
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High and fast: NMR protein–proton side-chain assignments at 160 kHz and 1.2 GHz†
Morgane
Callon‡
 *a,
Dominique
Luder
a,
Alexander A.
Malär‡§
a,
Thomas
Wiegand¶||
a,
Václav
Římal
a,
Lauriane
Lecoq
b,
Anja
Böckmann
b,
Ago
Samoson
c and
Beat H.
Meier
*a
*a,
Dominique
Luder
a,
Alexander A.
Malär‡§
a,
Thomas
Wiegand¶||
a,
Václav
Římal
a,
Lauriane
Lecoq
b,
Anja
Böckmann
b,
Ago
Samoson
c and
Beat H.
Meier
*a
aPhysical Chemistry, ETH Zürich, 8093 Zürich, Switzerland. E-mail: morgane.callon@phys.chem.ethz.ch; beme@ethz.ch
bMolecular Microbiology and Structural Biochemistry (MMSB) UMR 5086, CNRS, Université de Lyon, Labex Ecofect, 7 passage du Vercors, 69367 Lyon, France
cInstitute of Cybernetics, Spin Design Laboratory, Tallinn University of Technology, Tallinn, Estonia
First published on 20th September 2023
Abstract
The NMR spectra of side-chain protons in proteins provide important information, not only about their structure and dynamics, but also about the mechanisms that regulate interactions between macromolecules. However, in the solid-state, these resonances are particularly difficult to resolve, even in relatively small proteins. We show that magic-angle-spinning (MAS) frequencies of 160 kHz, combined with a high magnetic field of 1200 MHz proton Larmor frequency, significantly improve their spectral resolution. We investigate in detail the gain for MAS frequencies between 110 and 160 kHz MAS for a model sample as well as for the hepatitis B viral capsid assembled from 120 core-protein (Cp) dimers. For both systems, we found a significantly improved spectral resolution of the side-chain region in the 1H–13C 2D spectra. The combination of 160 kHz MAS frequency with a magnetic field of 1200 MHz, allowed us to assign 61% of the aliphatic protons of Cp. The side-chain proton assignment opens up new possibilities for structural studies and further characterization of protein–protein or protein–nucleic acid interactions.
Introduction
Fast magic-angle spinning (MAS) at rotational frequencies νr around 100 kHz, combined with proton detection (1H-MAS-NMR), has recently brought significant improvements in resolution and sensitivity1,2 to solid-state NMR spectroscopy. In addition, the increase in mass sensitivity by a factor of about 50 thanks to proton detection at fast MAS with rotors of 0.7 mm diameter compared to carbon detection in 3.2 mm ones,3 now allows the study of complex proteins that are only available in small amounts.4–6 The best spectral resolution is usually observed for the backbone amide (HN) and the Hα protons while the side-chain protons are typically not well-resolved even at the highest available magnetic fields.7 They feature a small chemical-shift dispersion. Therefore, most dipole-coupled protons are in an intermediate or strong coupling regime,7,8 where the homonuclear dipolar line broadening, as described by the higher order average Hamiltonian terms, is at least comparable to the chemical-shift separation at the magnetic fields currently used for protein NMR. This induces spectral overlap of these resonances. They are however crucial notably for structure determination, since, especially in the hydrophobic core, they form most of the essential contacts that define the protein fold. Proton spectra can also be important for the characterization of biomolecular interactions, as protons are localized at protein–protein interfaces, and can thus provide information on dimer contacts in multimers, or between a protein and a ligand or nucleic acid. Resolving these resonances requires a further improvement in spectral resolution, which can be achieved either by reducing their spectral line width and/or increasing the chemical-shift dispersion in the NMR spectra. Selective isotopic dilution of side-chain protons has been developed, and partial labelling of side-chains, particularly methyl groups, has enabled the recording of side-chain 1H–1H distances by solid-state NMR.9–15 In these studies, however, side-chain protons were not systematically assigned, and often methyl groups were exclusively used. Indeed, while methyl groups play an important role in defining the hydrophobic core of proteins, residues not carrying such protons are equally involved in protein–protein interactions, such as aromatic residues involved in π stacking, polar residues in side-chain hydrogen bonds, or cation–π interactions.16 Thus, working on fully protonated proteins will open up for investigations of a greater variety of structural interactions, and first attempts in this direction have recently been made on model systems17–20 as well as fully protonated membrane proteins21–26 at MAS frequencies of 100–111 kHz.In solid-state NMR spectroscopy, the proton line width consists of a homogeneous contribution, an inhomogeneous one and a chemical exchange part: Δtot = Δhomo + Δinhomo + Δexch. In the following we assume that the possible contribution from chemical exchange27 is negligible here. We also neglect the contribution of 1H–1H J-couplings which are typically below 15 Hz but can give rise to a (non-resolved) multiplet pattern that contributes to the linewidth.
The inhomogeneous line width is due to magnetic-field heterogeneity and inherent sample heterogeneities, and is largely independent of the spinning frequency, but increases at higher magnetic fields due to the higher chemical shift dispersion. The homogeneous line width consists of a coherent and an incoherent part (Δhomo = Δcoh + Δincoh, assuming Lorentzian lines). The incoherent part is due to transverse relaxation caused by protein dynamics or to chemical exchange, and is acting, albeit differently, both in solution and in solid-state NMR experiments. The coherent contribution, on the other hand, is specific to solid-state NMR, and arises from the anisotropic interactions (dipolar coupling and cross terms with chemical shift anisotropy, CSA) that are not fully averaged by MAS at finite MAS frequencies, as predicted by average Hamiltonian theory (AHT).28–30 Thus, in order to reduce the homogeneous line width to become similar to the “natural” line width given by the transverse relaxation, one must decrease the coherent contribution. This contribution was experimentally found to decrease with the inverse spinning frequency in a polynomial manner, containing a linear and a quadratic contribution.31–33 For the amide protons of a fully protonated protein, a 19% reduction of the line width was shown when spinning 26% faster (from 100 to 126 kHz MAS), and a further reduction by 25% when increasing the spinning frequency to 150 kHz.31,33 The side-chain protons, which are in a relatively strong coupling regime, exhibit a larger homogeneous line width at a given spinning frequency than Hα or HN. Higher magnetic field has the potential to further reduce their line width by increasing the chemical-shift differences leading to a weaker coupling regime.7 We show how the combination of high magnetic field, at 1200 MHz proton Larmor frequency, and fast MAS at 160 kHz frequency, positively affects the proton line widths, and allows proton side-chain resonance assignments of the HBV capsid core protein (Cp149). We first evaluate this gain in resolution on a model system, the phosphorylated amino acid O-phospho-L-serine. Then, we investigate the line widths of the side-chain resonances of Cp149. While the 1HN,13C,15N assignments have been determined previously,34 we assigned, combining fast spinning at 160 kHz and high magnetic field, 61% of the Hα and side-chain proton resonances using three-dimensional 1H-detected spectra.
Material and methods
Sample preparation
O-Phospho-L-serine has been purchased from Sigma Aldrich and was used without further processing. The sample is fully protonated and contains predominantly 14N and 12C. Protonated and uniformly [13C–15N]-labeled (UL) Cp149 empty capsids (not containing nucleic acids) were produced and purified as described in Lecoq et al.34 In brief, Cp149 protein was expressed in E. coli BL21(DE3)*CP strain, grown at 37 °C in LB medium containing 1 g L−1 of 15NH4Cl and 2 g L−1 of 13C-glucose in H2O and supplemented with 100 μg mL−1 ampicillin and 34 μg mL−1 chloramphenicol. When the OD600 reached 0.7, the expression was induced with 1 mM IPTG and the culture was further incubated for 6 h at 25 °C. Harvested cells were resuspended in 15 mL of lysis buffer (50 mM Tris pH 7.5, 300 mM NaCl, 5 mM DTT) and incubated on ice for 45 min with 1 mg mL−1 of chicken lysozyme, 1× of protease inhibitor cocktail solution and 0.5% of Triton-X-100, then mixed with 4 μL of Benzonase nuclease for 30 min at room temperature. Cells were broken by sonication and centrifuged for 1 h at 8000g to remove cell debris. UL-Cp149 capsids in the supernatant were purified on a stepwise sucrose gradient from 10 to 60% (m/v) sucrose (buffered in 50 mM Tris pH 7.5, 300 mM NaCl) which was centrifuged at 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800g for 3 h at 4 °C. The fractions containing UL-Cp149 were further purified by (NH4)2SO4 precipitation (up to 35% saturation) and finally dialyzed in the solid-state-NMR buffer (50 mM Tris pH 7.5, 5 mM DTT) overnight at 4 °C. 20 μL of saturated 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) solution were added to the protein for chemical-shift referencing prior to the sedimentation step. Sub-milligram amounts of capsids were filled into a 0.5 mm rotor using home-made filling tools35 by centrifugation (200000g, 17 h, 4 °C).
800g for 3 h at 4 °C. The fractions containing UL-Cp149 were further purified by (NH4)2SO4 precipitation (up to 35% saturation) and finally dialyzed in the solid-state-NMR buffer (50 mM Tris pH 7.5, 5 mM DTT) overnight at 4 °C. 20 μL of saturated 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) solution were added to the protein for chemical-shift referencing prior to the sedimentation step. Sub-milligram amounts of capsids were filled into a 0.5 mm rotor using home-made filling tools35 by centrifugation (200000g, 17 h, 4 °C).
NMR spectroscopy, data processing and analysis
Solid-state NMR experiments were recorded at static magnetic-field strengths of 20.0 and 28.2 T (wide-bore 850 MHz Bruker Avance III and standard-bore 1200 MHz Bruker Avance NEO spectrometer, respectively). The spectra of O-phospho-L-ser were acquired using a 0.5 mm triple-resonance probe head (Ago Samoson, Darklands OÜ, Tallinn, Estonia). For the resonance-specific investigation of the homogeneous line width at 850 and 1200 MHz proton frequency, sixteen and eighteen 2D-hCH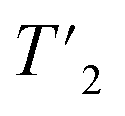 relaxation experiments with echo-delay time varied from 0.002 to 6 ms and 0.001 to 10 ms respectively were measured. No cooling was applied during the measurement of the O-phospho-L-ser spectra, thus the sample temperature increased with increasing MAS frequency according to the Fig. S1.†
relaxation experiments with echo-delay time varied from 0.002 to 6 ms and 0.001 to 10 ms respectively were measured. No cooling was applied during the measurement of the O-phospho-L-ser spectra, thus the sample temperature increased with increasing MAS frequency according to the Fig. S1.†
Solid-state NMR spectra of UL-Cp149 were recorded on capsids sedimented36,37 in H2O and internally referenced to DSS. The spectra were acquired using 0.5 mm triple-resonance probe heads and spinning frequencies of 110 and 160 kHz MAS. The sample temperature of around 25 °C was determined from the relation T (°C) = 455 − 90δH2O where δH2O denotes the supernatant water chemical shift in ppm.35 The sample temperature was kept constant at 25 °C at all MAS frequencies by using an air-cooling system which compensates the MAS sample heating due to fast sample rotation shown in Fig. S1.†
The 2D-hCH experiments were recorded with 800 μs cross-polarization (CP) transfers. For the residue-specific measurement of the homogeneous line width, eight 2D-hCH  relaxation experiments with echo-delay times up to 4.5 ms were measured at 110 and 160 kHz MAS frequency and 1200 MHz proton frequency. Bulk proton longitudinal relaxation times T1 (1H) have been determined using a saturation recovery sequence with 16 variable delays up to 7 and 8 s for the HN/aliphatic protons (1Hali) respectively. Bulk proton transverse relaxation times
relaxation experiments with echo-delay times up to 4.5 ms were measured at 110 and 160 kHz MAS frequency and 1200 MHz proton frequency. Bulk proton longitudinal relaxation times T1 (1H) have been determined using a saturation recovery sequence with 16 variable delays up to 7 and 8 s for the HN/aliphatic protons (1Hali) respectively. Bulk proton transverse relaxation times 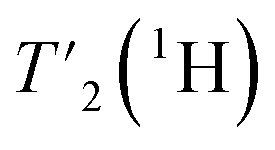 have been determined using a Hahn-echo sequence with 16 variable echo delays τecho from 1 μs to 6/7 ms for the HN/1Hali respectively. Bulk proton relaxation times under spin lock T1ρ(1H) have been determined with 10 variable spin lock times from 1 μs to 40 ms (1H spin lock rf-field strength of 13 kHz). Bulk 15N transverse relaxation times
have been determined using a Hahn-echo sequence with 16 variable echo delays τecho from 1 μs to 6/7 ms for the HN/1Hali respectively. Bulk proton relaxation times under spin lock T1ρ(1H) have been determined with 10 variable spin lock times from 1 μs to 40 ms (1H spin lock rf-field strength of 13 kHz). Bulk 15N transverse relaxation times  have been determined using a Hahn-echo sequence with 16 variable echo delays τecho from 1 μs to 35 ms. The 3D-hNCH experiment was recorded with 20 ms Cα-N cross-polarization (CP) transfers. The 3D-hCCH TOBSY experiment used a C957 block38 for the TOBSY mixing of 15 ms (see puse program in Fig. S2†) and was recorded using the simultaneous acquisition of a frequency reference (SAFR).39,44 As well, additional 3D-hCCH and 3D-HcCH TOBSY experiments were recorded using a WALTZ-16 bloc40 for the TOBSY transfer with 30 kHz rf amplitude and with a length of 11 ms. The inter-scan delay for the experiments has been set to the value of 1.27T1(1Hali) based on the measured bulk protein T1(1Hali) relaxation time. All experiments use the MISSISSIPPI scheme41 for solvent suppression, WALTZ-64 heteronuclear decoupling on 13C during 1H acquisition40,42 and frequency-sweep low-power TPPM decoupling on 1H during indirect time increments in 2D and 3D experiments.43 Detailed information about all acquisition parameters can be found in Tables S1–S4.†
have been determined using a Hahn-echo sequence with 16 variable echo delays τecho from 1 μs to 35 ms. The 3D-hNCH experiment was recorded with 20 ms Cα-N cross-polarization (CP) transfers. The 3D-hCCH TOBSY experiment used a C957 block38 for the TOBSY mixing of 15 ms (see puse program in Fig. S2†) and was recorded using the simultaneous acquisition of a frequency reference (SAFR).39,44 As well, additional 3D-hCCH and 3D-HcCH TOBSY experiments were recorded using a WALTZ-16 bloc40 for the TOBSY transfer with 30 kHz rf amplitude and with a length of 11 ms. The inter-scan delay for the experiments has been set to the value of 1.27T1(1Hali) based on the measured bulk protein T1(1Hali) relaxation time. All experiments use the MISSISSIPPI scheme41 for solvent suppression, WALTZ-64 heteronuclear decoupling on 13C during 1H acquisition40,42 and frequency-sweep low-power TPPM decoupling on 1H during indirect time increments in 2D and 3D experiments.43 Detailed information about all acquisition parameters can be found in Tables S1–S4.†
The spectra were processed using Topspin 4.0.6 (Bruker Biospin). The O-phospho-L-serine spectra were processed without apodization function and calibrated using an external calibration on the 13C spectra of solid adamantane recorded in the same probe head directly after the measurement. The UL-Cp149 spectra were processed with zero filling to the double amount of data points and a shifted sine-bell apodization function in direct and indirect dimensions with SSB = 2.5. Pre-existing 13C resonance assignments were transferred from Lecoq et al.34 (BMRB accession number 27317). The spectra were analyzed in CcpNmr Analysis 2.4.2.44,45
Relaxation and line width analysis
For O-phospho-L-serine, site-specific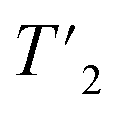 (1H) relaxation times were extracted from a series of 1D 1H spin-echo experiments, where the peak areas have been determined using the relaxation analysis tool in Topspin and exported to MATLAB (version 9.6.0). Relaxation curves were fitted with a mono-exponential decay function with two free parameters and functional form Aexp(−τecho/t) and are shown in Fig. S3 and S4.† For UL-Cp149, site-specific
(1H) relaxation times were extracted from a series of 1D 1H spin-echo experiments, where the peak areas have been determined using the relaxation analysis tool in Topspin and exported to MATLAB (version 9.6.0). Relaxation curves were fitted with a mono-exponential decay function with two free parameters and functional form Aexp(−τecho/t) and are shown in Fig. S3 and S4.† For UL-Cp149, site-specific 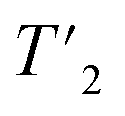 (1H) relaxation times were extracted from a series of 2D-hCH
(1H) relaxation times were extracted from a series of 2D-hCH 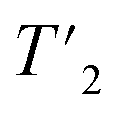 relaxation experiments, using the spectral fitting package INFOS.46 Relaxation curves were fitted with a mono-exponential fit and are shown for all resonances in Fig. S5 and S6.† Site-specific homogeneous line widths have been determined using the relation
relaxation experiments, using the spectral fitting package INFOS.46 Relaxation curves were fitted with a mono-exponential fit and are shown for all resonances in Fig. S5 and S6.† Site-specific homogeneous line widths have been determined using the relation  . Experimental errors σ have been determined using bootstrapping methods with 200 iterations for relaxation time data and subsequent Gaussian error propagation for the error on the corresponding homogeneous line widths. For O-phospho-L-serine, the MAS dependence of the homogeneous line widths Δhomo(1H) were fitted by a quadratic polynomial model of the form Δhomo(νr) = c(1) + c(2)/νr + c(3)/νr2, with the spinning frequency νr, forcing the y-axis intercept to go through the origin (c(1) = 0). For UL-Cp149 bulk relaxation measurement, relaxation times were extracted from a series of 1D hcH and hnH experiments, where the peak areas have been determined using the relaxation analysis tool in Topspin and exported to MATLAB. Relaxation curves were fitted with a mono-exponential decay function with two free parameters and functional form Aexp(−τecho/t) and are shown in Fig. S7.† For UL-Cp149, site-specific total line widths were extracted from series of 2D-hCH CP-based spectra using the spectral fitting package INFOS.46 For O-phospho-L-serine, the site-specific total line widths were determined using the line-shape integrated tool in Topspin (Bruker). The 1D spectra are fitted using a mixed Lorentzian and Gaussian function and the full-widths at half maximum (FWHM) corresponding to the total line width is extracted.
. Experimental errors σ have been determined using bootstrapping methods with 200 iterations for relaxation time data and subsequent Gaussian error propagation for the error on the corresponding homogeneous line widths. For O-phospho-L-serine, the MAS dependence of the homogeneous line widths Δhomo(1H) were fitted by a quadratic polynomial model of the form Δhomo(νr) = c(1) + c(2)/νr + c(3)/νr2, with the spinning frequency νr, forcing the y-axis intercept to go through the origin (c(1) = 0). For UL-Cp149 bulk relaxation measurement, relaxation times were extracted from a series of 1D hcH and hnH experiments, where the peak areas have been determined using the relaxation analysis tool in Topspin and exported to MATLAB. Relaxation curves were fitted with a mono-exponential decay function with two free parameters and functional form Aexp(−τecho/t) and are shown in Fig. S7.† For UL-Cp149, site-specific total line widths were extracted from series of 2D-hCH CP-based spectra using the spectral fitting package INFOS.46 For O-phospho-L-serine, the site-specific total line widths were determined using the line-shape integrated tool in Topspin (Bruker). The 1D spectra are fitted using a mixed Lorentzian and Gaussian function and the full-widths at half maximum (FWHM) corresponding to the total line width is extracted.
Results
Line width gain at higher field and faster MAS
We first investigated a small model system, the amino acid O-phospho-L-serine47–50 (for the chemical structure see Fig. 1) The spinning frequency dependence of its proton line widths between 40 and 160 kHz MAS at 850 and 1200 MHz proton Larmor frequency is shown in Fig. 1. Over the entire range of spinning frequencies and for both magnetic-field strengths, we observe a clear decrease in line width with increasing spinning frequency, most prominently for the CH2 resonances. This is also shown in Tables S5a and b† summarizing the total proton line widths (the sum of homogeneous and inhomogeneous contributions) measured at 850 and 1200 MHz proton frequency respectively. Improvements at higher magentic field by the expected factor of 1.4 (representing the ratio of the two magnetic fields) is observed for the H′ and Hγ spins (nomenclature in Fig. 1a), while for the remaining spins, fits of the line shapes by Lorentzian/Gaussian lines are poor as the line shapes are complex due to the higher-order terms in the AHT expansion (Fig. S8†). | ||
| Fig. 1 Proton line width as a function of MAS frequency for O-phospho-L-serine. Comparison between 1D 1H spectra recorded at MAS frequencies from 40 to 160 kHz and at (a) 850 MHz proton frequency and (b) 1200 MHz proton frequency. The proton resonances are labeled according to the chemical structure of O-phospho-L-serine in the figure. Note that the 1D spectra were not fully relaxed, explaining the intensity difference. (c) Resonance-specific homogeneous line width (Δhomo(1H)) as a function of the rotor period τr = 1/νr and corresponding quadratic fits of the experimental data measured at 850 MHz (dashed lines) and 1200 MHz (solid lines) proton Larmor frequency at MAS frequencies of 60 to 160 and 100 to 160 kHz MAS frequency respectively (fits shown in Fig S3 and S4†). The MAS frequency is shown on the upper axis. Error bars are calculated as described in the Materials and methods section and given as 2σ. At 60 kHz MAS frequency and 850 MHz proton frequency, the two protons of the CH2 group are not resolved enough to be measured individually. A zoom on the proton resonances of the CH2 group (Hβ2 and Hβ3) is shown in (d) to illustrate the similar line width obtained by spinning 20 kHz faster or going to a higher magnetic field. | ||
To evaluate the homogeneous contribution to the total line width, 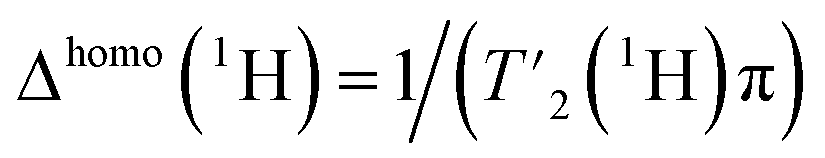 was extracted from the Hahn-echo 1D 1H spectra. Fig. 1c shows the Δhomo(1H) dependence with τr = 1/νr and the homogeneous line widths at 850 and 1200 MHz are given respectively in Tables 1 and 2. For both magnetic fields, the proton line widths always benefit from a faster MAS frequency. The homogenous line width improvement when going from 100 to 160 kHz spinning frequency is shown to be greatest for the CH2 group protons (Hβ2 and Hβ3, more than a factor of two). Indeed, at MAS frequencies of 160 kHz, their homogeneous line width becomes comparable to that of the H and Hα protons (see Table 2 at 1200 MHz). Fig. 1c shows that, for all protons, the homogeneous line width, Δhomo(1H), is always lower at 1200 MHz (solid lines) than at 850 MHz (dotted lines) (see also Table 1). This effect is more pronounced for the protons of the CH2 group.
was extracted from the Hahn-echo 1D 1H spectra. Fig. 1c shows the Δhomo(1H) dependence with τr = 1/νr and the homogeneous line widths at 850 and 1200 MHz are given respectively in Tables 1 and 2. For both magnetic fields, the proton line widths always benefit from a faster MAS frequency. The homogenous line width improvement when going from 100 to 160 kHz spinning frequency is shown to be greatest for the CH2 group protons (Hβ2 and Hβ3, more than a factor of two). Indeed, at MAS frequencies of 160 kHz, their homogeneous line width becomes comparable to that of the H and Hα protons (see Table 2 at 1200 MHz). Fig. 1c shows that, for all protons, the homogeneous line width, Δhomo(1H), is always lower at 1200 MHz (solid lines) than at 850 MHz (dotted lines) (see also Table 1). This effect is more pronounced for the protons of the CH2 group.
| MAS frequ./kHz | Δ homo(H′)/Hz | Δ homo(Hγ)/Hz | Δ homo(H)/Hz | Δ homo(Hα)/Hz | Δ homo(Hβ2)/Hz | Δ homo(Hβ3)/Hz |
|---|---|---|---|---|---|---|
| 160 | 32 ± 2 | 26 ± 2 | 84 ± 4 | 125 ± 8 | 124 ± 6 | 94 ± 6 |
| 150 | 36 ± 2 | 30 ± 2 | 92 ± 3 | 119 ± 7 | 133 ± 7 | 105 ± 6 |
| 140 | 39 ± 2 | 34 ± 2 | 98 ± 4 | 131 ± 7 | 146 ± 7 | 121 ± 7 |
| 130 | 44 ± 2 | 38 ± 2 | 107 ± 4 | 146 ± 8 | 163 ± 7 | 139 ± 7 |
| 120 | 49 ± 2 | 44 ± 2 | 118 ± 4 | 160 ± 30 | 184 ± 8 | 159 ± 7 |
| 110 | 58 ± 3 | 52 ± 2 | 130 ± 4 | 194 ± 18 | 214 ± 9 | 188 ± 8 |
| 100 | 62 ± 3 | 57 ± 3 | 144 ± 5 | 203 ± 10 | 241 ± 9 | 220 ± 8 |
| 80 | 84 ± 3 | 80 ± 3 | 185 ± 5 | 267 ± 12 | 337 ± 11 | 317 ± 11 |
| 60 | 114 ± 4 | 115 ± 3 | 248 ± 7 | 415 ± 16 | — | — |
| MAS frequ./kHz | Δ homo (H′)/Hz | Δ homo (Hγ)/Hz | Δ homo (H)/Hz | Δ homo (Hα)/Hz | Δ homo (Hβ2)/Hz | Δ homo (Hβ3)/Hz |
|---|---|---|---|---|---|---|
| 160 | 36 ± 2 | 30 ± 2 | 86 ± 4 | 105 ± 5 | 104 ± 7 | 74 ± 4 |
| 150 | 39 ± 2 | 33 ± 2 | 93 ± 3 | 112 ± 5 | 120 ± 7 | 80 ± 5 |
| 140 | 43 ± 3 | 36 ± 3 | 100 ± 3 | 115 ± 5 | 129 ± 8 | 91 ± 5 |
| 130 | 47 ± 2 | 40 ± 3 | 108 ± 4 | 130 ± 6 | 144 ± 9 | 106 ± 6 |
| 120 | 52 ± 2 | 44 ± 3 | 118 ± 4 | 136 ± 6 | 162 ± 9 | 123 ± 7 |
| 110 | 57 ± 3 | 49 ± 3 | 130 ± 4 | 155 ± 7 | 179 ± 10 | 148 ± 7 |
| 100 | 64 ± 3 | 57 ± 3 | 145 ± 4 | 164 ± 9 | 204 ± 11 | 175 ± 9 |
Finally, a comparison of the MAS-frequency dependence shows that it is possible to obtain a similar proton homogeneous line width at 850 MHz as at 1200 MHz, by spinning about 20 kHz faster. This is illustrated in Fig. 1d, which shows that for the proton resonances of the CH2 group, the homogeneous line widths at 160 kHz and 850 MHz for Hβ2 and Hβ3 are 124 ± 6 Hz and 94 ± 6 Hz, respectively, close to what would be obtained at 140 kHz and 1200 MHz, 129 ± 8 Hz and 91 ± 5 kHz, respectively (Tables 1 and 2).
Taken together, the proton line widths benefit from both a faster MAS frequency and a higher magnetic field. In addition, the experimental CH2 proton homogeneous line width remains to be described over the extended range up to 160 kHz MAS by a sum of a linear and a quadratic function of 1/νr. A further linewidth improvement is expected for MAS frequencies above 160 kHz as, indeed, for the two CH2 resonances at 1200 MHz, the total line width (118 ± 7 and 128 ± 12 Hz, Table S5†) is still dominated by the homogeneous contribution (74 ± 7 and 104 ± 7 Hz, Table 2).
The gain in line width in a protein sample
Next, we investigated the assembly domain of the HBV capsid core protein (Cp149),51,52 for which amide-proton,3 as well as 13C and 15N assignments34 have been previously reported, but side-chain assignments are still missing. In order to compare line widths, we recorded 2D-hCH CP-based spectra of protonated and uniformly [13C,15N] labeled (UL) Cp149 at MAS frequencies of 110 and 160 kHz, at 850 MHz and 1200 MHz proton frequency. The obtained spectra are shown in Fig. 2, where the aliphatic backbone and side-chain proton–carbon correlations are displayed.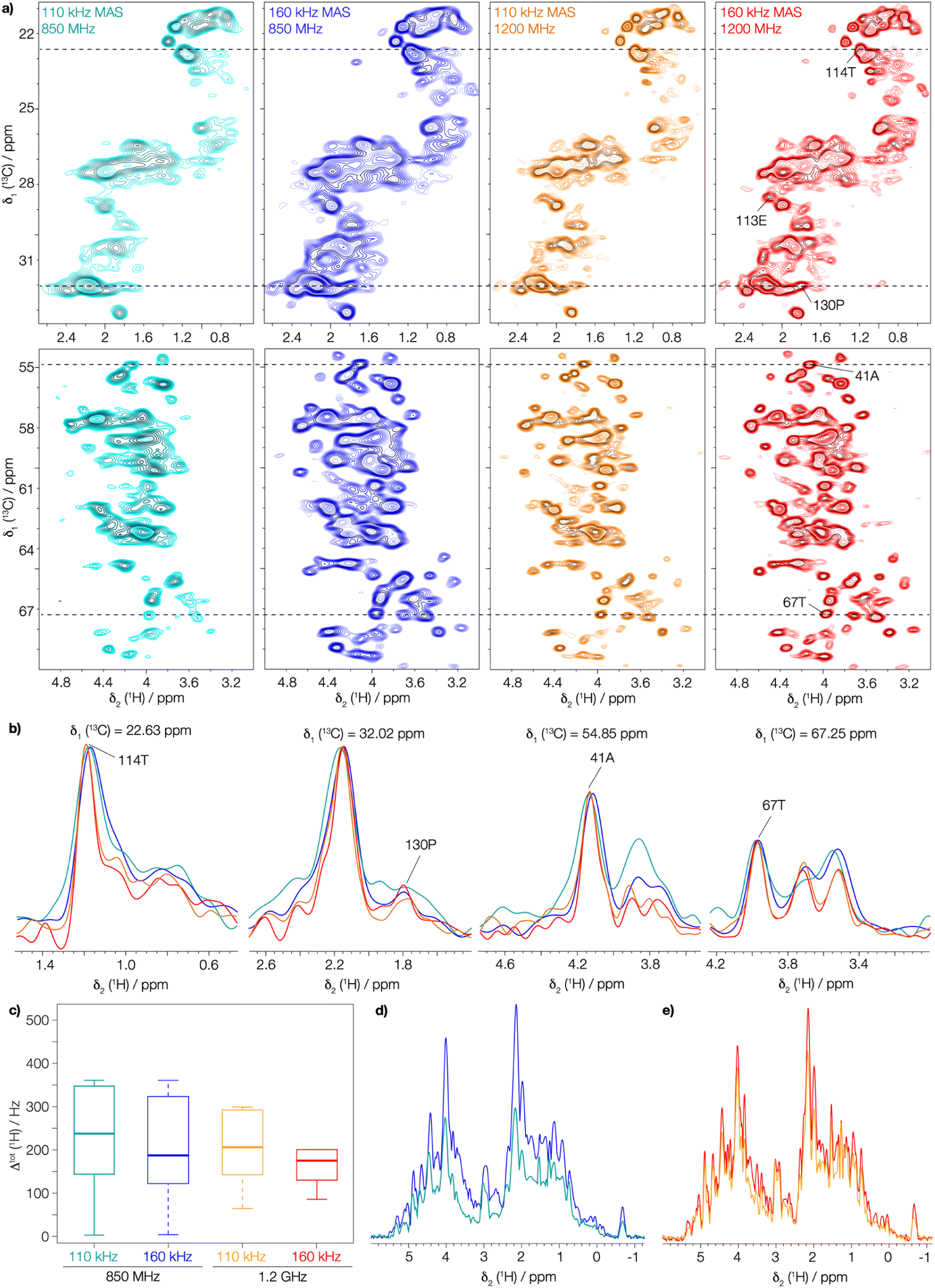 | ||
| Fig. 2 The gain in resolution at faster spinning and higher field in UL-Cp149 spectra. (a) Aliphatic regions of 2D-hCH UL-Cp149 spectra recorded at 160 kHz (red) and 110 kHz (orange) MAS frequency and 1200 MHz proton frequency and 160 kHz (blue) and 110 kHz (cyan) MAS frequency and 850 MHz proton frequency. (b) Corresponding 1D traces at the positions indicated in the 2D spectra with dashed lines. (c) Corresponding boxplot statistics of the site-specific line width of 87 peaks from the spectra in (a). The 87 peaks (Fig. S9†) were selected for evaluation are resolved in all four spectra. The thick lines in each box represent the median total proton line width (Δtot(1H)), the box areas range from the 25th to the 75th percentile and the error bars cover the 95% confidence interval. (d) 1D-1H projections of the 2D-hCH spectra recorded at a magnetic field of 850 MHz and (e) 1D-1H projections of the 2D-hCH spectra recorded at 1200 MHz proton frequency. | ||
Already the inspection of the spectra in Fig. 2a allows one to appreciate the gain in resolution obtained by faster spinning and higher field, which is also clearly visible in the corresponding extracted 1D traces in Fig. 2b. To quantify this gain, we extracted the total proton line width from a selection of peaks picked in the 2D-hCH CP-based spectra. We selected 87 peaks that are resolved in all four spectra (peaks shown in Fig. S9†). The MAS frequency dependence of the line width is illustrated by the median total proton line width (Δtot(1H)) which improves at 1200 MHz from 207 ± 9 Hz at 110 kHz to 176 ± 7 Hz at 160 kHz MAS frequency (thick lines in Fig. 2c). At 850 MHz, Δtot(1H) improves from 237 ± 14 Hz at 110 kHz to 186 ± 8 Hz at 160 kHz MAS frequency. Thus, between 110 and 160 kHz, we observe an improvement in the total proton line width of 18% at 1200 MHz and 27% at 850 MHz. In addition, the gain in sensitivity can be seen by comparing the 1D 1H projections of the 2D-hCH spectra (Fig. 2d and e). This is due to the reduction in line width at faster spinning, but is also offset by the reduction in CP transfer performance at higher MAS frequency.53 At 1200 MHz, a gain in SNR of a factor of 1.16 is observed by spinning at 160 kHz compared to 110 kHz, while at 850 MHz the gain is 1.5. Moving to a higher magnetic field at constant spinning frequency (160 kHz) also improves the spectral resolution, as shown by comparing Δtot((H)1H), from 186 ± 8 Hz at 850 MHz to 176 ± 7 Hz at 1200 MHz. The combination of faster spinning and higher magnetic field made it possible to achieve a significant reduction in the total proton line width, by a total of 35%. This is likely to enable for proton side-chain resonance assignments in many large proteins, and highlights the added benefit of combining faster MAS with higher magnetic fields.
Relaxation times increase with faster spinning
We have seen in O-phospho-L-serine that the homogeneous line width decreases with increasing magnetic field and MAS frequency. Therefore, the proton transverse relaxation time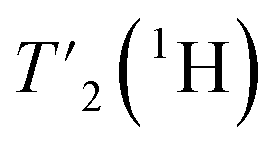 , which is a measure of the homogenous line width, should increase. This is indeed the case for the bulk
, which is a measure of the homogenous line width, should increase. This is indeed the case for the bulk 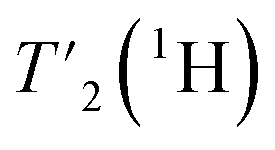 of UL-Cp149, as shown in Fig. S7 and Table S6.† We measured in addition bulk rotating-frame proton relaxation times T1ρ((H)1H), as well as longitudinal relaxation T1(1H). Both relaxation times lengthen with increasing MAS frequency and magnetic field, as expected, for amide and aliphatic protons (Fig. S7 and Table S6†).
of UL-Cp149, as shown in Fig. S7 and Table S6.† We measured in addition bulk rotating-frame proton relaxation times T1ρ((H)1H), as well as longitudinal relaxation T1(1H). Both relaxation times lengthen with increasing MAS frequency and magnetic field, as expected, for amide and aliphatic protons (Fig. S7 and Table S6†).
To go further, we measured site-specific transverse relaxation times of the aliphatic protons of UL-Cp149 using 2D-hCH 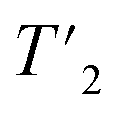 relaxation experiments at MAS frequencies of 110 and 160 kHz at 1200 MHz proton frequency. We recorded eight 2D-hCH
relaxation experiments at MAS frequencies of 110 and 160 kHz at 1200 MHz proton frequency. We recorded eight 2D-hCH 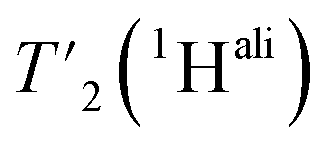 spectra at different echo times, with the spectra recorded at 0.001 ms shown in Fig. S5 and S6† for 160 kHz and 110 kHz, respectively. The 51 residues shown in blue in Fig. S9† were selected because they are resolved and assigned in both spectra. For each of the peaks, the relaxation decay was fitted with a mono-exponential function (shown in Fig. S5 and S6†). The site-specific
spectra at different echo times, with the spectra recorded at 0.001 ms shown in Fig. S5 and S6† for 160 kHz and 110 kHz, respectively. The 51 residues shown in blue in Fig. S9† were selected because they are resolved and assigned in both spectra. For each of the peaks, the relaxation decay was fitted with a mono-exponential function (shown in Fig. S5 and S6†). The site-specific 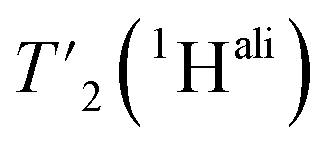 are listed in Table S7† and the median relaxation times summarized in Table 3. Comparing the median
are listed in Table S7† and the median relaxation times summarized in Table 3. Comparing the median 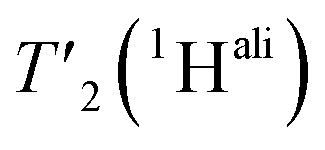 between 110 and 160 kHz reveals an increase in relaxation times of about 50% when going to faster spinning. The longest
between 110 and 160 kHz reveals an increase in relaxation times of about 50% when going to faster spinning. The longest 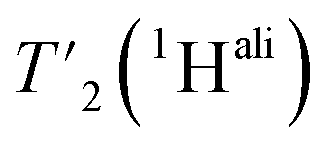 are found for methyl protons (CH3) and Hα protons.
are found for methyl protons (CH3) and Hα protons.
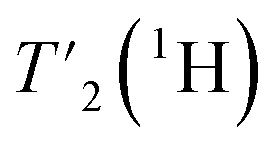 relaxation times and Δhomo(1H) of UL-Cp149 aliphatic protons recorded at 1200 MHz proton frequency and spinning frequencies of 110 and 160 kHz. The spectral regions are defined by type of protons, with the CH3 spectral region between δ(13C) = 16.8–26.0 ppm, the CH2 spectral region between δ(13C) = 26.0–51.4 ppm and the Hα spectral region between δ(13C) = 51.4–70.0 ppm. Experimental errors (σ) are calculated as described in the Materials and methods section
relaxation times and Δhomo(1H) of UL-Cp149 aliphatic protons recorded at 1200 MHz proton frequency and spinning frequencies of 110 and 160 kHz. The spectral regions are defined by type of protons, with the CH3 spectral region between δ(13C) = 16.8–26.0 ppm, the CH2 spectral region between δ(13C) = 26.0–51.4 ppm and the Hα spectral region between δ(13C) = 51.4–70.0 ppm. Experimental errors (σ) are calculated as described in the Materials and methods section
| 110 kHz MAS | 160 kHz MAS | |||
|---|---|---|---|---|
|
|
Δ homo(1Hali)/Hz |
|
Δ homo(1Hali)/Hz | |
| H all | 4.3 ± 1.1 | 77 ± 16 | 6.6 ± 1.0 | 50 ± 7 |
| H α | 4.5 ± 1.1 | 71 ± 17 | 6.3 ± 1.1 | 50 ± 7 |
| CH2 | 3.4 ± 0.6 | 94 ± 16 | 5.3 ± 0.7 | 60 ± 7 |
| CH3 | 4.9 ± 1.1 | 64 ± 13 | 8.1 ± 1.7 | 39 ± 6 |
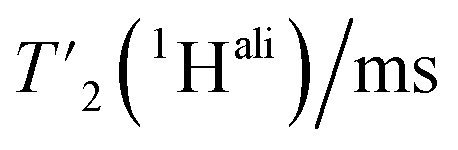
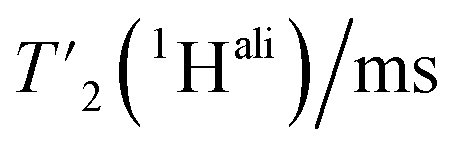
We then extracted the site-specific homogeneous line width of the aliphatic protons, 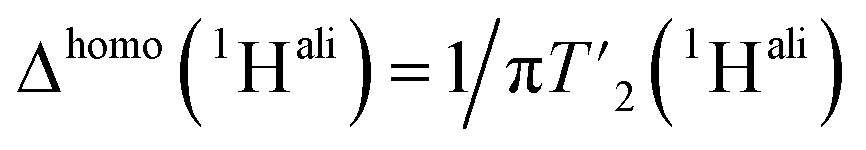 , from the measured
, from the measured 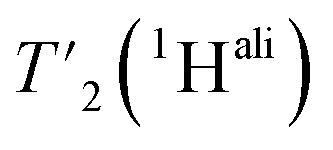 relaxation times of the 51 residues, at 160 and 110 kHz MAS frequency. The site-specific homogeneous proton line widths are shown in Fig. 3a and b respectively and listed in Table S7.† As for the model compound, the median homogeneous line width decreases at higher MAS frequencies, from 77 ± 16 at 110 to 50 ± 7 Hz at 160 kHz MAS frequency, corresponding to a reduction of a factor 1.5. This is close to the expected improvement which corresponds to the inverse ratio of the spinning frequencies, 160 kHz/110 kHz = 1.4.
relaxation times of the 51 residues, at 160 and 110 kHz MAS frequency. The site-specific homogeneous proton line widths are shown in Fig. 3a and b respectively and listed in Table S7.† As for the model compound, the median homogeneous line width decreases at higher MAS frequencies, from 77 ± 16 at 110 to 50 ± 7 Hz at 160 kHz MAS frequency, corresponding to a reduction of a factor 1.5. This is close to the expected improvement which corresponds to the inverse ratio of the spinning frequencies, 160 kHz/110 kHz = 1.4.
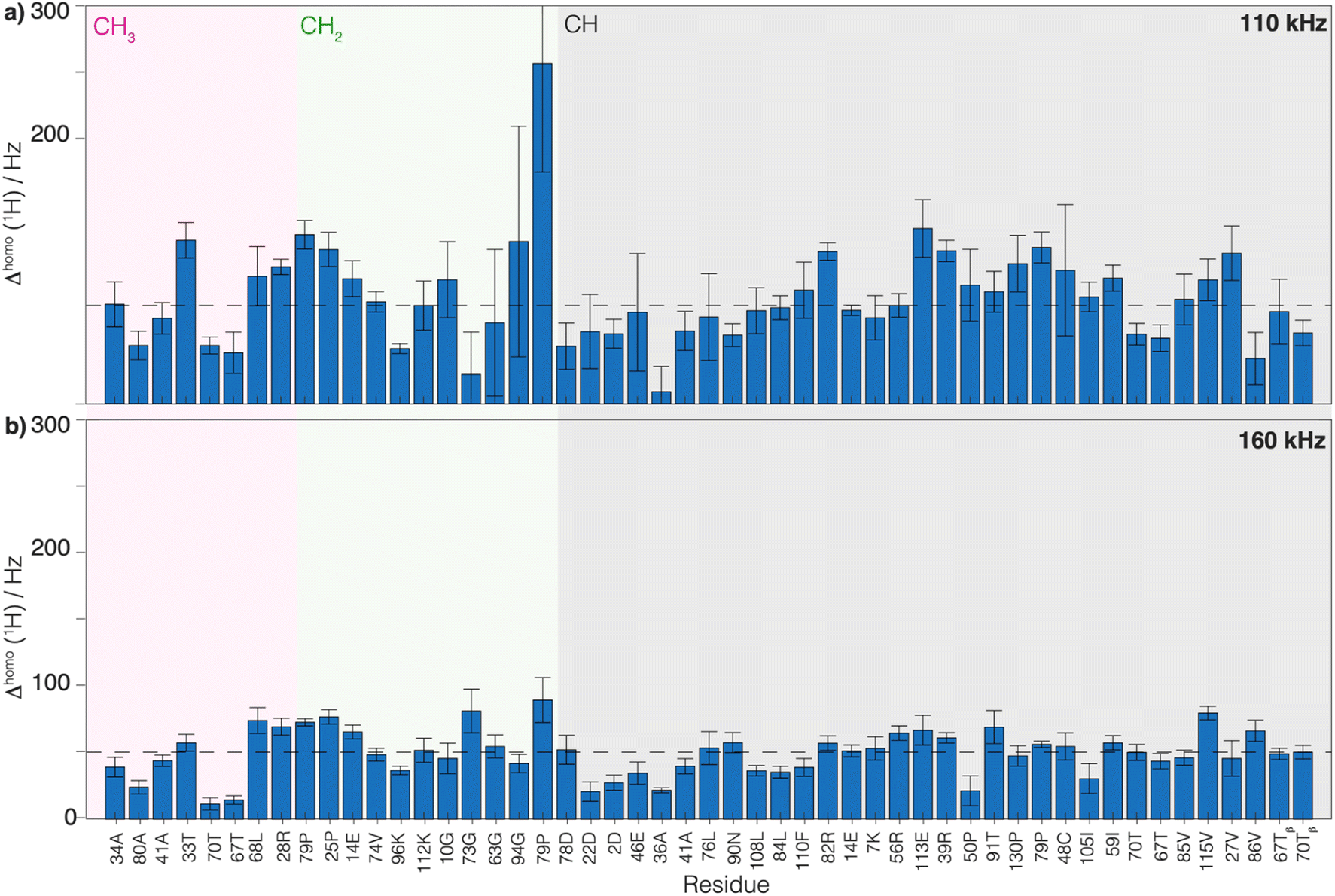 | ||
| Fig. 3 Comparison of the homogeneous proton line widths measured at 1200 MHz proton frequency at 160 and 110 kHz MAS. Site-specific homogeneous proton line width of UL-Cp149 extracted from 2D-hCH CP-based spectra measured at (a) 110 kHz and (b) 160 kHz MAS frequency and at 1200 MHz proton frequency. In both plots, the dotted line represents the median homogeneous proton line width. The spectral regions are defined as follows: the CH3 spectral region between δ(13C) = 16.8–26.0 ppm (grey area), the CH2 spectral region between δ(13C) = 26.0–51.4 ppm (green area) and the CH spectral region between δ(13C) = 51.4–70.0 ppm (pink area). The error bars are calculated according to the description in the Material and methods section and given as 2σ. The residues labeled with a star in the CH region are CH2 groups. | ||
Thus, the reduction of the homogeneous line width is higher than the reduction of the total line width (reduction by a factor of about 1.2, see above). This is due to the larger inhomogeneous than homogeneous contribution to the total line width, which is independent of the spinning frequency. Thus, we conclude that the dominant contribution to the total line width at 160 kHz comes from the inhomogeneous contributions (median Δinhomo = Δtot − Δhomo = 126 Hz, for the selected 51 residues). It accounts in UL-Cp149 for two-thirds of the total line width and limits the amount of improvement that can be achieved by spinning even faster. One should note that Cp149 has four different molecules in the asymmetric unit, resulting in some resonances being split into four resolved peaks3 and, for other resonances, unresolved peak quadrupling might contribute to, or even dominate, the inhomogeneous line width in this sample. Note that the homogeneous line width is larger for the CH2 spins than for the Hα or CH3 (Table 3) due to the stronger dipolar coupling they experience. For these spin systems, the advantage of going to a higher field and spinning faster is more pronounced.
Aliphatic proton assignments at 1200 MHz and 160 kHz MAS
Using fastest spinning and highest magnetic field, we recorded backbone and side-chain spectra allowing to assign the aliphatic protons. We first used a 3D-hNCAHA19 spectrum of UL-Cp149 recorded at 160 kHz MAS and 1200 MHz proton frequency (shown in Fig. S10†) to assign the Hα resonances based on the 15N and 13C assignments (Lecoq et al.34) 51% of the Hα protons with a 13C assignment from Lecoq et al.34 could be assigned from this spectrum. The assigned residues are labeled in Fig. S10,† where the right panel shows a single 3D plane extracted at δ1(15N) = 114.67 ppm as an example. To further assign the aliphatic side-chain protons, we recorded a 3D-hCCH total through bond correlation spectroscopy (TOBSY) spectrum.10,19,20,38,54,55 We used as mixing sequence a C957 mixing block38 to maximize the 13C–13C polarization transfer efficiency (pulse sequence shown in Fig. S2†). A mixing time of 15 ms, slightly longer than 1/2JCC ∼ 11 ms, was used as a compromise to obtain both one- and two-bond carbon J-coupling transfers. A 10 ms z-filter allowed to remove unwanted coherences from the spectrum without significant magnetization loss. Note that, 3D-hCCH and -HcCH TOBSY experiments were also recorded using a WALTZ-16 block during the TOBSY transfer with an irradiation amplitude of 30 kHz (giving the strongest signals in the 1D and 2D versions of the 3D-hCCH TOBSY used for optimization, see Fig. S11 and S12†). The 3D-hCCH TOBSY-C9 was further used for side-chain proton assignments, since it resulted in more cross peaks than the WALTZ-16 versions.Fig. 4 shows extracted planes of the 3D-hCCH TOBSY-C9 spectrum of UL-Cp149 recorded at 1200 MHz proton frequency and 160 kHz MAS frequency. The assignment strategy for assigning the aliphatic protons using the 3D-hCCH spectrum is shown for residue 22D. Based on the known 13C assignments from Lecoq et al.34 the resonance in the CC plane resulting from one-bond TOBSY magnetization transfer could be assigned, identifying the bound aliphatic proton. Following this strategy, the Hα (4.62 ppm), Hβ1 (3.02 ppm) and Hβ2 (2.65 ppm) resonances of 22D could be assigned thanks to their corresponding carbon assignment (Fig. 4).
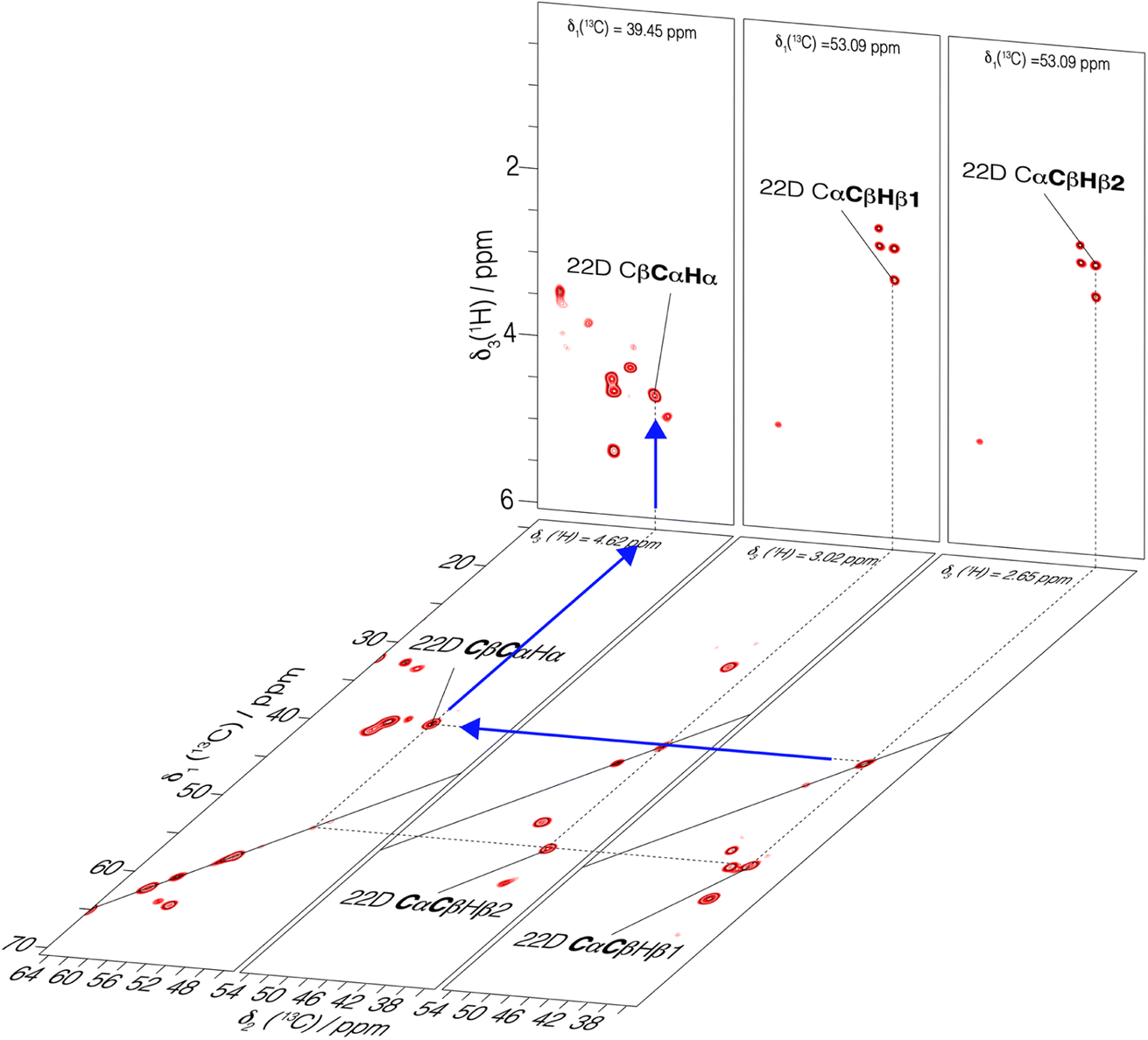 | ||
| Fig. 4 3D-hCCH TOBSY-C9 spectrum of UL-Cp149 recorded at 160 kHz MAS frequency and 1200 MHz proton frequency. Selected 2D-hCCh and hcCH planes are shown. An example of assignment of Hα of residue 22D is represented with blue arrows where the known carbon chemical shifts34 served as basis to assign the bound protons. | ||
In total, 61% of the aliphatic protons could be assigned, including 81% of Hα and 52% of the CH2 and CH3 groups (see Fig. S13†). Note that the protons marked with an asterisk in Fig. S13† could not be assigned, because their directly bonded carbon had not been assigned previously. They are however taken into account in calculating the percentages. Furthermore, the fastest spinning allows to resolve the two diastereotopic proton resonances in about half of the assigned CH2 groups. This is shown as an example in Fig. 4, where the two beta proton resonances of 22D (Hβ1 and Hβ2) are resolved in the hcCH plane of the 3D-hCCH TOBSY spectrum. The total assignment is shown in the 2D-hCH spectrum of UL-Cp149 in Fig. 5 below.
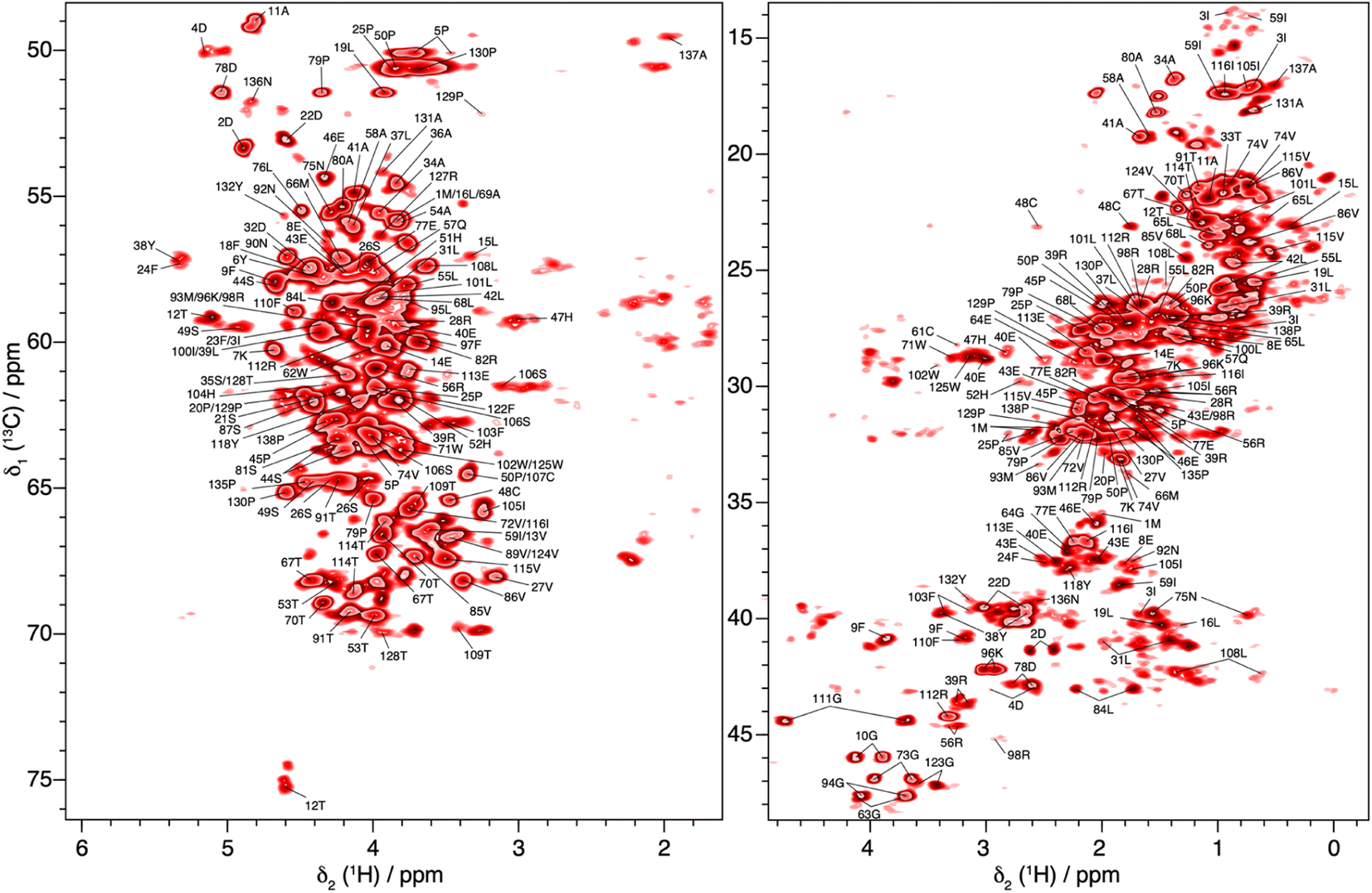 | ||
| Fig. 5 2D-hCH spectrum of UL-Cp149 measured at 160 kHz MAS frequency and 1200 MHz proton frequency, with aliphatic proton assignments obtained from the 3D-hNCH and hCCH experiments. | ||
Discussion and conclusion
We describe the benefits of fast magic-angle spinning at 160 kHz, combined with high magnetic field, to proton-detected solid-state NMR. The basic aspects are described semi-quantitatively for a model system, and the findings are confirmed for the hepatitis B viral capsid protein, in which we achieve a large part of the side-chain assignments using proton-detected MAS solid-state NMR spectroscopy at 160 kHz MAS and 1200 MHz. We quantify the gain in resolution obtained at this spinning speed compared to the more standard 110 kHz MAS and observe an improvement in the total proton line width of 18% at 1200 MHz and 27% at 850 MHz. Subsequently, the gain in SNR is found to be 1.16 and 1.5 respectively. The reduction of the homogeneous line width and consequently of the total line width by faster spinning, together with the use of a higher static magnetic field of 1200 MHz, leads to an improvement of the total line width of 35%. Thus, a gain in resolution is achieved, notably of the side-chain protons, which suffer most from signal overlap at 110 kHz. The methylene protons benefit the most from faster spinning because they are involved in a stronger coupling regime compared to amide or Hα protons. Indeed, they are not only close in chemical shift to their neighbors but also often surrounded by numerous additional protons with similar chemical shift, forming a strong dipolar coupling network.The combination of spinning at 160 kHz MAS frequency combined with high magnetic field of 1200 MHz proton Larmor frequency allowed us to assign 61% of the aliphatic protons of the HBV capsid protein and 67% of aliphatic protons having a directly bonded carbon assigned.
This information is crucial as these protons are often located at protein interfaces and their observation will further allow to characterize interactions between proteins, or to nucleic acids or small ligands. We conclude that 1H-detected MAS-NMR at spinning frequencies of 160 kHz and faster allows the resolution of protein side-chain resonances in fully protonated systems which carry crucial information through the contacts they make at protein–protein, protein–drug or protein–RNA (DNA) interfaces.
Data availability
The aliphatic proton assignment of the Cp149 capsid protein has been deposited in the BioMagResBank (BMRB) under the accession number 52014. The original contributions presented in the study are included in the article/ESI,† further inquiries are available from the corresponding authors upon reasonable request.Author contributions
Conceptualization: BHM, MC, AB, TW, AS; AAM, BHM, AAM; formal analysis: MC, DL, AAM, TV, VR; funding acquisition: BHM, AB; investigation: MC, DL, AAM, TW, VR, LL AB, AS, BHM; methodology: BHM, MC, AB, TW, AS; project administration: BHM; resources: BHM, AB, AS; supervision: BHM, AB, AS; writing – original draft: MC, AAM, TW; writing – review & editing: all authors.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was supported by an ERC Advanced Grant (B. H. M., grant number 741863, FASTER), by the Swiss National Science Foundation (B. H. M., grant number 200020_188711), the French ANRS (A. B., ECTZ205154, ECTZ205074, ECTZ71388 and ECTZ100488) and the CNRS (A. B. and L. L., CNRS-Momentum 2018). A. S. acknowledges support by the Estonian Research Agency (ETAg, PRG1832).References
- A. Böckmann, M. Ernst and B. H. Meier, Spinning proteins, the faster, the better?, J. Magn. Reson., 2015, 253, 71–79 CrossRef PubMed.
- T. Le Marchand, et al., 1H-Detected Biomolecular NMR under Fast Magic-Angle Spinning, Chem. Rev., 2022, 122, 9943–10018 CrossRef CAS PubMed.
- L. Lecoq, et al., 100 kHz MAS Proton-Detected NMR Spectroscopy of Hepatitis B Virus Capsids, Front. Mol. Biosci., 2019, 6, 58 CrossRef CAS PubMed.
- G. David, et al., Structural Studies of Self-Assembled Subviral Particles: Combining Cell-Free Expression with 110 kHz MAS NMR Spectroscopy, Angew. Chem., Int. Ed., 2018, 57, 4787–4791 CrossRef CAS PubMed.
- V. Jirasko, et al., Dimer organization of membrane-associated NS5A of hepatitis C virus as determined by highly sensitive 1H-detected solid-state NMR, Angew. Chem., Int. Ed., 2021, 60, 5339–5347 CrossRef CAS PubMed.
- D. Stöppler, et al., Insight into small molecule binding to the neonatal Fc receptor by X-ray crystallography and 100 kHz magic-angle-spinning NMR, PLoS Biol., 2018, 16, e2006192 CrossRef PubMed.
- M. Callon, et al., Biomolecular solid-state NMR spectroscopy at 1200 MHz: the gain in resolution, J. Biomol. NMR, 2021, 75, 255–272 CrossRef CAS PubMed.
- A. A. Malär, et al., Quantifying proton NMR coherent linewidth in proteins under fast MAS conditions: a second moment approach, Phys. Chem. Chem. Phys., 2019, 21, 18850–18865 RSC.
- V. Agarwal, A. Diehl, N. Skrynnikov and B. Reif, High Resolution 1H Detected 1H,13C Correlation Spectra in MAS Solid-State NMR using Deuterated Proteins with Selective 1H,2H Isotopic Labeling of Methyl Groups, J. Am. Chem. Soc., 2006, 128, 12620–12621 CrossRef CAS PubMed.
- V. Agarwal and B. Reif, Residual methyl protonation in perdeuterated proteins for multi-dimensional correlation experiments in MAS solid-state NMR spectroscopy, J. Magn. Reson., 2008, 194, 16–24 CrossRef CAS PubMed.
- S. Asami, K. Szekely, P. Schanda, B. H. Meier and B. Reif, Optimal degree of protonation for 1H detection of aliphatic sites in randomly deuterated proteins as a function of the MAS frequency, J. Biomol. NMR, 2012, 54, 155–168 CrossRef CAS PubMed.
- S. Asami and B. Reif, Accessing Methyl Groups in proteins via 1 H-detected MAS solid-state NMR Spectroscopy employing Random protonation, Sci. Rep., 2019, 9, 15903 CrossRef PubMed.
- D. Mance, et al., An Efficient Labelling Approach to Harness Backbone and Side-Chain Protons in 1H-Detected Solid-State NMR Spectroscopy, Angew. Chem., Int. Ed., 2015, 54, 15799–15803 CrossRef CAS PubMed.
- D. Nand, A. Cukkemane, S. Becker and M. Baldus, Fractional deuteration applied to biomolecular solid-state NMR spectroscopy, J. Biomol. NMR, 2012, 52, 91–101 CrossRef CAS PubMed.
- T. Sinnige, M. Daniëls, M. Baldus and M. Weingarth, Proton Clouds to Measure Long-Range Contacts between Nonexchangeable Side Chain Protons in Solid-State NMR, J. Am. Chem. Soc., 2014, 136, 4452–4455 CrossRef CAS PubMed.
- J. P. Gallivan and D. A. Dougherty, Cation-π interactions in structural biology, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9459–9464 CrossRef CAS PubMed.
- S. Ahlawat, et al., Assignment of aromatic side-chain spins and characterization of their distance restraints at fast MAS, J. Struct. Biol.: X, 2023, 7, 100082 CAS.
- A. Klein, S. K. Vasa, B. Söldner, K. Grohe and R. Linser, Unambiguous Side-Chain Assignments for Solid-State NMR Structure Elucidation of Nondeuterated Proteins via a Combined 5D/4D Side-Chain-to-Backbone Experiment, J. Phys. Chem. Lett., 2022, 13, 1644–1651 CrossRef CAS PubMed.
- J. Stanek, et al., NMR Spectroscopic Assignment of Backbone and Side-Chain Protons in Fully Protonated Proteins: Microcrystals, Sedimented Assemblies, and Amyloid Fibrils, Angew. Chem., Int. Ed., 2016, 55, 15504–15509 CrossRef CAS PubMed.
- P. Paluch, et al., NMR Assignment of Methyl Groups in Immobilized Proteins Using Multiple-Bond 13C Homonuclear Transfers, Proton Detection, and Very Fast MAS, Front. Mol. Biosci., 2022, 9, 828785 CrossRef CAS PubMed.
- D. Lalli, et al., Proton-Based Structural Analysis of a Heptahelical Transmembrane Protein in Lipid Bilayers, J. Am. Chem. Soc., 2017, 139, 13006–13012 CrossRef CAS PubMed.
- T. Schubeis, J. Stanek and G. Pintacuda, Backbone assignment of crystalline E. coli maltose binding protein, Biomol. NMR Assignments, 2021, 15, 317–322 CrossRef CAS PubMed.
- T. Schubeis, et al., Resonance assignment of the outer membrane protein AlkL in lipid bilayers by proton-detected solid-state NMR, Biomol. NMR Assignments, 2020, 14, 295–300 CrossRef CAS PubMed.
- J. Struppe, et al., Expanding the horizons for structural analysis of fully protonated protein assemblies by NMR spectroscopy at MAS frequencies above 100 kHz, Solid State Nucl. Magn. Reson., 2017, 87, 117–125 CrossRef CAS PubMed.
- A. Klein, et al., Atomic-resolution chemical characterization of (2x)72-kDa tryptophan synthase via four- and five-dimensional 1H-detected solid-state NMR, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2114690119 CrossRef PubMed.
- L. B. Andreas, et al., Structure of fully protonated proteins by proton-detected magic-angle spinning NMR, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 9187–9192 CrossRef CAS PubMed.
- D. Marion, D. F. Gauto, I. Ayala, K. Giandoreggio-Barranco and P. Schanda, Microsecond Protein Dynamics from Combined Bloch-McConnell and Near-Rotary-Resonance R1p Relaxation-Dispersion MAS NMR, ChemPhysChem, 2019, 20, 276–284 CrossRef CAS PubMed.
- P. Schanda and M. Ernst, Studying dynamics by magic-angle spinning solid-state NMR spectroscopy: Principles and applications to biomolecules, Prog. Nucl. Magn. Reson. Spectrosc., 2016, 96, 1–46 CrossRef CAS PubMed.
- M. Chávez, T. Wiegand, A. A. Malär, B. H. Meier and M. Ernst, Residual dipolar line width in magic-angle spinning proton solid-state NMR, Magn. Reson., 2021, 2, 499–509 CrossRef.
- B. Simões De Almeida, P. Moutzouri, G. Stevanato and L. Emsley, Theory and simulations of homonuclear three-spin systems in rotating solids, J. Chem. Phys., 2021, 155, 84201 CrossRef PubMed.
- M. Schledorn, et al., Protein NMR Spectroscopy at 150 kHz Magic-Angle Spinning Continues To Improve Resolution and Mass Sensitivity, ChemBioChem, 2020, 21, 2540–2548 CrossRef CAS PubMed.
- U. Sternberg, et al., 1H line width dependence on MAS speed in solid state NMR – Comparison of experiment and simulation, J. Magn. Reson., 2018, 291, 32–39 CrossRef CAS PubMed.
- S. Penzel, et al., Spinning faster: protein NMR at MAS frequencies up to 126 kHz, J. Biomol. NMR, 2019, 73, 19–29 CrossRef CAS PubMed.
- L. Lecoq, et al., Solid-state [13C–15N] NMR resonance assignment of hepatitis B virus core protein, Biomol. NMR Assignments, 2018, 12, 205–214 CrossRef CAS PubMed.
- A. Böckmann, et al., Characterization of different water pools in solid-state NMR protein samples, J. Biomol. NMR, 2009, 45, 319–327 CrossRef PubMed.
- I. Bertini, et al., Solid-state NMR of proteins sedimented by ultracentrifugation, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 10396–10399 CrossRef CAS PubMed.
- C. Gardiennet, et al., A Sedimented Sample of a 59 kDa Dodecameric Helicase Yields High-Resolution Solid-State NMR Spectra, Angew. Chem., Int. Ed., 2012, 51, 7855–7858 CrossRef CAS PubMed.
- K. O. Tan, et al., Efficient low-power TOBSY sequences for fast MAS, Solid State Nucl. Magn. Reson., 2018, 89, 27–34 CrossRef CAS PubMed.
- V. Římal, et al., Correction of field instabilities in biomolecular solid-state NMR by simultaneous acquisition of a frequency reference, Magn. Reson., 2022, 3, 15–26 CrossRef.
- A. J. Shaka, J. Keeler, T. Frenkiel and R. Freeman, An improved sequence for broadband decoupling: WALTZ-16, J. Magn. Reson., 1983, 52, 335–338 CAS.
- D. H. Zhou and C. M. Rienstra, High-performance solvent suppression for proton detected solid-state NMR, J. Magn. Reson., 2008, 192, 167–172 CrossRef CAS PubMed.
- Z. Zhou, et al., A new decoupling method for accurate quantification of polyethylene copolymer composition and triad sequence distribution with 13C NMR, J. Magn. Reson., 2007, 187, 225–233 CrossRef CAS PubMed.
- R. S. Thakur, N. D. Kurur and P. K. Madhu, Swept-frequency two-pulse phase modulation for heteronuclear dipolar decoupling in solid-state NMR, Chem. Phys. Lett., 2006, 426, 459–463 CrossRef CAS.
- W. F. Vranken, et al., The CCPN data model for NMR spectroscopy: Development of a software pipeline, Proteins, 2005, 59, 687–696 CrossRef CAS PubMed.
- T. J. Stevens, et al., A software framework for analysing solid-state MAS NMR data, J. Biomol. NMR, 2011, 51, 437–447 CrossRef CAS PubMed.
- A. A. Smith, INFOS: spectrum fitting software for NMR analysis, J. Biomol. NMR, 2017, 67, 77–94 CrossRef CAS PubMed.
- A. A. Malär, et al., Temperature-Dependent Solid-State NMR Proton Chemical-Shift Values and Hydrogen Bonding, J. Phys. Chem. B, 2021, 125, 6222–6230 CrossRef PubMed.
- L. Duma, D. Abergel, P. Tekely and G. Bodenhausen, Proton chemical shift anisotropy measurements of hydrogen-bonded functional groups by fast magic-angle spinning solid-state NMR spectroscopy, Chem. Commun., 2008, 2361, 10.1039/b801154k.
- M. J. Potrzebowski, et al., An Experimental and Theoretical Study of the 13C and 31P Chemical Shielding Tensors in Solid O-Phosphorylated Amino Acids, J. Am. Chem. Soc., 2003, 125, 4223–4232 CrossRef CAS PubMed.
- A. Iuga and E. Brunner, Phosphorylated amino acids: model compounds for solid-state 31P NMR spectroscopic studies of proteins, Magn. Reson. Chem., 2004, 42, 369–372 CrossRef CAS PubMed.
- A. Gallina, et al., A recombinant hepatitis B core antigen polypeptide with the protamine-like domain deleted self-assembles into capsid particles but fails to bind nucleic acids, J. Virol., 1989, 63, 4645–4652 CrossRef CAS PubMed.
- F. Birnbaum and M. Nassal, Hepatitis B virus nucleocapsid assembly: primary structure requirements in the core protein, Virol. J., 1990, 64, 3319–3330 CrossRef CAS PubMed.
- A. Šmelko, J. Blahut, B. Reif and Z. Tošner, Performance of the cross-polarization experiment in conditions of radiofrequency field inhomogeneity and slow to ultrafast magic angle spinning (MAS), Magn. Reson., 2023, 4, 199–215 CrossRef.
- M. Baldus and B. H. Meier, Total Correlation Spectroscopy in the Solid State. The Use of Scalar Couplings to Determine the Through-Bond Connectivity, J. Magn. Reson., 1996, 121, 65–69 CrossRef CAS.
- E. H. Hardy, R. Verel and B. H. Meier, Fast MAS total through-bond correlation spectroscopy, J. Magn. Reson., 2001, 148, 459–464 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03539e |
| ‡ These authors contributed equally. |
| § Present address: Fraunhofer Headquarters, Hansastr. 27c, 80686 Munich, Germany. |
| ¶ Present address: Institute of Technical and Macromolecular Chemistry, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany. |
| || Present address: Max-Planck-Institute for Chemical Energy Conversion, Stiftstr. 34-36, 45470 Mülheim an der Ruhr, Germany. |
| This journal is © The Royal Society of Chemistry 2023 |