
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A binuclear guanidinate yttrium carbyne complex: unique reactivity toward unsaturated C–N, C–O and C–S bonds†
Wen
Jiang‡
a,
Feng
Kong‡
 a,
Iker del
Rosal
b,
Meng
Li
a,
Kai
Wang
a,
Laurent
Maron
*b and
Lixin
Zhang
*a
a,
Iker del
Rosal
b,
Meng
Li
a,
Kai
Wang
a,
Laurent
Maron
*b and
Lixin
Zhang
*a
aDepartment of Chemistry, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Fudan University, 2005 Songhu Road, Jiangwan Campus, Shanghai, 200438, P. R. China. E-mail: lixinzh@fudan.edu.cn
bLPCNO, Université de Toulouse, 31077 Toulouse, France. E-mail: maron@irsamc.ups-tlse.fr
First published on 3rd August 2023
Abstract
A guanidinato-stabilized binuclear yttrium carbyne complex [(PhCH2)2NC(NC6H3iPr2-2,6)2]2Y2(μ2-Me)(AlMe3)2(μ4-CH) (1) was synthesized via C–H bond activation and its versatile reactivities were investigated. Complex 1 underwent σ-bond metathesis with PhSSPh and nucleophilic addition with PhCN to form the corresponding yttrium thiolate complex 3 and aza–allyl complex 4 respectively. Additionally, the rare yttrium carbide complex 5 was also prepared by treatment of complex 1 with S8. Interestingly, in the reaction with PhNCS, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S double bond was cleaved, followed by C–H bond activation to give the yttrium sulfide complex 7 with a ketenimine dianion ligand. Unexpectedly, the reaction of complex 1 with CO (1 atm) resulted in deoxygenative coupling of CO, to afford mono- or dioxo-yttrium complexes at different temperatures. The mechanism of the possible formation processes of complexes 3 and 9 was elucidated by DFT calculations.
S double bond was cleaved, followed by C–H bond activation to give the yttrium sulfide complex 7 with a ketenimine dianion ligand. Unexpectedly, the reaction of complex 1 with CO (1 atm) resulted in deoxygenative coupling of CO, to afford mono- or dioxo-yttrium complexes at different temperatures. The mechanism of the possible formation processes of complexes 3 and 9 was elucidated by DFT calculations.
Introduction
Since the first typical metal carbyne M![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR (M = Cr, Mo, W) was reported,1 transition-metal alkylidyne complexes have been extensively investigated because of their unique structural features and great potential in alkyne metathesis and stoichiometric reactions.2–4 In stark contrast, the studies on the well-defined rare-earth-metal alkylidyne complexes are relatively limited.5–8 This might be because the empty orbitals of rare-earth-metals are very high-energy d-orbitals and highly contracted f-orbitals, preventing the formation of strong covalent bonds with carbon atom orbitals. To date, four types of rare-earth-metal carbyne complexes have been successfully isolated and fully characterized. Firstly, Anwander et al. reported successfully a pentanuclear carbyne complex La5Al9(CH)6(CH3)30 (A) and tetranuclear carbyne complexes [{(C5Me5)Y(μ2-Me)2AlMe}2(μ2-Me)(μ4-CH)]2 (B), La4Al8(CH)4 (CH2)2(CH3)20(PMe3) (C), and La4Al8(C)(CH)2(CH2)2(CH3)22(toluene) (D) via donor-induced alkylaluminate cleavage of heterobimetallic Ln–(CH3)–Al linkages.6 Subsequently, Mitzel and co-workers isolated the mononuclear Sm derivative [(TCyTAC)SmIII(μ4-CH)(AlMe3)3]7a,b (TCyTAC = 1,3,5-tricyclohexyl-1,3,5-triazacyclo-hexane) (F) and Pr derivative (η3-TMTAC)Pr(η4-{CH(AlMe3)3})7c (TMTAC = 1,3,5-trimethyl-1,3,5-triazacyclohexane) (G) through bulky base cyclic triamine initiated multiple C–H activation.
CR (M = Cr, Mo, W) was reported,1 transition-metal alkylidyne complexes have been extensively investigated because of their unique structural features and great potential in alkyne metathesis and stoichiometric reactions.2–4 In stark contrast, the studies on the well-defined rare-earth-metal alkylidyne complexes are relatively limited.5–8 This might be because the empty orbitals of rare-earth-metals are very high-energy d-orbitals and highly contracted f-orbitals, preventing the formation of strong covalent bonds with carbon atom orbitals. To date, four types of rare-earth-metal carbyne complexes have been successfully isolated and fully characterized. Firstly, Anwander et al. reported successfully a pentanuclear carbyne complex La5Al9(CH)6(CH3)30 (A) and tetranuclear carbyne complexes [{(C5Me5)Y(μ2-Me)2AlMe}2(μ2-Me)(μ4-CH)]2 (B), La4Al8(CH)4 (CH2)2(CH3)20(PMe3) (C), and La4Al8(C)(CH)2(CH2)2(CH3)22(toluene) (D) via donor-induced alkylaluminate cleavage of heterobimetallic Ln–(CH3)–Al linkages.6 Subsequently, Mitzel and co-workers isolated the mononuclear Sm derivative [(TCyTAC)SmIII(μ4-CH)(AlMe3)3]7a,b (TCyTAC = 1,3,5-tricyclohexyl-1,3,5-triazacyclo-hexane) (F) and Pr derivative (η3-TMTAC)Pr(η4-{CH(AlMe3)3})7c (TMTAC = 1,3,5-trimethyl-1,3,5-triazacyclohexane) (G) through bulky base cyclic triamine initiated multiple C–H activation.
More recently, Cheng et al. discussed a rigid trinuclear scandium carbyne complex [(C5Me5)Sc(μ2-Br)]3(μ3-CH) (E) via methyl/halogenido exchange protocol of [(C5Me5)Sc(Me2)]2 with tBuBr.8 However, a significant knowledge gap in the area involving binuclear rare-earth-metal alkylidyne complexes was incontrovertible. Additionally, compared to transition-metal alkylidyne complexes, the reactivity exploration of rare-earth-metal alkylidyne complexes remains scarce.8
In the last few decades, investigations on the synthesis and reactivity of rare-earth-metal alkylidene complexes have been documented widely.9–15 Our previous studies on amidinate rare-earth-metal carbene derivatives have also shown the unique reactivity of μ3-CH2 dianions.16,17 The results indicate that there are significant ligand effects on the structure and reactivity of these complexes. However, attempts to prepare guanidinate rare-earth-metal methylidene complexes by a similar synthetic process for the synthesis of amidinate methylidene complexes were unsuccessful, and an unexpected binuclear carbyne complex was isolated. Interestingly, this complex shows high and intriguing reactivity under mild conditions, which is distinct from those already reported for other transition-metal carbyne complexes.
Results and discussion
Synthesis and structural characterization
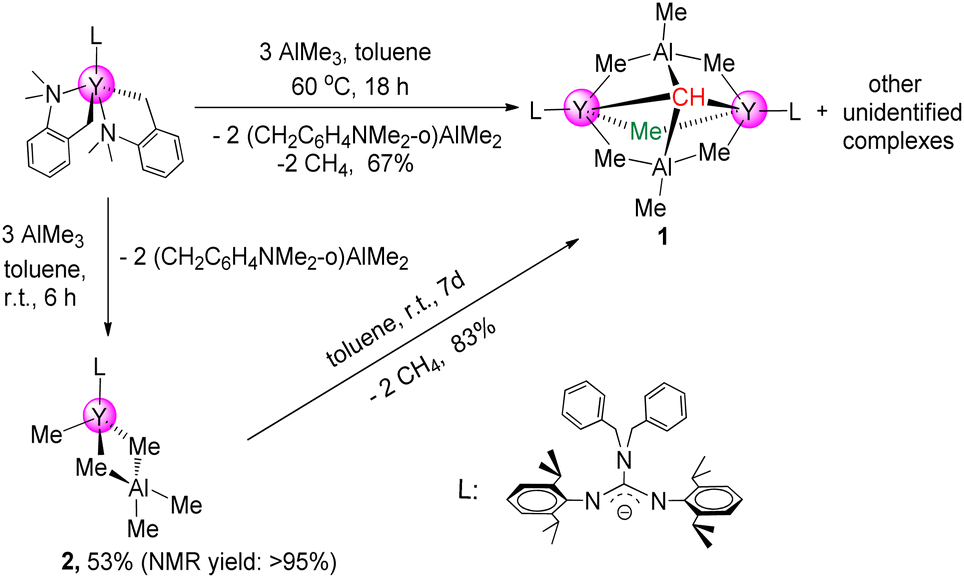
The first binuclear yttrium carbyne complex [(PhCH2)2NC(NC6H3iPr2-2,6)2]2Y2(μ2-Me)(AlMe3)2(μ4-CH) (1) was obtained in 67% yield via a metal-assisted multiple C–H bond activation process when three equivalents of AlMe3 were added into a stirred toluene solution of the guanidinate yttrium bis(o-aminobenzyl) complex18 at 60 °C for 18 h (Scheme 1). In order to get more insights into the formation process of the yttrium carbyne complex, the same reaction was carried out at room temperature for 6 h which gave a heterobimetallic Y/Al complex (PhCH2)2NC(NC6H3iPr2-2,6)2Y(Me)(AlMe4) (2) in 53% yield. It is interesting that complex 2 could turn into complex 1 in high yield in toluene at room temperature for 7 days with the release of CH4 (ESI Fig. S6†). This is sufficient to show that complex 2 is an intermediate in the formation of complex 1. | ||
| Scheme 1 Rare-earth-metal carbyne complexes. | ||
The single crystal X-ray diffraction analysis of complex 1 shows that two distorted octahedral yttrium centers are linked by μ4-CH and μ2-Me units to form a distorted square while two AlMe3 groups work as bulky ligands to stabilize this binuclear carbyne complex (Fig. 1). The average Y–C1(μ2-Me) bond length (2.535(5) Å) is similar to that in [(η-C5H5)2YMe]2 (2.537(9) Å).19a It is noticeable that the Y–C2(μ4-CH) bond lengths of 2.398(5) Å and 2.418(5) Å are shorter than those in [{(C5Me5)Y(μ2-Me)2AlMe}2(μ2-Me)(μ4-CH)]2 (2.444(3)–2.464(3) Å). A similar trend is observed for the Al–C2(μ4-CH) bond lengths (1.970(5) Å and 1.974(5) Å) versus 1.984(3)–1.993(3) Å for Al–C(μ4-CH),6a probably due to the steric hindrance. In the 1H NMR spectrum, the resonance of the [CH]3− group is observed as a singlet at δ = 2.36 (benzene-d6) ppm, which is different to the proton signal at δ = 12.16 ppm in [(C5Me5)Sc(μ2-Br)]3(μ3-CH).8 The metal-bonded Y2CHAl2 methine carbon atom is shown at δ = 90.2 ppm based on a comprehensive analysis of the 1H, 13C{1H} and two-dimensional HMQC NMR spectra (Fig. S1–S3†). The bridged μ2-Me(Y–Me–Y) and AlMe3 are shown as two singlets at δ = −0.26 and 0.34 ppm, respectively.
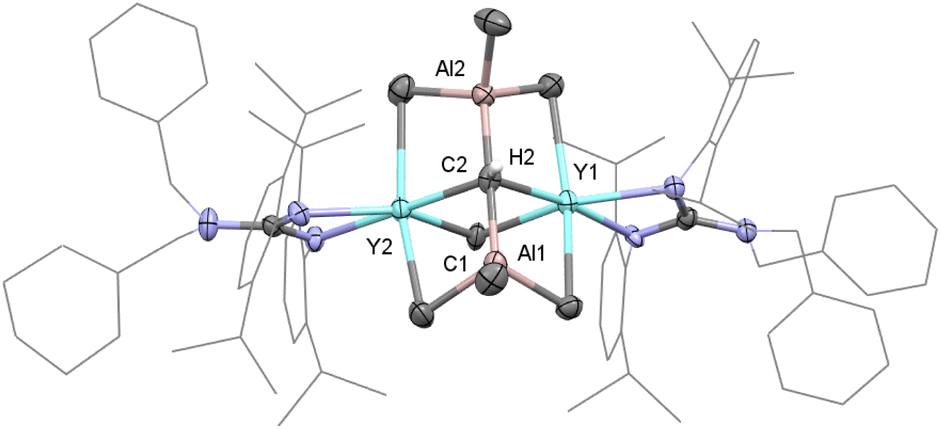 | ||
| Fig. 1 Molecular structure of complex 1 with thermal ellipsoids at 30% probability except for the 2,6-(iPr)2C6H3 groups and benzyl groups in the guanidinate ligand. All of the hydrogen atoms (except for H2) are omitted for clarity. Selected bond distances (Å) and angles (°): Y(1)–C(1) 2.528(6), Y(1)–C(2) 2.418(5), Y(2)–C(1) 2.543(5), Y(2)–C(2) 2.398(5), C(2)–Al(1) 1.974(5), and C(2)–Al(2) 1.970(5); C(1)–Y(1)–C(2) 85.47(15), C(1)–Y(2)–C(2) 85.55(15), Y(1)–C(1)–Y(2) 91.29(17), and Y(1)–C(2)–Y(2) 97.65(16). | ||
Complex 1 has good thermodynamic stability and multiple potential reactive sites (e.g. μ2-methyl and [CH]3− groups) which makes this complex suitable for reactivity exploration. Complex 1 underwent a σ-bond metathesis reaction towards one equivalent of phenyl disulfide at room temperature, electron reduction of PhSSPh to form a (SPh)− anion moiety and release PhSMe, affording the yttrium thiolate-bridged carbyne complex [(PhCH2)2NC(NC6H3iPr2-2,6)2]2Y2(μ2-SPh)(AlMe3)2(μ4-CH) (3) in 86% isolated yield (Scheme 2). The molecular structure of 3 is depicted in ESI Fig. S26.†. Complex 3 is a dimer containing a μ2-SPh ligand, and the two Y–S bond lengths are 2.799(12) Å and 2.821(13) Å respectively, which are slightly shorter than those in [(C5Me5)2Y(μ-SPh)]2,20 ranging from 2.893(6) to 2.903(6) Å, probably due to the steric hindrance. In the 1H NMR spectrum, the distinct singlet at δ = 2.00 (benzene-d6) ppm is assigned to the [CH]3− group, which is shifted to a high field compared to 1. The methyl protons in the AlMe3 unit split into two groups at δ = 0.89 and −0.50 ppm at room temperature, while they become one broad peak at δ = 0.51 ppm at 60 °C (ESI Fig. S8†).19b Interestingly, the 1H NMR monitoring of this reaction reveals that the chemoselectivity of the reaction of 1 with PhSSPh is independent of the amount of PhSSPh used, and no further reaction was observed even with an excess of PhSSPh.
 | ||
| Scheme 2 Synthesis of binuclear yttrium carbyne complex 1. | ||
To develop a synthetic methodology of metal N-heterocycle complexes from nitriles,21 benzonitrile (2 equiv.) was added into the toluene solution of complex 1 at room temperature, obtaining a diinsertion product [(PhCH2)2NC(NC6H3iPr2-2,6)2]2Y2[μ3-η1:η2:η5-HC(CPhN)2](AlMe3)(μ2-Me) (4) in excellent yield as red blocks (Scheme 2). The X-ray single crystal diffraction study shows the structure of complex 4 in which the tri-anionic amino-alkenyl [CH(CPhN)2]3− unit acts as a pentadentate bridge ligand to connect two yttrium atoms in two bonding models (η5 and η2). In the NMR spectra, the [CH]3− group appears as a singlet at δ = 7.13 ppm (13C: 96.5 ppm), which is shifted to a low field compared to 1 and 3. The average C–N bond length of 1.319(5) Å is longer than the normal CN double bond distance (1.26 Å),22 while the C–C distance (av. 1.439(6) Å) is between the values observed for a C–C single bond and a CC double bond, indicating that the three negative charges are delocalized partially in the [CH(CPhN)2]3− unit. It is interesting that the nucleophilic addition has only taken place on the single [CH]3− group, and the bridged μ2-Me(Y–Me–Y) group is inert even if an excess amount of benzonitrile was used under the same conditions, or even heated up to 60 °C for 12 h. The results are clearly different from those of rare-earth-metal methylidene complexes, in which only monoinsertion products [PhC(NC6H3iPr2-2,6)2]3Ln3(μ2-Me)3(μ3-Me)[μ-η1:η1:η3-CH2C(Ph)N] (Ln = Y, Lu) were yielded.17c Additionally, the present reaction is also different from that of the scandium carbyne complex with PhCN, which afforded the monoaddition product [(C5Me5)Sc(μ2-OMe)]3[μ-η2:η2:η3-CHC(Ph)N]8 possibly due to the steric hindrance and rigid structure.
Amazingly, the reaction of complex 1 with sulfur resulted in carbyne deprotonation to afford the rare carbide complex [(PhCH2)2NC(NC6H3iPr2-2,6)2]2Y2(μ2-SMe)(AlMe3)2(μ5-C)(AlMe2) (5), along with the formation of the tetramethylaluminate complex [(PhCH2)2NC(NC6H3iPr2-2,6)2]Y(AlMe4)2 (6) (Scheme 3). The single crystal structure of complex 5 reveals that the stabilization of the carbide unit (C4−) is achieved by the alkylaluminate ligands. The carbide carbon atom features a unique distorted trigonal bipyramidal geometry, in which three Al atoms lie in the equatorial plane and two Y atoms occupy the axial positions (ESI Fig. S28†). The bond length of Y–C7(μ5-C) (av. 2.434(3) Å) is shorter than the Y–C(μ6-C) in {[(TMTAC)Y][Y2(μ2-CH3)]}[{(μ6-C)[Al(μ2-CH3)2(CH3)]3}{(μ3-CH2)(μ2-CH3)Al(CH3)2}2] (2.696(6) Å),23 possibly due to higher coordination numbers of the carbide atoms in latter. The results suggest that the elemental sulfur initiates a bimolecular reaction where the [CH]3− group was deprotonated by the methyl in the AlMe3 ligand to form a more stable carbide complex with the release of CH4.
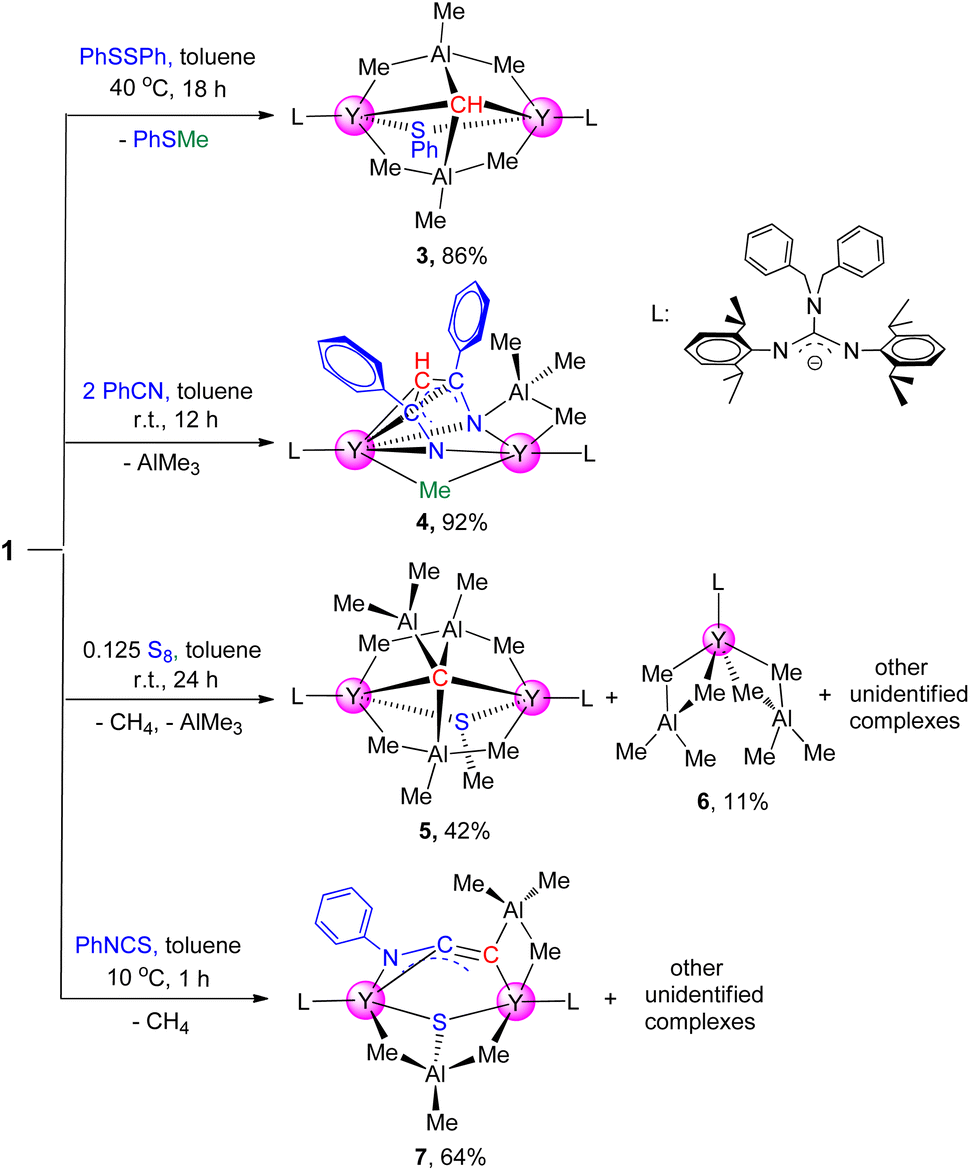 | ||
| Scheme 3 Reactions of complex 1 with benzonitrile, phenyl disulfide, sulfur and phenyl isothiocyanate. | ||
Additionally, in order to expand the application of complex 1 as a nucleophile in the synthesis of rare-earth derivatives, the reaction of complex 1 with PhNCS was carried out. This reaction yields a yttrium sulfide complex [(PhCH2)2NC(NC6H3iPr2-2,6)2]2Y2(μ3-η1:η1:η2-CCNPh)(AlMe3)2(μ3-S) (7) with a bridged ketenimine dianion unit (Scheme 3). The single crystal structure of complex 7 reveals that the N1–C1–C2 moiety displays a delocalized electronic framework, in which the C1–N1 (1.329(8) Å) bond length is intermediate between those of typical single and double bonds while the bond length of C1–C2 (1.221(8) Å) is slightly longer than the normal CC triple bond distance. And the average bond length of Y–S (2.660(17) Å) is longer than those in [(C5Me5)2Y]2(μ-S) (2.54(5) Å)24 probably due to the coordination of the AlMe3 unit. The mechanism of this reaction is proposed to begin with PhNCS molecule insertion into the Y–C(μ4-CH) bond, followed by isomerization to form a CC double bond, and then C–S single bond cleavage takes place to give the final reductive coupling product 7via C–H bond activation (ESI Fig. S33†). This result reveals the unique reactivity of the [CH]3− group towards PhNCS, which is in sharp contrast with that of the cubane-type lutetium methylidene complex with PhNCS, which gave the corresponding ethylene amido thiolate/methylidene product through addition of a Lu–methylidene bond with a less sterically demanding CS bond followed by isomerization.11b The present reaction is also different from previous rare-earth-metal alkylidene complexes in cases where the fragments formed after the cleavage of the CS double bond in PhNCS participate in the construction of the free coupled organic molecules.25
Carbon monoxide (CO), important C1 feedstock, insertion into metal–carbon bonds and its subsequent transformations were thought to play important roles in organometallic chemistry and industrial catalytic processes (Fischer–Tropsch synthesis).26 However, the mechanistic details have not been fully understood at the molecular level; in consequence, the reaction of complex 1 with CO (1 atm) was examined at room temperature. Unfortunately, a complicated mixture is detected by using the 1H NMR spectrum (ESI Fig. S22†). It is interesting that when the mixture was further heated to 50 °C for 18 h, an unexpected dioxo yttrium complex {[(PhCH2)2NC(NC6H3iPr2-2,6)2]Y(μ3-O)(AlMe3)}2 (8) was isolated in high yield as colorless crystals (Scheme 4). In the NMR spectra of 8, only one singlet at δ = −0.16 ppm (13C = −2.6 ppm) was ascribed to AlMe3 (ESI Fig. S18 and S19†). The X-ray single crystal diffraction study shows that two yttrium centers are linked by two oxygen atoms to form a distorted square in the dimeric system (ESI Fig. S31†), and the Y–O bond lengths (ranging from 2.122(3) to 2.207(2) Å) are comparable to those in (TpMe2)3Y3(μ3-O)(μ2-O)(μ2-OCH2CHCH2)(μ2-H) (from 2.127(2) to 2.268(19) Å).27 To date, the reaction of CO with rare-earth-metal alkylidene complexes generated only rare-earth-metal ketene complexes through nucleophilic addition of an alkylidene group to CO.11b,15f
 | ||
| Scheme 4 Reactions of complex 1 with CO. | ||
Although the CO deoxygenative coupling reaction induced by rare-earth-metal phosphinidene and polyhydrido complexes has been well documented,27,28 the well-defined rare-earth-metal alkyl complex induced deoxygenative coupling reaction of CO was unprecedented. In order to offer more insight into the formation process of complex 8, the same reaction was carried out in toluene at 10 °C for 6 h. To our delight, the yttrium mono-oxo complex [(PhCH2)2NC(NC6H3iPr2-2,6)2]2Y2(μ4-O)(AlMe3)2(μ2-Me)(μ2-CCH) (9) was obtained in 84% isolated yield as colorless crystals (Scheme 4). According to single crystal X-ray diffraction, the C2–C3 bond length (1.065(12) Å) reveals a substantial CC triple bond character (Fig. 2). And the NMR spectra of 9 show a singlet at δ = 1.80 ppm (13C{1H} = 112.4 ppm), which is regarded as bridged ethynyl (Fig. S17 and S18†). It's worth noting that complex 9 could turn into complex 8 almost quantitatively in toluene at 50 °C for 10 h, indicating that complex 9 is an intermediate in the formation of complex 8. These reactions of CO may offer a convenient route to rare-earth-metal oxo complexes, a class of complexes that are of considerable current interest but still remain limited.27–29
 | ||
| Fig. 2 Molecular structure of complex 9 with thermal ellipsoids at 30% probability except for the 2,6-(iPr)2C6H3 groups and benzyl groups in the guanidinate ligand. All of the hydrogen atoms (except for H3) are omitted for clarity. Selected bond distances (Å) and angles (°): Y(1)–C(1) 2.493(6), Y(1)–C(2) 2.537(6), Y(1)–O(1) 2.331(4), and C(2)–C(3) 1.065(12); Y(1)–C(1)–Y(2) 81.99(17), Y(1)–C(2)–Y(2) 84.13(18), Y(1)–O(1)–Y(2) 91.44(12), and Y(1)–C(2)–C(3) 118.1(7). | ||
DFT calculations
The formation of complex 3 from complex 1 was investigated computationally at the DFT level (B3PW91 functional including dispersion). As can be seen from Fig. 3, the dispersion corrections play an important role to account for the reactivity due to the size of the substrate PhSSPh. Indeed, the coordination of the substrate is crucial in this case and implies the rearrangement of the μ2-Me(Y–Me–Y) to the terminal to allow the coordination of the substrate, which is only favored because of the dispersion forces (stabilization of 27.0 kcal mol−1). This is allowed to reach TS5 where the methyl groups attack the sulfur inducing S–S bond breaking with an accessible barrier of 29.0 kcal mol−1. This barrier is in line with the experimental observation (18 h at 40 °C). Following the intrinsic reaction coordinate, it yields complex 3 whose formation is thermodynamically favorable (−59.4 kcal mol−1). The reaction cannot occur on the carbyne side as moving the carbyne to the top position is too unstable. | ||
| Fig. 3 Computed (DFT) enthalpy pathway of complex 3 at room temperature with (black) and without (blue) dispersion correction. The Gibbs free energies are given between brackets. | ||
In addition, the formation of complex 9 from complex 1 was also investigated computationally. The computed enthalpy profile (Fig. 4) begins by the coordination of CO to one of the yttrium centers. This coordination is slightly exothermic by 17.2 kcal mol−1. From this adduct (Int1), the migratory insertion of CO onto the Y–CH bond is observed viaTS1. The associated barrier is very low (6.4 kcal mol−1) in line with a facile insertion. Following the intrinsic reaction coordinate, it yields the insertion product (Int2) whose formation is exothermic by 10.0 kcal mol−1 with respect to complex 1. This intermediate will undergo an O–Al bond formation which is concomitant with C–Al bond breaking viaTS2. The associated barrier is 6.5 kcal mol−1 from Int2 (13.7 kcal mol−1 from complex Int1) and corresponds to the rate-determining step of the entire transformation. This barrier height fits nicely the experimental conditions. TS2 connects with Int3, whose formation is favorable (−29.9 kcal mol−1). Int3 undergoes easy C–O bond breaking (TS3) with an enthalpy barrier of 0.2 kcal mol−1. This reaction yields a vinylidyne complex (Int4), with an extra stabilization of 29.6 kcal mol−1 with respect to Int3. A rotamer of Int4 (Int5) was located on the Potential Energy Surface (PES) with an enthalpy gain of 7.0 kcal mol−1. In the final step, the system undergoes C–Al bond formation with concomitant O–Al bond formation viaTS4. The associated barrier is 8.6 kcal mol−1 and allows the formation of complex 9. The latter is found to be the most stable species of the whole reaction profile (−83.1 kcal mol−1). Th addition of CO on the μ2-Me(Y–Me–Y) side was also computed and is higher in energy (ESI Fig. S34†).
 | ||
| Fig. 4 Computed (DFT) enthalpy pathway of complex 9 at room temperature with (black) and without (blue) dispersion correction. The Gibbs free energies are given between brackets. | ||
Conclusions
In summary, the first example of a well-defined binuclear carbyne complex 1 has been demonstrated. Complex 1 revealed diverse reactivity towards various substrates, such as nucleophilic addition with benzonitrile and phenyl isothiocyanate to form aza–allyl complex 4 and sulfide complex 7, σ-bond metathesis towards phenyl disulfide to give thiolate complex 3, and insertion of sulfide into the Y–C(μ2-Me) bond along with carbyne deprotonation to the afford carbide complex 5. Unexpectedly, the rare-earth-metal carbyne complex induced deoxygenative coupling reaction of CO was also presented for the first time. The results have afforded some evidence to suggest that unsaturated substrates are attacked preferentially by the methine rather than the methyl. The observed rich reactivity of this binuclear carbyne complex could provide new insight into the nature of rare-earth-metal carbyne complexes. We are currently probing the reactivity of complex 1 with other molecules.Data availability
The data that support the findings of this study are available in the ESI† of this article.Author contributions
Li, Meng designed the carbyne complex. Jiang, Wen and Kong, Feng performed the synthetic experiment and X-ray diffraction measurements. Rosal, Iker del and Maron, Laurent performed the DFT calculations. Zhang, Lixin and Wang, Kai reviewed/revised the manuscript. All authors contributed to the analysis of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 21871052 and 21672038). We thank Prof. Zhou Xigeng and Prof. Wang Huadong for their suggestions. The authors acknowledge the HPCs CALcul en Midi-Pyrénées (CALMIP-EOS grant 1415).Notes and references
- E. O. Fischer, G. Kreis, C. G. Kreiter, J. Moller, G. Huttner and H. Lorenz, Angew. Chem., Int. Ed. Engl., 1973, 12, 564–565 CrossRef.
- For examples of transition-metal alkylidyne complexes, see: (a) J. L. Polse, R. A. Andersen and R. G. Bergman, J. Am. Chem. Soc., 1998, 120, 13405–13414 CrossRef CAS; (b) R. R. Schrock, Chem. Rev., 2002, 102, 145–179 CrossRef CAS PubMed; (c) R. R. Schrock and A. H. Hoveyda, Angew. Chem., Int. Ed., 2003, 42, 4592–4633 CrossRef CAS PubMed; (d) B. C. Bailey, A. R. Fout, H. Fan, J. Tomaszewski, J. C. Huffman and D. J. Mindiola, Angew. Chem., Int. Ed., 2007, 46, 8246–8249 CrossRef CAS PubMed.
- (a) M. Kamitani, B. Pinter, K. Searles, M. G. Crestani, A. Hickey, B. C. Manor, P. J. Carroll and D. J. Mindiola, J. Am. Chem. Soc., 2015, 137, 11872–11875 CrossRef CAS PubMed; (b) J. Hillenbrand, M. Leutzsch and A. Fgrstner, Angew. Chem., Int. Ed., 2019, 58, 15690–15696 CrossRef CAS PubMed; (c) A. Haack, J. Hillenbrand, M. Leutzsch, M. van Gastel, F. Neese and A. Fgrstner, J. Am. Chem. Soc., 2021, 143, 5643–5648 CrossRef CAS PubMed; (d) S. Trzmiel, J. Langmann, D. Werner, C. Maichle-Mçssmer, W. Scherer and R. Anwander, Angew. Chem., Int. Ed., 2021, 60, 20049–20054 CrossRef CAS PubMed.
- (a) P. F. Engel and M. Pfeffer, Chem. Rev., 1995, 95, 2281–2309 CrossRef CAS; (b) M. J. Benedikter, F. Ziegler, J. Groos, P. M. Hauser, R. Schowner and M. R. Buchmeiser, Coord. Chem. Rev., 2020, 415, 213315 CrossRef CAS; (c) M. Cui, H. H. Y. Sung, I. D. Williams and G. Jia, J. Am. Chem. Soc., 2022, 144, 6349–6360 CrossRef CAS PubMed; (d) M. Cui and G. Jia, J. Am. Chem. Soc., 2022, 144, 12546–12566 CrossRef CAS PubMed.
- P. Zatsepin, E. Lee, J. Gu, M. R. Gau, P. J. Carroll, M. Baik and D. J. Mindiola, J. Am. Chem. Soc., 2020, 142, 10143–10152 CrossRef CAS PubMed.
- (a) H. M. Dietrich, H. Grove, K. W. Törnroos and R. Anwander, J. Am. Chem. Soc., 2006, 128, 1458–1459 CrossRef CAS PubMed; (b) L. C. H. Gerber, E. Le Roux, K. W. Törnroos and R. Anwander, Chem.–Eur. J., 2008, 14, 9555–9564 CrossRef CAS PubMed.
- (a) D. Bojer, A. Venugopal, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2010, 49, 2611–2614 CrossRef CAS PubMed; (b) D. Bojer, B. Neumann, H.-G. Stammler and N. W. Mitzel, Chem.–Eur. J., 2011, 17, 6239–6247 CrossRef CAS PubMed; (c) D. Bojer, B. Neumann, H.-G. Stammler and N. W. Mitzel, Eur. J. Inorg. Chem., 2011, 3791–3796 CrossRef CAS.
- P. Deng, X. Shi, X. Gong and J. Cheng, Chem. Commun., 2021, 57, 6436–6439 RSC.
- (a) K. Aparna, M. Ferguson and R. G. Cavell, J. Am. Chem. Soc., 2000, 122, 726–727 CrossRef CAS; (b) D. P. Mills, L. Soutar, W. Lewis, A. J. Blake and S. T. Liddle, J. Am. Chem. Soc., 2010, 132, 14379–14381 CrossRef CAS PubMed; (c) M. Fustier, X. F. Le Goff, P. Le Floch and N. Mézailles, J. Am. Chem. Soc., 2010, 132, 13108–13110 CrossRef CAS PubMed; (d) S. T. Liddle, D. P. Mills and A. J. Wooles, Chem. Soc. Rev., 2011, 40, 2164–2176 RSC; (e) M. Fustier, X. F. Le Goff, M. Lutz, J. C. Slootweg and N. Mézailles, Organometallics, 2015, 34, 63–72 CrossRef CAS.
- (a) H. M. Dietrich, K. W. Törnroos and R. Anwander, J. Am. Chem. Soc., 2006, 128, 9298–9299 CrossRef CAS PubMed; (b) M. Zimmermann, J. Takats, G. Kiel, K. W. Törnroos and R. Anwander, Chem. Commun., 2008, 612–614 RSC; (c) M. Zimmermann, D. Rauschmaier, K. Eichele, K. W. Törnroos and R. Anwander, Chem. Commun., 2010, 46, 5346–5348 RSC; (d) C. O. Hollfelder, L. N. Jende, H. M. Dietrich, K. Eichele, C. Maichle-Mössmer and R. Anwander, Chem.–Eur. J., 2019, 25, 7298–7302 CrossRef CAS PubMed; (e) D. Barisic, D. Diether, C. Maichle-Mössmer and R. Anwander, J. Am. Chem. Soc., 2019, 141, 13931–13940 CrossRef CAS PubMed; (f) C. O. Hollfelder, M. Zimmermann, C. Spiridopoulos, D. Werner, K. W. Törnroos, C. Maichle-Mössmer and R. Anwander, Molecules, 2019, 24, 3703–3730 CrossRef CAS PubMed; (g) D. A. Buschmann, L. Schumacher and R. Anwander, Chem. Commun., 2022, 58, 9132–9135 RSC.
- (a) W. Zhang, Z. Wang, M. Nishiura, Z. Xi and Z. Hou, J. Am. Chem. Soc., 2011, 133, 5712–5715 CrossRef CAS PubMed; (b) T. Li, M. Nishiura, J. Cheng, Y. Li and Z. Hou, Chem.–Eur. J., 2012, 18, 15079–15085 CrossRef CAS PubMed.
- (a) J. Scott, H. J. Fan, B. F. Wicker, A. R. Fout, M. H. Baik and D. J. Mindiola, J. Am. Chem. Soc., 2008, 130, 14438–14439 CrossRef CAS PubMed; (b) R. Litlabø, M. Zimmermann, K. Saliu, J. Takats, K. W. Törnroos and R. Anwander, Angew. Chem., Int. Ed., 2008, 47, 9560–9564 CrossRef PubMed; (c) P. Zatsepin, E. Lee, J. Gu, M. R. Gau, P. J. Carroll, M. Baik and D. J. Mindiola, J. Am. Chem. Soc., 2020, 142, 10143–10152 CrossRef CAS PubMed.
- (a) S. Li, M. Wang, B. Liu, L. Li, J. Cheng, C. Wu, D. Liu, J. Liu and D. Cui, Chem.–Eur. J., 2014, 20, 15493–15498 CrossRef CAS PubMed; (b) T. Li, G. Zhang, J. Guo, S. Wang, X. Leng and Y. Chen, Organometallics, 2016, 35, 1565–1572 CrossRef CAS; (c) F. Yan, S. Li, L. Li, W. Zhang, D. Cui, M. Wang and Y. Dou, Eur. J. Inorg. Chem., 2019, 2277–2283 CrossRef CAS.
- (a) W. Ma, C. Yu, Y. Chi, T. Chen, L. Wang, J. Yin, B. Wei, L. Xu, W. Zhang and Z. Xi, Chem. Sci., 2017, 8, 6852–6856 RSC; (b) Y. Zheng, C. Cao, W. Ma, T. Chen, B. Wu, C. Yu, Z. Huang, J. Yin, H. Hu, J. Li, W. Zhang and Z. Xi, J. Am. Chem. Soc., 2020, 142, 10705–10714 CrossRef CAS PubMed; (c) Z. Lv, Z. Chai, M. Zhu, J. Wei and W. Zhang, J. Am. Chem. Soc., 2021, 143, 9151–9161 CrossRef CAS PubMed; (d) Z. Lv, W. Liu, M. Zhu, Z. Chai, J. Wei and W. Zhang, Chem.–Eur. J., 2021, 27, 16498–16504 CrossRef CAS PubMed.
- (a) D. J. Mindiola and J. Scott, Nat. Chem., 2011, 3, 15–17 CrossRef CAS PubMed; (b) W. Mao, L. Xiang, C. A. Lamsfus, L. Maron, X. Leng and Y. Chen, J. Am. Chem. Soc., 2017, 139, 1081–1084 CrossRef CAS PubMed; (c) W. Mao, L. Xiang, L. Maron, X. Leng and Y. Chen, J. Am. Chem. Soc., 2017, 139, 17759–17762 CrossRef CAS PubMed; (d) C. Wang, W. Mao, L. Xiang, Y. Yang, J. Fang, L. Maron, X. Leng and Y. Chen, Chem.–Eur. J., 2018, 24, 13903–13917 CrossRef CAS PubMed; (e) W. Q. Mao, L. Xiang, C. A. Lamsfus, L. Maron, X. Leng and Y. Chen, Chin. J. Chem., 2018, 36, 904–908 CrossRef CAS; (f) W. Mao, Y. Wang, L. Xiang, Q. Peng, X. Leng and Y. Chen, Chem.–Eur. J., 2019, 25, 10304–10308 CrossRef CAS PubMed.
- J. Hong, L. Zhang, X. Yu, M. Li, Z. Zhang, P. Zheng, M. Nishiura, Z. Hou and X. Zhou, Chem.–Eur. J., 2011, 17, 2130–2137 CrossRef CAS PubMed.
- (a) J. Hong, L. Zhang, K. Wang, Y. Zhang, L. Weng and X. Zhou, Chem.–Eur. J., 2013, 19, 7865–7873 CrossRef CAS PubMed; (b) K. Wang, G. Luo, J. Hong, X. Zhou, H. Weng, Y. Luo and L. Zhang, Angew. Chem., Int. Ed., 2014, 53, 1053–1056 CrossRef CAS PubMed; (c) J. Hong, Z. Li, Z. Chen, L. Weng, X. Zhou and L. Zhang, Dalton Trans., 2016, 45, 6641–6649 RSC; (d) J. Hong, H. Tian, L. Zhang, X. Zhou, I. del Rosal, L. Weng and L. Maron, Angew. Chem., Int. Ed., 2018, 57, 1062–1067 CrossRef CAS PubMed; (e) H. Tian, J. Hong, K. Wang, I. Rosal, L. Maron, X. Zhou and L. Zhang, J. Am. Chem. Soc., 2018, 140, 102–105 CrossRef CAS PubMed.
- F. Kong, M. Li, X. Zhou and L. Zhang, RSC Adv., 2017, 7, 29752–29761 RSC.
- (a) J. Holton, M. F. Lappert, D. G. H. Ballard, R. Pearce, J. L. Atwood and W. E. Hunter, J. Chem. Soc., Dalton Trans., 1979, 54–61 RSC; (b) R. Litlabø, M. Zimmermann, K. Saliu, J. Takats, K. W. Törnroos and R. Anwander, Angew. Chem., Int. Ed., 2008, 47, 9560–9564 CrossRef PubMed.
- W. J. Evans, B. M. Schmiege, S. E. Lorenz, K. A. Miller, T. M. Champagne, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, J. Am. Chem. Soc., 2008, 130, 8555–8563 CrossRef CAS PubMed.
- (a) R. A. Michelin, M. Mozzon and R. Bertani, Coord. Chem. Rev., 1996, 147, 299–338 CrossRef CAS; (b) V. Y. Kukushkin and A. J. L. Pombeiro, Chem. Rev., 2002, 102, 1771–1802 CrossRef CAS PubMed; (c) H. Wu and N. K. Devaraj, Acc. Chem. Res., 2018, 51, 1249–1259 CrossRef CAS PubMed.
- G. Häfelinger, MO-π-Bindungsordnung-Bindungslängen-Beziehungen-für Hetero-π-Systeme, I. π-Systememit CN-Bindungen, Chem. Ber., 1970, 103, 2902–2921 CrossRef.
- A. Venugopal, I. Kamps, D. Bojer, R. J. F. Berger, A. Mix, A. Willner, B. Neumann, H.-G. Stammler and N. W. Mitzel, Dalton Trans., 2009, 5755–5765 RSC.
- M. E. Fieser, C. W. Johnson, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, Organometallics, 2015, 34, 4387–4393 CrossRef CAS.
- (a) D. P. Mills, L. Soutar, O. J. Cooper, W. Lewis, A. J. Blake and S. T. Liddle, Organometallics, 2013, 32, 1251–1264 CrossRef CAS; (b) D. P. Mills, W. Lewis, A. J. Blake and S. T. Liddle, Organometallics, 2013, 32, 1239–1250 CrossRef CAS.
- (a) C. K. Rofer-Depoorter, Chem. Rev., 1981, 81, 447–474 CrossRef CAS; (b) J. P. Collmann, L. S. Hegedus, J. R. Norton and R. G. Finke, Principles and Applications of Organotransition Metal Chemistry, University Science Book, Mill Valley, CA, 1987 Search PubMed; (c) P. L. Arnold and Z. R. Turner, Nat. Rev. Chem., 2017, 1, 0002 CrossRef CAS.
- J. Cheng, M. J. Ferguson and J. Takats, J. Am. Chem. Soc., 2010, 132, 2–3 CrossRef CAS PubMed.
- (a) T. Shima and Z. Hou, J. Am. Chem. Soc., 2006, 128, 8124–8125 CrossRef CAS PubMed; (b) Y. Takenaka, T. Shima, J. Baldamus and Z. Hou, Angew. Chem., Int. Ed., 2009, 48, 7888–7891 CrossRef CAS PubMed; (c) Y. Lv, C. E. Kefalidis, J. Zhou, L. Maron, X. Leng and Y. Chen, J. Am. Chem. Soc., 2013, 135, 14784–14796 CrossRef CAS PubMed.
- (a) W. J. Evans, J. W. Grate, I. Bloom, W. E. Hunter and J. L. Atwood, J. Am. Chem. Soc., 1985, 107, 405–409 CrossRef CAS; (b) O. Tardif, D. Hashizume and Z. Hou, J. Am. Chem. Soc., 2004, 126, 8080–8081 CrossRef CAS PubMed; (c) J. Scott, H. Fan, B. F. Wicker, A. R. Fout, M. H. Baik and D. J. Mindiola, J. Am. Chem. Soc., 2008, 130, 14438–14439 CrossRef CAS PubMed; (d) M. Fustier, X. F. Le Goff, P. Le Floch and N. Mézailles, J. Am. Chem. Soc., 2010, 132, 13108–13110 CrossRef CAS PubMed; (e) P. L. Arnold, E. Hollis, F. J. White, N. Magnani, R. Caciuffo and J. B. Love, Angew. Chem., Int. Ed., 2011, 50, 887–890 CrossRef CAS PubMed; (f) W. Zhang, Z. Wang, M. Nishiura, Z. Xi and Z. Hou, J. Am. Chem. Soc., 2011, 133, 5712–5715 CrossRef CAS PubMed; (g) P. L. Damon, G. Wu, N. Kaltsoyannis and T. W. Hayton, J. Am. Chem. Soc., 2016, 138, 12743–12746 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental procedures, spectra, and analytical data. CCDC 2189939, 2189941–2189946 and 2262169. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03483f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |