
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControllable synthesis of a Na-enriched Na4V2(PO4)3 cathode for high-energy sodium-ion batteries: a redox-potential-matched chemical sodiation approach†
Mingli
Xu
a,
Fengxue
Zhang
b,
Yanhui
Zhang
b,
Chen
Wu
a,
Xue
Zhou
c,
Xinping
Ai
 a and
Jiangfeng
Qian
*a
a and
Jiangfeng
Qian
*a
aHubei Key Laboratory of Electrochemical Power Sources, College of Chemistry and Molecular Sciences, Wuhan University, Wuhan, Hubei 430072, China. E-mail: jfqian@whu.edu.cn
bHubei Baijierui Advanced Materials Co., Ltd, Wuhan, Hubei 430072, China
cCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan, Hubei 430072, China
First published on 25th October 2023
Abstract
Exploring a sodium-enriched cathode (i.e. Na4V2(PO4)3, which differs from its traditional stoichiometric counterpart Na3V2(PO4)3 that can provide extra endogenous sodium reserves to mitigate the irreversible capacity loss of the anode material (i.e. hard carbon), is an intriguing presodiation method for the development of high energy sodium-ion batteries. To meet this challenge, herein, we first propose a redox-potential-matched chemical sodiation approach, utilizing phenazine-sodium (PNZ-Na) as the optimal reagent to sodiate the Na3V2(PO4)3 precursor into Na-enriched Na4V2(PO4)3. The spontaneous sodiation reaction enables a fast reduction of one-half V ions from V3+ to V2+, followed by the insertion of one Na+ ion into the NASICON framework, which only takes 90 s to obtain the phase-pure Na4V2(PO4)3 product. When paired with a hard carbon anode, the resulting Na4VP‖HC full cell exhibits a high energy density of 251 W h kg−1, which is 58% higher than that of 159 W h kg−1 for the Na3VP‖HC control cell. Our chemical sodiation methodology provides an innovative approach for designing sodium-rich cathode materials and could serve as an impetus to the development of advanced sodium-ion batteries.
Introduction
Lithium-ion batteries (LIBs) are currently the dominant energy storage technology for portable and automotive applications; however, the high cost and geographical constraints of lithium resources make it impossible to sustain and meet the exploding market demands.1 As an alternative to LIBs, sodium-ion batteries (SIBs) are gaining increased recognition as next generation low cost energy storage devices, owing to the natural abundance of sodium resources (∼2.5% in the Earth's crust) and their similar electrochemical properties to LIBs.2,3 A series of high-performance sodium storage electrode materials have been investigated to pursue the ever-growing demand for higher energy density.4 Cathode materials such as layered oxides,5 polyanionic compounds,6,7 and Prussian blue analogues,8 as well as anode materials such as hard carbons,9 conversion materials,10 and alloy compounds,11,12 have all demonstrated high specific capacity and excellent cycle stability.However, the low initial coulombic efficiency (ICE), an intrinsic problem of anode materials, is generally ignored by researchers but seriously impedes the improvement of the energy density of SIBs.13,14 The low ICE signifies that a large number of active sodium ions are irreversibly consumed to form a solid electrolyte interphase (SEI) layer on the anode during the first cycle.15 This adverse effect is not as strongly felt in half cells, since the Na metal counter electrode can provide ample Na sources. However, as for Na-ion full cells, where the Na-containing cathode is the only reserve of active sodium ions, such a considerable Na+ loss dramatically reduces the overall energy density and shortens the lifespan of full cells. For instance, hard carbon (HC) anodes usually have a low ICE of only 60–80%, which leads to nearly one-third of capacity loss during the initial charge process.16
Numerous research efforts have been committed to resolving this issue by offering an additional Na source to counteract the initial capacity loss (ICL) of anodes, known as presodiation.17,18 In the early stage, presodiation has been carried out by the operation of highly active Na mental. Examples include direct contact of the anode electrodes with Na metal disks19 or assembling them into a primary cell.20 In these cases, the irreversible capacity can be readily dismissed via the self-discharge or electrochemical discharge mechanism. Another effective approach is to incorporate sacrificial sodium salts such as NaN3,21 Na2C2O4,22 and Na2C4O4,23 and Na3C6H5O7,24 into the cathode side by using their irreversible charging capacity through the electrochemical decomposition mechanism with undesirable impurity generation. Developing a sodium-enriched cathode that can provide extra endogenous sodium ions (beyond its normal stoichiometric ratio) is also an intriguing presodiation method. For instance, the Na3V2(PO4)3 (Na3VP) cathode can safely accommodate over-stoichiometric Na+ to form novel Na-enriched materials of Na4V2(PO4)3 (Na4VP) and Na5V2(PO4)3 (Na5VP), which correspond to distinct sodiation plateaus at approximately ∼1.6 V and ∼0.3 V vs. Na+/Na, respectively.25–29 Each excess Na+ in the formula theoretically provides an additional 58.8 mA h g−1 charging capacity. Li et al.27 prepared a Na4VP electrode by electrochemically discharging Na3VP to 1.0 V vs. Na+/Na. The intermediate product Na4VP is then used as a cathode and an extra Na source to sodiate hard carbon, after which it reverts to Na3VP with no residue left in the cell configurations. The resulting Na4VP‖HC full cell exhibits a high energy density of 265 W h kg−1, which is 76% higher than that of 151 W h kg−1 for the Na3VP‖HC control cell. Nevertheless, the electrochemical presodiation procedure is complicated and time-consuming, and thus not suitable for industrial application due to the multiple assembling-dissembling steps.
Alternatively, chemical presodiation using hyperactive arene-sodium reagents is more promising from the perspective of application considering its high efficiency, straightforward operation, and easily scalable feature.30,31 Many aromatic sodium complexes, such as biphenyl sodium (Biph-Na) and naphthalene sodium (Naph-Na), with redox potentials of 0.16 and 0.09 V (vs. Na+/Na), have been widely investigated in presodiation for anode materials.32–36 Liu and coworkers used Biph-Na as a liquid sodium source and obtained Na4VP electrodes after immersing Na3VP for 30 s. In anode-free Na4VP‖Al batteries and Na4VP‖HC full cells, the efficiency of the presodiation strategy has been reconfirmed with indicators of significantly increased specific capacity and lifespan.26 However, it should be pointed out that Biph-Na adopted by Liu et al. may not be a suitable reagent for Na4VP generation because of their potential mismatch (which will be discussed in detail below), that might cause oversodiation to form Na5VP. Besides, a series of parasite reactions including structure degradation and interface damage will be triggered during the over-reduction process, which negatively affect electrochemical performance.37–39 In conclusion, the key to cathode presodiation is to design sodiation reagents with modest reducing ability that are specifically designed for cathode materials.
Herein, we first developed a phenazine-mediated ambient chemical sodiation strategy to controllably synthesize a Na-enriched Na4V2(PO4)3 cathode guided by the principle of redox-potential matching. Phenazine-sodium (PNZ-Na) is elaborately screened as the optimal sodiation reagent because it possesses an appropriate reducing potential of 1.56 V vs. Na+/Na, which is just below the sodiation potential of Na3VP/Na4VP (1.67 V vs. Na+/Na), but much higher than that of the over-sodiation reaction turning into Na5VP (0.28 V vs. Na+/Na). The crystal structure, sodiation reaction mechanism and electrochemical performance of the as-obtained Na4VP have been verified by multiple characterization techniques, including XRD, XPS, TEM, ssNMR, and electrochemical tests. Benefiting from excess Na+ in the formula, the Na4VP cathode completely compensated the ICL from the anode without any exogenous impurities, resulting in a Na4VP‖HC full cell exhibiting higher reversible capacity and energy density than the control Na3VP‖HC full cell.
Results and discussion
Screening appropriate sodiation reagents guided by the potential matching principle
Na3V2(PO4)3 with a NASICON (Na super ionic conductor) structure is formed by the corner-sharing of the PO4 tetrahedron and VO6 octahedron, providing plenty of large channels for Na+ diffusion.40 As depicted in Fig. 1a, its crystal structure contains three isolated sodium storage sites with different oxygen surroundings, namely occupied Na1 and Na2 sites and a vacant Na3 site. More specifically, two Na+ ions at the Na2 site (0.67 occupation) exhibit electrochemical activity, while one Na+ ion at the Na1 site (1.0 occupation) is electrochemically inert. The remaining empty sites enable the lattice to accommodate over-stoichiometric Na+ and further generate Na-enriched phases.41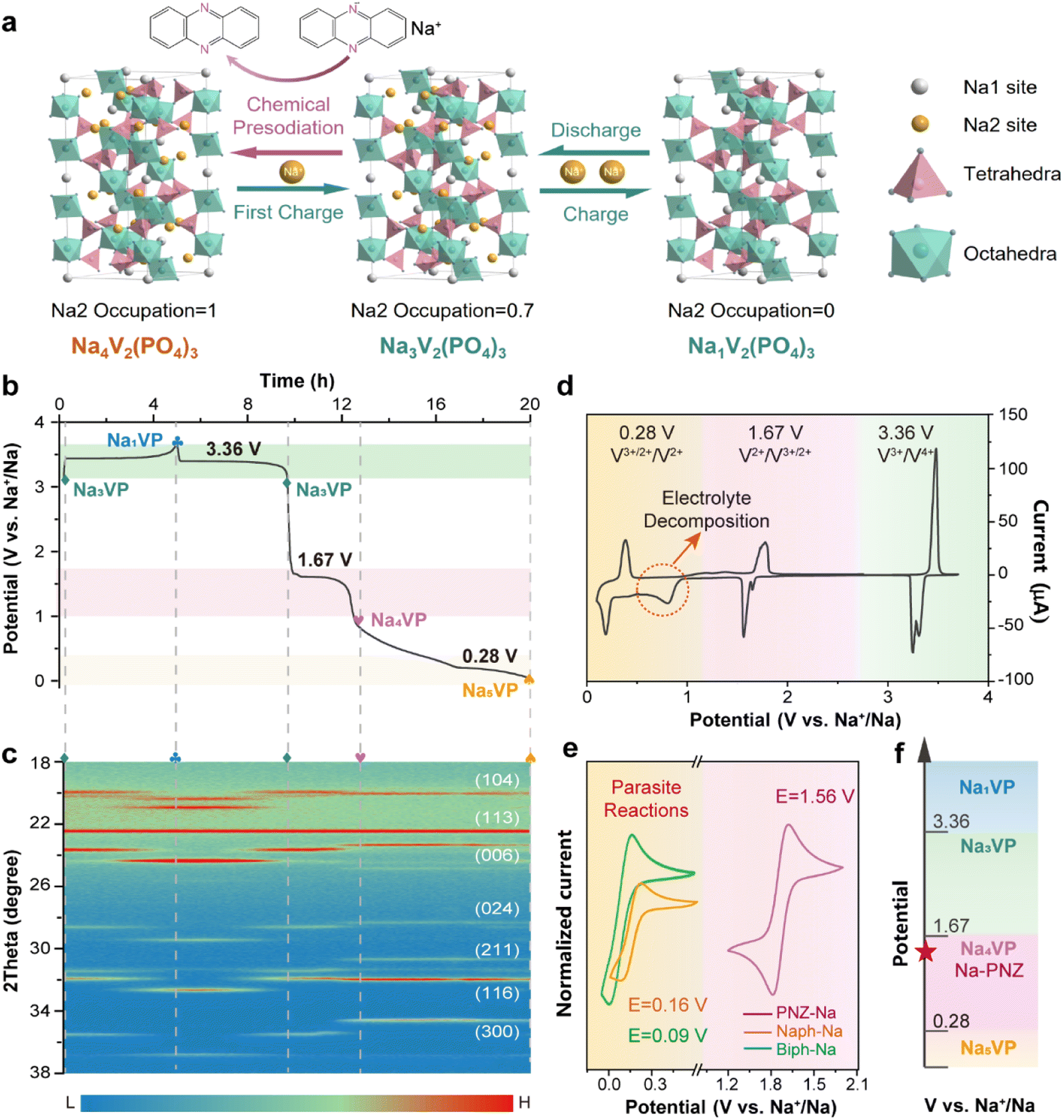 | ||
| Fig. 1 (a) Schematic illustration of chemical presodiation and the subsequent cycling process of the Na4VP cathode. (b) Initial charge–discharge profile of Na3VP and (c) the corresponding in situ XRD counter map. (d) Cyclic voltammogram profiles of the Na3VP cathode in the potential range of 0.1–3.7 V vs. Na+/Na. (e) Cyclic voltammogram profiles of different sodiation reagents: phenazine-sodium (PNZ-Na), naphthalene-sodium (Nap-Na), and biphenyl-sodium (Biph-Na) at a scan rate of 50 mV s−1. (f) Illustration diagram of the potential matching principle for chemical sodiation of Na3VP. | ||
As the cathode, Na3VP undergoes a typical biphase transition to form Na1V2(PO4)3 (Na1VP) and completely removes two Na+ from the Na2 site accompanied by two V3+ oxidizing to V4+, resulting in a theoretical capacity of 117.6 mA h g−1 and a voltage plateau at 3.36 V vs. Na+/Na (Fig. 1b). Recent investigations revealed that the V2+/V3+ redox couple in Na3VP can also be activated by controlling the sodiation depth, viz., the potential window of charge/discharge measurement.25 In the over-discharging process (Fig. 1b), two V3+ in Na3VP can be reduced to V2+ respectively and over-stoichiometric Na+ is embedded in the lattice to balance the charge. The first Na+ is inserted into the empty Na2 site to generate the Na4V2(PO4)3 phase (Na4VP, 1.67 V vs. Na+/Na), followed by the second Na+ in the vacant Na3 site to generate the Na5V2(PO4)3 phase (Na5VP, 0.28 V vs. Na+/Na). Of particular note, deep-sodiated Na5VP is energetically unfavorable due to the intense coulombic repulsion, thus leading to an extraordinarily low voltage plateau.
The structure evolution of Na3VP during the desodiation/sodiation process is intensively investigated by in situ XRD (Fig. 1c and S1†). Initially, all diffraction peaks can be assigned to Na3VP, showing a rhombohedral structure with a space group of R-3c.25 When charging (desodiation) to 3.7 V, the diffraction peaks of Na3VP diminish gradually while the peaks of Na1VP increase, presenting continuous phase transformation. The peak shift to higher angles demonstrates lattice shrinkage resulting from the removal of Na+. During the discharge (sodiation) process, an opposite trend is observed from Na1VP to Na3VP. Afterwards, over-discharging leads to the appearance of the Na4VP new phase, as indicated by a plateau at around 1.67 V (vs. Na+/Na) and emerging diffraction peaks at 23.5°, 28.5°, 30.8°, and 34.7° with gradually increasing intensity, which is in good agreement with previous reports.27,41 This demonstrates that the two phases coexist and correspond directly to the voltage plateau. Upon further sodiation, a new plateau appears at about 0.28 V (vs. Na+/Na) while diffraction peaks at 23.5° and 28.5° fade away gradually, accompanying the formation of Na5VP.28 Operando XRD confirms the feasibility of the presodiation strategy to produce sodium-enriched phases from Na3VP and reveal the corresponding structure information.
The cyclic voltammogram (CV) profile (Fig. 1d) obtained between 0.1 and 3.7 V (vs. Na+/Na) reconfirms the accessibility of Na4VP and Na5VP. Three pairs of cathodic/anodic peaks located at ∼0.20/0.36, ∼1.56/1.78 and ∼3.30/3.48 V are in good agreement with plateaus observed in the galvanostatic charge/discharge profiles (eqn (1)–(3)). Notably, a broad irreversible peak is observed between 0.3 and 0.9 V, implying aggressive electrolyte decomposition. During the cycles (Fig. S2†), all the peak currents prominently diminish and the polarization gradually increases, owing to the structure deterioration of active materials and the accumulation of the SEI on the electrode surface. In contrast, the CV curves between 1.0 and 4.0 V almost completely overlapped after the initial cycle (Fig. S3†), indicating the highly reversible phase transition among Na1VP, Na3VP, and Na4VP. The split of the cathodic peak in the first cycle may be ascribed to the minor structure rearrangement of the active material.42 The narrower CV peaks also imply a faster Na+ insertion process for Na4VP than Na5VP. It is important to note that Na5VP exhibits low inital coulombic efficiency (ICEcathode is defined as the ratio of sodiation capacity to desodiation capacity of a cathode), rendering it incompatible with HC anodes (Fig. S4†). As mentioned above, the typical ICEanode (defined as the ratio of desodiation capacity to sodiation capacity of an anode) of HC anodes is only 60–80%. When Na5VP with an ICE of merely 40% matches with HC anodes, surplus Na+ will be introduced into the full cells and form uncontrollable sodium dendrites on the anode side, resulting in safety concerns. Therefore, Na4VP has a suitable ICE (∼64%) and excellent structural stability, denoting its superiority for active sodium compensation in full cells.
As displayed in Fig. 1e, currently reported aromatic sodium reagents, such as Biph-Na and Naph-Na, demonstrate ultra-low redox potentials of 0.16 V and 0.09 V vs. Na+/Na for cathode materials (lower than the Na5VP formation potential of 0.28 V vs. Na+/Na). In order to prevent over-sodiation from yielding Na5VP or other parasite reactions, the treatment duration with sodium reagents must be strictly controlled, which is not conducive to industrial batch application. Therefore, it is of utmost importance to develop appropriate sodium reagents to achieve a refined precise and controllable preparation of Na4VP.
Na+ insertion/extraction reaction of NaxVP:
| Na1VP + 2e− + 2Na+ ⇌ Na3VP E = 3.36 V | (1) |
| Na3VP + e− + Na+ ⇌ Na4VP E = 1.67 V | (2) |
| Na4VP + e− + Na+ ⇌ Na5VP E = 0.28 V | (3) |
Preparation of the PNZ-Na reagent:
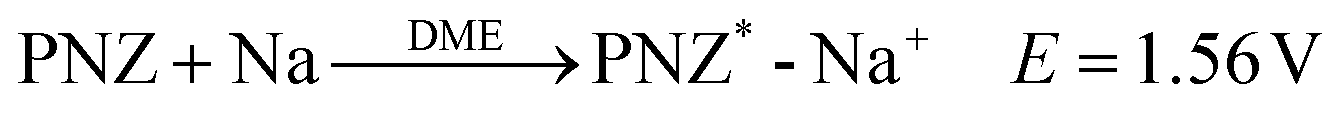 | (4) |
Precise chemical sodiation of Na3VP enabled by the PNZ-Na reagent:
| PNZ-Na + Na3VP → PNZ + Na4VP | (5) |
In accordance with the potential-matched chemical sodiation strategy (Fig. 1f), phenazine sodium (PNZ-Na) has been elaborately chosen as a suitable sodiation reagent due to its appropriate potential. The PNZ-Na reagent displays a pair of reversible redox peaks at 1.51 and 1.62 V (vs. Na+/Na, Fig. 1e), corresponding to the PNZ/PNZ− redox couple (eqn (4)). PNZ-Na exhibits the highest potential as a sodiation reagent reported so far,26,32,33,43 benefiting from the strong electron-withdrawing effect of nitrogen atoms and the efficient charge dispersion provided by the additional six-membered ring.44,45 Hence, the end-point of the reaction is effectively clamped by the PNZ-Na reagent potential, thus preventing over-sodiation. During the chemical presodiation process, reductive PNZ-Na concurrently donates Na+ and e− for Na3VP while also leading to the recovery of intrinsic phenazine molecules (eqn (5)). The sodium ions diffuse into the Na3VP lattice and locate at the partially occupied Na2 site to form a pure Na4VP phase.
Controllable synthesis of Na-enriched Na4VP via the chemical sodiation strategy
The chemical presodiation of the Na3VP cathode is conducted simply by immersing the electrode into PNZ-Na solution for a particular time, followed by DME rinse to remove the residual PNZ-Na. Phenazine and sodium metal are mixed in dimethoxyethane (DME) solvent in equal amounts to obtain the PNZ-Na reagent, as described in eqn (4). The strong electron affinity of heterocyclic aromatics causes the outermost electrons of Na metal to transfer spontaneously to the conjugated phenazine rings, producing grape-purple colored PNZ− radicals (Fig. 2a left).46,47 Once the Na3VP electrode is immersed in the PNZ-Na solution, the sodiation reaction proceeds through electron transfer from the PNZ− radical to Na3VP along with Na+ accommodation to form Na4VP (eqn (5)). Following the reaction, the purple PNZ− radical dissolved in DME is converted to a pristine PNZ molecule, resulting in the PNZ-Na solution fading (Fig. 2a right).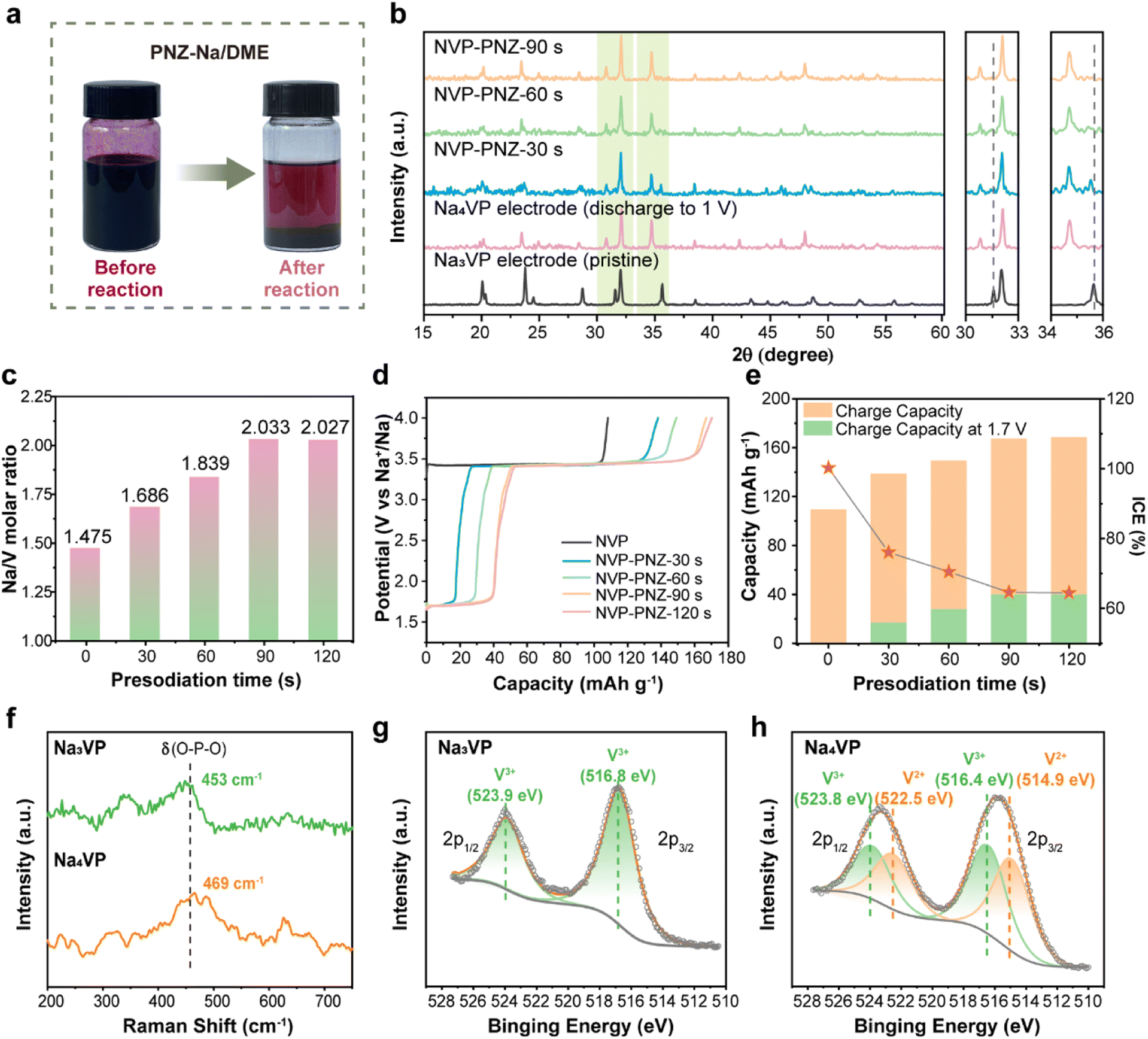 | ||
| Fig. 2 (a) Optical photographs of the phenazine sodium reagent before and after reaction with Na3VP. (b) XRD patterns, (c) ICP results, (d) initial charge curve, and (e) initial charge capacity and ICE of the NaxVP electrodes with different presodiation times. (f) Raman spectroscopy spectra of Na3VP and Na4VP. XPS V 2p spectra of (g) Na3VP and (h) Na4VP. | ||
To probe the reaction pathways, we prepared presodiated NaxVP (3 ≤ x ≤ 4) electrodes with various immersion times of 30–120 s. As the reaction time increases, the emergence of new diffraction peaks at 30.8° and 34.7°, along with growing intensity, is attributable to the Na4VP phase, while the peaks of Na3VP at 31.6° (116) and 35.6° (300) weaken (Fig. 2b). This outcome reveals that two phases, Na3VP and Na4VP, coexist during the presodiation process (30 s and 60 s), which is consistent with in situ XRD and prior reports.26,28 Notably, Na3VP is completely converted to Na4VP after presodiation for more than 90 s. (Fig. S5†) The XRD pattern of chemically presodiated Na4VP is identical to that of electrochemically sodiated Na4VP, implying a rapid Na+ migration in the cathode and a uniform sodiation process achieved by chemical presodiation. The inductively coupled plasma atomic emission spectroscopy (ICP-OES) tests accurately determine the Na content in different samples (Fig. 2c). The samples with treatment time of 30, 60, and over 90 s are accordingly denoted as Na3.37VP, Na3.68VP, and Na4VP.
The presodiation degree of NaxVP achieved via chemical treatment is also evaluated using electrochemical performance in sodium half-coin cells. The initial charge curves of NaxVP, including ICE and plateau capacity at about 1.67 V that correspond to Na4VP, are displayed in Fig. 2d and e. The open circuit voltage (OCV vs. Na+/Na) dramatically drops during the biphasic process from Na3VP to Na4VP, after which it remains stable at around 1.67 V. The presodiated NaxVP exhibits a continuous increase in initial charge capacity (i.e., Na extraction), whereas the ICE decreases with increasing sodiation time, indicating a sustained Na+ insertion reaction (Fig. S6†). Consequently, the chemical presodiation process continues until Na3VP diminishes, as reflected by a constant ICE of ∼64% and an increased specific capacity of ∼56 mA h g−1, respectively. Notably, no unnecessary parasitic reactions are introduced due to the identical charge–discharge curves and ICE between chemical and electrochemical sodiation (Fig. S7a†). The redox potentials between the reductive agent and Na3VP are well-matched, thereby avoiding the issues of over-sodiation, inhomogeneity, and inconsistency during scaling up for mass production. Even with excessive treatment time (30 min) and reagent dosage, there is no structural damage to the cathode materials, demonstrating the success of our chemical presodiation strategy based on the redox-potential matched principle (Fig. S7b†). The sodiation reaction self-terminates once all Na3VP transforms into sodium-rich Na4VP due to the elaborately screened sodiation reagents. We further conducted the in situ XRD test of the PNZ-Na presodiated Na4VP electrode (Fig. S8 and S9†). The variation of diffraction peaks is consistent with the electrochemical sodiation process, and there is no impurity phase introduced by presodiation treatment. It is a highly reversible transformation between Na4VP and Na3VP and reconfirms that presodiation has no adverse effect on the lattice structure. Therefore, the reliability of the chemical presodiation strategy is firmly authenticated.
Raman spectra and X-ray photoelectron spectroscopy (XPS) are further employed in order to explore the effect of presodiation treatment on the structure of the Na3VP cathode. The Raman spectra shown in Fig. 2f indicate that the symmetric bending vibration peak of the PO4 tetrahedron in Na3VP at 453 cm−1 is slightly blue-shifted after presodiation treatment. This result suggests the enhancement of P–O bond energy and boosting ionicity of the Na4VP lattice.48 The valence states of vanadium ions are verified by the XPS results. The survey XPS spectrum (Fig. S10†) confirms the presence of the same chemical composition elements in Na3VP and Na4VP. The binding energy of P 2p shifts toward the higher energy direction (from 133.4 eV to 133.7 eV), reconfirming the stronger P–O bonds (Fig. S11†). As seen in Fig. 2g and h, the V 2p peaks broaden after presodiation and reveal the coexistence of mixed V3+ and V2+ ions in the lattice. The two peaks of Na3VP at 516.8 eV and 523.9 eV coincide with V 2p3/2 and V 2p1/2 of V3+, which means only trivalent vanadium is observed in the pristine electrode.49 Except for the peaks above, new peaks at 514.9 eV and 522.5 eV can be ascribed to the contribution of V2+ in Na4VP.26 Further quantitative analysis of the V 2p spectrum indicates a V3+/V2+ ratio of 0.49/0.51 based on the integral area of deconvoluted peaks, which agrees well with the valence distribution for Na4VP. All the characterization studies above demonstrate that the PNZ-Na solution can readily and controllably presodiate the Na3VP cathode in a relatively short time.
The morphological evolution of the Na3VP and Na4VP samples is investigated by scanning electron microscopy (SEM), focused ion beam (FIB), and high-resolution transmission electron microscopy (HRTEM). The results show that both Na3VP and Na4VP exhibit a regular spherical morphology, with a diameter ranging from 5–15 μm, consisting of primary particles that are densely packed to form secondary particles (Fig. 3a and d). Apparently, the morphology characteristic of sodiated Na4VP is not affected by the chemical treatment and helps to retain the electrochemical performance (Fig. S12†). The energy-dispersive X-ray (EDX) analysis reconfirms the Na/V ratios of Na3VP and Na4VP to be 1.49 and 1.99, respectively (Fig. S13†). Cross sections are obtained by FIB-SEM to monitor microstructural damage, while no microcracks are observed after presodiation treatment (Fig. 3b and e). It should be noted that the internal voids in the particles are formed by primary particle agglomeration rather than structure degradation. HRTEM images in Fig. 3c reveal well-defined lattice fringes of 0.373 nm ascribed to the (113) planes in the Na3VP lattice. Likewise, we observe characteristic lattice fringes of 0.378 nm (Fig. 3f), corresponding to the emerging 23.5° diffraction peak in the Na4VP lattice (Fig. 2b).
 | ||
| Fig. 3 (a–c) SEM image, cross section and HR-TEM image of Na3VP, respectively. (d–f) SEM image, cross section and HR-TEM image of Na4VP, respectively. | ||
23Na/31P solid state nuclear magnetic resonance (ssNMR) offers in-depth insights into local structural transformation while probing the oxidation states of the neighboring vanadium ions. For 31P spectra, the signals are caused by the unpaired electronic spin transfer from TM d orbitals to the 31P nuclei through chemical bonds.50 Fig. S14† presents the room temperature 31P spectra for Na3VP and Na4VP. In the Na3VP lattice, a broad asymmetric peak at 2000–3500 ppm reflects a twisted environment for the 31P nuclei.51 While in Na4VP, the corresponding peak with decreasing intensity reveals the reducing dipolar interaction between unpaired electron spins of V and P nuclei. In contrast, a new broad peak at 3500–5000 ppm is observed in Na4VP, which is attributed to the complicated interaction between V2+ and adjacent phosphate groups.50 This phenomenon is because the V2+ (t2g3eg0) owning three unpaired electrons is more paramagnetic than V3+ (t2g2eg0) and generally causes a larger Fermi contact shift. Similar results are also observed in 23Na spectra.
Turning to the 23Na NMR spectra, the signals of Na4VP are recorded at 8, 10, and 12 kHz to distinguish the isotropic signals and spinning side bands denoted by asterisks (Fig. 4a). A typical 23Na spectrum of Na3VP is presented in Fig. 4b and c, which includes two significant signals at 83 and 18 ppm with very different magnitudes and a set of spinning side bands. As previously reported by Hu et al.,52 the oxidation state of vanadium centers coordinated with Na at Na1 sites is higher than those coordinated with Na at Na2 sites, leading to a more significant 23Na shift at the Na2 site in contrast with that at the Na1 site. Similar paramagnetic effects have also been reported in LIBs on 7Li NMR shifts.53 Based on these results, the signals at 83 and 18 ppm are assigned to the partially occupied Na2 site and fully filled Na1 site, respectively. Only Na at the Na2 site has electrochemical activity on account of the relatively weak bonding. The signal peaks shrink and slightly shift to the higher direction with the increasing sodium content of these samples (Fig. 4b). This phenomenon suggests a more ordered environment of Na, whereas more V2+ leads to a higher shift. We speculate that the shrinkage of peaks from Na3VP to Na4VP is due to a decrease in Na+ motion. Because of the partially occupied Na2 site, Na3VP has more Na vacancies than the fully occupied Na4VP. We further investigate Na+ diffusion kinetics by the CV technique at varied scan rates (Fig. S15 and S16†). The Na+ diffusion coefficients of Na4VP decrease as we suspected, indicating the reduced mobility of sodium ions. The fitted spectra of samples with different sodiated degrees are presented in Fig. 4c and d and we conclude that the Na2/Na1 peak area ratio gradually increases from 1.371 to 2.225. In other words, excess sodium ions that Na3VP receives from the PNZ-Na solution are preferentially accommodated in the Na2 site, making it close to completely occupied in the Na4VP sample.
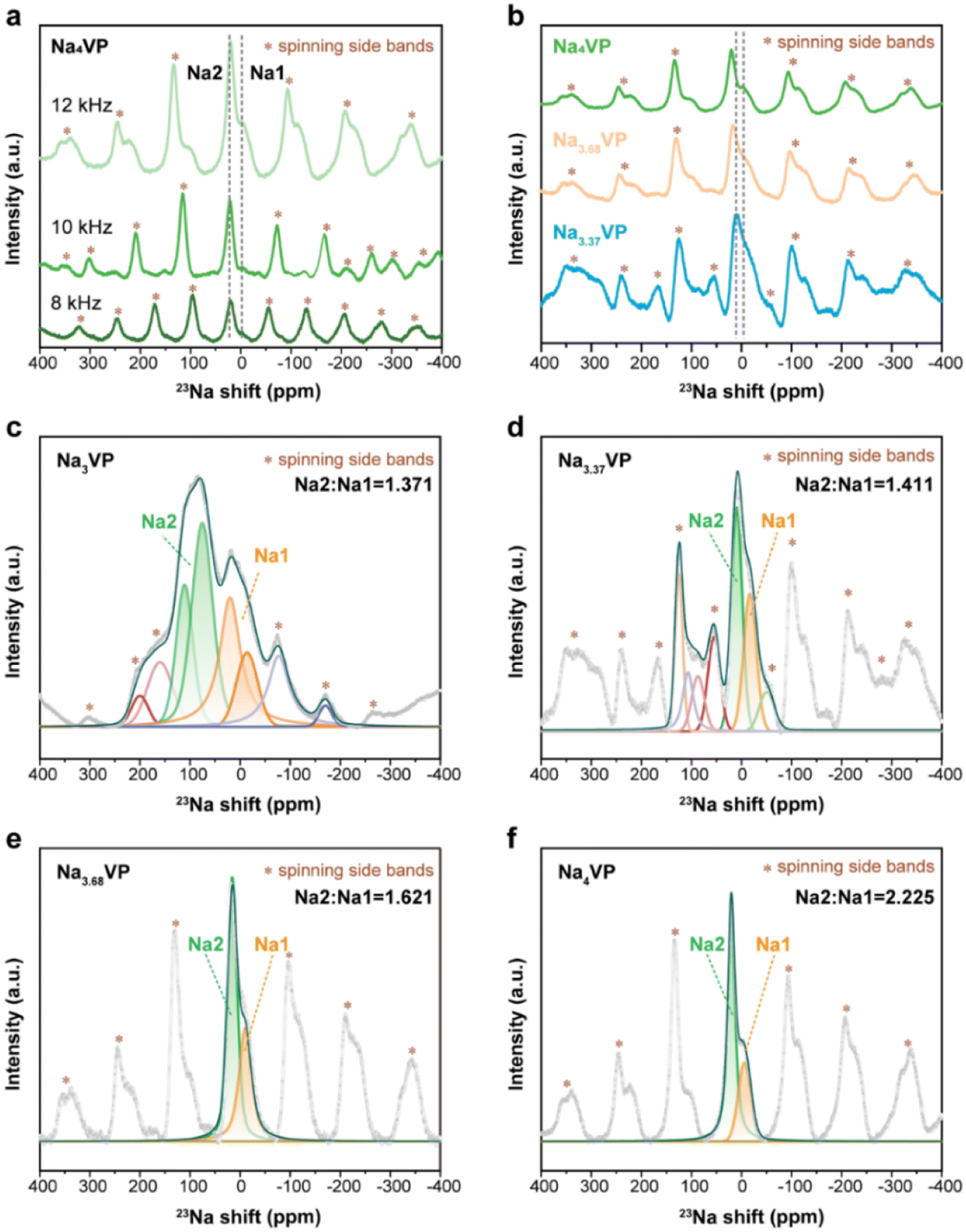 | ||
| Fig. 4 (a) 23Na ssNMR spectra of Na4VP at 8, 10, and 12 kHz. (b) 23Na ssNMR spectra of NaxVP with different sodiation depths at 12 kHz. The fitted 23Na ssNMR spectra of (c) Na3VP, (d) Na3.37VP, (e) Na3.68VP, and (f) Na4VP. | ||
The electrochemical performance of the Na-enriched Na4VP cathode
The sodium storage capability of the sodiatied samples is investigated through galvanostatic charge–discharge tests in half-cells with a Na-metal anode. All of the NaxVP electrodes with different treatment times manifest the same cycling stability at 0.5C for 100 cycles as pristine Na3VP (Fig. S17†). Even extending the treatment time to 30 min, no discernible capacity decay is observed, suggesting that the electrochemical performance is completely retained after presodiation treatment. Our presodiation strategy is not strict with processing time and is expected to achieve large-scale production. In the following discussion, we primarily focus on the comparison between pristine Na3VP and the sodiated Na4VP. Fig. 5a displays the CV curves of Na4VP at a scan rate of 0.1 mV s−1. The OCV of the Na4VP sample is close to 1.6 V which is related to the average valence of V ions, in line with previous discussion. During the first cathodic scan, the first oxidation peak appears at around 1.7 V corresponding to the first distinct plateau in the charge profile of Na4VP (Fig. 5b). These features, which are absent in Na3VP electrodes, are because of the oxidation V2+/V3+ in the Na-enriched phase and the extraction of the pre-doped sodium ions. When all pre-doped sodium is removed, the Na-enriched Na4VP entirely transforms into Na3VP and then presents a typical voltage plateau of 3.4 V attributed to the reversible V3+/V4+ redox couple, validated by well-overlapped CV profiles. In brief, active sodium compensation during cycling is achieved by the irreversible oxidation of V2+, viz., by controlling the voltage window. These results reconfirm that the preparation of the Na4VP cathode to offer charge compensation and an additional Na reservoir has been successfully implemented. The rate capability and long-term cycling stability of the two samples are analysed as shown in Fig. 5c–e in a voltage window of 2.0–4.0 V (vs. Na+/Na). The Na4VP electrode exhibits reversible desodiation capacities of 109.4, 107.5, 106.1, 101, 98.1, and 92.8 mA h g−1 at rates of 0.2, 0.5, 1, 2, 3, 4, and 5C, respectively, slightly higher than that of the Na3VP electrode. When the rate comes back to 0.2C, a reversible capacity of about 108 mA h g−1 is achieved, suggesting the excellent reversibility of the Na4VP electrode. The Na4VP electrode also retains an outperformed cycling stability with high capacity retentions of 95.3% at 0.5C after 200 cycles and 91.8% at 1C after 600 cycles.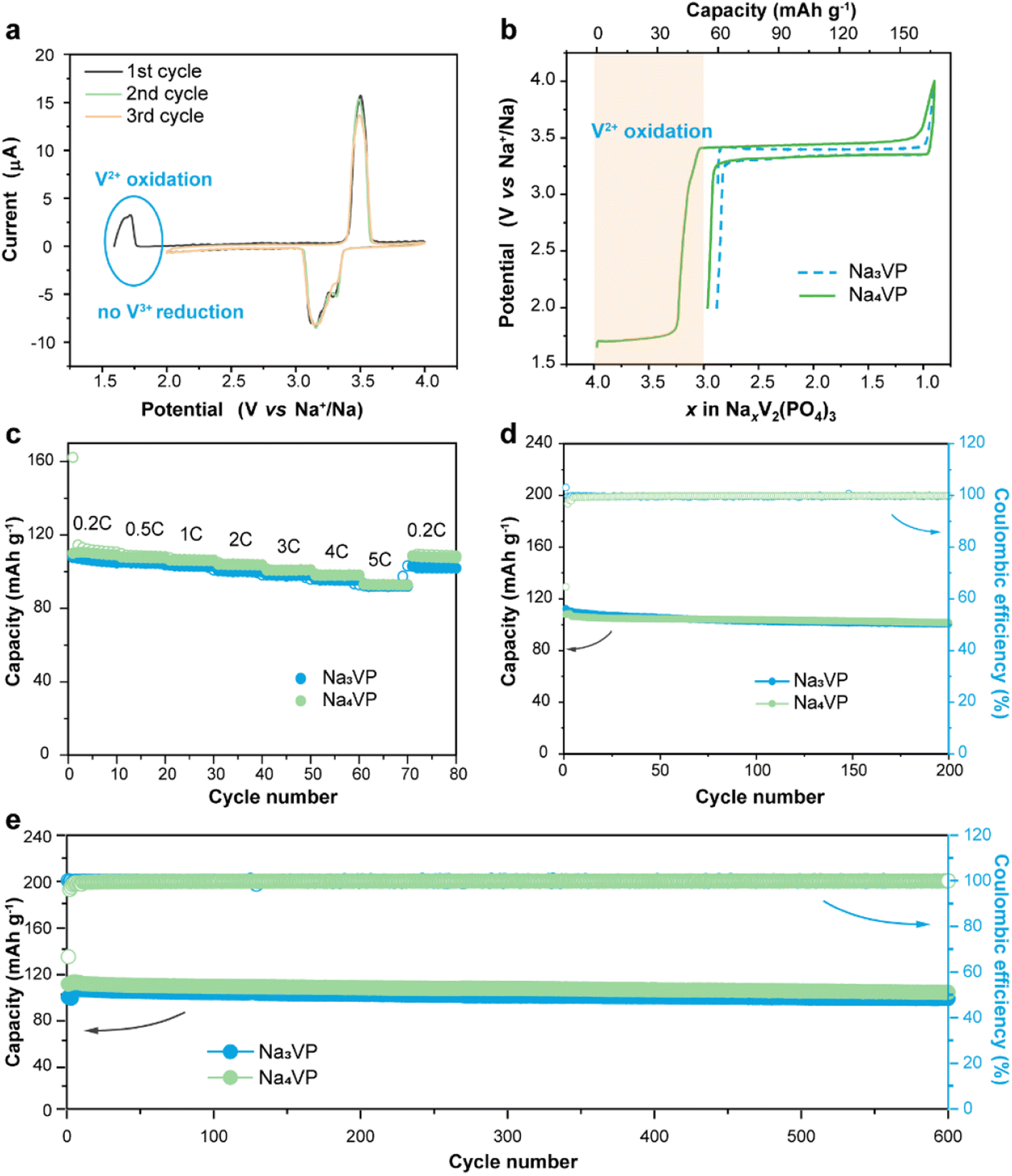 | ||
| Fig. 5 (a) Cyclic voltammogram profiles of the Na4VP cathode at a scan rate of 0.1 mV s−1. (b) Initial charge–discharge curves of Na3VP and Na4VP electrodes between 2.0 and 4.0 V at 0.2C. (c) Rate performance, (d) cycling stability at 0.5C, and (e) long-term cycling stability at 1C of Na3VP and Na4VP electrodes. | ||
Na4VP‖HC full cells are assembled to demonstrate the application feasibility of Na4VP as both the cathode and extra Na reservoir in enhancing the energy density of SIBs, and a control cell Na3VP‖HC is also constructed for comparison. For the fabrication of full cells, the mass matching between the cathode and anode is based on their capacity balance, which is determined by the charge capacity of the cathode and the discharge capacity of the anode. As displayed in Fig. 6a and b, during the first charge, both the irreversible sodium storage sites of the anode and the formation of the SEI will consume considerable active sodium. More Na3VP cathode is commonly to be matched with HC and in a Na-poor state in the following cycles, namely Na3−xVP. As an alternative, the Na4VP electrode used for full cells enables a 35.2 wt% reduction in cathode mass compared to the Na3VP electrode and ensures that the cathode as Na3VP without Na loss participates in subsequent cycles.
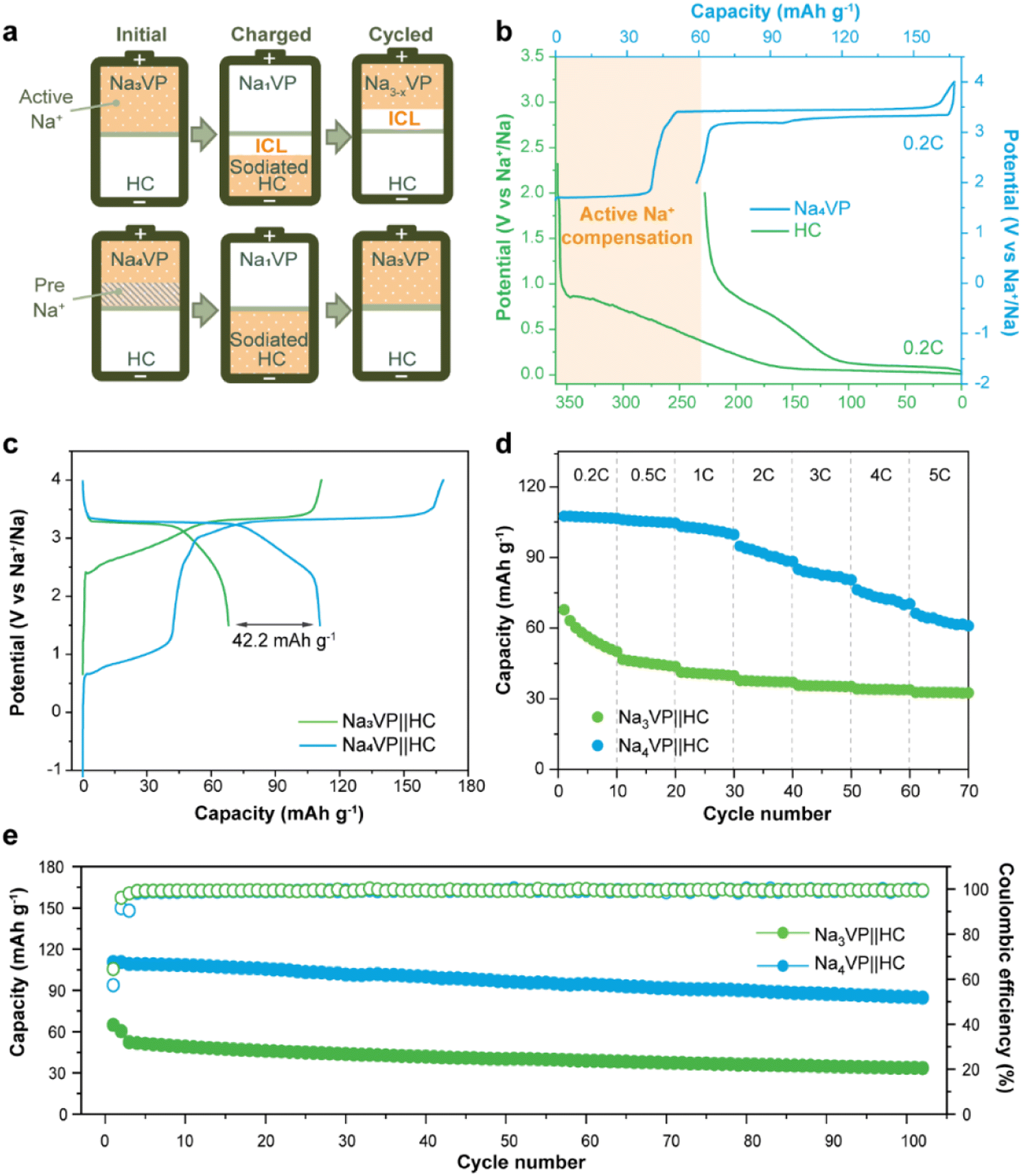 | ||
| Fig. 6 (a) Schematic illustration of the presodiation process by the Na-enriched Na4VP cathode in full cells. (b) Half-cell voltage profiles of the Na4VP cathode and the HC anode at 0.2C. (c) Initial voltage profiles, (d) rate performance, and (e) cycling stability at 1C of Na3VP‖HC and Na4VP‖HC full cells. | ||
As shown in Fig. 6c, the Na3VP‖HC and Na4VP‖HC full cells deliver a charge capacity of 111 and 168 mA h g−1, respectively, matching well with the values in half cell configuration. However, the reversible discharge capacity of the Na3VP‖HC full cell is merely 66.6 mA h g−1 with 39.3% capacity loss. This result can be attributed to the failed Na+ reinsertion on the cathode side in the absence of extra endogenous Na+. In comparison, the full cell with the Na4VP cathode shows an initial discharge capacity of 109.7 mA h g−1 and an ICE of 65%, which are equivalent to that of the Na3VP half-cell, demonstrating extremely high utilization of active materials. Similar to other published results (Table S1†), the low ICE is ascribed to irreversible Na compensation during the initial charge cycle, but subsequent average coulombic efficiency is higher than that of Na3VP‖HC without presodiation (Fig. S18†). The Na4VP‖HC full cell shows comparable cycling stability with the control Na3VP‖HC full cell, but shows markedly enhanced reversible capacity and excellent rate capability (Fig. 6d, e and S19). Fig. 6d demonstrates the rate capability of both Na4VP‖HC and Na3VP‖HC full cells from 0.2C to 5C. Na4VP‖HC displays a respectable rate performance benefiting from the presodated Na4VP cathode. Discharge capacities of 107.5, 106, 103.2, 94.8, 84.9, 76.3, and 66.3 mA h g−1 are delivered at current densities of 0.2, 0.5, 1, 2, 3, 4, and 5C, respectively. Even at a current density of 5C, corresponding to a charging duration as short as 12 min, Na4VP‖HC still delivers 61.7% capacity retention of the discharge capacity at 0.2C. In contrast, Na3VP‖HC exhibits much lower reversible capacity due to active sodium loss. Overall, Na4VP‖HC exhibits an impressive discharge capacity of 111 mA h g−1 with an ICE of 66%, and an energy density of 251.1 W h kg−1 based on the total mass of cathode and anode active materials. This is far superior to those of the control cell using the pristine Na3VP cathode (ICE of 60% and an energy density of 159.4 W h kg−1). The Na4VP‖HC full cell shows a decent cycling stability of 78% capacity retention at 1C after 100 cycles and 57% capacity retention at 2C after 500 cycles (Fig. 6e and S19), which is comparable with that of the control Na3VP‖HC full cell, but shows markedly enhanced reversible capacity. Table S1 in the ESI† compares the recently published NVP-based full cells in terms of electrochemical performance and energy density. To the best of our knowledge, the energy density of PNZ-Na presodiated Na4VP‖HC is among the best of the reported values for NVP-based full cells. These discrepancies strongly suggest that our presodiation strategy can improve the overall energy density of SIBs while simultaneously maintaining decent cycling stability.
Conclusions
In summary, we have reported a facile and effective chemical presodiation approach to prepare a Na-enriched Na4VP cathode under the guidance of the redox-potential-matching principle. The phenazine-Na reagent with a redox potential of 1.56 V vs. Na+/Na was employed to pre-dope active Na+ into the Na3VP lattice for the Na4VP cathode, successfully retaining the structure stability and electrochemical performance. Na4VP initially serves as an excess Na reservoir and a cathode to eliminate the active Na+ loss on the anode side, allowing the full capacity of Na3VP to be used for the subsequent cycles. Our presodiation strategy not only enables rapid preparation of Na4VP and is expected to achieve batch production, but also doesn't bring about any external impurities after sodium compensation. The energy density of the Na4VP‖HC full cell is 251.1 W h kg−1, much greater than that of the Na3VP‖HC full cell (159.4 W h kg−1). Most importantly, this approach can be broadly applicable to different cathode materials and opens up new avenues for the design of high-energy SIBs.Experimental section
Materials synthesis
All the reagents were used as received without further treatment. Commercial Na3VP and hard carbon (BHC400) powders were purchased from Guangdong Canrd New Energy Technology Co., Ltd Phenazine, biphenyl, naphthalene and electron-grade N-methyl-2-pyrrolidone (NMP) were obtained from Aladdin Chem. Co., Ltd Anhydrous DME was purchased from JK Chemical. The phenazine-sodium (PNZ-Na) reagent was prepared by adding equal stoichiometric sodium metal and phenazine in DME and stirring for four hours in a glove box. Sodium-enriched Na4VP was synthesized simply by immersing the Na3VP powders or cathode sheet in 0.05 M PNZ-Na solution for a certain time (0–120 s) and then rinsing it with DME to quench the reaction and remove the residue, followed by vacuum drying for 15 min.Characterization
XRD patterns were collected on an X-ray diffractometer (Rigaku Smartlab SE) with Cu-Ka radiation at 40 kV. In situ XRD was performed on a live charge/discharge process to monitor the structure evolution of Na3VP. Every single pattern was collected for 7 min in the scan range of 18–38°. The morphologies and fine phase structure of samples were obtained by SEM (SEM, Zeiss Merlin Compact) and HRTEM (JEM-2100). The valent information was obtained by XPS (ESCALAB250Xi) with Al Ka radiation. Raman spectra (Bruker MultiRam) were measured to investigate the variation of chemical bonds before and after persodiation. The chemical compositions of samples were determined by ICP-OES (Agilent 730) and EDS (X-Max 50). FIB was used to observe the cross section of active material particles on Tescan SOLARIS equipment. The ssNMR spectra were recorded with a Bruker AVANCE NEO 400 MHz NMR spectrometer.Electrochemical measurements
The Na3VP cathode sheet was fabricated by mixing Na3VP, Super P and polyvinylidene fluoride at a mass ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10 in NMP and further spreading the mixture on the aluminum foil with a doctor blade. After vacuum drying, the electrode was cut into discs with a diameter of 12 mm for use. For half cells, CR-2032-type coin cells were assembled by using pieces of Na foil as the counter and reference electrodes, Na3VP or Na4VP as the working electrode, and glass fiber as a separator in an argon-filled glove box with H2O and O2 contents less than 0.1 ppm. The electrolyte was 1 M NaClO4 dissolved in ethylene carbonate/diethyl carbonate (1:1 v/v) solution with a 5% fluoroethylene carbonate (FEC) additive. Galvanostatic charge/discharge tests were conducted between 2.0 and 4.0 V vs. Na+/Na on a NEWARE CT4008 cycler battery cycler at room temperature. In particular, the nominal capacity of 1C is 117.6 mA g−1. For full cells, all procedures were identical to those for half-cell assembly with the exception of using a hard carbon electrode as the anode. The cathode/anode capacity ratio was set as 1:1 and the voltage range was set as 1.5–4.0 V vs. Na+/Na. The CV measurement was executed on an Admiral electrochemical workstation at a scan rate of 0.1 mV s−1. The energy density (E) of the full cell was calculated using the following formula:
:10:10 in NMP and further spreading the mixture on the aluminum foil with a doctor blade. After vacuum drying, the electrode was cut into discs with a diameter of 12 mm for use. For half cells, CR-2032-type coin cells were assembled by using pieces of Na foil as the counter and reference electrodes, Na3VP or Na4VP as the working electrode, and glass fiber as a separator in an argon-filled glove box with H2O and O2 contents less than 0.1 ppm. The electrolyte was 1 M NaClO4 dissolved in ethylene carbonate/diethyl carbonate (1:1 v/v) solution with a 5% fluoroethylene carbonate (FEC) additive. Galvanostatic charge/discharge tests were conducted between 2.0 and 4.0 V vs. Na+/Na on a NEWARE CT4008 cycler battery cycler at room temperature. In particular, the nominal capacity of 1C is 117.6 mA g−1. For full cells, all procedures were identical to those for half-cell assembly with the exception of using a hard carbon electrode as the anode. The cathode/anode capacity ratio was set as 1:1 and the voltage range was set as 1.5–4.0 V vs. Na+/Na. The CV measurement was executed on an Admiral electrochemical workstation at a scan rate of 0.1 mV s−1. The energy density (E) of the full cell was calculated using the following formula:| E = Q × V/(mcathode + manode) | (6) |
Data availability
All the data supporting this study are included in the manuscript and the ESI.† Example data sets are available on request.Author contributions
J. Q. and M. X. conceived the idea of the study. M. X. contributed to the design of the research and the experimental data analysis. F. Z. and Y. Z. conducted the XRD experiments. C. W. conducted the SEM experiments and related data analysis. X. Z. conducted the ssNMR experiments and the analysis of ssNMR results. X. A. and J. Q. supervised the work. The manuscript was written by M. X. and all authors participated in reviewing the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant no. 22279093, and 22075216), the Natural Science Foundation of Hubei Province, China (Grant no. 2022CFB096) and the Fundamental Research Funds for Central University (Grant no. 2042022gf0005 and 2042021kf0194). We would like to thank the Core Research Facility of CCMS (WHU) and Shiyan Laboratory for access to analytical equipment. We thank Xiaobin Zhou from Shiyanjia Lab (http://www.shiyanjia.com) for his support on XPS analysis.References
- F. Wu, J. Maier and Y. Yu, Chem. Soc. Rev., 2020, 49, 1569–1614 RSC.
- J. Deng, W.-B. Luo, S.-L. Chou, H.-K. Liu and S.-X. Dou, Adv. Energy Mater., 2018, 8, 1701428 CrossRef.
- Y. Tian, G. Zeng, A. Rutt, T. Shi, H. Kim, J. Wang, J. Koettgen, Y. Sun, B. Ouyang, T. Chen, Z. Lun, Z. Rong, K. Persson and G. Ceder, Chem. Rev., 2021, 121, 1623–1669 CrossRef CAS PubMed.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- C. Zhao, Q. Wang, Z. Yao, J. Wang, B. Sanchez-Lengeling, F. Ding, X. Qi, Y. Lu, X. Bai, B. Li, H. Li, A. Aspuru-Guzik, X. Huang, C. Delmas, M. Wagemaker, L. Chen and Y. S. Hu, Science, 2020, 370, 708–711 CrossRef CAS PubMed.
- P. Hu, T. Zhu, C. Cai, X. Wang, L. Zhang, L. Mai and L. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202219304 CrossRef CAS PubMed.
- C. Liu, Z.-X. Zhang, R. Tan, J.-W. Deng, Q.-H. Li and X.-C. Duan, Rare Met., 2021, 41, 806–813 CrossRef.
- J. Qian, M. Zhou, Y. Cao, X. Ai and H. Yang, Adv. Energy Mater., 2012, 2, 410–414 CrossRef CAS.
- L. F. Zhao, Z. Hu, W. H. Lai, Y. Tao, J. Peng, Z. C. Miao, Y. X. Wang, S. L. Chou, H. K. Liu and S. X. Dou, Adv. Energy Mater., 2020, 11, 2002704 CrossRef.
- H. He, Q. Gan, H. Wang, G.-L. Xu, X. Zhang, D. Huang, F. Fu, Y. Tang, K. Amine and M. Shao, Nano Energy, 2018, 44, 217–227 CrossRef CAS.
- J. Qian, X. Wu, Y. Cao, X. Ai and H. Yang, Angew. Chem., Int. Ed., 2013, 52, 4633–4636 CrossRef CAS PubMed.
- Y. Li, X. Lai, J. Qu, Q. Lai and T. Yi, Acta Phys. – Chim. Sin., 2022, 38, 2204049 Search PubMed.
- E. Goikolea, V. Palomares, S. Wang, I. R. Larramendi, X. Guo, G. Wang and T. Rojo, Adv. Energy Mater., 2020, 10, 2002055 CrossRef CAS.
- C.-X. Yu, Y. Li, Z.-H. Wang, X.-R. Wang, Y. Bai and C. Wu, Rare Met., 2022, 41, 1616–1625 CrossRef CAS.
- H. He, D. Sun, Y. Tang, H. Wang and M. Shao, Energy Storage Mater., 2019, 23, 233–251 CrossRef.
- Y. R. Yang, C. Wu, X. X. He, J. H. Zhao, Z. Yang, L. Li, X. Q. Wu, L. Li and S. L. Chou, Adv. Funct. Mater., 2023, 2302277 CrossRef.
- D. Dewar and A. M. Glushenkov, Energy Environ. Sci., 2021, 14, 1380–1401 RSC.
- K. Zou, W. Deng, P. Cai, X. Deng, B. Wang, C. Liu, J. Li, H. Hou, G. Zou and X. Ji, Adv. Funct. Mater., 2020, 31, 2005581 CrossRef.
- I. Moeez, H. G. Jung, H. D. Lim and K. Y. Chung, ACS Appl. Mater. Interfaces, 2019, 11, 41394–41401 CrossRef CAS.
- H. Wang, Y. Xiao, C. Sun, C. Lai and X. Ai, RSC Adv., 2015, 5, 106519–106522 RSC.
- J. Martinez De Ilarduya, L. Otaegui, J. M. López del Amo, M. Armand and G. Singh, J. Power Sources, 2017, 337, 197–203 CrossRef CAS.
- Y. B. Niu, Y. J. Guo, Y. X. Yin, S. Y. Zhang, T. Wang, P. Wang, S. Xin and Y. G. Guo, Adv. Mater., 2020, 32, e2001419 CrossRef PubMed.
- J. Martínez De Ilarduya, L. Otaegui, M. Galcerán, L. Acebo, D. Shanmukaraj, T. Rojo and M. Armand, Electrochim. Acta, 2019, 321 Search PubMed.
- R. Zhang, Z. Tang, D. Sun, R. Li, W. Yang, S. Zhou, Z. Xie, Y. Tang and H. Wang, Chem. Commun., 2021, 57, 4243–4246 RSC.
- Z. Jian, Y. Sun and X. Ji, Chem. Commun., 2015, 51, 6381–6383 RSC.
- Y. Liu, X. Wu, A. Moeez, Z. Peng, Y. Xia, D. Zhao, J. Liu and W. Li, Adv. Energy Mater., 2022, 13, 2203283 CrossRef.
- S. Mirza, Z. Song, H. Zhang, A. Hussain, H. Zhang and X. Li, J. Mater. Chem. A, 2020, 8, 23368–23375 RSC.
- J. X. Wu, C. Lin, Q. H. Liang, G. D. Zhou, J. P. Liu, G. M. Liang, M. Wang, B. H. Li, L. Hu, F. Ciucci, Q. Liu, G. H. Chen and X. L. Yu, Infomat, 2022, 4, e12288 CrossRef CAS.
- K. Lin, Q. Liu, Y. Zhou, H. Chen, J. Liu, J.-Z. Zhao and X. Hou, Chem. Eng. J., 2023, 463, 142464 CrossRef CAS.
- Y.-S. Su and J.-K. Chang, Batteries, 2022, 8, 99 CrossRef CAS.
- M. Xu, M. Liu, Z. Yang, C. Wu and J. Qian, Acta Phys. – Chim. Sin., 2022, 39, 2210043 Search PubMed.
- M. Liu, J. Zhang, S. Guo, B. Wang, Y. Shen, X. Ai, H. Yang and J. Qian, ACS Appl. Mater. Interfaces, 2020, 12, 17620–17627 CrossRef CAS.
- Y. Cao, T. Zhang, X. Zhong, T. Zhai and H. Li, Chem. Commun., 2019, 55, 14761–14764 RSC.
- Y. Shen, J. Qian, H. Yang, F. Zhong and X. Ai, Small, 2020, 16, e1907602 CrossRef PubMed.
- M. Liu, Z. Yang, Y. Shen, S. Guo, J. Zhang, X. Ai, H. Yang and J. Qian, J. Mater. Chem. A, 2021, 9, 5639–5647 RSC.
- X. Liu, Y. Tan, T. Liu, W. Wang, C. Li, J. Lu and Y. Sun, Adv. Funct. Mater., 2019, 29, 1903795 CrossRef CAS.
- X. Liu, Y. Tan, W. Wang, C. Li, Z. W. Seh, L. Wang and Y. Sun, Nano Lett., 2020, 20, 4558–4565 CrossRef CAS PubMed.
- C. Wu, J. Hu, H. Chen, C. Zhang, M. Xu, L. Zhuang, X. Ai and J. Qian, Energy Storage Mater., 2023, 60, 102803 CrossRef.
- X. Liu, T. Liu, R. Wang, Z. Cai, W. Wang, Y. Yuan, R. Shahbazian-Yassar, X. Li, S. Wang, E. Hu, X.-Q. Yang, Y. Xiao, K. Amine, J. Lu and Y. Sun, ACS Energy Lett., 2020, 6, 320–328 CrossRef.
- S. Park, Z. Wang, Z. Deng, I. Moog, P. Canepa, F. Fauth, D. Carlier, L. Croguennec, C. Masquelier and J.-N. Chotard, Chem. Mater., 2021, 34, 451–462 CrossRef.
- R. Thangavel, D. Han, B. Moorthy, B. K. Ganesan, M. Moorthy, Y. Park, K. W. Nam and Y. S. Lee, Small Methods, 2022, 6, e2100888 CrossRef PubMed.
- C. Wei, F. Luo, C. Zhang, H. Gao, J. Niu, W. Ma, Y. Bai and Z. Zhang, Ionics, 2019, 26, 2343–2351 CrossRef.
- H. Fang, S. Gao, M. Ren, Y. Huang, F. Cheng, J. Chen and F. Li, Angew. Chem., Int. Ed., 2023, 62, e202214717 CrossRef CAS.
- J. Jang, I. Kang, J. Choi, H. Jeong, K. W. Yi, J. Hong and M. Lee, Angew. Chem., Int. Ed., 2020, 59, 14473–14480 CrossRef CAS PubMed.
- G. Wang, B. Huang, D. Liu, D. Zheng, J. Harris, J. Xue and D. Qu, J. Mater. Chem. A, 2018, 6, 13286–13293 RSC.
- B. Paduszek and M. K. Kalinowski, Electrochim. Acta, 1983, 28, 639–642 CrossRef CAS.
- M. Fujita, A. Ishida, T. Majima and S. Takamuku, J. Phys. Chem., 1996, 100, 5382–5387 CrossRef CAS.
- Z. Y. Gu, J. Z. Guo, Z. H. Sun, X. X. Zhao, W. H. Li, X. Yang, H. J. Liang, C. D. Zhao and X. L. Wu, Sci. Bull., 2020, 65, 702–710 CrossRef CAS.
- C. Xu, J. Zhao, Y. A. Wang, W. Hua, Q. fu, X. Liang, X. Rong, Q. Zhang, X. Guo, C. Yang, H. Liu, B. Zhong and Y. S. Hu, Adv. Energy Mater., 2022, 12, 2200966 CrossRef CAS.
- T. Broux, T. Bamine, F. Fauth, L. Simonelli, W. Olszewski, C. Marini, M. Ménétrier, D. Carlier, C. Masquelier and L. Croguennec, Chem. Mater., 2016, 28, 7683–7692 CrossRef CAS.
- G. Yan, S. Mariyappan, G. Rousse, Q. Jacquet, M. Deschamps, R. David, B. Mirvaux, J. W. Freeland and J. M. Tarascon, Nat. Commun., 2019, 10, 585 CrossRef CAS PubMed.
- Z. Jian, C. Yuan, W. Han, X. Lu, L. Gu, X. Xi, Y.-S. Hu, H. Li, W. Chen, D. Chen, Y. Ikuhara and L. Chen, Adv. Funct. Mater., 2014, 24, 4265–4272 CrossRef CAS.
- S. C. Yin, H. Grondey, P. Strobel, H. Huang and L. F. Nazar, J. Am. Chem. Soc., 2003, 125, 326–327 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03498d |
| This journal is © The Royal Society of Chemistry 2023 |