DOI:
10.1039/D3SC03806H
(Edge Article)
Chem. Sci., 2023,
14, 13944-13950
Group 13 ion coordination to pyridyl breaks the reduction potential vs. hydricity scaling relationship for dihydropyridinates†
Received
22nd July 2023
, Accepted 15th November 2023
First published on 16th November 2023
Abstract
The relationship Epvs. ΔGH– correlates the applied potential (Ep) needed to drive organohydride formation with the strength of the hydride donor that is formed: in the absence of kinetic effects Epvs. ΔGH– should be linear but it would be more energy efficient if Ep could be shifted anodically using kinetic effects. Biological hydride transfers (HT) performed by cofactors including NADH and lactate racemase do occur at low potentials and functional modeling of those processes could lead to low energy HT reactions in electrosynthesis and to accurate models for cofactor chemistry. Herein we probe the influence of N-alkylation or N-metallation on ΔGH– for dihydropyridinates (DHP−) and on Ep of the DHP− precursors. We synthesized a series of DHP− complexes of the form (pz2HP−)E via hydride transfer from their respective [(pz2P)E]+ forms where E = AlCl2+, GaCl2+ or Me+. Relative ΔGH– for the (pz2HP−)E series all fall within 1 kcal mol−1, and ΔGH– for (pz2HP)CH3 was approximated as 47.5 ± 2.5 kcal mol−1 in MeCN solution. Plots of Epvs. ΔGH– including [(pz2P)E]+ suggest kinetic effects shift Ep anodically by ∼215 mV.
Introduction
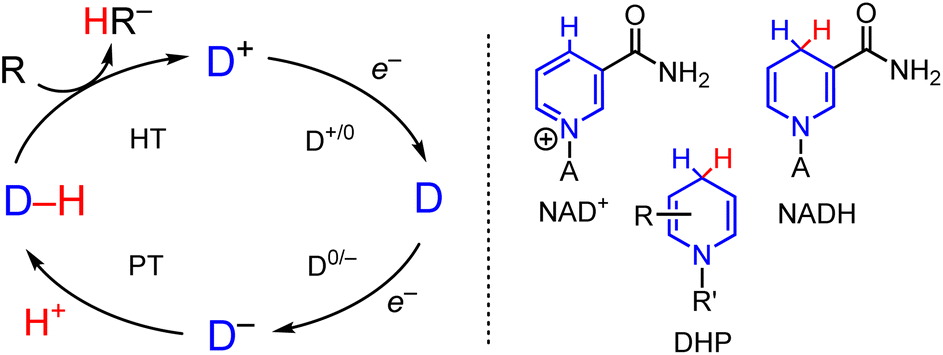
Reduction reactions involving hydride transfer (HT, formally two electrons and a proton) are important in transformations ranging from CO2 reduction1,2 to selective reductions of carbonyls, imines, and alkenes.3–5 In enzymes, tremendous selectivity and efficiency in reduction reactions involving HT is achieved by cofactors such as NAD+/NADH and the nickel pincer nucleotide (NPN) cofactor in lactate racemase, in a carefully tailored environment.6,7 Synthetic chemists have harnessed this control in systems which incorporate enzymes for highly efficient reactions in the presence of catalytic NAD+,8,9 and synthetic chemists aspire to similar reaction control as in the asymmetric co-catalysis of enantioselective organocatalytic hydride reduction.10,11 Alongside these reaction conditions, a wide array of both metal hydride (M–H) and organohydride (C–H) transfer agents has been developed, and ongoing efforts have various directions: improved selectivity for desired products,12–14 and functional group tolerance,15,16 are just two examples.
In the growing area of electrosynthesis and electrocatalysis additional considerations need to be made for the design of HT reagents that can be effectively (re)generated electrochemically. These include choice of proton source for hydride regeneration and catalytic turnover and design of precursors to the hydride which lower the applied potential required for the reduction steps that (re)generate the hydride reagent in the catalytic cycle (Scheme 1).17–19 The lowered potential is needed to save energy, and minimize side reactions including proton reduction to H2 and decomposition of catalyst or substrate. It is well-known that the half wave potential, E1/2(D+/0) for the hydride precursor (D+ in Scheme 1) scales with the free energy change associated with loss of hydride ion from a metal- or organo-hydride as in eqn (1):20,21
| | | D–H → D+ + H− free energy: ΔGH– | (1) |
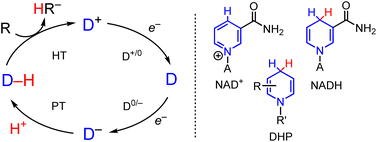 |
| | Scheme 1 (Left) Outline of electron and proton steps to generic cationic hydride precursor, D+, leading to hydride formation of a general hydride donor D–H and subsequent HT to substrate. (Right) Line drawing of NAD+, NADH and dihydropyridinate, DHP. A = Adenine dinucleotide. | |
This free energy change is also called hydricity (ΔGH–). Experimentally, hydricity is determined using thermochemical cycles that are well described in the literature,22,23 and cycles relevant to this work are included below (vide infra).
Hydricity can be a useful predictor for the overall driving force for an HT reaction and influence reaction selectivity, where a smaller ΔGH– value indicates a stronger thermodynamic driving force for HT, i.e. a stronger hydride donor. For example, electrocatalytic reduction of CO2 to HCOO− is optimized when the hydride donor is sufficiently hydridic to make CO2 reduction thermodynamically favorable but not so strong as to undergo competitive H2 evolution via reaction of the hydride with protons.24–27 Both E1/2 and ΔGH– are intrinsic thermodynamic properties of the hydride donor reagent.28,29 In practice, reduction of D+ is often coupled with proton transfer (PT) reactions and then the CV waveform manifests as an irreversible redox event where both thermodynamic and kinetic contributions to the D+/0 and D0/− redox couples and PT influence the necessary applied potential (Ep) that is required to generate D–H.30 Metal ion coordination is known qualitatively to lower Ep for organic molecules including DHP−s but not enough data has been reported to draw any correlations between structure and function.31,32
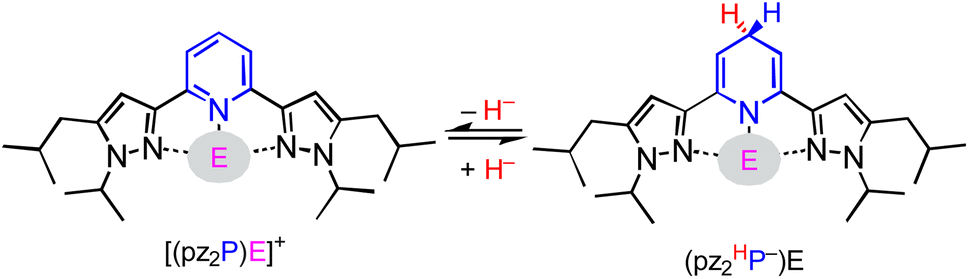
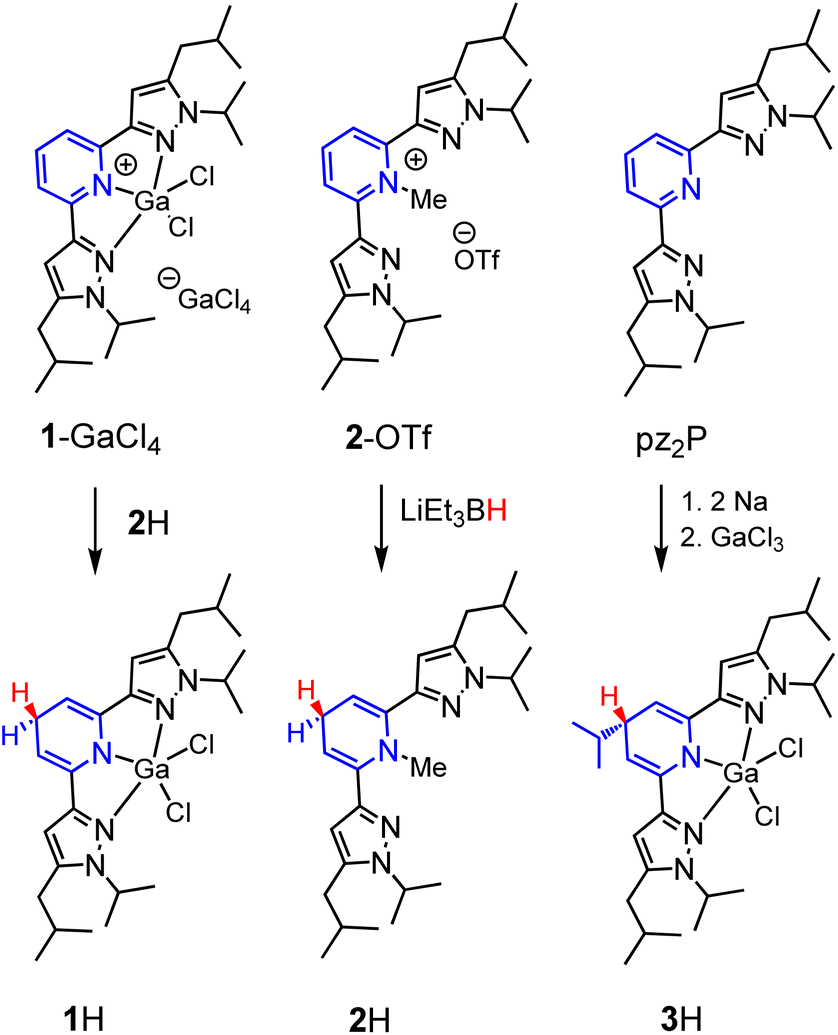
In this report, we investigate structural tuning of dihydropyridinates (DHP−) and their precursors via metal ion effects. To assess the results of metal ion coordination we considered the scaling relationship between Epvs. ΔGH– using extensive data reported in the literature, and as discussed below.21,33,36 The dipyrazolylpyridine ligand platform (pz2P) supports complexes with the form [(pz2P)E]+ where E = GaCl2+, CH3+ and AlCl2+, and those compounds are denoted herein as 1+, 2+ and 4+ respectively.30,36 In this report the syntheses of organohydrides (pz2HP−)E, are reported for E = GaCl2+ and CH3+, via a formal addition of hydride (two electrons and one proton) to [(pz2P)E]+ (Scheme 2). Characterization of the geometric and electronic structures of 1H, 2H and 4H was performed and the hydride donor ability for each compound benchmarked. A comparison of the properties of 1H and 2H with those of previously reported organohydride reagents reveals reduction potential is lowered by + 215 mV for 1H and does not deviate within error for 2H relative to Ep expected according to the Epvs. ΔGH– scaling relationship. The origin of this effect is discussed.
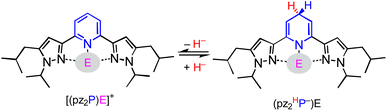 |
| | Scheme 2 Interconversion of (pz2P)E+ with (pz2HP−)E via HT, where E = AlCl2+, GaCl2+, and CH3+. | |
Results and discussion
Preparation of compounds
We targeted the 1,4-DHP−‘s derived from [(pz2P)GaCl2]+ (1+) and [(pz2P)CH3]+ (2+) (Scheme 3). In an attempt to generate [(pz2HP−)]GaCl2 (1H), two equivalents of Na were added to a solution of one equivalent pz2P in THF at room temperature and the clear solution turned deep red following consumption of the Na metal over 24 h.35 Addition of one equivalent of GaCl3 resulted in a further colour change to dark purple. The 1H NMR spectrum of the isolated product showed no resonances corresponding to protons on an aromatic pyridine ring but instead resonances at 5.21 and 3.57 ppm were observed. These resonances are consistent with a 1,4-DHP− ligand form coordinated to GaCl2+, and consistent with our previous report of [(pz2HP−)]AlCl2, 4H,36 where the resonances were observed at 4.99 and 3.50 ppm. However, integration of the resonance at 3.57 ppm gives a value of 1H atom for the Cp–H proton (see Fig. 1 for carbon atom labelling scheme), but integration of 2H atoms is expected for formation of 1H. A multiplet at 1.76 and doublet at 1.12 ppm corresponding to integrations of 1H atom and 6H atoms, respectively, suggest substitution of an iPr group at the Cp carbon (Fig. S1†), and this was confirmed by single crystal X-ray diffraction which identified [(pz2iPrHP−)]GaCl2 (3H), (vide infra). Presumably, a reductive cleavage of N-isopropyl generates isopropylsodium, which, in turn, reacts with the pyridine ring to form 3H.37
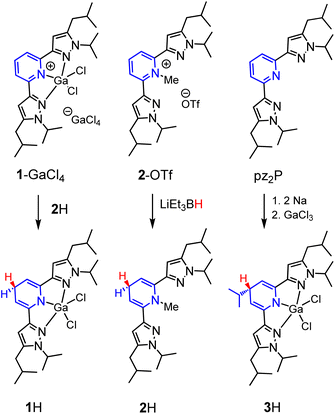 |
| | Scheme 3 Syntheses of 1H, 2H and 3H. | |
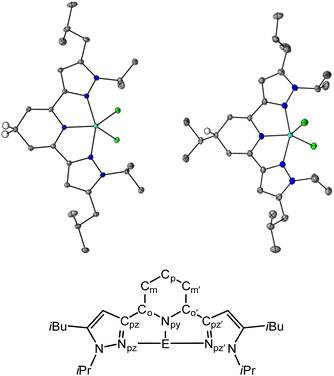 |
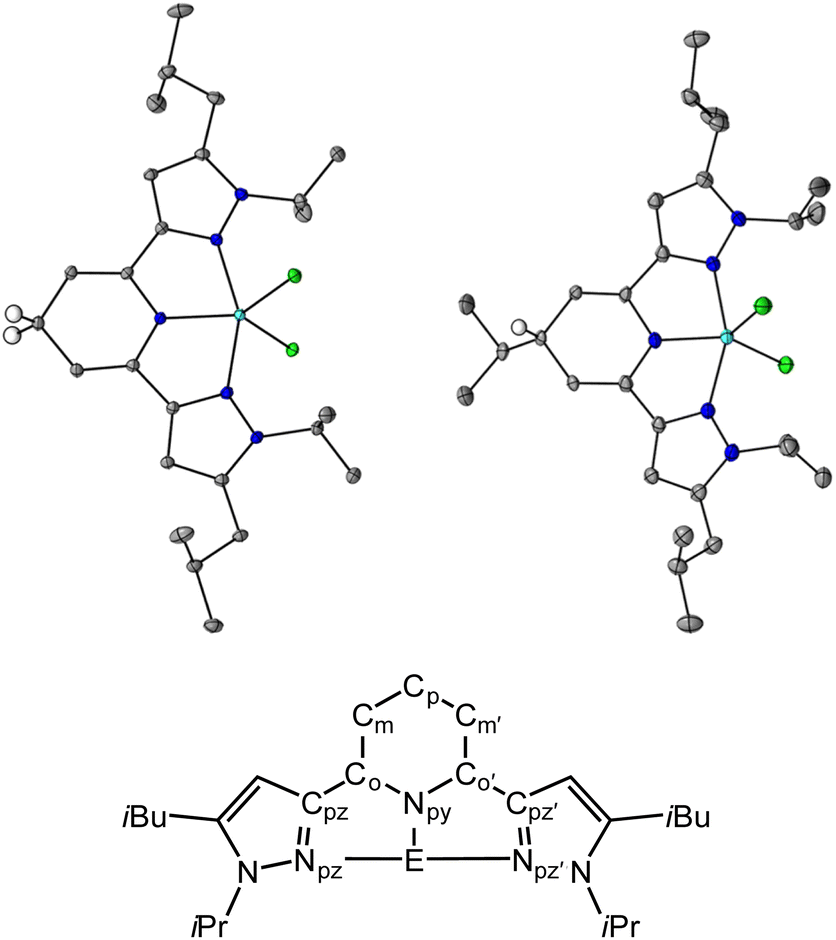
| | Fig. 1 Solid state structures of 1H (top left) and 3H (top right) and pz2P atom naming convention used throughout (bottom). Blue, light blue, green, gray ellipsoids, and white circles represent N, Ga, Cl, C, and H atoms, respectively. H atoms except Cp–H omitted for clarity. The thermal ellipsoids are shown at 30% probability. | |
Synthesis of 1H was also attempted via several additional routes
Reduction of [(pz2P)GaCl2]GaCl4 (1-GaCl4) was attempted via reactions with sodium metal, sodium napthalenide, and decamethylcobaltocene, but these reactions did not yield clean isolable products. Reactions of 1-GaCl4 with hydride donors NaBH4, LiAlH4, and LiEt3BH also did not yield 1H. Ultimately, we obtained 1H in 55% yield as crystals formed from a concentrated solution of 1-GaCl4 and 2H in MeCN (the synthesis of 2H is described in the next paragraph). The 1H NMR spectrum of 1H showed no aromatic resonances, but the Cp–H resonance is observed as a triplet at 3.48 ppm, and this is consistent with DHP− formation as observed in 4H, 2H, and 3H which have Cp–H resonances at 3.50, 3.62 and 3.57 ppm, respectively. The pyrazole ring proton resonance is a singlet, shifted upfield, from 7.01 in 1+ to 5.92 ppm in 1H (Fig. S2†). The composition of 1H was further confirmed by X-ray diffraction and combustion analysis.
The synthesis of 1,4-DHP−s from N-alkylated pyridinium salts has been reported, using sodium dithionate,38 or a metal hydride donor as reductant.39 For the preparation of 2H we found that addition of 1.3 equivalents of lithium triethyl borohydride, LiEt3BH, to a colourless solution of 2-OTf in THF resulted in an instant colour change to a yellow solution. After stirring for 5 minutes the solution was concentrated to a residue which was dissolved in hexane and filtered. The yellow oily filtrate yielded pz2(N-MeHP), 2H, in 27% yield following workup. The proton NMR spectrum of 2H is consistent with the 1,4-DHP− structure and with NMR resonances at 5.62 and 3.24 ppm (Fig. S3†). The composition of 2H was confirmed using HRMS.
Solid state structures
Crystals suitable for single crystal X-ray diffraction of the 1,4-DHP− ligand compounds, 1H and 3H, were obtained from saturated solutions of MeCN and hexane respectively over a period of three days, and were obtained as purple, and colorless block shaped crystals, respectively (Tables S1 and S2,†Fig. 1). We were not able to obtain crystals of 2H despite many attempts. For 1H, the average bond lengths of the Npy–Co, Co–Cm and Cp–Cm bonds are 1.387(6), 1.343(7) and 1.507(7) Å, respectively, and for 3H these are 1.433(6), 1.33(2) and 1.507(8) Å, respectively (see Fig. 1 for pz2P atom naming). The increased bond lengths for Cp–Cm in both 1H and 3H relative to 1+, are characteristic of 1,4-DHP− structures. Upon formation of the 1,4-DHP−, the geometry around the Ga(III) center becomes closer to trigonal bipyramidal with a τ5 value of 0.76 for both 1H and 3H.40 There is also an increase in the Npz–Ga–Npz’ bond angle from 154.9(1)° in 1+ to 156.47(3)° and 156.51(8)° in 1H and 3H respectively.30 The ligand twists to accommodate the distorted trigonal bipyramidal geometry as indicated by the torsion angle between the two coordinating pyrazine arms Cpz![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NpzCpz′Npz’ which increase by 2.89° and 1.91° for 1H and 3H respectively relative to 1+. There is no significant difference in the bond distances within the pyrazole rings between all compounds discussed (Tables S3 and S4†).
NpzCpz′Npz’ which increase by 2.89° and 1.91° for 1H and 3H respectively relative to 1+. There is no significant difference in the bond distances within the pyrazole rings between all compounds discussed (Tables S3 and S4†).
Hydride transfer reactions of 1H, 2H and 4H
There are several possible experiments that can be used to obtain ΔGH-.22 We found hydride transfer best suited for 1H, 2H and 4H where chemical equilibria were established between 1H, 2H and 4H, and organohydride molecules of known ΔGH-. Using this method, direct HT from a donor (D–H) to acceptor (A+) is observed and then an equilibrium constant can be determined which establishes the difference in ΔGH- between the donor and acceptor. The relationship between the hydride donor ability of D–H and A–H, and the hydride transfer between D+ and A+ is described by eqn (2)–(5):22| | | D–H + A+ ⇌ D+ + A–H K(2) | (2) |
| | | A–H ⇌ A+ + H− ΔGH−(A–H) | (3) |
| | | D–H ⇌ D+ + H− ΔGH–(D–H) | (4) |
where ΔGH–(4) = ΔGH–(2) – ΔGH–(3)
and
| | ΔGH–(2) = –RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(K(2)) ln(K(2)) | (5) |
We first set out to determine the hydricity of 2H, because 2H is the easiest of the hydrides to make, with a series of hydride acceptors of known hydricity. Reaction of 2-OTf with one equivalent of 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole, BnHPh (ΔGH– = 50 kcal mol−1 in MeCN, (Chart 1))19 resulted in HT which was observed by 1H NMR in CD3CN after 2 days and approached equilibrium after two weeks, as in eqn (6):
| | | BnHPh + 2+ ⇌ BnPh+ + 2H | (6) |
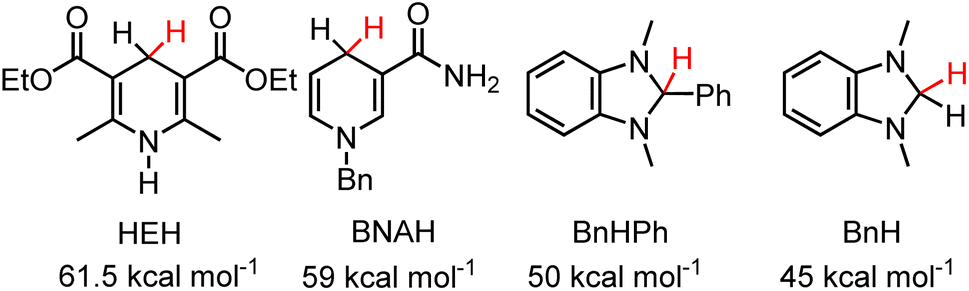
HT from 2H to one equivalent of 1,3-dimethyl-1H-benzimidazolium, Bn+ (ΔGH– = 45 kcal mol−1 in MeCN (Chart 1))29 was also observed by 1H NMR after 3 days and approached equilibrium after two weeks (eqn (7), Fig. S4 and S5†). The equilibria were heavily reactant favoured suggesting that the hydricity of 2H lies close to the middle of the range from 45–50 kcal mol−1 in MeCN solution. Control experiments run in parallel consisting of the starting materials 2-OTf, 2H, 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole and 1,3-dimethyl-1H-benzimidazolium iodide in CD3CN with trimethoxybenzene as an internal standard show no changes after 2 weeks (Fig. S6–S9†). We, therefore, estimate for 2H that ΔGH- = 47.5 ± 2.5 kcal mol−1. These measurements put the ΔGH– of 2H on the lower side (stronger hydride donor) of values reported for 1,4-DHPs which have been reported from 73–43 kcal mol−1.21 As a reference point to commonly employed DHPs, the Hantsch ester (HEH) has ΔGH– = 61.5 kcal mol−1 and BNAH (1-benzyl-1,4-dihydronicotinamide) has ΔGH– = 59 kcal mol−1. An established trend is that DHP−s are more hydridic when they are functionalized with electron donating groups. Both the Hantzsch ester and BNAH feature electron withdrawing groups (Chart 1), whereas 2H has two moderately electron donating pyrazole groups. The hydricity of 2H is similar to methyl substituted N-alkylated DHP−s.21
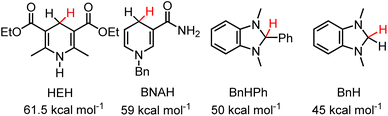 |
| | Chart 1 1,4-DHPs and benzimidazoles discussed in the text and their reported hydricities in MeCN.21 | |
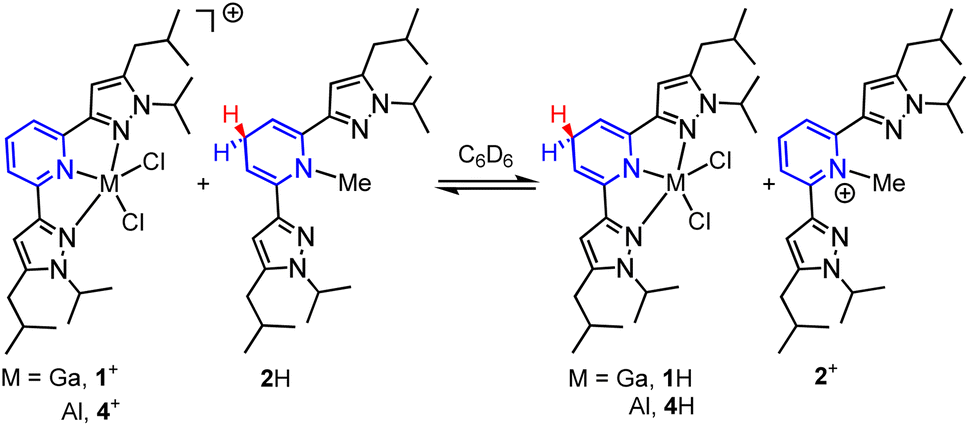
Having established the hydricity of 2H, we endeavoured to determine if HT from 2H would be observed to either 1+ or 4+. Reactions of a 1:1 molar ratio of 1-GaCl4 with 2H, and of [(pz2P)AlCl2]AlCl4, 4-AlCl4, with 2H were monitored by 1H NMR in C6D6 in order to determine the reaction equilibria (Scheme 4).1,41 The back reaction in these equilibria corresponds to eqn (2) and were used in calculations of ΔGH–. Integration of 1H NMR signals (Cp–H of 4+/4H and 1+/1H and Npy–CH3 of 2+/2H) relative to the internal standard trimethoxybenzene showed equilibria were established after 4 days for reaction of 1+ and 2H and after 3 days for the reaction of 4+ and 2H (see ESI for full experimental details†). We recognize that solvents such as DMSO, MeCN or H2O, which have higher dielectric constants and a wealth of reported hydricity data, would be a better choice for monitoring these equilibria,23 but 1-GaCl4 is insoluble in DMSO, 1H is sparingly soluble in MeCN, and both 4+ and 1+ are unstable in protic solvents. Equilibrium constants (K) were calculated as 0.83 for the back reaction of 1-GaCl4 with 2H and 0.24 for the back reaction of 4-AlCl4 with 2H. These values correspond to a difference in ΔGH- between 1H and 2H of 0.1 ± 0.1 kcal mol−1 and a difference in ΔGH- between 4H and 2H of 0.8 ± 0.1 kcal mol−1. Given that the values of 1H, 2H and 4H were all found to be within 1 kcal mol−1 in benzene, which has a dielectric constant over 15 times lower than MeCN, we estimate that the hydricity values of 1H and 4H in MeCN would differ from 2H by no more than 1 kcal mol−1.25,28,42 Minor differences in ΔGH- might arise from the varied Lewis acidity of the cations.
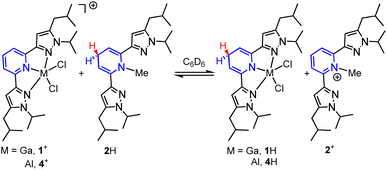 |
| | Scheme 4 HT equilibria between 1-GaCl4 and 2H, and 4-AlCl4 and 2H. Counter ions omitted for clarity. | |
Metal ion effects on the scaling relationship between Ep and ΔGH– for organohydrides
Linear correlations between hydricity (ΔGH–) of a transition metal hydride and the first reversible redox couple, E1/2(D+/0) in Scheme 1, of a parent transition metal complex are well established within several classes of metal complexes.24,43,44 Concomitant reports by the groups of Kubiak,20 and Glusac,21 showed that a more general correlation exists across several classes of transition metal and ligand sets, and across structurally diverse organohydrides.
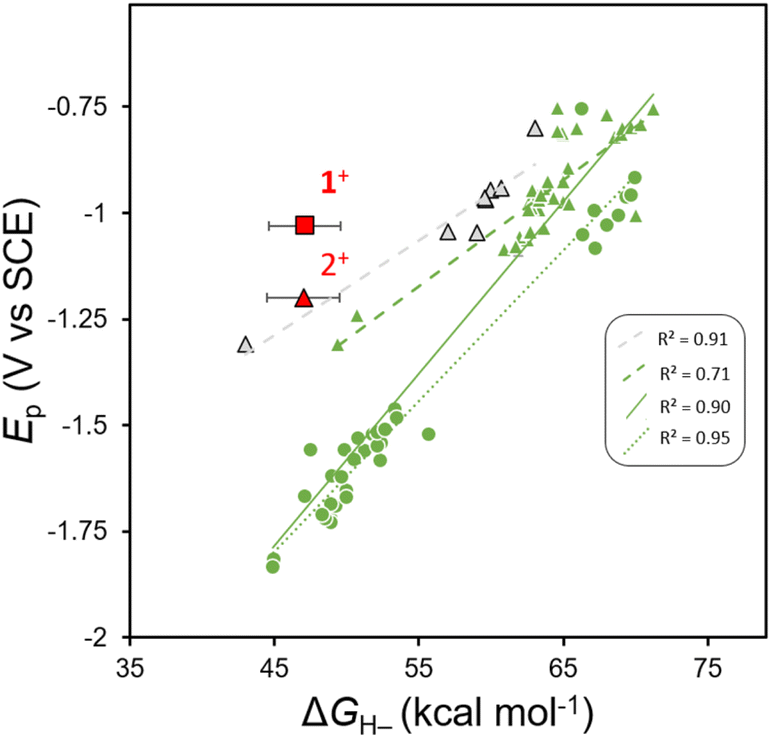
It is known that many oxidized organohydride precursors display irreversible reduction events, Ep, in CV experiments and we wondered if there is any correlation between a plot of Epvs. ΔGH– for those compounds and whether the plot might highlight a variation in kinetic contributions to reduction that are causing the irreversibility. To construct a plot of Epvs. ΔGH– we used reported, computationally obtained ΔGH– values for N-containing heterocyclic organohydrides in MeCN solution,21 with their reported irreversible cathodic peak potentials (Ep) obtained using CV in MeCN solution.33,36 We compiled Ep values for various imidazoles,33 and substituted 1,4-DHPs;36 and all Ep values were converted to V vs. SCE. Linear regression provides a linear relationship (Fig. 2). We notice that when fit independently, predicted Ep values for 1,4-DHPs are at more positive potentials than benzimidazoles with comparable ΔGH– within the reported ΔGH– range. Furthermore, another anodic shift in predicted Ep values is observed from fits of experimental ΔGH– values of 1,4-DHPs,45–48 compared to those reported from computational methods. Experimentally measured values of ΔGH– (for 1H and 2H) and of Ep (for 1+ and 2+) were added to the plot of Epvs. ΔGH– for comparison (red symbols, Fig. 2). Relative to the correlation line, 1+ is more easily reduced: Ep(1+) is +215 mV more anodic than predicted. The Al-supported DHP compound 4H has a similar Ep to 2H when both are measured in THF, and this suggests that both AlCl2+ and GaCl2+ similarly break the Epvs. ΔGH– scaling relationship, but there is not enough data available in THF to illustrate this point on a plot as we do for MeCN in Fig. 2. All the organohydrides plotted have cationic oxidized forms so the overall charge on the molecules should not be a factor contributing to Ep.
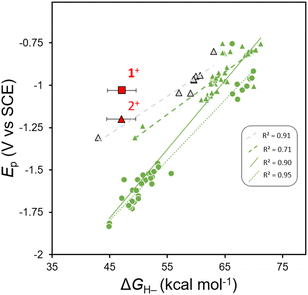 |
| | Fig. 2 Plot of Epvs. ΔGH– for benzimidazoles (circles), 1,4-DHPs (triangles) 2+ and those previously published and complexe 1+. Black outlines around markers represent experimentally determined ΔGH– values, green markers indicate computational ΔGH– values and red markers corresponds to compounds reported herein. The figure key displays R2 values for linear fits to: 1,4-DHPs with experimentally determined ΔGH– values (dashed grey line).36,45–48 1,4-DHPs with computationally determined ΔGH– values (dashed green line),21,36 1,4-DHPs and benzimidazoles with computationally determined ΔGH– values (solid green line),21,33,36 and benzimidazoles with computationally determined ΔGH– values (dotted green line).21,334+ is not included on the plot since it is not stable in MeCN. | |
The combined data for 1H, 2H (and 1+, 2+) and the deviation of 1+ from the Epvs. ΔGH– correlation lines are consistent with differences in reorganization energy that can be rationalized by the structural changes between the 1+ and 1H pair. We expect no large structural changes upon conversion to 1H and so it is reasonable to expect that kinetic contributions to Ep might be relatively low. In contrast, there are obvious structural differences between the 2+/2H pair. For 2+ we know that the flanking pyrazolyl rings of pz2P have N-donor atoms oriented in toward the cationic N-Me-pyridyl group, and in 2H those N-donor atoms of the pyrazolyl rings rotate away since the py ring is no longer cationic, as in the known structure of pz2P where the pyrazolyl N atoms rotate out.36 An additional effect of the Group 13 cation may be to stabilize the DHP−via bonding interactions between the Npy π-electrons and pz orbital of the metal: electron donation from Npy to group 13 3+ cations have been observed in other complexes of tridentate pyridyl-centered ligands,49 and may stabilize the DHP−.50
Conclusion
New compounds 1H and 2H were prepared by a direct reaction of a hydride donor with the neutral ligand complexes 1+, and 2+, respectively: this hydride transfer reaction is a formal two-electron reduction and single protonation. Measurements of the Ep and ΔGH– for 1+, 2+, and 4+, show that 1+ and 4+ do not follow the Epvs. ΔGH– scaling relationship. Both 1+ and 4+ have N-pyridyl coordination to Group 13 3+ cations, whereas 2+ is N-alkylated. These data were collected in THF and MeCN where possible, and the work in MeCN additionally permitted some quantification of this effect, where 2+ is shifted anodically from the Epvs. ΔGH– relationship by 215 mV (Fig. 2). Some of the observed anodic shift may arise from the cationic nature of [(pz2P)E]+ but it is unlikely that the full 215 mV anodic shift can be attributed to a single positive charge given that all of the model organohydrides on the plot have equivalent cationic charge. Based on these results, we propose that N-coordination of a Group 13 3+ cation to pyridyl offers a strategy for kinetic lowering of Ep. Future work on well-designed N-metallated DHP−s will target electrochemically-driven HT reaction chemistry.
Author contributions
Leo Parsons collected and analyzed the data, and was involved in writing the manuscript. James Fettinger was involved in refinement of solid state structural X-ray data. Louise Berben was responsible for project design and management, and writing the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This manuscript is based on work supported by the National Science Foundation with award CHE-2054529.
Notes and references
- R. B. Weerasooriya, J. L. Gesiorski, A. Alherz, S. Ilic, G. N. Hargenrader, C. B. Musgrave and K. D. Glusac, Kinetics of Hydride Transfer from Catalytic Metal-Free Hydride Donors to CO2, J. Phys. Chem. Lett., 2021, 12, 2306–2311 CrossRef CAS PubMed.
- D. Ghosh, K. Kobayashi, T. Kajiwara, S. Kitagawa and K. Tanaka, Catalytic Hydride Transfer to CO2 Using Ru-NAD-Type Complexes under Electrochemical Conditions, Inorg. Chem., 2017, 56, 11066–11073 CrossRef CAS PubMed.
- C. Zheng and S.-L. You, Transfer Hydrogenation with Hantzsch Esters and Related Organic Hydride Donors, Chem. Soc. Rev., 2012, 41, 2498–2518 RSC.
- Q.-X. Xie, L.-X. Liu, Z.-H. Zhu, C. Yu and Y.-G. Zhou, Asymmetric Transfer Hydrogenation of 2,3-Disubstituted Flavanones through Dynamic Kinetic Resolution Enabled by RetroOxa-Michael Addition: Construction of Three Contiguous Stereogenic Centers, J. Org. Chem., 2022, 87, 7521–7530 CrossRef CAS PubMed.
- B. Gao, W. Meng, X. Feng and H. Du, Regenerable Dihydrophenanthridine via Borane-Catalyzed Hydrogenation for the Asymmetric Transfer Hydrogenation of Benzoxazinones, Org. Lett., 2022, 24, 3955–3959 CrossRef CAS PubMed.
- F. Hollmann, D. J. Opperman and C. E. Paul, Biocatalytic Reduction Reactions from a Chemist's Perspective, Angew. Chem., Int. Ed., 2021, 60, 5644–5665 CrossRef CAS PubMed.
- B. Desguin, J. Urdiain-Arraiza, M. Da Costa, M. Fellner, J. Hu, R. P. Hausinger, T. Desmet, P. Hols and P. Soumillion, Uncovering a superfamily of nickel-dependent hydroxyacid racemases and epimerases, Sci. Rep., 2020, 10, 18123 CrossRef CAS PubMed.
- F. G. Mutti, T. Knaus, N. S. Scruttion, M. Breuer and N. J. Turner, Conversion of Alcohols to Enantiopure Amines Through Dual-Enzyme Hydrogen-Borrowing Cascades, Science, 2015, 349, 1525–1529 CrossRef CAS PubMed.
- S. L. Montgomer, J. Magnas-Sanchez, M. P. Thompson, G. A. Aleku, B. Dominguez and N. J. Turner, Direct Alkylation of Amines with Primary and Secondary Alcohols through Biocatalytic Hydrogen Borrowing, Angew. Chem., Int. Ed., 2017, 56, 10491–10494 CrossRef PubMed.
- S. G. Ouellet, J. B. Tuttle and D. W. C. MacMillan, Enantioselective Organocatalytic Hydride Reduction, J. Am. Chem. Soc., 2005, 127, 32–33 CrossRef CAS PubMed.
- A. M. F. Phillips and A. J. L. Pomberio, Recent Advances in Organocatalytic Enantioselectrive Transfer Hydrogenation, Org. Biomol. Chem., 2017, 15, 2307–2340 RSC.
- Z.-H. Zhu, Y.-X. Ding and Y.-G. Zhou, Transfer-Catalyst-Free Biomimetic Asymmetric Reduction of 3-Sulfonyl Coumarins with a Regenerable NAD(P)H Model, Chem. Commun., 2022, 58, 3973–3976 RSC.
- Y. Gu, J. R. Norton, F. Salahi, V. G. Lisnyak, Z. Zhou and S. A. Snyder, Highly Selective Hydrogenation of C-C Bonds Catalyzed by a Rhodium Hydride, J. Am. Chem. Soc., 2021, 143, 9657–9663 CrossRef CAS PubMed.
- J. Wang, Z.-H. Zhu, M.-W. Chen, Q.-A. Chen and Y.-G. Zhou, Catalytic Biomimetic Asymmetric Reduction of Alkenes and Imines Enabled by Chiral and Regenerable NAD(P)H Models, Angew. Chem., Int. Ed., 2019, 58, 1813–1817 CrossRef CAS PubMed.
- A. B. Shabade, D. M. Sharma, P. Bajpai, R. G. Gonnade, K. Vanka and B. Punji, Room Temperature Chemoselective Hydrogenation Of C=C, C=O and C=N Bonds by Using a Well-defined Mixed Donor Mn(I) Pincer Catalyst, Chem. Sci., 2022, 13, 13764–13773 RSC.
- A. Marrone and R. H. Fish, Bioorganometallic Chemistry at the Interface with Biocatalysis: Chemoselective Reduction of Biomimetic NAD+ Cofactors with [Cp*Rh(bpy)H]+, Tandem Catalysis with 1,4-NADH-Dependent Enzymes, Chiral Synthesis, Organotin Metabolites, and DFT Mechanism Studies, Organometallics, 2023, 42, 288–306 CrossRef CAS.
- P. Karak, S. Kumar, K. Mandal and J. Choudhury, Bis-Imidazolium-Embedded Heterohelicene: A Regenerable NADP+ Cofactor Analogue for Electrocatalytic CO2 Reduction, J. Am. Chem. Soc., 2023, 145, 7230–7241 CrossRef CAS PubMed.
- S. Ilic, A. Alherz, C. B. Musgrave and K. D. Glusac, Importance of Proton-Coupled Electron Transfer in Cathodic Regeneration of Organic Hydrides, Chem. Commun., 2019, 55, 5583–5586 RSC.
- S. Ilic, U. P. Kadel, Y. Basdogan, J. A. Keith and K. D. Glusac, Thermodynamic Hydricities of Biomimetic Organic Hydride Donors, J. Am. Chem. Soc., 2018, 140, 4569–4579 CrossRef CAS PubMed.
- K. M. Waldie, A. L. Ostericher, M. H. Reineke, A. F. Sasayama and C. P. Kubiak, Hydricity of Tansition-Metal Hydrides: Thermodynamic Considerations for CO2 Reduction, ACS Catal., 2018, 8, 1313–1324 CrossRef CAS.
- S. Ilic, A. Alherz, C. B. Musgrave and K. D. Glusac, Thermodynamic and Kinetic Hydricities of Metal-Free Hydrides, Chem. Soc. Rev., 2018, 47, 2809–2836 RSC.
- E. S. Wiedner, M. B. Chambers, C. L. Pitman, M. R. Bullock, A. J. M. Miller and A. M. Appel, Thermodynamic hydricity of transition metal hydrides, Chem. Rev., 2016, 116, 8655–8692 CrossRef CAS PubMed.
- K. R. Brereton, N. E. Smith, N. Hazari and A. Miller, Thermodynamic and kinetic hydricity of transition metal hydrides, Chem. Soc. Rev., 2020, 49, 7929–7984 RSC.
- N. D. Loewen, T. V. Neelakantan and L. A. Berben, Renewable Formate from C−H Bond Formation with CO2: Using Iron Carbonyl Clusters as Electrocatalysts, Acc. Chem. Res., 2017, 50, 2362–2370 CrossRef CAS PubMed.
- A. Taheri and L. A. Berben, Tailoring Electrocatalysts for Selective CO2 or H+ Reduction: Iron Carbonyl Clusters as a Case Study, Inorg. Chem., 2016, 55, 378–385 CrossRef CAS PubMed.
- B. M. Caballos and J. Y. Yang, Directing the Reactivity of Metal Hydrides for Selective CO2 Reduction, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 12686–12691 CrossRef PubMed.
- J. M. Barlow and J. Y. Yang, Thermodynamic Considerations for Optimizing Selective CO2 Reduction by Molecular Catalysts, ACS Cent. Sci., 2019, 5, 580–588 CrossRef CAS PubMed.
- Y. Matsubara, E. Fujita, M. D. Doherty, J. T. Muckerman and C. Creutz, Thermodynamic and Kinetic Hydricity of Ruthenium(II) Hydride Complexes, J. Am. Chem. Soc., 2012, 134, 15743–15757 CrossRef CAS PubMed.
- W. W. Ellis, J. W. Raebiger, C. J. Curtis, J. W. Bruno and D. L. DuBois, Hydricities of BzNADH, C5H5Mo(PMe3)(CO)2H, and C5Me5Mo(PMe3)(CO)2H in Acetonitrile, J. Am. Chem. Soc., 2004, 126, 2738–2743 CrossRef CAS PubMed.
- L. W. T. Parsons, J. C. Fettinger and L. A. Berben, Group 13 ion coordination to pyridyl models NAD+ reduction potentials, Chem. Commun., 2023, 59, 9110–9113 RSC.
- E. J. Thompson and L. A. Berben, Electrocatalytic Hydrogen Production by an Aluminum(III) Complex: Ligand-Based Proton and Electron Transfer, Angew. Chem., Int. Ed., 2015, 54, 11642–11646 CrossRef CAS PubMed.
- K. T. Horak, D. G. VanderVelde and T. Agapie, Tuning of Metal Complex Electronics and Reactivity by Remote Lewis Acid Binding to π-Coordinated Pyridine Diphosphine Ligands, Organometallics, 2015, 34, 4753–4765 CrossRef CAS.
- X.-Q. Zhu, M.-T. Zhang, A. Yu, C.-H. Wang and J. P. Cheng, Hydride, Hydrogen Atom, Proton, and Electron Transfer Driving Forces of Various Five-Membered Heterocyclic Organic Hydrides and Their Reaction Intermediates in Acetonitrile, J. Am. Chem. Soc., 2008, 130, 2501–2516 CrossRef CAS PubMed.
- X.-Q. Zhu, Y. Tan and C.-T. Cao, Thermodynamic Diagnosis of the Properties and Mechanism of Dihydropyridine-Type Compounds as Hydride Source in Acetonitrile with “Molecule ID Card”, J. Phys. Chem. B, 2010, 114, 2058–2075 CrossRef CAS PubMed.
- T. W. Myers and L. A. Berben, A sterically demanding iminopyridine ligand affords redox-active complexes of aluminum(III) and gallium(III), Inorg. Chem., 2012, 51, 1480–1488 CrossRef CAS PubMed.
- T. J. Sherbow, J. C. Fettinger and L. A. Berben, Control of Ligand pKa Values Tunes the
Electrocatalytic Dihydrogen Evolution Mechanism in a Redox-Active Aluminum(III) Complex, Inorg. Chem., 2017, 56, 8651–8660 CrossRef CAS PubMed.
- G. Zhang, J. Wu, S. Zheng, M. C. Neary, J. Mao, M. Flores, R. J. Trovitch and P. A. Dub, Redox-noninnocent ligand-supported vanadium catalysts for the chemoselective reduction of CX (X = O, N) functionalities, J. Am. Chem. Soc., 2019, 141, 15230–15239 CrossRef CAS PubMed.
- D. Mauzerall and F. H. Westheimer, 1-Benzyldihydronicotinamide – a model for reduced DPN, J. Am. Chem. Soc., 1955, 77, 2261–2264 CrossRef CAS.
- J. Zhang, J.-D. Yang and J.-P. Cheng, A Nucleophilicity scale for the reactivity of diazaphospholenium hydrides: structural insights and synthetic applications, Angew. Chem., Int. Ed., 2019, 58, 5983–5987 CrossRef CAS PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijin and G. C. Verschoor, Synthesis, Structure, and spectroscopic properties of copper(II) compounds containing nitrogen-sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2’-yl)-2,6-dithiaheptane]copper(II) perchlorate, J. Chem. Soc., Dalton Trans., 1984, 7, 1349–1356 RSC.
- A. Kobayashi, H. Konno, K. Sakamoto, A. Sekine, Y. Ohashi, M. Iida and O. Isitani, Transition metal complexes coordinated by an NAD(P)H model compound and their enhanced hydride-donating abilities in the presence of a base, Chem. Eur. J., 2005, 11, 4219–4226 CrossRef CAS PubMed.
- C. Creutz and M. H. Chou, Rapid transfer of hydride ion from a ruthenium complex to C1 species in water, J. Am. Chem. Soc., 2007, 129, 10108–10109 CrossRef CAS PubMed.
- D. E. Berning, B. C. Noll and D. L. DuBois, Relative Hydride, Proton, and Hydrogen Atom Transfer Abilities of [HM(diphosphine)2]PF6 Complexes (M = Pt, Ni), J. Am. Chem. Soc., 1999, 121, 11432–11447 CrossRef CAS.
- N. Sarker and J. W. Bruno, Thermodynamic and Kinetic Studies of Hydride Transfer for a Series of Molybdenum and Tungsten Hydrides, J. Am. Chem. Soc., 1999, 121, 2174–2180 CrossRef CAS.
- Y. Matsubara, F. Etsuko, M. D. Doherty, J. T. Muckerman and C. Creutz, J. Am. Chem. Soc., 2012, 134, 15743–15757 CrossRef CAS PubMed.
- S. Ilic, U. P. Kadel, Y. Basdogan, J. A. Keith and K. D. Glusac, J. Am. Chem. Soc., 2018, 140, 4569–4579 CrossRef CAS PubMed.
- W. W. Ellis, J. W. Raebiger, C. J. Curtis, J. W. Bruno and D. L. DuBois, J. Am. Chem. Soc., 2004, 126, 2738–2743 CrossRef CAS PubMed.
- T.-Y. Cheng, B. S. Brunschwig and R. M. Bullock, J. Am. Chem. Soc., 1998, 120, 13121–13137 CrossRef CAS.
- T. M. Bass, C. R. Carr, T. J. Sherbow, J. C. Fettinger and L. A. Berben, Syntheses of Square Planar Gallium Complexes and a Proton NMR Correlation Probing Metalloaromaticity, Inorg. Chem., 2020, 59, 13517–13523 CrossRef CAS PubMed.
- E. Radcliffe, J. J. Dunsford, J. Cid, V. Fasano and M. J. Ingleson, N-Heterocycle-Ligated Borocations as Highly Tunable Carbon Lewis Acids, Organometallics, 2017, 36, 4952–4960 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of compounds, 1H and 13C NMR, tables of crystallographic data, crystallographic data (CIF). CCDC 2278349 and 2278350. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03806h |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  and
Louise A.
Berben
and
Louise A.
Berben