. We have shown that bulk crystalline powders and single crystals of these layered hybrids across a series of ten different organic ligands can be isolated according to either intercalation- or templation-based approaches, including using mild solution-state reductants in water at temperatures as low as 80 °C, and crucially, that we can control their degree of mixed valency. Through a suite of characterization techniques, we have demonstrated that the extended metal–oxygen orbital overlap in hybrid molybdenum and tungsten bronzes ensures the population of electrons introduced into the inorganic layers through chemical doping is relatively delocalized, similar to the all-inorganic Mo and W bronzes. Indeed, room temperature four-point dc transport measurements on pressed pellets of templated hybrid bronzes reveal conductivity as high as 8.4 × 10−2 S cm−1 that is comparable to or exceeds H0.33MoO3 under the same conditions. The structure–property insights gained herein have laid the groundwork for the creation of hybrid bronze design principles. These results, coupled with what are likely direct band gaps based on measured diffuse reflectance spectra and reports for the fully oxidized hybrid metal oxides,32 suggest that hybrid bronzes are poised as potentially transformational next-generation materials for energy-related applications.Data availability
Crystallographic data for (azp)0.5MoO3, (pyz)H0.3MoO3, and (4,4′-bipy)0.5H0.3WO3 have been deposited at the CCDC under deposition numbers 2282209, 2282218, and 2282208, respectively.Author contributions
A. J., W. L. D., and R. T. C. conceived the project. W. L. D., R. T. C., B. P. S., and E. M. W. synthesized and structurally characterized the samples. W. L. D. and B. P. S. performed the electrochemistry. W. L. D. and R. T. C. carried out all other experimentation. All authors contributed to the analysis and interpretation of data. A. J., W. L. D., and R. T. C. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank ND Energy for support of W. L. D. through the Patrick and Jana Eilers Graduate Student Fellowship for Energy Related Research and for support of R. T. C. through the Forgash Fellowship for Graduate Student Research in Solar Energy. We are grateful to NDnano for support of B. P. S. through the NDnano Undergraduate Research Fellowship. Single-crystal and powder X-ray diffraction studies were performed at the Notre Dame Molecular Structure Facility. We thank the ND Energy Materials Characterization Facility (MCF) for the use of the UV-visible spectrometer and X-ray photoelectron spectrometer to acquire diffuse reflectance and XPS measurements, respectively. The MCF is funded by the Sustainable Energy Initiative (SEI), which is part of the Center for Sustainable Energy at Notre Dame (ND Energy). Variable temperature conductivity measurements were performed at the Analytical Science and Engineering at Notre Dame (ASEND) Core Facility. We thank Dr Allen Oliver for assistance with X-ray diffraction studies, Dr Ian Lightcap and Anna Matzner for assistance with XPS and diffuse reflectance spectroscopy, and Dr Karl Cronberger for assistance with variable temperature conductivity studies. We also thank William Webb for experimental assistance.Notes and references
- A. R. West, Solid state chemistry and its applications, John Wiley & Sons, Inc., Chichester, West Sussex, U.K., 2nd edn, 2014 Search PubMed.
- M. Greenblatt, Chem. Rev., 1988, 88, 31–53 CrossRef CAS.
- E. Grabowska, Appl. Catal., B, 2016, 186, 97–126 CrossRef CAS.
- P. G. Dickens and M. S. Whittingham, Q. Rev., Chem. Soc., 1968, 22, 30–44 RSC.
- X. Yu, T. J. Marks and A. Facchetti, Nat. Mater., 2016, 15, 383–396 CrossRef CAS PubMed.
- P. M. Marley, G. A. Horrocks, K. E. Pelcher and S. Banerjee, Chem. Commun., 2015, 51, 5181–5198 RSC.
- N. A. Chernova, M. Roppolo, A. C. Dillon and M. S. Whittingham, J. Mater. Chem., 2009, 19, 2526–2552 RSC.
- Z. Zhang, J. Liu, J. Gu, L. Su and L. Cheng, Energy Environ. Sci., 2014, 7, 2535–2558 RSC.
- E. V. Miu, J. R. McKone and G. Mpourmpakis, J. Am. Chem. Soc., 2022, 144, 6420–6433 CrossRef CAS PubMed.
- S. Noriyuki, F. Takuya, E. Kazuo, K. Masakazu and N. Masahito, Chem. Lett., 1999, 28, 593–594 CrossRef.
- S. Adams, J. Solid State Chem., 2000, 149, 75–87 CrossRef CAS.
- T. Yamauchi, M. Isobe and Y. Ueda, Solid State Sci., 2005, 7, 874–881 CrossRef CAS.
- B. S. Guiton, M. Stefik, V. Augustyn, S. Banerjee, C. J. Bardeen, B. M. Bartlett, J. Li, V. López-Mejías, L. R. MacGillivray, A. Morris, E. E. Rodriguez, A. C. S. Samia, H. Sun, P. Sutter and D. R. Talham, MRS Bull., 2020, 45, 951–964 CrossRef.
- L. S. Xie, G. Skorupskii and M. Dincă, Chem. Rev., 2020, 120, 8536–8580 CrossRef CAS PubMed.
- T. Li and J. E. Goldberger, Chem. Mater., 2015, 27, 3549–3559 CrossRef CAS.
- G.-E. Wang, S. Luo, T. Di, Z. Fu and G. Xu, Angew. Chem., Int. Ed., 2022, 61, e202203151 CrossRef CAS PubMed.
- B. Saparov and D. B. Mitzi, Chem. Rev., 2016, 116, 4558–4596 CrossRef CAS PubMed.
- A. Nag, Nano Lett., 2021, 21, 8529–8531 CrossRef CAS PubMed.
- N. R. Wolf, B. A. Connor, A. H. Slavney and H. I. Karunadasa, Angew. Chem., Int. Ed., 2021, 60, 16264–16278 CrossRef CAS PubMed.
- J. W. Johnson, A. J. Jacobson, S. M. Rich and J. F. Brody, J. Am. Chem. Soc., 1981, 103, 5246–5247 CrossRef CAS.
- Y. Zhang, R. C. Haushalter and A. Clearfield, Inorg. Chem., 1996, 35, 4950–4956 CrossRef CAS PubMed.
- T. Chirayil, P. Y. Zavalij and M. S. Whittingham, Chem. Mater., 1998, 10, 2629–2640 CrossRef CAS.
- P. J. Hagrman, D. Hagrman and J. Zubieta, Angew. Chem., Int. Ed., 1999, 38, 2638–2684 CrossRef PubMed.
- N. Guillou and G. Férey, J. Solid State Chem., 1999, 147, 240–246 CrossRef CAS.
- P. J. Hagrman, R. L. LaDuca, H. J. Koo, R. Rarig, R. C. Haushalter, M.-H. Whangbo and J. Zubieta, Inorg. Chem., 2000, 39, 4311–4317 CrossRef CAS PubMed.
- B. Yan, Y. Xu, N. K. Goh and L. S. Chia, Chem. Commun., 2000, 2169–2170 RSC.
- Y. Liang, M. Hong, R. Cao, D. Sun and J. Weng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2001, 57, m159–m161 CrossRef CAS.
- B. Yan and P. A. Maggard, Inorg. Chem., 2007, 46, 6640–6646 CrossRef CAS PubMed.
- Islah-u-din, M. R. Fox, H. Martin, G. J. Gainsford, J. Kennedy, A. Markwitz, S. G. Telfer, G. B. Jameson and J. L. Tallon, Chem. Commun., 2010, 46, 4261–4263 RSC.
- S. V. Chong and J. L. Tallon, J. Phys. Chem. Solids, 2010, 71, 303–308 CrossRef CAS.
- M. Bujoli-Doeuff, R. Dessapt, P. Deniard and S. Jobic, Inorg. Chem., 2012, 51, 142–149 CrossRef CAS PubMed.
- X. Zhang, M. Hejazi, S. J. Thiagarajan, W. R. Woerner, D. Banerjee, T. J. Emge, W. Xu, S. J. Teat, Q. Gong, A. Safari, R. Yang, J. B. Parise and J. Li, J. Am. Chem. Soc., 2013, 135, 17401–17407 CrossRef CAS PubMed.
- T. R. Amarante, P. Neves, A. A. Valente, F. A. A. Paz, M. Pillinger and I. S. Gonçalves, J. Catal., 2016, 340, 354–367 CrossRef CAS.
- A. B. Lysenko, G. A. Senchyk, K. V. Domasevitch, M. Kobalz, H. Krautscheid, J. Cichos, M. Karbowiak, P. Neves, A. A. Valente and I. S. Gonçalves, Inorg. Chem., 2017, 56, 4380–4394 CrossRef CAS PubMed.
- R. Schöllhorn, T. Schulte-Nölle and G. Steinhoff, J. Less-Common Met., 1980, 71, 71–78 CrossRef.
- N. Kinomura, K. Mizumoto and N. Kumada, J. Solid State Chem., 1997, 128, 256–260 CrossRef CAS.
- R. Wang, X. Lu, L. Hao, W. Jiao, W. Liu, J. Zhang, F. Yuan and F. Yang, J. Mater. Chem. C, 2017, 5, 427–433 RSC.
- D. Ji, S. Cai, T. R. Paudel, H. Sun, C. Zhang, L. Han, Y. Wei, Y. Zang, M. Gu, Y. Zhang, W. Gao, H. Huyan, W. Guo, D. Wu, Z. Gu, E. Y. Tsymbal, P. Wang, Y. Nie and X. Pan, Nature, 2019, 570, 87–90 CrossRef CAS PubMed.
- X. Feng, R. Cheng, L. Yin, Y. Wen, J. Jiang and J. He, Adv. Mater., 2023, 2304708 CrossRef PubMed.
- O. Glemser and G. Lutz, Z. Anorg. Allg. Chem., 1951, 264, 17–33 CrossRef CAS.
- J. J. Birtill and P. G. Dickens, Mater. Res. Bull., 1978, 13, 311–316 CrossRef CAS.
- H. Cheng, M. Wen, X. Ma, Y. Kuwahara, K. Mori, Y. Dai, B. Huang and H. Yamashita, J. Am. Chem. Soc., 2016, 138, 9316–9324 CrossRef CAS PubMed.
- S. Noriyuki, E. Kazuo, S. Masayasu and T. Sadao, Bull. Chem. Soc. Jpn., 1989, 62, 903–907 CrossRef.
- P. G. Dickens and S. J. Hibble, Solid State Ionics, 1986, 22, 69–73 CrossRef CAS.
- K. Ara, H. Tagaya, T. Ogata, K. Matsushita, J.-i. Kadokawa, M. Karasu and K. Chiba, Mater. Res. Bull., 1996, 31, 283–294 CrossRef CAS.
- J. Z. Ou, J. L. Campbell, D. Yao, W. Wlodarski and K. Kalantar-zadeh, J. Phys. Chem. C, 2011, 115, 10757–10763 CrossRef CAS.
- A. B. Lysenko, G. A. Senchyk, J. Lincke, D. Lässig, A. A. Fokin, E. D. Butova, P. R. Schreiner, H. Krautscheid and K. V. Domasevitch, Dalton Trans., 2010, 39, 4223–4231 RSC.
- I. D. Brown, The Chemical Bond in Inorganic Chemistry: The Bond Valence Model, Oxford University Press, 2006 Search PubMed.
- I. D. Brown, Chem. Rev., 2009, 109, 6858–6919 CrossRef CAS PubMed.
- M. B. Robin and P. Day, in Advances in Inorganic Chemistry and Radiochemistry, ed. H. J. Emeléus and A. G. Sharpe, Academic Press, 1968, vol. 10, pp. 247–422 Search PubMed.
- D. Tinet, P. Canesson, H. Estrade and J. J. Fripiat, J. Phys. Chem. Solids, 1980, 41, 583–589 CrossRef CAS.
- B. Y. Zhang, A. Zavabeti, A. F. Chrimes, F. Haque, L. A. O'Dell, H. Khan, N. Syed, R. Datta, Y. Wang, A. S. R. Chesman, T. Daeneke, K. Kalantar-zadeh and J. Z. Ou, Adv. Funct. Mater., 2018, 28, 1706006 CrossRef.
- D. O. Scanlon, G. W. Watson, D. J. Payne, G. R. Atkinson, R. G. Egdell and D. S. L. Law, J. Phys. Chem. C, 2010, 114, 4636–4645 CrossRef CAS.
- F. Y. Xie, L. Gong, X. Liu, Y. T. Tao, W. H. Zhang, S. H. Chen, H. Meng and J. Chen, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 112–118 CrossRef CAS.
- N. Suzuki, K. Yabuki, N. Oshimura, S. Yoshio and K. Adachi, J. Phys. Chem. C, 2022, 126, 15436–15445 CrossRef CAS.
- Q. Qu, W.-B. Zhang, K. Huang and H.-M. Chen, Comput. Mater. Sci., 2017, 130, 242–248 CrossRef CAS.
- F. Wang, C. Di Valentin and G. Pacchioni, J. Phys. Chem. C, 2011, 115, 8345–8353 CrossRef CAS.
- T. Sekikawa, J. L. Tallon, S. V. Chong and Y. Ōno, J. Phys. Soc. Jpn., 2023, 92, 023702 CrossRef.
- J. Tauc, Mater. Res. Bull., 1968, 3, 37–46 CrossRef CAS.
- Y. Kuwahara, Y. Yoshimura, K. Haematsu and H. Yamashita, J. Am. Chem. Soc., 2018, 140, 9203–9210 CrossRef CAS PubMed.
- N. S. Hush, Electrochim. Acta, 1968, 13, 1005–1023 CrossRef CAS.
- N. S. Hush, Coord. Chem. Rev., 1985, 64, 135–157 CrossRef CAS.
- O. F. Schirmer and E. Salje, Solid State Commun., 1980, 33, 333–336 CrossRef CAS.
- D. Emin, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13691–13702 CrossRef CAS PubMed.
- O. F. Schirmer, V. Wittwer, G. Baur and G. Brandt, J. Electrochem. Soc., 1977, 124, 749–753 CrossRef CAS.
- T. He and J. Yao, J. Photochem. Photobiol., C, 2003, 4, 125–143 CrossRef CAS.
- S. Raj, T. Sato, S. Souma, T. Takahashi, D. D. Sarma and P. Mahadevan, Mod. Phys. Lett. B, 2009, 23, 2819–2846 CrossRef CAS.
- J. Yin, H. Li, D. Cortecchia, C. Soci and J.-L. Brédas, ACS Energy Lett., 2017, 2, 417–423 CrossRef CAS.
- A. J. Neukirch, W. Nie, J.-C. Blancon, K. Appavoo, H. Tsai, M. Y. Sfeir, C. Katan, L. Pedesseau, J. Even, J. J. Crochet, G. Gupta, A. D. Mohite and S. Tretiak, Nano Lett., 2016, 16, 3809–3816 CrossRef CAS PubMed.
- X. Y. Zhu and V. Podzorov, J. Phys. Chem. Lett., 2015, 6, 4758–4761 CrossRef CAS PubMed.
- E. R. Dohner, A. Jaffe, L. R. Bradshaw and H. I. Karunadasa, J. Am. Chem. Soc., 2014, 136, 13154–13157 CrossRef CAS PubMed.
- M. D. Smith, A. Jaffe, E. R. Dohner, A. M. Lindenberg and H. I. Karunadasa, Chem. Sci., 2017, 8, 4497–4504 RSC.
- K. Morita, M. J. Golomb, M. Rivera and A. Walsh, Chem. Mater., 2023, 35, 3652–3659 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, diffraction patterns, spectra, and supplemental discussion. CCDC 2282208, 2282209, and 2282218. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03828a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |