
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent-dependent self-assembly of N-annulated perylene diimides. From dimers to supramolecular polymers†
Cristina
Naranjo‡
a,
Azahara
Doncel-Giménez‡
b,
Rafael
Gómez
a,
Juan
Aragó
 *b,
Enrique
Ortí
*b and
Luis
Sánchez
*a
*b,
Enrique
Ortí
*b and
Luis
Sánchez
*a
aDepartamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, 28040 Madrid, Spain. E-mail: lusamar@quim.ucm.es
bInstituto de Ciencia Molecular (ICMol), Universidad de Valencia, C/Catedrático José Beltrán, 2, 46980 Paterna, Spain. E-mail: juan.arago@uv.es; enrique.orti@uv.es
First published on 29th August 2023
Abstract
The synthesis and self-assembling features of the N-annulated perylene diimide (NPBI) 1 in different solvents are reported. Compound 1 possesses two chiral linkers, derived from (S)-(+)-alaninol, that connect the central aromatic NPBI segment and the peripheral trialkoxybenzamide units. The Ala-based linker has been demonstrated to strongly favor the formation of intramolecularly H-bonded seven-membered pseudocycles. NPBI 1 shows a strong tendency to self-assemble even in a good solvent like CHCl3 and the formation of chiral dimers is detected in this good solvent. Both experimental techniques and theoretical calculations reveal that the intramolecular H-bonded pseudocycles are very robust and the formation of chiral dimers is driven by the π-stacking of two units of the NPBI core. Unexpectedly, an efficient transfer of the asymmetry of the point chirality at the linker to the aromatic moiety is observed in the molecularly dissolved state. Changing the solvent to more apolar methylcyclohexane modifies the self-assembly process and the formation of chiral supramolecular polymers is detected. The supramolecular polymerization of 1 is demonstrated to follow an isodesmic mechanism unlike previous referable systems. In the formation of the supramolecular polymers of 1, the combination of experimental and computational data indicates that the H-bonded pseudocycles are also present in the aggregated state and the rope-like, columnar aggregates formed by the self-assembly of 1 rely on the π-stacking of the NPBI backbones.
Introduction
Solvent–solute interactions are key in natural and synthetic self-assembly and also in conditioning chemical reactivity. The helical structure of DNA or the folding of proteins are strongly influenced by the presence of water.1 In synthetic assemblies, the morphology, the functionality and the dimensionality of the aggregated species can be finely modulated by the nature of the solvent that strongly affects the binding constant of the interacting constitutive fragments.2 The role of the solvent is also unquestionable in the area of supramolecular polymers, macromolecules built up by the non-covalent interactions of monomeric units.3 The chemical structure of a vast majority of the reported supramolecular polymers consists in discotic aromatic monomers, poorly soluble in a variety of solvents. To enhance the solubility of the monomeric and aggregated species, these discotic segments are decorated with flexible side chains and, in many cases, with functional groups able to form H-bonding arrays. The synergy of the H-bonding interaction between such functional groups and the π-stacking of the aromatic backbones yields the final supramolecular polymers. Very often, the combination of these non-covalent forces generates a large dipole moment along the columnar stack that finally results in a cooperative supramolecular polymerization mechanism, featuring an initial nucleation process followed by an elongation regime.3,4 The lack of directional forces, like H-bonding interactions, however, usually yields isodesmic supramolecular polymerizations controlled by a single binding constant. In both cooperative and isodesmic aggregation mechanisms, the solvent nature plays a pivotal role reinforcing or weakening the non-covalent interactions that dictate the thermodynamics and length of the supramolecular polymer. Indeed, a solvent denaturation model has been developed to investigate the supramolecular polymerization mechanism in highly stable aggregated species.5 In this model, the combination of a good solvent, which favors the molecularly dissolved state, and a bad solvent, which facilitates the formation of the supramolecular species, is required to achieve a complete disassembly or assembly, respectively.Solvents also play a crucial role acting as additives that strongly modify the final properties of the supramolecular polymers6 or to accomplish bisignated processes in which highly stable supramolecular polymers are obtained by heating7 or dilution.8 These counterintuitive phenomena are justified by competitive solvent–solute interactions and bring to light the influence that these interactions exert on the final outcome for supramolecular polymerizations. Solvent–solute interactions have also been reported to switch relevant features of self-assembled systems like the supramolecular polymerization mechanism. Thus, the N,N′-diethylhexylureidotoluenes reported by Bouteiller et al.2a or the naphthalene-diimides reported by Kar et al.,9 in which the nature of the solvent controls the final ordering of the corresponding supramolecular polymers, are remarkable examples.
In the field of supramolecular polymers, the achievement of homochiral aggregated species that resemble the natural helical structures is a very active topic.10 The handedness of such homochiral, helical supramolecular polymers, generated from chiral, non-racemic monomers, is dictated by the element(s) of asymmetry present in the monomeric structure. Although very often this element of asymmetry is a stereogenic centre located at the solubilizing, peripheral side chains,10 homochiral supramolecular polymers can also be achieved by utilizing non-planar, chiral aromatic (atropisomers, helicenes, etc), units.11 In some cases, the handedness of the final helical aggregates can also be tuned by the solvent, and the solvent–solute interactions have been used to bias the formation of enantioenriched supramolecular polymers. Typical examples are the utilization of chiral solvents to shift the formation of a racemic polymeric mixture from two helical enantiomers to the formation of enantioenriched M- or P-type homopolymers.6b,12 Interestingly, there are also examples in which the helical inversion of a supramolecular polymer is provoked by changing the achiral solvent solubilizing the final aggregated species. This is the case of the coronene diimide reported by Kulkarni et al.13 or the N-annulated perylene diimides (NPBIs) investigated by our research groups.14
Inspired by nature, some helical polymers are constructed by using amino acids giving rise to fascinating examples of functional supramolecular polymers15 and to uncommon supramolecular polymerizations like living self-assembly16 or to supramolecular polymorphism.17 However, a detailed investigation of the influence of the amino acid on the final supramolecular polymerization mechanism, that would allow the understanding of the chiroptical features shown by the resulting aggregate and would contribute to establish clear structure–property rules, are very scarce in literature. Of special interest is the perylene diimide (PBI) functionalized at the imide groups with alanine linking benzylamine residues.18 The proximity of the stererogenic centre to the PBI core favors the formation of dimers in a good solvent like CHCl3. Changing the solvent to more apolar methylcyclohexane (MCH) results in an anti-cooperative supramolecular polymerization, in which the above mentioned dimers stack on top of each other to form larger oligomers thus demonstrating the influence of solvent–solute interactions in the achievement of dimers or oligomers.18
Herein, we report on the synthesis of a N-annulated perylene diimide (NPBI) decorated in its periphery with solubilizing trialkoxybenzamide moieties linked to the central imide units by a chiral fragment prepared from alanine (compound 1 in Fig. 1a). The synergy between experimental data and theoretical calculations reveals the strong influence exerted by the solvent and the stereogenic centre present in the central linker both on the supramolecular polymerization mechanism and the optical asymmetry shown by the monomeric and aggregated species. The alanine-based linkers present in 1 determine a strong trend of the monomeric units to form seven-membered H-bonded pseudocycles in all the solvents investigated (CHCl3, toluene (Tol), and MCH, Fig. 1a). The formation of such pseudocycles cancels the conformational flexibility of the central tether affording optical activity for the monomeric species due to the transfer of asymmetry from the point chirality of the linker to the aromatic core, a unique feature for molecularly dissolved species (Fig. 1b). Theoretical calculations show that the intramolecularly H-bonded species can readily form dimers with optical activity in which the periphery is coiled by the aliphatic side chains (Fig. 1c). Concentration dependent UV-vis experiments in CHCl3, a good solvent often used to achieve molecularly dissolved species,5 confirm the dimerization of 1 and allows deriving the corresponding dimerization constant KD. The utilization of MCH as bad solvent favors the formation of helical supramolecular aggregates. In these conditions, the ability of the NPBIs to self-assemble without the operation of intermolecular H-bonding interactions results in the formation of H-type aggregates following an isodesmic mechanism in MCH. Experimental data and theoretical calculations show that the intramolecularly H-bonded species remain unaltered in the supramolecular polymer, and the peripheral trialkoxyphenyl groups are coiling the columnar arrangement of the NPBI moieties (Fig. 1d). The results presented herein shed light on the effect that solvent and minute structural changes exert in the self-assembling features of functional systems thus contributing to establish structure/function relationships.
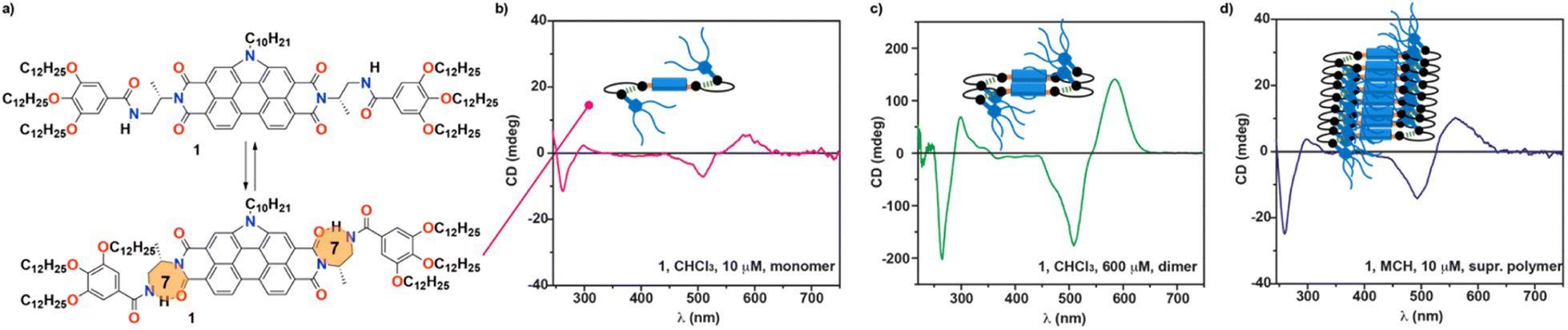 | ||
| Fig. 1 (a) Chemical structure of the chiral NPBI 1 and the formation of the intramolecularly H-bonded, seven membered pseudocycles. Circular dichroism (CD) spectra of the (b) monomeric, (c) dimeric and (d) aggregated chiral species of 1. The inset in (b–d) shows a cartoon of the monomer, dimer and supramolecular polymer of 1. | ||
Results and discussion
Synthesis and dimerization of NPBI 1
The investigated NPBI 1 was prepared by a multistep protocol starting from commercially available (S)-(+)-2-amino-1-propanol 2 and perylene-3,4,9,10-tetracarboxylic dianhydride 7 (Scheme S1†). Compound 2 is transformed into the trialkoxybenzamide 6 by following a N-BOC protection, a subsequent Mitsunobu SN2 reaction with phtalimide to yield compound 4 that, upon amidation reaction with previously reported trialkoxybenzoic acid 5 and acidic deprotection with TFA, yields benzamide 6.17 The condensation reaction of 6 with the previously reported dianhydride 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 in the presence of zinc acetate and imidazole and under microwave irradiation affords the target NPBI 1 in 42% yield. Importantly, the presence of the pyrrolic ring at the PBI core strongly favors the aggregation of referable NPBIs by a charge-transfer (CT) mediated process in which the operation of H-bonding interactions is not required.14,19 This CT-mediated process has not been demonstrated for perylenediimides in which the formation of intermolecular H-bonds is required to yield supramolecular polymers.17,18
19 in the presence of zinc acetate and imidazole and under microwave irradiation affords the target NPBI 1 in 42% yield. Importantly, the presence of the pyrrolic ring at the PBI core strongly favors the aggregation of referable NPBIs by a charge-transfer (CT) mediated process in which the operation of H-bonding interactions is not required.14,19 This CT-mediated process has not been demonstrated for perylenediimides in which the formation of intermolecular H-bonds is required to yield supramolecular polymers.17,18
The spectroscopic characterization of compound 1 already shows the strong tendency of 1 to self-assemble since all the resonances in the 1H NMR spectrum in CDCl3, both the aromatic and aliphatic protons, appear as very broad signals (Fig. S1†). Even by using solvents such as Tol-d8 or THF-d8 that favors the solvation of the aromatic backbones and the rupture of the H-bonding interactions, respectively, very broad resonances are found (Fig. S1†). Heating up a diluted solution of 1 (total concentration, cT = 1 mM, in Tol-d8 at 70 °C) produces a slight sharpening of some of the resonances that, however, are still broad due to the marked tendency of 1 to self-assemble (Fig. S2†). The broad resonances observed in any of the deuterated solvents utilized to characterize compound 1 prevents to perform more detailed analysis by applying techniques like NOESY, ROESY or DOSY.
The FTIR spectrum of 1 was registered in different solvents (cT = 1 mM, Fig. 2a and b) to further investigate the self-assembling features. In CHCl3, a good solvent that favors the molecularly dissolved state, the FTIR spectrum shows the stretching NH band of the amide group at 3395 cm−1 (Fig. 2a), a wavenumber that is often ascribed to the formation of intramolecularly H-bonded pseudocycles (Fig. 1a).17,18,20 However, a very similar value of 3406 cm−1 has been reported by Würthner and coworkers for the formation of dimers through the operation of two intermolecular H-bonding interactions between the amide functional groups in a PBI functionalized with the same chiral linker that tethers the PBI moiety and the peripheral side chains.18 Noteworthy, the band at ∼3450 cm−1, ascribable to the stretching NH bands for free amides is not observed.17,18,20 The stretching Amide I band appears at ∼1649 cm−1 in all the solvents utilized (Fig. 2b). This wavenumber value is closer to the values reported previously for intermolecular H-bonding interactions in the formation of dimers.17 These findings are diagnostic of the stability of either the seven-membered pseudocycles formed between the carbonyls of the imides and the NH of the peripheral amide functional groups, as sketched in Fig. 1a, or the intermolecular H-bonded dimers previously reported by Würthner and coworkers.18 Similar findings are also observed when registering the FTIR in Tol as solvent (Fig. 2a and b).
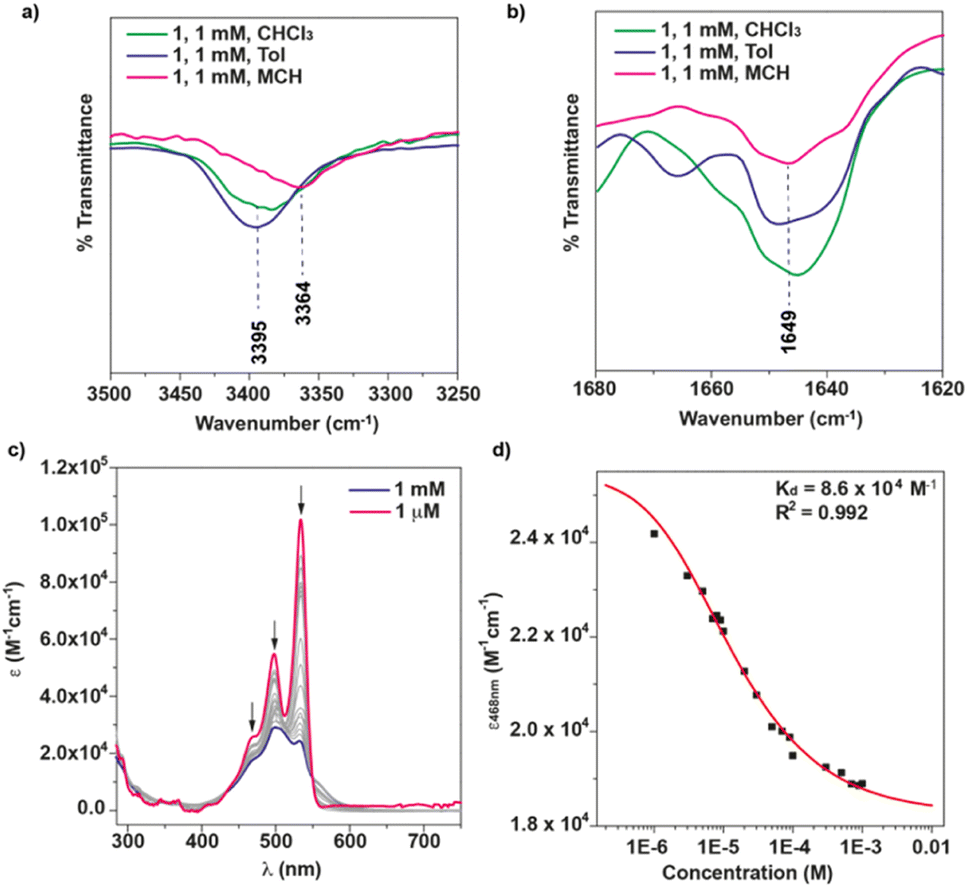 | ||
| Fig. 2 Partial FTIR spectra (a and b) of 1 in different solvents showing the stretching NH and Amide I bands (cT = 1 mM). (c) UV-vis spectra of 1 in CHCl3 at different concentrations. The arrows show the changes in the absorption maxima upon increasing the concentration. (d) Plot of the changes in the molar absorptivity of 1 in CHCl3 upon increasing the concentration. The red line depicts the fit to the dimerization model. | ||
To unravel the nature of the aggregated species formed in the good solvent CHCl3, we first registered the UV-vis spectrum of 1 at different concentrations. At highly diluted conditions (cT = 1 and 10 μM), the UV-vis spectrum 1 exhibits the typical absorption band of molecularly dissolved PBIs with a particular vibrational pattern increasing in intensity from shorter to longer wavelengths (Fig. 2c and S3†). This is in line with the shoulder at 468 nm and the two well-resolved peaks at 499 and 534 nm, respectively corresponding to the A0–2, A0–1 and A0–0 transitions of the vibronic progression.21 The monomeric dissolved nature of the species present in these diluted conditions is corroborated by the emission spectrum that shows a small Stokes shift and the presence of two maxima at 546 and 585 nm and a shoulder at 631 nm (Fig. S4†). These features are identical for some other good solvents like dioxane (Fig. S4†).
Increasing the concentration of 1 in CHCl3 (cT = 1 mM) results in a clear hypo- and hypsochromic effect in the UV-vis spectrum that now shows maxima absorption peaks at 504 and 531 nm and a shoulder at 563 nm, diagnostic of a dimerization process (Fig. 2c). In fact, a dimerization constant KD of 8.6 × 104 M−1 is derived by fitting the variation of the molar absorptivity of 1 upon increasing the concentration (c0) to eqn (1) in which εM and εD are the molar absorptivity for the monomer and dimer, respectively (Fig. 2d).22
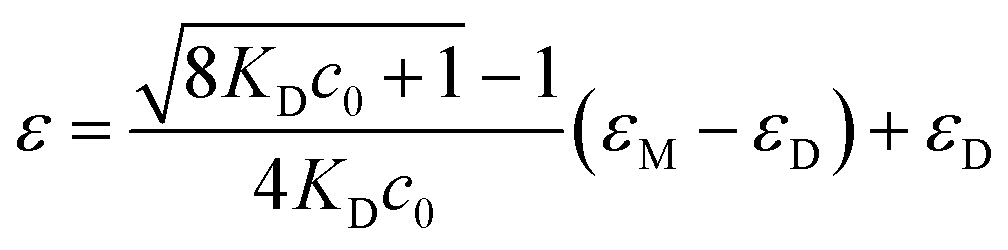 | (1) |
Unfortunately, and stated before, the broad resonances observed in all deuterated solvents utilized for the characterization of compound 1 prevents the use of 2D techniques (DOSY) to elucidate the dimensions of the dimers formed by 1 in solution. On the other hand, the strong absorption of both the monomeric and dimeric species formed by 1 in CHCl3 impedes measuring the hydrodynamic radii of the dimers by utilizing dynamic or static light scattering.
To shed light on the dimerization process of 1, the molecular structure of the monomeric and dimeric species of this NPBI were investigated by using quantum and classical theoretical calculations (see the ESI† for full computational details). We first tried to identify the most stable monomeric conformers and those optimal for self-assembly using a simplified molecular model, in which the peripheral dodecyloxy side chains were replaced by H atoms and the decyl tail attached to the central nitrogen of the NPBI core was substituted by a methyl group. Fig. S5† displays a variety of conformers (1a–1g) obtained using the current CREST technique23 at the semiempirical GFN2-xTB level24 and CHCl3 as solvent (see the ESI†). The structure of the conformers mainly differs in the orientation of the peripheral benzamide moieties that are pointing toward the same side (syn orientation, 1a–1d) or opposite sides (anti orientation, 1e) of the NPBI backbone or adopt an extended disposition giving rise to more planar situations (1f and 1g). Conformers 1f and 1g enable the formation of the seven-membered pseudocycles sketched in Fig. 1a due to the H-bond between the NH of the amide and one of the NPBI imide carbonyls showing N–H⋯O distances in the 1.85–1.90 Å range. The main difference between conformers 1f and 1g is that the former forms the intramolecular H-bonded pseudocycle only on one side of the NPBI core and the latter on both sides. This determines the higher relative stability of 1g compared to 1f (Fig. S5†).
After the pre-screening with the simplified NPBI model, the most stable conformers (1d and 1e) and those prone to promote the supramolecular growth (1f and 1g) were reoptimized at the GFN2-xTB level in CHCl3 but now incorporating all the peripheral side chains to get a more realistic description. A larger energy difference between these four conformers is now obtained. As for the simplified model, the most stable conformer corresponds to structure 1d, the other structures being significantly higher in energy (Fig. 3a and S6†). However, despite their higher energies, conformers 1f and 1g present favorable dispositions for an optimal self-assembly and may give rise to dimers and oligomers. In contrast, the disposition of the trialkoxybenzamide units covering the NPBI moiety limits the possibilities of supramolecular growth for conformers 1d and 1e. For 1d, only π-stacked dimers between the free core sides of two NPBI moieties are expected, whereas for conformer 1e the two sides of the NPBI are unapproachable thus impeding the supramolecular growth.
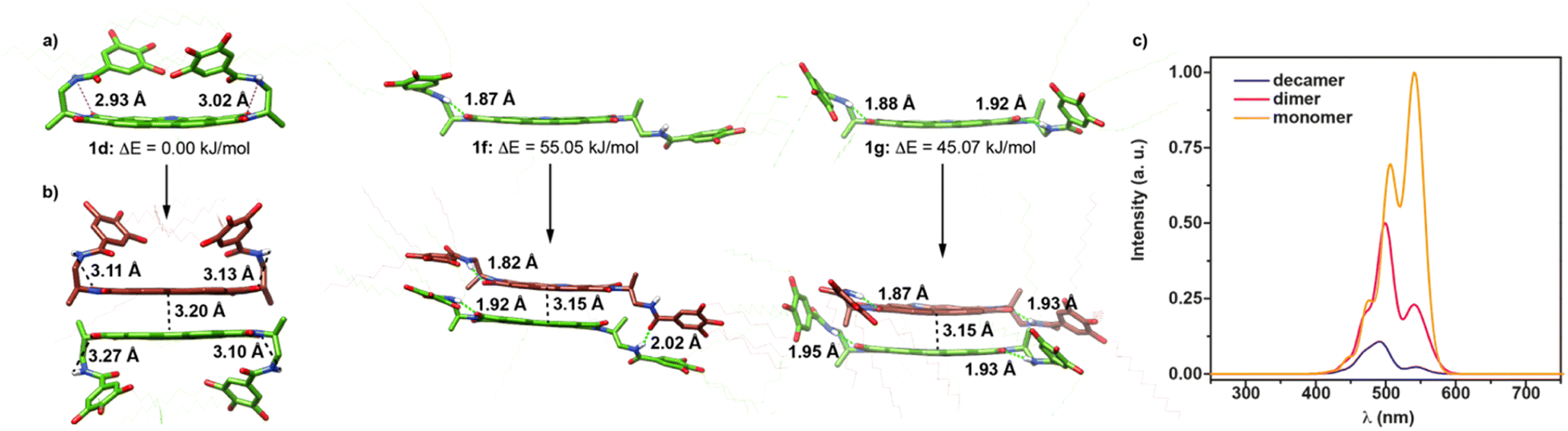 | ||
| Fig. 3 Minimum-energy structures, with their relative energy indicated, computed at the GFN2-xTB level in CHCl3 for selected conformers of monomer 1 (1d, 1f, and 1g) (a) and the three dimers (D1d, D1f, and D1g) resulting from the supramolecular interaction of the respective monomers. (b) For clarity, all non-relevant hydrogen atoms have been omitted and the side chains depicted in an almost transparent way. (c) Simulated UV-vis spectra calculated for the monomer, dimer, and decamer models (c) using the vibronic Hamiltonian (see the ESI† for additional details). | ||
With this in mind, supramolecular dimers D1d, D1f, and D1g were built up from the respective monomer structures and were computed at the GFN2-xTB level of theory by considering that the pyrrolic rings are pointing to the same direction, as it has been previously demonstrated in referable NPBIs (Fig. 3b and S7–S10†).14,19 All the three dimers D1d, D1f, and D1g present short π–π contacts between the NPBI cores (Fig. 3b). Dimer D1d (Fig. S7†) is formed by two stacked units of conformer 1d approximately rotated by 31° along the axis perpendicular to the NPBI cores which are separated by an intermolecular distance of 3.20 Å. In line with the monomeric unit, dimer D1d does not present seven-membered H-bonded pseudocycles. In dimer D1g (Fig. S8†), the NPBI cores are separated by 3.18 Å and are approximately rotated by 40°. In contrast to D1d, all the four imide groups in dimer D1g form seven-membered pseudocycles with intramolecular H-bonds in the 1.87–1.95 Å range. It is worth mentioning that, in this dimer, the peripheral side chains are pointing outwards leaving the NPBI conjugated core free to interact with other monomers and thus favoring the further growing into larger aggregates by changing the solvent conditions (see below). The less stable dimer D1f (Fig. S9†) also involves short intermolecular contacts between the π-conjugated cores (∼3.15 Å) and a rotation of the monomeric units of approximately 27° along the stacking axis (Fig. 3b). This dimer displays an asymmetric arrangement in terms of H-bonding interactions. Whereas the two 1f monomers preserve their intramolecular H-bonds on one end of the NPBI units, an intermolecular H-bond is formed between the amide groups of the other end. Note that a new dimer D1h similar to that proposed by Würthner and coworkers with four intermolecular H-bonds and the pyrrolic rings pointing in opposite directions was also built and optimized (Fig. S10†).17,18 Nevertheless, this dimer D1h is found to be rather unstable (>80 kJ mol−1) compared to the other ones (D1d, D1f and D1g) and is then discarded in the further discussion here following.
As a third step to elucidate the structure of the supramolecular dimers, classical molecular dynamics (MD) simulations were performed in CHCl3 starting from the GFN2-xTB-minimized D1d, D1f and D1g dimers to investigate if their structures are preserved or interconverted along the dynamics. The evolution of the distances of the intra- and intermolecular H-bonds was analyzed to understand the changes in the supramolecular structure along the trajectories. Starting from dimer D1d, MD simulations show that this dimer undergoes a significant structural change, along the dynamics, in which the peripheral moieties adopt a more extended disposition pointing outwards and, as a consequence, seven-membered pseudocycles are formed after 0.5 ns due to the intramolecular H-bond between the imide oxygens and the amide groups (Fig. S11†). Dimer D1d therefore evolves to a supramolecular D1g-like arrangement. For D1f, the intermolecular hydrogen bond between the monomeric units is broken during the first nanosecond, allowing the peripheral ethyl benzamide units to change the orientation regarding to the NPBI core and to establish a second intramolecular H-bond on each NPBI monomer (seven-membered pseudocycles). The resulting structure also resembles that of dimer D1g in line with the evolution of the computed H-bond distances (Fig. S11†). In contrast, the MD simulation of D1g shows that the distances of the four intramolecular H-bonds barely change along the trajectory and the structure of D1g is preserved (Fig. S11†). Therefore, dimers D1d and D1f evolve to form dimer D1g but the reverse did not occur. The theoretical results then suggest that dimer D1g, in which the two monomers present intramolecular H-bonds forming seven-membered pseudocycles and mainly interact through π–π contacts between the NPBI cores and also between the terminal phenyl rings, is likely to be the most plausible dimeric unit for further self-assembly.
The optical properties of 1 in its monomeric (1d) and dimeric (D1g) forms were also theoretically investigated by means of time-dependent density functional theory (TD-DFT) calculations along with a vibronic excitonic model including only local excitations (Frenkel-type states) (see the ESI† for details).25 In good accord with the experimental UV-vis spectrum at cT = 1 μM (Fig. 2c), the simulated absorption spectra calculated for the monomer shows a shoulder at 470 nm and two peaks growing in intensity at 500 and 550 nm (Fig. 3c). These bands are due to the A0–2, A0–1 and A0–0 vibronic transitions, respectively, of the lowest-energy S0 → S1 electronic excitation. The simulated spectrum of the dimer changes significantly and displays maxima at 495 nm and 545 nm, the latter being much less intense in good agreement with the UV-vis spectrum registered at cT = 1 mM (Fig. 2c).
MCH as solvent: from dimers to supramolecular polymers
To evaluate the influence of the solvent on the self-assembling features of NPBI 1, the FTIR spectrum was registered in more apolar solvents (Fig. 2a and b). To our surprise, the FTIR spectrum of 1 in toluene presents NH and Amide I bands at similar wavenumbers to those registered in CHCl3. In MCH, the stretching NH and Amide I bands appear at lower wavenumbers (∼3364 and ∼1649 cm−1, respectively) but still resembling those in the good solvent CHCl3. Therefore, these values indicate the unforeseen formation of seven-membered H-bonded pseudocycles in these nonpolar solvents that usually are reported to induce the formation of intermolecular H-bonding arrays leading to columnar supramolecular polymers.17,18,20To further investigate the self-assembling features in MCH, the UV-vis spectrum of 1 was registered in this solvent at diluted conditions (cT = 10 μM). At these experimental conditions, the UV-vis spectrum displays absorption maxima at 501 and 546 nm, the former being significantly more intense (Fig. 4a and S12a†). This absorption pattern is similar to that registered in CHCl3 at high concentration (cT > 500 μM) although the intensity ratio of the absorption maxima is not exactly the same (Fig. S12b†). This difference in the ratio of the absorption maxima has been previously reported on related PBIs functionalized at the imide groups with linked alanine and benzylamine residues, and it has been accounted for by the operation of an anti-cooperative supramolecular polymerization mechanism in which the dimers initially formed grow up to form longer aggregated species.18 However, the aggregates formed by 1 in MCH are highly stable as the minute changes observed in the UV-Vis spectra of 1 in MCH upon heating at 90 °C demonstrate (Fig. S13†). Heating up the MCH solution of 1, even for one hour, indeed results only in the partial disassembly of 1 as suggested by the intensity ratio of the characteristic A0–1/A0–0 absorption maxima (Fig. S13†).
To solve this drawback, we utilized toluene as solvent. Once again, the UV-vis spectrum of 1 in this solvent at highly diluted conditions (cT = 5 μM) shows an absorption pattern similar to that registered in MCH (Fig. S14a†). However, and in contrast to that found in MCH, heating up this diluted solution to 100 °C results in a complete disassembly showing the typical absorption pattern of monomeric PBIs with maxima at 528 and 492 nm (Fig. S14a†). Plotting the variation of the absorbance upon decreasing the temperature yields a sigmoidal curve that presents a good fitting to the one-component equilibrium (EQ) model (Fig. S14b†).26 However, the total supramolecular polymerization at low temperatures seems to be uncompleted. To discard a possible kinetic effect, the corresponding heating curve was also registered and showed no hysteresis thus ruling out a kinetic effect (Fig. S14b†). Interestingly, and despite the absorption pattern ascribable to an aggregated species of 1, the emission spectrum of this toluene solution at cT = 5 μM reveals the presence of a broad band centred at λ = 640 nm, also observed in the PL spectrum of 1 in MCH and ascribable to the formation of H-type supramolecular polymers, together with the emission pattern characteristic of the monomeric species observed in CHCl3 at diluted conditions (Fig. S4†). This emission pattern therefore supports the partial supramolecular polymerization of 1 in toluene that accounts for the uncompleted aggregation curve.
Taking into account the stability of the aggregated species formed by 1 in MCH and the partial aggregation of 1 in Tol, the solvent denaturation (SD) protocol was employed to derive the thermodynamic parameters associated to the supramolecular polymerization of NPBI 1.27 The application of this model requires the mixing of two solutions of NPBI 1 in a good solvent, CHCl3, and a bad solvent, MCH, keeping constant the total concentration of the sample (Fig. 4a). The balance between these two solvents conditions the stability of the supramolecular polymer and, consequently, the free Gibbs energy in a pure solvent is linearly correlated with the volume fraction of the good solvent, X, by a factor m according to eqn (2):
| ΔG′ = ΔG + mX | (2) |
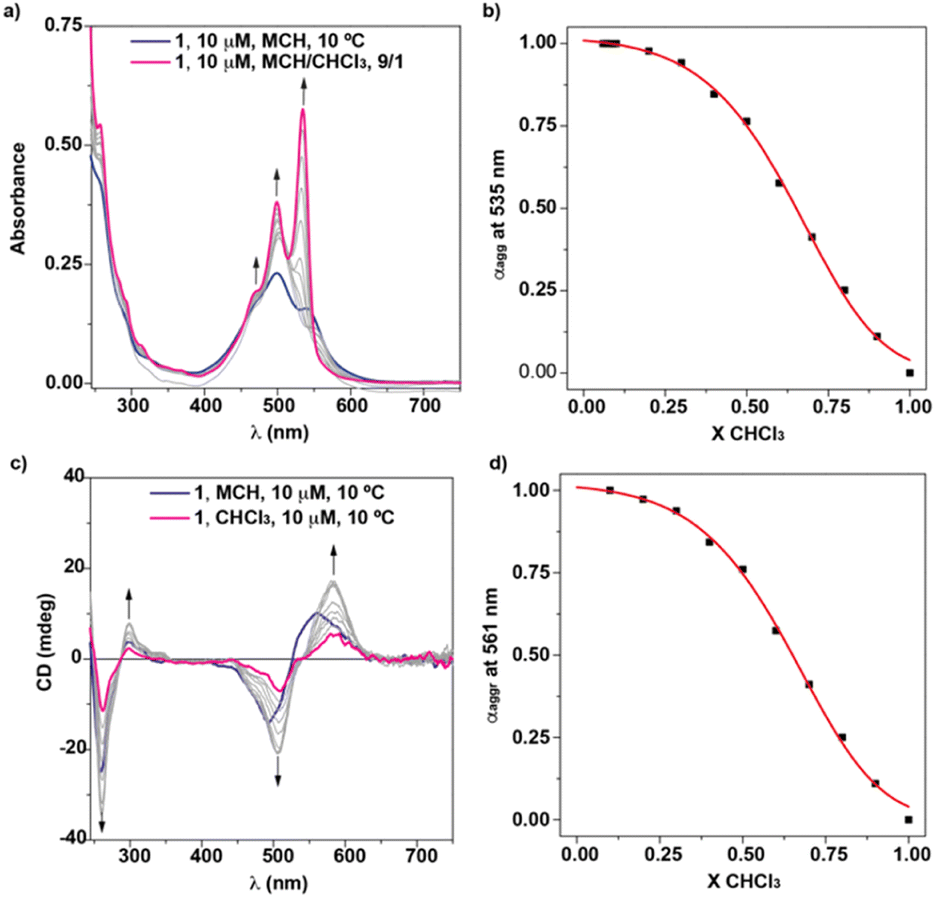 | ||
| Fig. 4 UV-vis (a) and CD (c) spectra and the respective denaturation curves of 1 in MCH/CHCl3 mixtures (cT = 10 μM) (b and d). The red line in panels (b and d) depicts the fit to the SD model. Arrows in panels (a) and (c) indicate the changes in the spectra upon increasing the amount of CHCl3. | ||
Plotting the variation of the degree of aggregation (αagg), calculated from the variation of the absorbance of 1 in MCH at λ = 535 nm upon the addition of increasing amounts of the solution of 1 in CHCl3, results in a sigmoidal curve that accurately fits to an isodesmic mechanism with values for ΔG′ = −39.8 kJ mol−1, m = 17.1, and the degree of cooperativity σ = 1 (Fig. 4b). From this ΔG′, and considering a null molar fraction of CHCl3, a value of 9.4 × 106 M−1 is inferred for the binding constant K. Similar results are obtained by using MCH as a bad solvent and dioxane as good solvent (Fig. S15†). In this case, the SD model for the isodesmic supramolecular polymerization of 1 affords values for ΔG′ = −35.5 kJ mol−1, K = 1.6 × 106 M−1, m = 39.1 and the degree of cooperativity σ = 1.
To further investigate the supramolecular polymerization of NPBI 1, its chiroptical properties were investigated by registering the circular dichroism (CD) spectra in different experimental conditions. The CD spectrum of 1 in MCH at cT = 10 μM demonstrates an efficient transfer of asymmetry from the stereogenic centre located at the lateral linker and shows a double ± bisignated Cotton effect with maxima at 561, 494, 298 and 261 nm and zero-crossing points at 526 and 287 nm (Fig. 4c). These chiroptical features are indicative of the formation of P-type helical structures. Therefore, the supramolecular polymerization of 1 constitutes one of the scarce examples of helical aggregates formed by monomeric units that self-assemble by following an isodesmic mechanism.4 These examples encompass benzenetricarboxamides (BTAs) endowed with lateral bipyridine fragments,28C3-symmetric oligo(phenylene ethynylene)tricarboxamides functionalized with Phe residues29 and NPBIs functionalized at the imide functional groups with trialkoxyphenyl units.14
Unexpectedly, the transfer of asymmetry from the point chirality of the tether groups to the central NPBI moiety is also observed in the good solvent CHCl3 (Fig. 4c). The CD spectrum of 1 in CHCl3 at diluted conditions also displays a double ± bisignated Cotton effect with maxima at 583, 507, 298, and 261 nm and zero-crossing points at 541 and 287 nm (Fig. 4c). Considering that, in these experimental conditions, NPBI 1 is in a molecularly dissolved state, this is a quite unique phenomenon of transfer of asymmetry from the stereocentre to the aromatic backbone. As far as we are aware, this effect is uncommon since those monomeric self-assembling units endowed with peripheral stereocentres do not show any transfer of asymmetry in the molecularly dissolved state.10,12–15,30 In this monomeric species, the formation of the pseudocycles could reduce the conformational flexibility of the peripheral side chains, which favors the efficient transfer of the asymmetry of the stereogenic centres at the linker to the aromatic moiety even in the molecularly dissolved state.
The dissimilar chiroptical features of 1 in MCH and CHCl3 prompted us to perform a denaturation experiment by using CD spectroscopy. The addition of minute amounts of the CHCl3 solution of 1 at cT = 10 μM results in CD spectra that resemble that registered in pristine CHCl3 but of higher intensity (Fig. 4c, 1b–d and S16a†). The UV-vis spectra of these solutions coincide with the UV-vis spectra of large aggregates (pristine MCH), dimers (mixtures MCH/CHCl3) and monomers (pristine CHCl3) (Fig. S16b†). In good analogy with the SD experiment carried out by using UV-vis spectroscopy, the data obtained in the denaturation experiment performed by CD spectroscopy can be also fitted to an isodesmic mechanism. Thus, the variation of the dichroic response at λ = 561 nm against the molar fraction of CHCl3 present a sigmoidal shape with thermodynamic values in the same range to those estimated previously (ΔG′ = −39.7 kJ mol−1, m = 17.0, and σ = 1) (Fig. 4d). To corroborate the dimerization of 1 in CHCl3, we also registered the CD spectra of 1 in this solvent by increasing the concentration. The dichroic pattern is identical for all the concentrations utilized, which supports the formation of the dimer from the chiral monomeric species (Fig. 1b, c and S17†).
The influence of the solvent to bias the self-assembling features of NPBI 1 was also visualized by atomic force microscopy (AFM). Spin-coating a solution of 1 in CHCl3 at cT = 50 μM onto a surface of highly oriented pyrolytic graphite (HOPG) results in a dense network of nanoparticles of ∼15 nm height (Fig. 5a and S18a–c†). The high volatility of CHCl3 may induce an increase of the local concentration that results in the interaction of the supramolecular dimers present in this solvent to yield nanoparticles whose dimensions are larger to those computed for the dimers. In contrast, the AFM images of a spin-coated solution of 1 in MCH at cT = 10 μM onto HOPG show the formation of isolated, rope-like fibers of several micrometers length and typical heights of 5 nm in agreement with the formation of supramolecular polymers in this solvent (Fig. 5b and S18d–f†).
 | ||
| Fig. 5 Height AFM images of the nanoparticles and isolated fibers formed upon self-assembly of 1 in CHCl3 (a) and MCH (b), respectively. Experimental conditions: HOPG as surface, cT = 50 and 10 μM for CHCl3 and MCH, respectively, 20 °C. | ||
The experimental findings on the self-assembling features of 1 demonstrate a dissimilar behaviour of this NPBI in comparison to the PBIs endowed with Ala-based chiral tethers reported by Würthner and coworkers.17,18 To disentangle the differences found in the self-assembling features, the formation of large aggregates of 1 was theoretically investigated. As stated above, based on MD calculations, dimer D1g appears to be the most plausible arrangement for further supramolecular polymerization in MCH. In this regard, we have built a long oligomer (16-mer model) based on D1g and fully optimized its structure at the GFN2-xTB level in solution (see the ESI† for additional details). In this minimized columnar 16-mer, the monomeric units are separated by 3.40 Å and are approximately rotated by 40° along the stacking axis to form a P-type helix (Fig. 6a–c). The self-assembling driving force in this polymeric model is the π-stacking of the NPBI cores and also between the outer phenyl groups of vicinal chains since the intramolecular H-bonds between amide and imide groups are preserved in the final structure (distances of ca. 2.00 Å). To shed light on the supramolecular polymerization mechanism, oligomers of increasing size (n = 2–12) were extracted from the central part of the previously-optimized 16-mer to avoid terminal effects. The binding energy per interacting pair of molecules (ΔEbind,n−1) was estimated by performing single-point energy calculations on those oligomers as explained in the ESI.† Calculations reveal that ΔEbind,n−1 barely changes with the oligomer size (Fig. S19†), which is indicative of an isodesmic supramolecular polymerization mechanism in good accord with the experimental evidence.
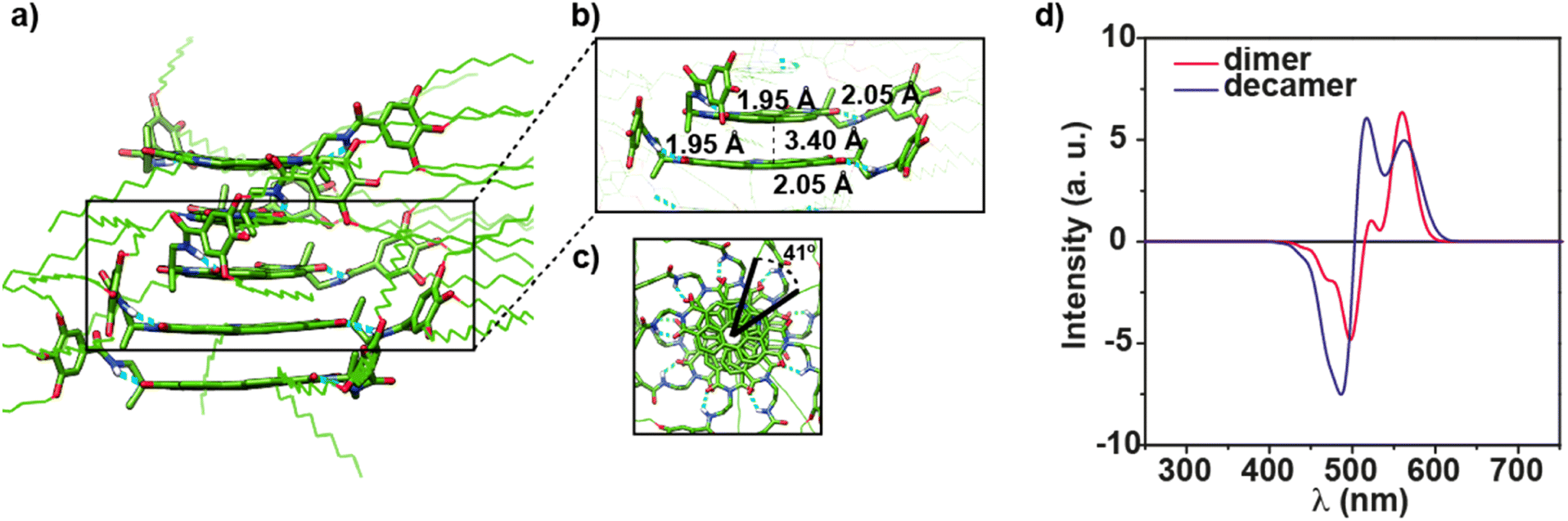 | ||
| Fig. 6 a) Minimum-energy structure calculated for a pentamer of 1 at the GFN2-xTB level. (b) Close-up picture (side view) of a dimer within the pentamer to visualize the relevant interactions. (c) Reduced top view representation of pentamer 1 to highlight the helical structure of the supramolecular aggregate. (d) Calculated CD spectra for the supramolecular dimer and decamer models of 1. | ||
Therefore, the N-alkyl group bridging the naphthalene units in NPBI 1 has indeed important structural effects in the formation of dimers and aggregates compared to the PBIs endowed with Ala-based chiral tethers reported by Würthner and coworkers.17,18 As discussed above on the basis of theoretical calculations, the N-alkyl group forces a rotation of the NPBI cores in adjacent stacked molecules that favors the formation of intramolecular H-bonds against intermolecular H-bonds. This trend is clearly supported by the relative energies theoretically predicted for dimers of NPBI 1 (Fig. S7–S10†). Dimer D1g, bearing four intramolecular H-bonds (Fig. S8†), is indeed predicted to be approximately 100 kJ mol−1 more stable than dimer D1h, showing four intermolecular H-bonds (Fig. S10†) similarly to the dimer proposed for the PBI derivative reported by Würthner.17 The prevalence of intramolecular H-bonds in the aggregated species of 1 is furthermore supported by classical molecular dynamics (MD) simulations performed in both CHCl3 and MCH (Fig. S11†), which clearly show that dimers D1d and D1f evolve along the dynamics to dimer D1g and the reverse did not occur. Therefore, the formation of intramolecular H-bonds giving rise to seven-membered pseudocycles is highly favoured for 1 and supports the self-assembly of 1 in helical π-stacked supramolecular polymers following an isodesmic mechanism. This is not the case for the PBI-based supramolecular polymer reported by Würthner and coworkers,17 for which intermolecular H-bonds are favoured and the supramolecular polymer formed follows an anticooperative supramolecular polymerization mechanism.17
The UV-vis and CD spectra of large aggregates of 1 were also theoretically investigated by simulating the spectra of a supramolecular decamer using a vibronic excitonic model (see the ESI† for details). The computed spectrum presents one maximum at 490 nm and a less intense band at 535 nm (Fig. 6d), in very good correlation with the experimental UV-vis spectrum of 1 registered in MCH with maxima at 501 and 546 nm (Fig. 4a and S12a†). This agreement supports the formation of H-type aggregated species for 1 in MCH.14
Taking into account the good correlation between the experimental and theoretical UV-vis spectra of all the species involved in this study, we also computed the CD spectra corresponding to the monomeric, dimeric and aggregated states. The CD spectra of the simplified model of conformers 1d and 1e were first calculated at the ωB97XD/6-31G** level (Fig. S20†). The CD spectrum computed for both conformers has a similar pattern and show two ± Cotton effects with maxima at 448, 378, 313 and 288 nm and zero-crossing points at 401 and 299 nm for 1d. However, the intensity ratio between the different dichroic bands in conformer 1d matches better the experimentally CD spectrum registered in CHCl3 at diluted conditions (compare Fig. 1b or 4c with S20†). The divergence in the position of the dichroic bands between the experimental and theoretical CD spectra is justified by the influence of the solvent.
Finally, the vibronic model was used to calculate the CD spectra for dimer D1g and a decamer model removing in both cases the peripheral side chains to reduce the computational cost. The simulated CD spectrum of the dimer present a ± bisignated Cotton effect with maxima at 559 and 496 nm, a zero-crossing point at 515 nm, a small band at 522 nm, and a shoulder at 465 nm (Fig. 6d). This calculated CD spectrum matches the CD spectrum experimentally registered in CHCl3 at high concentration (cT = 1 mM) thus supporting the formation of dimers at these experimental conditions (Fig. 1c and S17†). In the case of the decamer, the band at 520 nm grows in intensity at the expense of the band at 560 nm, and the negative band also increases its intensity and shifts hypsochromically to 480 nm (Fig. 6d). All these changes agree with those experimentally observed for 1 in MCH (Fig. 4c) and support the fact that this NPBI derivative can self-assemble forming the proposed P-type helical aggregate.
Conclusions
Natural and synthetic self-assembly exemplify the relevance of solvent–solute interactions that are especially relevant in the field of supramolecular polymers. Solvents have demonstrated to induce unexpected effects in the final outcome of this type of self-assembly that yields complex and functional supramolecular structures. Herein, we describe the synthesis of NPBI 1 endowed with two trialkoxybenzamide segments that are tethered by a chiral linker derived from (S)-(+)-alaninol that strongly favors the formation of intramolecularly H-bonded, seven-membered pseudocycles. The self-assembling features of 1 are strongly conditioned by the solvent utilized. The trend of 1 to self-assemble is so high that even in a good solvent like CHCl3, amply used to achieve the molecularly dissolved state, the formation of chiral dimers is favored. The synergy of experimental techniques and theoretical calculations reveals that the intramolecular H-bonded pseudocycles are very robust and the formation of the dimer is driven by the π-stacking of two units of the NPBI 1. Importantly, seven-membered H-bonded pseudocycles restrict the conformational flexibility of the peripheral side chains, which results in an efficient transfer of the asymmetry of the stereogenic centres at the linker to the aromatic moiety even in the molecularly dissolved state. Changing the solvent to more apolar MCH modifies the self-assembly process and the formation of chiral supramolecular polymers is detected. Unlike previous reports on the anti-cooperative supramolecular polymerization of structurally close PBI-based referable systems, the supramolecular polymerization mechanism of 1 has been demonstrated to be isodesmic in nature. Once again, the combination of experimental and computational data indicates that H-bonded pseudocycles are also formed and the rope-like, columnar aggregates formed by the self-assembly of 1 relies on the π-stacking of the NPBI backbones. The results presented in this manuscript reveal that small structural changes exert a strong impact on the self-assembling features of dye-based monomeric units that readily form supramolecular polymers. In addition, the cooperation between experiments and theoretical calculations is key to elaborate more general structure/function relationships useful in predicting relevant features of supramolecular polymers.Data availability
All data associated with this article have been included in the main text and ESI.†Author contributions
All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the MCIN/AEI of Spain (projects CNS2022-135187, PID2021-128569NB-I00, PID2020-113512GB-I00; TED2021-130285B-I00, and CEX2019-000919-M, funded by MCIN/AEI/10.13039/501100011033, by “ERDF A way of making Europe” and by the “European Union NextGenerationEU/PRTR”), the Generalitat Valenciana (PROMETEO/2020/077 and MFA/2022/017) and the Comunidad de Madrid (S2018/NMT-4389) is acknowledged. The MFA/2022/017 project forms part of the Advanced Materials programme supported by MCIN with funding from European Union NextGenerationEU (PRTR-C17.I1) and by Generalitat Valenciana. J. A. is indebted to the MCIN/AEI for his Ramón-y-Cajal (RyC-2017-23500) fellowship funded by MCIN/AEI/10.13039/501100011033 and by “ESF Investing in your future”. A. D.-G. acknowledges the Generalitat Valenciana for her I + d + i predoctoral fellowship.Notes and references
- (a) W. Fuller, T. Forsyth and A. Mahendrasingam, Philos. Trans. R. Soc., B, 2004, 359, 1237 CrossRef CAS PubMed; (b) T. Siebert, B. Guchhait, Y. Liu, B. P. Fingerhut and T. Elsaesser, J. Phys. Chem. Lett., 2016, 7, 3131 CrossRef CAS PubMed; (c) K. A. Dill, S. Bromberg, K. Yue, H. S. Chan, K. M. Ftebig, D. P. Yee and P. D. Thomas, Protein Sci., 1995, 4, 561 CrossRef CAS PubMed.
- (a) L. Bouteiller, O. Colombani, F. Lortie and P. Terech, J. Am. Chem. Soc., 2005, 127, 8893 CrossRef CAS PubMed; (b) H.-J. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924 CrossRef CAS PubMed; (c) K. Takaishi, K. Iwachido and T. Ema, J. Am. Chem. Soc., 2020, 142, 1774 CrossRef CAS PubMed; (d) J. S. Valera, H. Arima, C. Naranjo, T. Saito, N. Suda, R. Gómez, S. Yagai and L. Sánchez, Angew. Chem., Int. Ed., 2022, 61, e202114290 CrossRef CAS PubMed.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS PubMed.
- C. Kulkarni, S. Balasubramanian and S. J. George, ChemPhysChem, 2013, 14, 661 CrossRef CAS PubMed.
- P. A. Korevaar, C. Schaefer, T. F. A. De Greef and E. W. Meijer, J. Am. Chem. Soc., 2012, 134, 13482 CrossRef CAS PubMed.
- (a) G. Ghosh, A. Chakraborty, P. Pal, B. Jana and S. Ghosh, Chem. - Eur. J., 2022, 28, e202201082 CrossRef CAS PubMed; (b) M. L. Ślȩczkowski, M. F. J. Mabesoone, P. Ślȩczkowski, A. R. A. Palmans and E. W. Meijer, Nat. Chem., 2021, 13, 200 CrossRef PubMed; (c) S. Kotha, M. F. J. Mabesoone, D. Srideep, R. Sahu, S. K. Reddy and K. V. Rao, Angew. Chem., Int. Ed., 2021, 60, 5459 CrossRef CAS PubMed.
- (a) F. Helmich, C. C. Lee, M. M. L. Nieuwenhuizen, J. C. Gielen, P. C. M. Christianen, A. Larsen, G. Fytas, P. E. L. G. Lecler02CBe, A. P. H. J. Schenning and E. W. Meijer, Angew. Chem., Int. Ed., 2010, 49, 3939 CrossRef CAS PubMed; (b) L. Su, J. Mosquera, M. F. J. Mabesoone, S. M. C. Schoenmakers, C. Muller, M. E. J. Vleugels, S. Dhiman, S. Wijker, A. R. A. Palmans and E. W. Meijer, Science, 2022, 377, 213 CrossRef CAS PubMed.
- (a) K. V. Rao, D. Miyajima, A. Nihonyanagi and T. Aida, Nat. Chem., 2017, 9, 1133 CrossRef PubMed; (b) K. V. Rao, M. F. J. Mabesoone, D. Miyajima, A. Nihonyanagi, E. W. Meijer and T. Aida, J. Am. Chem. Soc., 2020, 142, 598 CrossRef CAS PubMed.
- H. Kar, G. Ghosh and S. Ghosh, Chem. - Eur. J., 2017, 23, 10536 CrossRef CAS PubMed.
- (a) A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2007, 46, 8948 CrossRef CAS PubMed; (b) Y. Dorca, E. E. Greciano, J. S. Valera, R. Gómez and L. Sánchez, Chem. - Eur. J., 2019, 25, 5848 CrossRef CAS PubMed.
- (a) Z. Xie, V. Stepanenko, K. Radacki and F. Würthner, Chem. - Eur. J., 2012, 18, 7060 CrossRef CAS PubMed; (b) J. Buendía, E. E. Greciano and L. Sánchez, J. Org. Chem., 2015, 80, 12444 CrossRef PubMed; (c) M. Wehner, M. I. S. Röhr, V. Stepanenko and F. Würthner, Nat. Commun., 2020, 11, 5460 CrossRef CAS PubMed; (d) R. Rodríguez, C. Naranjo, A. Kumar, P. Matozzo, T. K. Das, Q. Zhu, N. Vanthuyne, R. Gómez, R. Naaman, L. Sánchez and J. Crassous, J. Am. Chem. Soc., 2022, 144, 7709 CrossRef PubMed; (e) J. Guilleme, M. J. Mayoral, J. Calbo, J. Aragó, P. M. Viruela, E. Ortí, T. Torres and D. González-Rodríguez, Angew. Chem., Int. Ed., 2015, 54, 2543 CrossRef CAS PubMed; (f) M. J. Mayoral, J. Guilleme, J. Calbo, J. Aragó, F. Aparicio, E. Ortí, T. Torres and D. González-Rodríguez, J. Am. Chem. Soc., 2020, 142, 21017 CrossRef CAS PubMed.
- (a) S. J. George, Z. Tomovic, A. P. H. J. Schenning and E. W. Meijer, Chem. Commun., 2011, 47, 3451 RSC; (b) V. Stepanenko, X. Li, J. Gershberg and F. Würthner, Chem. - Eur. J., 2013, 19, 4176 CrossRef CAS PubMed; (c) A. K. Mondal, M. D. Preuss, M. L. Ślȩczkowski, T. K. Das, G. Vantomme, E. W. Meijer and R. Naaman, J. Am. Chem. Soc., 2021, 143, 7189 CrossRef CAS PubMed.
- C. Kulkarni, P. A. Korevaar, K. K. Bejagam, A. R. A. Palmans, E. W. Meijer and S. J. George, J. Am. Chem. Soc., 2017, 139, 13867 CrossRef CAS PubMed.
- M. A. Martínez, A. Doncel-Giménez, J. Cerdá, J. Calbo, R. Rodríguez, J. Aragó, J. Crassous, E. Ortí and L. Sánchez, J. Am. Chem. Soc., 2021, 143, 13281 CrossRef PubMed.
- (a) Z. Álvarez, A. N. Kolberg-Edelbrock, I. R. Sasselli, J. A. Ortega, R. Qiu, Z. Syrgiannis, P. A. Mirau, F. Chen, S. M. Chin, S. Weigand, E. Kiskinis and S. I. Stupp, Science, 2021, 374, 848 CrossRef PubMed; (b) Z. Wang, A. Hao and P. Xing, Angew. Chem., Int. Ed., 2020, 59, 11556 CrossRef CAS PubMed; (c) J. D. Tovar, Acc. Chem. Res., 2013, 46, 1527 CrossRef CAS PubMed.
- (a) S. Ogi, K. Matsumoto and S. Yamaguchi, Angew. Chem., Int. Ed., 2018, 57, 2339 CrossRef CAS PubMed; (b) N. Fukaya, S. Ogi, M. Kawashiro and S. Yamaguchi, Chem. Commun., 2020, 56, 12901 RSC.
- M. Wehner, M. I. S. Röhr, M. Bühler, V. Stepanenko, W. Wagner and F. Würthner, J. Am. Chem. Soc., 2019, 141, 6092 CrossRef CAS PubMed.
- J. Gershberg, F. Fennel, T. H. Rehm, S. Lochbrunner and F. Würthner, Chem. Sci., 2016, 7, 1729 RSC.
- E. E. Greciano, J. Calbo, E. Ortí and L. Sánchez, Angew. Chem., Int. Ed., 2020, 59, 17517 CrossRef CAS PubMed.
- (a) C. Naranjo, S. Adalid, R. Gómez and L. Sánchez, Angew. Chem., Int. Ed., 2023, 63, e202218572 Search PubMed; (b) E. E. Greciano, S. Alsina, G. Gosh, G. Fernández and L. Sánchez, Small Methods, 2020, 4(1–8), 1900715 CrossRef CAS.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawata and D. Schmidt, Chem. Soc. Rev., 2016, 116, 962 CrossRef PubMed.
- A. Löhr, M. Grüne and F. Würthner, Chem. - Eur. J., 2009, 15, 3691 CrossRef PubMed.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169 RSC.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652 CrossRef CAS PubMed.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069 CrossRef CAS PubMed.
- H. M. M. ten Eikelder, A. J. Markvoort, T. F. A. de Greef and T. P. A. J. Hilbers, J. Phys. Chem. B, 2012, 116, 5291 CrossRef CAS PubMed.
- P. A. Korevaar, C. Schaefer, T. F. A. De Greef and E. W. Meijer, J. Am. Chem. Soc., 2012, 134, 13482 CrossRef CAS PubMed.
- A. R. A. Palmans, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Angew Chem. Int. Ed. Engl., 1997, 36, 2648 CrossRef CAS.
- J. Buendía, F. García, B. Yélamos and L. Sánchez, Chem. Commun., 2016, 52, 8830 RSC.
- X. Shang, I. Song, H. Ohtsu, J. Tong, H. Zhang and J. H. Oh, Sci. Rep., 2017, 7(1–10), 5508 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details, characterization and additional spectroscopic measurements, additional AFM images, theoretical calculations details and supporting references including Fig. S1–S20, (PDF). See DOI: https://doi.org/10.1039/d3sc03372d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |