
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ortho-Selective amination of arene carboxylic acids via rearrangement of acyl O-hydroxylamines†
James E.
Gillespie
,
Nelson Y. S.
Lam
 and
Robert J.
Phipps
*
and
Robert J.
Phipps
*
Yusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: rjp71@cam.ac.uk
First published on 18th August 2023
Abstract
Direct amination of arene C–H bonds is an attractive disconnection to form aniline-derived building blocks. This transformation presents significant practical challenges; classical methods for ortho-selective amination require strongly acidic or forcing conditions, while contemporary catalytic processes often require bespoke directing groups and/or precious metal catalysis. We report a mild and procedurally straightforward ortho-selective amination of arene carboxylic acids, arising from a facile rearrangement of acyl O-hydroxylamines without requiring precious metal catalysts. A broad scope of benzoic acid substrates are compatible and the reaction can be applied to longer chain arene carboxylic acids. Mechanistic studies probe the specific requirement for trifluoroacetic acid in generating the active aminating agent, and suggest that two separate mechanisms may be operating in parallel in the presence of an iron catalyst: (i) an iron-nitrenoid intermediate and (ii) a radical chain pathway. Regardless of which mechanism is followed, high ortho selectivity is obtained, proposed to arise from the directivity (first) or attractive interactions (second) arising with the carboxylic acid motif.
Introduction
Ortho-Amino arene carboxylic acids are prevalent motifs in bioactive natural products and medicinal agents, present either explicitly as a discrete unit, or implicitly embedded within the structure of heterocycles. Ortho-Aminobenzoic acids (i.e. anthranilic acids) are a particularly versatile building block present in a wide range of heterocycles, and can be identified within the core of a number of prominent medicinal compounds (Fig. 1A).1 By the same retrosynthetic logic, 2-oxindoles2 and dihydroquinolin-2(1H)-ones,3 among other heterocycles, can be analogously synthesised from their corresponding higher order ortho-aminoalkyl benzoic acid derivatives.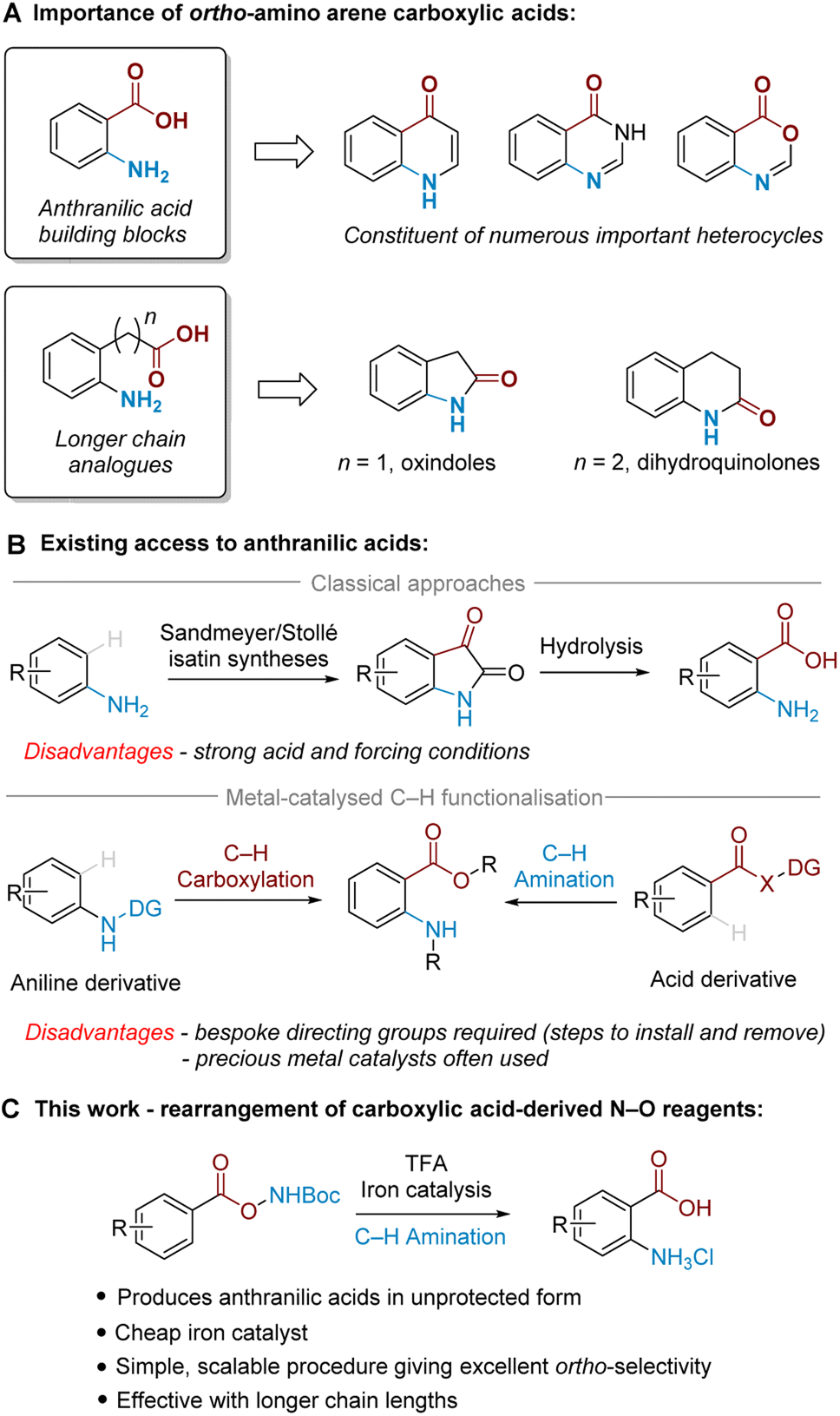 | ||
| Fig. 1 Background and summary of the present work. | ||
While many methods for ortho-amino arene carboxylic acid synthesis are known, few exist that can access them via direct C–H amination from the widely available parent arene carboxylic acid. For anthranilic acids, these are classically accessed by oxidative cleavage of isatins, which can be made by Sandmeyer4 or Stollé5 methodology via multistep synthesis from aniline (Fig. 1B, upper).6 Alternatively, nitration of benzoic acid followed by reduction is also known;7 this typically requires harsh conditions and only provides the required ortho relationship under specific substrate-imposed circumstances.8 More modern synthetic approaches utilise transition metal-catalysed ortho-selective C–H amination of benzoic acid derivatives9 or C–H carboxylation of aniline derivatives10 (Fig. 1B, lower). These methods, however, are often poorly generalisable to higher order arene carboxylic acids, and typically require precious metals and/or bespoke directing groups (DGs), meaning that separate DG installation/removal steps are needed to access the desired product and accessing unprotected anthranilic acids in many cases can be lengthy.11
We have been particularly interested in overcoming the regioselectivity challenges associated with arene amination using hydroxylamine-derived aminating agents.12–16 To this end, we recently developed an ortho-selective amination of aniline-derived sulfamate salts,17 and further discovered that sulfonyl O-hydroxylamines could undergo an aminative rearrangement to access ortho-sulfonyl anilines.18 In those reactions we believe that the ortho selectivity is dictated by attractive non-covalent interactions (NCIs) between the arenesulfonate anion and a putative cationic ammonium radical generated upon cleavage of the weak N–O bond.19,20
In the present work, we disclose the finding that arene carboxylic acid-derived N–O reagents can undergo a highly ortho-selective aminative rearrangement upon simple treatment with trifluoroacetic acid, with reactivity enhanced by the addition of only 1 mol% of an iron catalyst (Fig. 1C). This is surprising because benzoic acid-derived N–O reagents have been used with increasing frequency in recent years as sources of electrophilic nitrogen, and their ability to ‘self-aminate’ under mild conditions with high regioselectivity has not yet been reported. Complementing the toolkit of contemporary amination methods, we believe that this mild and straightforward ortho-amination protocol will be of significant practical utility for the following reasons: it produces valuable ortho-aminated acids in unprotected form, is scalable with precursors accessed in a single step from readily available materials, is generalisable to longer chain lengths and does not require precious metal catalysts. This study also probes the mechanism of the process, including the role of the iron catalyst as well as the unique role of TFA.
Results and discussion
Optimisation and scope investigation
We commenced our investigation with Boc-protected benzoyl O-hydroxylamine (1a), readily accessed in a single step from the corresponding benzoic acid. Treatment of 1a with five equivalents of TFA in CH2Cl2 at 40 °C resulted in product 2a, albeit in only 16% NMR yield (Table 1, entry 1). A solvent survey (see ESI† for full details) indicated that TFE was optimal in terms of yield and reproducibility (entry 2). The addition of 1 mol% of FeSO4·7H2O gave a further increase in the reaction yield (72%, entry 3), in line with our previous observations.18 A brief exploration of alternative acids indicated that TFA was singularly effective (entries 4–6; vide infra for discussion), and we note that the ortho-aminated product was the only product isomer observed.|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Acid | Catalyst | Yield (%) |
| a Yields determined by 1H NMR using 1,2-dimethoxyethane (DME) as an internal standard. | ||||
| 1 | CH2Cl2 | TFA | — | 16 |
| 2 | TFE | TFA | — | 68 |
| 3 | TFE | TFA | FeSO 4 ·7H 2 O | 72 |
| 4 | TFE | AcOH | FeSO4·7H2O | 0 |
| 5 | TFE | pTsOH | FeSO4·7H2O | 0 |
| 6 | TFE | TfOH | FeSO4·7H2O | 2 |
We proceeded to evaluate the substrate scope of the aminative rearrangement of benzoic acid derivatives to give substituted anthranilic acids. Evaluation of a small selection of substrates with and without an iron catalyst gave mixed results in which some substrates benefitted significantly from the presence of iron whilst others did not (see the ESI†). For this reason, we elected to perform the substrate scope in the presence of the iron catalyst to give the best consistency. A practical benefit of our process is that the majority of products could be easily purified through simple precipitation as the corresponding hydrochloride salt, and no special precautions were needed to exclude air or moisture from the reaction.
A series of alkyl-substituted benzoic acid-derived N–O reagents smoothly generated the desired products (Scheme 1A, 2a–2i). Similarly, halogen-substituted variants encompassing fluoride (2j–2l), also chloride (2m–2q), bromide (2r–2u) and iodide (2v) substituents worked in moderate to good yields, and poly-halogenated substrates (2w–2y) were also competent. These results showcase the orthogonality of our method to C–N bond formation through cross-coupling approaches, while preserving useful reactive handles for subsequent diversification. Electronically diverse aryl substituents were similarly competent; the reaction tolerates electron donating alkoxy groups (2z–2ac), as well as electron withdrawing trifluoromethyl (2ad, 2ae) and difluoromethoxy groups (2af). Pleasingly, alkyl substituents bearing a potentially labile benzylic chloride (2ag) or bromide (2ah) also undergo the rearrangement and could act as handles for further functionalisation. In cases where the substrate has two inequivalent ortho positions, the product was obtained as a mixture of regioisomers and by precipitation it was unfortunately not possible to separate these regioisomers. In these cases, there seems to be little sensitivity to sterics. For example, good reactivity was seen with 2g and 2i, which could be due to the small size of the putative aminium electrophile. In cases where yields were low, the remaining mass balance typically consists of the corresponding benzoic acid, where the N–O bond has been cleaved but productive arene amination has not occurred, rather than the presence of other regioisomers.
 | ||
| Scheme 1 Substrate scope. Yields are isolated. All reactions conducted with no exclusion of air or moisture. Ratio (r.r.) refers to regioisomeric ratio of aminated product, where (*) denotes minor regioisomer. Ratio in parenthesis refers to r.r from crude material prior to purification.a Isolated as free amine. | ||
Next, we questioned whether mono-alkylated amines could be transferred to give N-alkyl anthranilic acids.12f Pleasingly, NHMe transfer proved to be viable when the reaction was performed using HFIP as the solvent (Scheme 1B). In comparison with NH2 transfer, NHMe transfer typically required more electron rich substrates to obtain productive reactivity, the exact reason for which remains to be determined.21 Substrates bearing one (4a) or two (4b) methoxy groups on the aromatic ring were well tolerated, giving the desired products in excellent yields. Methoxy groups in combination with chlorine (4c), bromine (4d) and methyl (4e) substituents also proceeded smoothly. Monoalkylated substrates (4f) as well as substrates bearing an alkyl group alongside a chlorine atom (4g) worked in good isolated yield and two alkyl groups (4h–4i) were also tolerated.
We considered whether the reaction would tolerate the addition of a methylene unit between the carboxylate group and aromatic ring, which, after in situ cyclisation, should allow direct access to 2-oxindole products. We were pleased to find that a number of substrates proved amenable; the unsubstituted substrate could undergo the rearrangement followed by cyclisation to give the oxindole in 56% isolated yield (6a). α-Mono- and di-alkylated substrates were also amenable (6b–6d). Chloride (6e), methyl (6f) and trifluoromethyl (6g) substituents were tolerated at the arene ortho-position. A 3,5-dimethyl substrate (6h) as well as a substrate bearing a bromine atom in the meta position (6i) also gave the desired products. Extending the chain between the arene and the carboxylic acid further to include two methylene units, we found that the reaction still proceeds, now affording the corresponding 3,4-1H-dihydroquinolinone after in situ cyclisation (6j–6k). Interestingly, in the case of a para-methyl substituted substrate, an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of ortho:meta selectivity in the amination was observed, evidenced by isolation of dihydroquinolinone 6l and meta-aminated product 6m (Scheme 1C, inset box, see later for discussion).
:1 ratio of ortho:meta selectivity in the amination was observed, evidenced by isolation of dihydroquinolinone 6l and meta-aminated product 6m (Scheme 1C, inset box, see later for discussion).
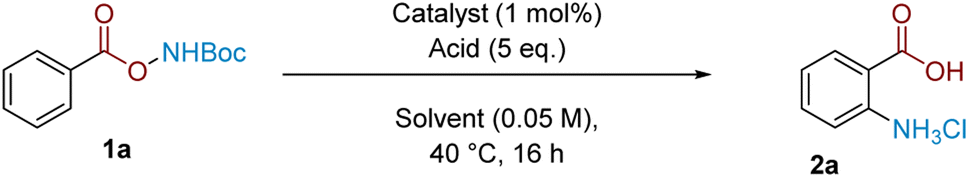
A strength of this method lies in its procedural ease and that it leads directly to unprotected amines.22 We further demonstrate that substrate synthesis from benzoic acid can be successfully telescoped with the rearrangement protocol: coupling of N-Boc hydroxylamine followed by rearrangement gave anthranilic acid 2a in 58% isolated yield (Scheme 2A). The rearrangement protocol performs well on a 5 mmol scale and 2a could be isolated in 79% yield (Scheme 2B). The crude reaction mixture following amination could be telescoped with electrophilic reagents to generate related benzo-fused heterocycles (Scheme 2C); treatment of crude 2a with CH(OMe)3 and NH4OAc cleanly afforded quinazolinone 7 (see the ESI† for more quinazolinone derivatisations), while the corresponding treatment with p-toluoyl chloride under basic conditions afforded substituted benzoxazinone 8.
 | ||
| Scheme 2 Synthetic applications. | ||
Probing inter-vs. intramolecularity of the rearrangement
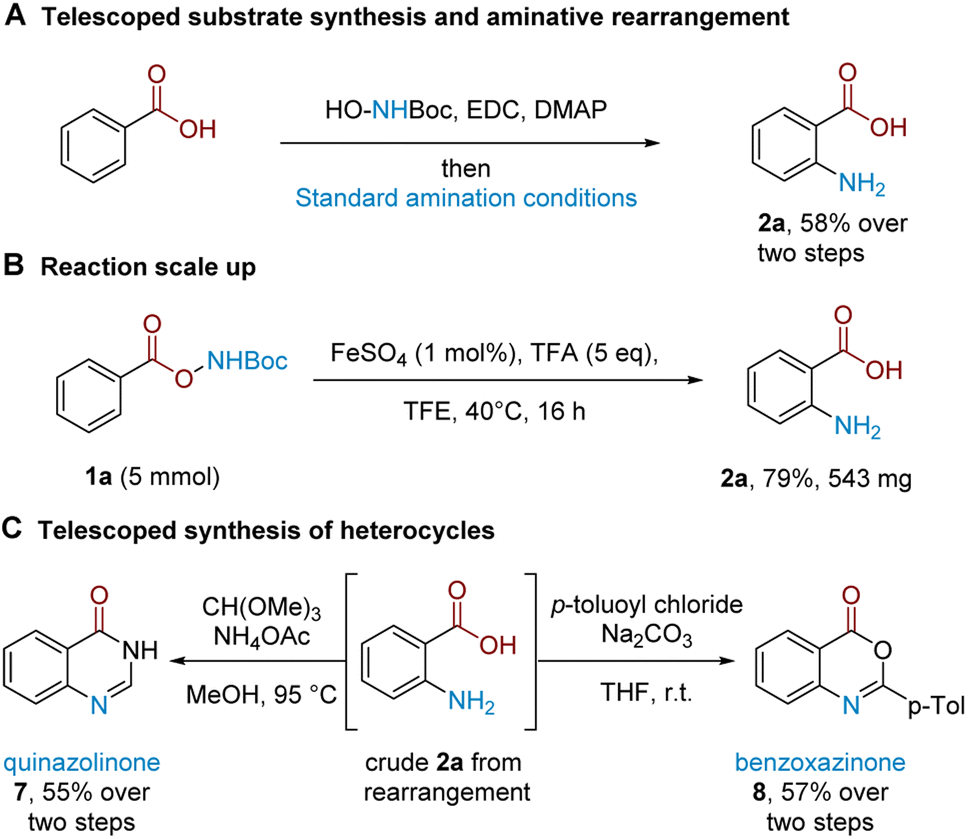
Having established the synthetic viability and scope, we sought to gain insight into its mechanism. Together with our previously-reported aminative rearrangement of sulfonyl N–O reagents, several questions remain outstanding relating to (i) the seemingly unique ability of TFA to promote the reaction amongst other Brønsted acids, (ii) the role of the iron catalyst, and (iii) whether the rearrangement proceeds via an intramolecular or intermolecular mechanism.In our previous work, we established that the related aminative rearrangement of sulfonyl O-hydroxylamines proceeded via an intermolecular mechanism, shown by extensive scrambling in crossover experiments.18,23 In contrast, analogous experiments in the present system—i.e. subjecting carboxyl substrates 1b (delivering NH2) and 3i (delivering NHMe) afforded no product crossover, which suggested that the reaction might proceed via an intramolecular mechanism (Scheme 3A). This was further confirmed with an alternative electronically-differentiated substrate pair (1ac, 3i; Scheme 3B). A further competition experiment was next conducted where an equimolar amount of an electronically-similar (Scheme 3C) or electronically-different (Scheme 3D) benzoic acid was exogenously added to the rearrangement reaction of 1b.24 This revealed that crossover only occurred when arenes of different electronics were used to give 2ac as the crossover product (Scheme 3D; 9% with Fe, 20% without; see the ESI† for details). This suggests that an intermolecular amination mechanism can occur, but only for electronically-activated substrates that can out-compete against a more favourable proximity-driven intramolecular/self-amination process. This outcome reconciles observations seen during our scope exploration, where we noted that the para-methyl substituted substrate 5l with a longer tether gave a 1:1 mixture of ortho and meta aminated products (Scheme 1C inset box; see the ESI† for further examples and discussion).
 | ||
| Scheme 3 (A and B): Two pairs of crossover experiments. (C and D): Same-substrate class competition experiments indicating crossover only for arenes with different electronics. | ||
Probing the role of trifluoroacetic acid
A full survey of Brønsted acids in the rearrangement indicated that reactivity was decoupled from acid pKa values and that carboxylic acids bearing inductively withdrawing groups were uniquely enabling (Table 2, entries 4–7). Weaker carboxylic acids were evidently inferior at Boc deprotection giving little or no conversion to 2a (entries 8 and 9). Meanwhile, stronger acids (MsOH, pTsOH, TfOH) are presumed to cleave the Boc group, but this did not result in product formation, with only benzoic acid observed in crude reaction mixtures (entries 1–3). This very particular requirement mirrors the prominence of TFA in related aminations that utilise N–O reagents though the reasons for this remain unclear.15b,18,25|
|
||||
|---|---|---|---|---|
| Entry | Acid | Yield 2a | Yield 1a | Yield BzOH |
| a Yields determined by 1H NMR using 1,2-DME as internal standard. | ||||
| 1 | TfOH | 2 | 0 | 63 |
| 2 | MsOH | 0 | 0 | 100 |
| 3 | pTsOH | 0 | 0 | 60 |
| 4 | TFA | 64 | 0 | 22 |
| 5 | TCA | 56 | 0 | 29 |
| 6 | CF 2 HCO 2 H | 34 | 17 | 34 |
| 7 | C 2 F 5 CO 2 H | 56 | 0 | 19 |
| 8 | CF3CH2CO2H | 4 | 70 | 14 |
| 9 | AcOH | 0 | 33 | 12 |
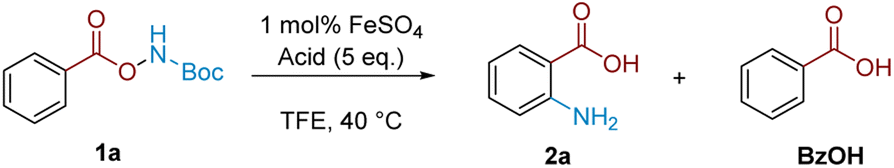
To gain insight into the reasons for this strict acid dependence, we next investigated the role of hydroxylamine protonation on reactivity. Anticipating that deprotection of the Boc group might complicate investigation of the role of acid in the subsequent mechanistic steps, we commenced the investigation using benzoyl O-hydroxylammonium triflate (9a) (Scheme 4). Subjecting 9a to TFA or TCA (5 eq.) did not give product formation. However, subsequent in situ treatment of the above reaction mixtures with Et3N (1 eq.) led to product formation in good yield (Path 1). Analogously, product formation was observed in similarly good yield when the sequence was reversed—1 eq. Et3N addition to 9a to form the free base, followed by addition of 5 eq. of TFA (Path 2). Hypothesising that formation of the trifluoroacetate anion was crucial for reactivity, we performed the reciprocal reaction by treating triflate salt 9a with 5 eq. of either sodium trifluoroacetate (NaTFA) or sodium trichloroacetate (NaTCA). In both cases, product formation was observed in good yields (Path 3), whereas none was observed when the reaction mixture was pretreated with Et3N to deprotonate the starting material prior to addition of NaTFA/NaTCA (Path 4). These results were corroborated using neutral benzoyl O-hydroxylamine (9b), where reactivity was only observed using TFA; acids that were more acidic but lacked carboxyl motifs (TfOH) or not acidic enough but contained carboxyl motifs (AcOH) did not react (Path 5). The result also showed that the free base by itself was not competent in the reaction. These observations pointed to the importance of acyl O-hydroxylamine protonation, as well as the carboxylate motif for productive reaction.
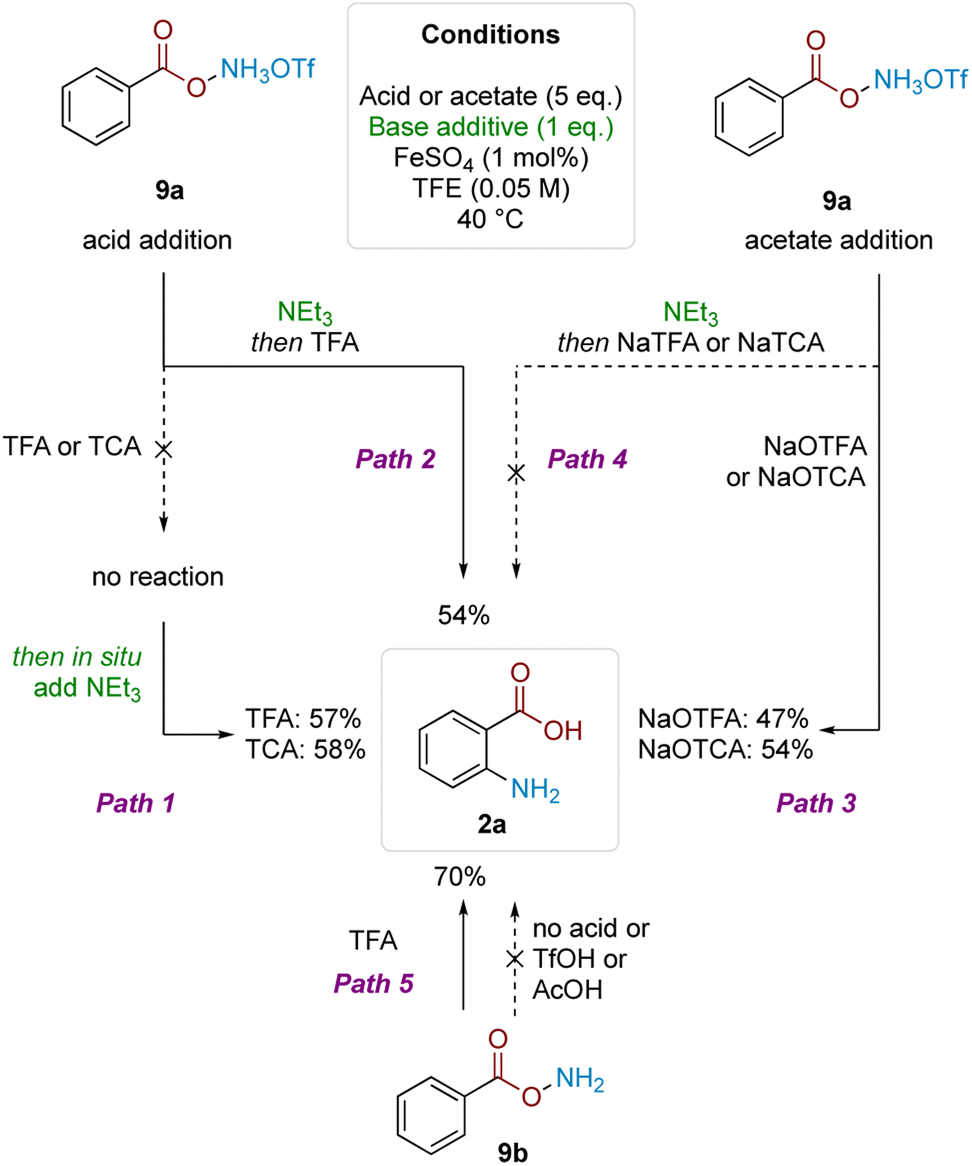 | ||
| Scheme 4 Studies to probe role of acid. | ||
The results previously outlined (Scheme 4) indicate that the crucial role of TFA must occur after Boc deprotection. We sought to identify the point in the reaction pathway downstream of Boc deprotection (i.e. N–O bond cleavage, arene amination or deprotonation/rearomatisation) where these features in TFA were crucial for reactivity. Reaction order analysis indicated a positive order in NaTFA when starting from ammonium triflate salt 9a, leading us to conclude that TFA is likely involved in the RDS (Fig. 2A, see ESI† for detailed discussion). Supporting experiments were next conducted to position the RDS in the reaction pathway. In brief, no kinetic isotope effect was observed,12c,d,16 suggesting against deprotonation-rearomatisation as rate determining (see ESI† for details).26 Additionally, modulation of arene electronics did not meaningfully alter the rate of product formation, suggesting against arene amination (Fig. 2B). Altogether, these experiments position the RDS—where TFA plays a vital role—at the N–O bond cleavage step.27
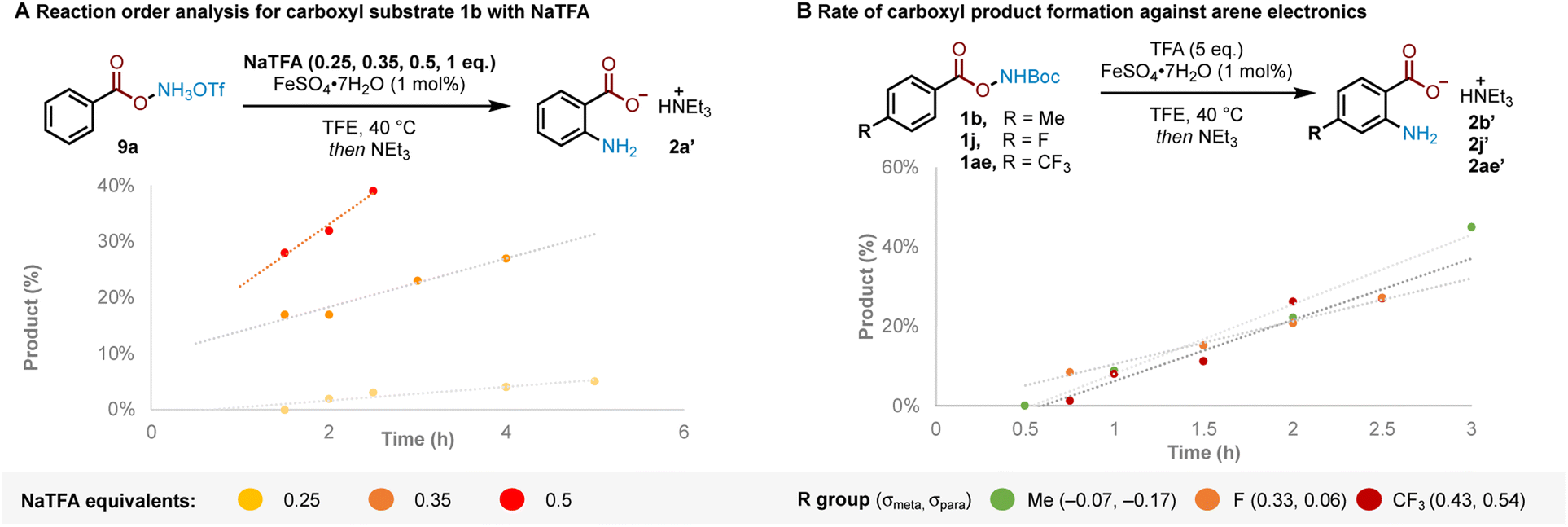 | ||
| Fig. 2 (A) Reaction order analysis in NaTFA for triflate salt 9a. (B) Rate-determining step (RDS) inference from rate of product formation with varying arene electronics. Yields determined by 1H NMR using 1,2-DME as an internal standard after quenching with Et3N and filtering through silica. | ||
Probing the role of the iron catalyst
Examining the reaction profile for 1b with and without iron catalysis revealed two quite different time course profiles (Fig. 3). Appreciable and consistent product formation was seen with Fe catalysis, whereas a clear induction period was observed in its absence, notably with an accumulation of the deprotected starting material during this period.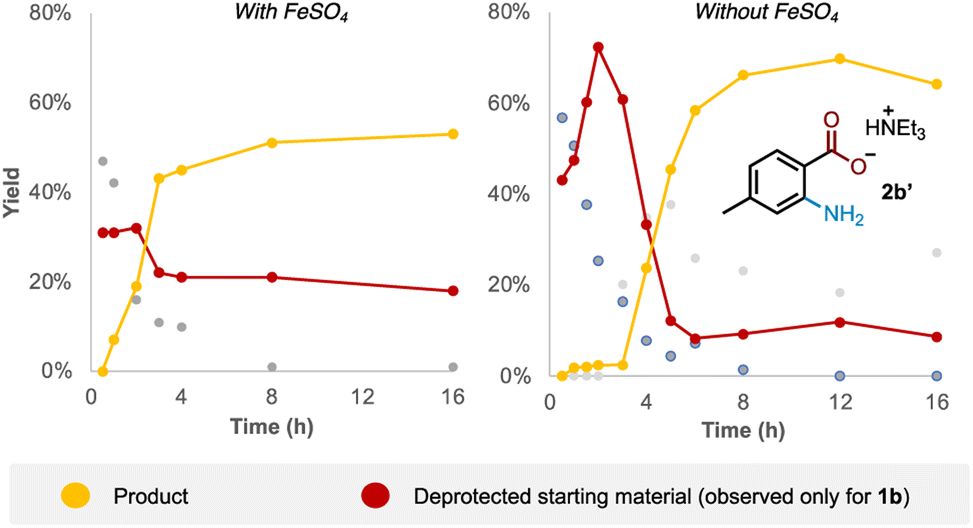 | ||
| Fig. 3 Time course reaction profile for 1b with and without iron catalysis. | ||
We next probed whether a radical mechanism was operative in the rearrangement of acyl O-hydroxylamines, which we had presumed to be the case for the corresponding sulfonyl substrates in our previous work. We subjected carboxyl substrate 1b to our standard Fe-catalysed and Fe-free aminative conditions with TEMPO as a stoichiometric additive, and compared the outcome by subjecting sulfonyl substrate 10 under the same conditions as a control. The addition of one equivalent of TEMPO heavily suppressed reactivity for the sulfonyl substrate 10 (Table 3; entries 1 and 2), as well as for the carboxyl substrate 1b without Fe catalysis (entry 3), strongly suggesting that a radical pathway is operating for these processes. However, reactivity was only modestly suppressed in the presence of Fe catalysis for 1b (entry 4). Furthermore there was no further reduction in product yield at superstoichiometric TEMPO loading (entry 5; 2 eq.). The latter experiment provides evidence against Fe solely serving as a radical initiator for a radical chain mechanism, although this possibility cannot be rigorously excluded at this stage.
|
|
|||
|---|---|---|---|
| Entry | Substrate | Catalyst (1 mol%) | Yielda (%) |
| a Yields determined by 1H NMR using 1,2-dimethoxyethane as an internal standard after quenching with Et3N and filtration over silica. b Two eq. TEMPO added. | |||
| 1 | 10 | — | 0% (82% without TEMPO) |
| 2 | 10 | FeSO4·7H2O | 6% (70% without TEMPO) |
| 3 | 1b | — | 6% (57% without TEMPO) |
| 4 | 1b | FeSO4·7H2O | 26% (54% without TEMPO) |
| 5b | 1b | FeSO4·7H2O | 25% (54% without TEMPO) |
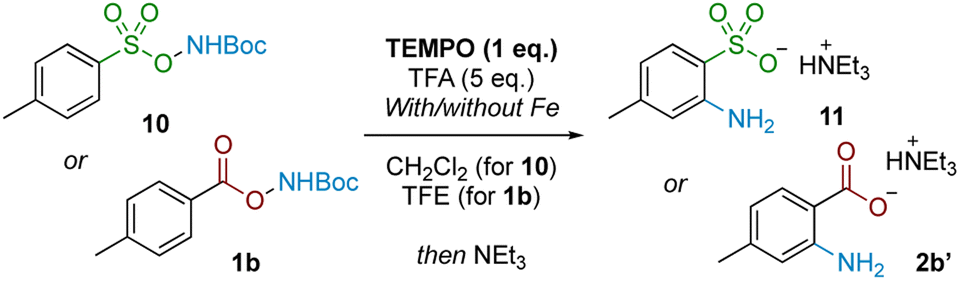
In conjunction with the divergent reaction profile, this observation raises the possibility that a radical mechanism may not fully account for reactivity under an Fe-catalysed regimen, and that a parallel mechanism may also be operating. Further evidence supporting an alternative, iron-catalysed pathway was seen from divergent reaction outcomes after subjecting styrenyl substrate 12 under Fe and Fe-free conditions. While no reaction was observed under iron-free conditions, under iron-catalysed conditions aminolactone 13 was isolated, potentially arising from nucleophilic attack from the neighbouring carboxylate motif into an arizidinyl intermediate 12a (Scheme 5). This correlates with the established reactivity of iron nitrenoid-mediated alkene aziridination, although an alternative mechanism involving a carbocationic intermediate cannot be rigorously excluded at this stage.27,28
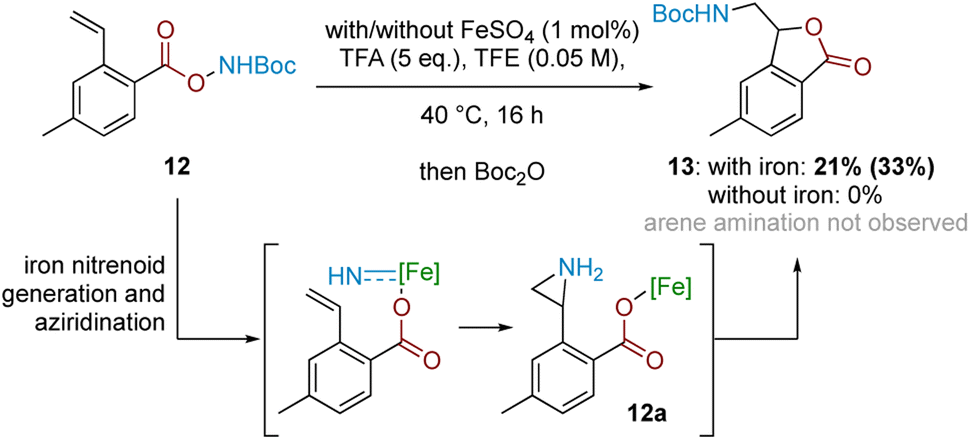 | ||
| Scheme 5 Mechanistic probe with styrenyl substrate 12. Bracketed yield denotes NMR yield. | ||
Summary of mechanistic findings
Combining findings from our above mechanistic experiments, we believe that, following Boc deprotection, the N–O reagent is protonated by TFA to give I. Experiments suggest that TFA is a crucial component of the rate-determining N–O cleavage step, potentially rendering the N–O bond more susceptible to cleavage and/or assisting in stabilising the formed intermediate (Scheme 6A).25,29,30 In an Fe-catalysed pathway, we speculate that the active aminating agent may involve an electrophilic iron nitrenoid species, based on known reactivity (see Scheme 5) and literature precedents,27,28,31 where Fe-assisted N–O bond reduction could generate a carboxylate-bound iron nitrenoid complex IIin situ.27 Several potential pathways (e.g. electrophilic amination, radical amination pathways etc.) could occur to deliver the product.28a Taking our crossover studies into account,32ortho selectivity is likely driven by the directing effect by the carboxylate group acting as a ligand for iron, where intramolecular arene amination occurs at the most proximal position. Accounting for the modest reduction in yield through TEMPO addition, as well as the fact that some substrates undergo conversion in the absence of a catalyst, a radical chain pathway likely occurs in parallel. This pathway would then become the sole productive pathway in the absence of Fe catalysis. For some substrates, this radical-chain pathway delivers high yields, though variability in initiation, aminative capture or unaccounted side-reactivity of the aminium radical could lead to poorer generalisability in this pathway (see ESI† for a comparison of various substrates with and without iron catalyst). From I, reductive N–O cleavage, most likely as part of a radical chain process,33 would generate a putative TFA-stabilised planar aminium radical cation (III, Scheme 6B). The observed selectivity most likely results from attractive NCIs (ion-pairing and hydrogen bonding) with the associated carboxyl motif. This would place the aminating agent proximal to the ortho position, in analogy to previously proposed related systems.17,18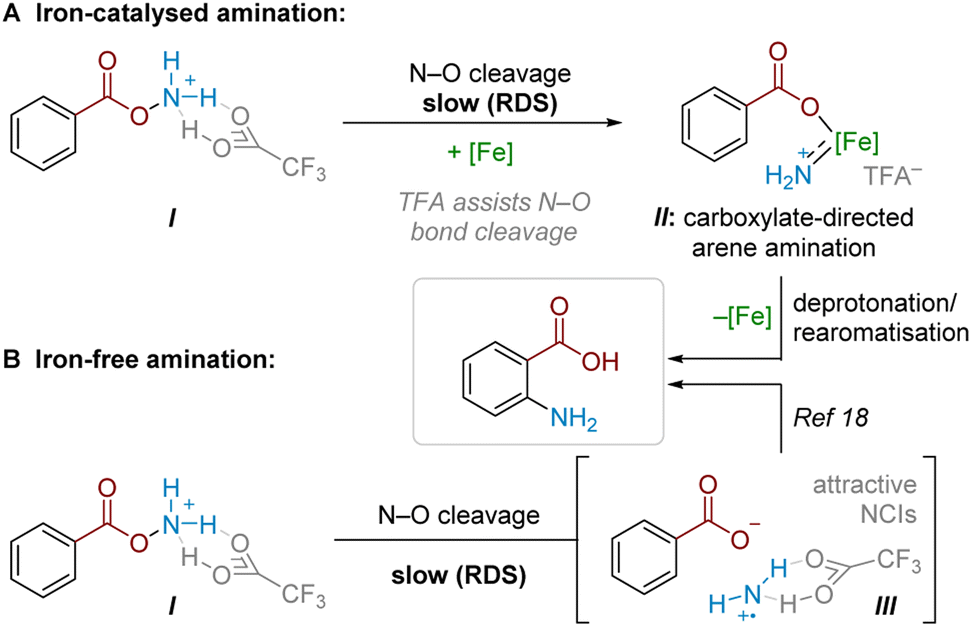 | ||
| Scheme 6 Proposed amination mechanisms. | ||
Conclusions
We have developed a practical method for the ortho-amination of benzoic and higher order arene carboxylic acid derivatives that proceeds via a facile rearrangement of acyl O-hydroxylamine derivatives. Notable for its efficacy on electron-deficient benzoic acids, this mild and procedurally straightforward method directly generates valuable anthranilic acids. The protocol is also applicable for higher order arene carboxylic acids, which undergo in situ cyclisation to generate heterocyclic products. Mechanistic studies suggest that the two mechanisms may be occurring in parallel, and we suggest that an iron catalyst may permit an iron nitrenoid mechanism to take place in concurrence with a radical-based amination pathway. The generally excellent ortho-selectivity is imagined to be dictated by the directing effect of the pendant carboxyl group for the former and attractive NCIs for the latter. Further investigations revealed the crucial role of TFA in this class of rearrangement, indicating that substrate protonation and activation via the non-innocent trifluoroacetate motif likely assists in the generation of the active aminating agent.This method complements the existing toolkit of site-selective C–N bond forming reaction, particularly on electron-deficient arenes, and represents a practical method to generate these ubiquitous motifs valuable in organic synthesis. Moreover, it has the potential be applied as a retrosynthetic strategy to expedite the synthesis of functionalised heterocyclic scaffolds from simple precursors. In addition, we hope that the mechanistic insights may be more broadly applicable to future reaction development of N–O reagents for electrophilic aminative processes.
Data availability
All data associated with this work can be found in the ESI.†Author contributions
The manuscript was written through the contributions of all authors and all authors have given approval to the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Royal Society for a PhD studentship (J. E. G.) and a University Research Fellowship (R. J. P.; URF\R\191003)). We also thank Trinity Hall, Cambridge for a Research Fellowship (N. Y. S. L.) and the ERC (Starting Grant NonCovRegioSiteCat, 757381) for funding.References
- (a) P. Wiklund and J. Bergman, Curr. Org. Synth., 2006, 3, 379 CrossRef CAS; (b) J. Schörghuber, L. Geist, M. Bisaccia, F. Weber, R. Konrat and R. J. Lichtenecker, J. Biomol. NMR, 2017, 69, 13 CrossRef PubMed; (c) P. Prasher and M. Sharma, Drug Dev. Res., 2021, 82, 945 CrossRef CAS PubMed; (d) N. Tezuka, K. Shimojo, K. Hirano, S. Komagawa, K. Yoshida, C. Wang, K. Miyamoto, T. Saito, R. Takita and M. Uchiyama, J. Am. Chem. Soc., 2016, 138, 9166 CrossRef CAS PubMed.
- (a) A. Millemaggi and R. J. K. Taylor, Eur. J. Org Chem., 2010, 2010, 4527 CrossRef; (b) M. Kaur, M. Singh, N. Chadha and O. Silakari, Eur. J. Med. Chem., 2016, 123, 858 CrossRef CAS PubMed; (c) Y. M. Khetmalis, M. Shivani, S. Murugesan and K. V. G. Chandra Sekhar, Biomed. Pharmacother., 2021, 141, 111842 CrossRef CAS PubMed.
- (a) H. Hayashi, Y. Miwa, I. Miki, S. Ichikawa, N. Yoda, A. Ishii, M. Kono and F. Suzuki, J. Med. Chem., 1992, 35, 4893 CrossRef CAS PubMed; (b) R. W. Carling, P. D. Leeson, K. W. Moore, J. D. Smith, C. R. Moyes, I. M. Mawer, S. Thomas, T. Chan and R. Baker, J. Med. Chem., 1993, 36, 3397 CrossRef CAS PubMed.
- T. Sandmeyer, Helv. Chim. Acta, 1919, 2, 234 CrossRef CAS.
- R. Stollé, Chem. Ber., 1914, 47, 2120 CrossRef.
- (a) W. C. Sumpter, Chem. Rev., 1944, 34, 393 CrossRef CAS; (b) S. Varun and R. Kakkar, MedChemComm, 2019, 10, 351 RSC.
- G. N. Walker, J. Am. Chem. Soc., 1955, 77, 3844 CrossRef CAS.
- J. V. Crivello, J. Org. Chem., 1981, 46, 3056 CrossRef CAS.
- (a) L. D. Tran, J. Roane and O. Daugulis, Angew. Chem., Int. Ed., 2013, 52, 6043 CrossRef CAS PubMed; (b) M. Shang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 3354 CrossRef CAS PubMed; (c) Z. Li, H. Yu and C. Bolm, Angew. Chem., Int. Ed., 2017, 56, 9532 CrossRef CAS PubMed; (d) M. Shang, Q. Shao, S.-Z. Sun, Y.-Q. Chen, H. Xu, H.-X. Dai and J.-Q. Yu, Chem. Sci., 2017, 8, 1469 RSC; (e) H. Kim, J. Heo, J. Kim, M.-H. Baik and S. Chang, J. Am. Chem. Soc., 2018, 140, 14350 CrossRef CAS PubMed; (f) L.-L. Xu, X. Wang, B. Ma, M.-X. Yin, H.-X. Lin, H.-X. Dai and J.-Q. Yu, Chem. Sci., 2018, 9, 5160 RSC; (g) H. Kim, K. Shin and S. Chang, J. Am. Chem. Soc., 2014, 136, 5904 CrossRef CAS PubMed; (h) J. Kim and S. Chang, Angew. Chem., Int. Ed., 2014, 53, 2203 CrossRef CAS PubMed; (i) T. Matsubara, S. Asako, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2014, 136, 646 CrossRef CAS PubMed; (j) Q. Yan, Z. Chen, W. Yu, H. Yin, Z. Liu and Y. Zhang, Org. Lett., 2015, 17, 2482 CrossRef CAS PubMed; (k) H.-Y. Thu, W.-Y. Yu and C.-M. Che, J. Am. Chem. Soc., 2006, 128, 9048 CrossRef CAS PubMed; (l) E. J. Yoo, S. Ma, T.-S. Mei, K. S. L. Chan and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 7652 CrossRef CAS PubMed; (m) C. Grohmann, H. Wang and F. Glorius, Org. Lett., 2012, 14, 656 CrossRef CAS PubMed; (n) K.-H. Ng, Z. Zhou and W.-Y. Yu, Org. Lett., 2012, 14, 272 CrossRef CAS PubMed; (o) K. Shin, Y. Baek and S. Chang, Angew. Chem., Int. Ed., 2013, 52, 8031 CrossRef CAS PubMed; (p) H.-W. Wang, Y. Lu, B. Zhang, J. He, H.-J. Xu, Y.-S. Kang, W.-Y. Sun and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 7449 CrossRef CAS PubMed; (q) M. Shang, S.-H. Zeng, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, Org. Lett., 2013, 15, 5286 CrossRef CAS PubMed; (r) M. R. Yadav, R. K. Rit and A. K. Sahoo, Org. Lett., 2013, 15, 1638 CrossRef CAS PubMed.
- (a) C. E. Houlden, M. Hutchby, C. D. Bailey, J. G. Ford, S. N. G. Tyler, M. R. Gagné, G. C. Lloyd-Jones and K. I. Booker-Milburn, Angew. Chem., Int. Ed., 2009, 48, 1830 CrossRef CAS PubMed; (b) R. Giri, J. K. Lam and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 686 CrossRef CAS PubMed; (c) Q. Li, S.-Y. Zhang, G. He, Z. Ai, W. A. Nack and G. Chen, Org. Lett., 2014, 16, 1764 CrossRef CAS PubMed.
- Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247 CrossRef CAS PubMed.
- For leading examples, see: (a) F. Minisci, R. Galli and M. Cecere, Tetrahedron Lett., 1965, 6, 4663 CrossRef; (b) A. Citterio, A. Gentile, F. Minisci, V. Navarrini, M. Serravalle and S. Ventura, J. Org. Chem., 1984, 49, 4479 CrossRef CAS; (c) L. Legnani, G. Prina Cerai and B. Morandi, ACS Catal., 2016, 6, 8162 CrossRef CAS; (d) J. Liu, K. Wu, T. Shen, Y. Liang, M. Zou, Y. Zhu, X. Li, X. Li and N. Jiao, Chem. - Eur. J., 2017, 23, 563 CrossRef CAS PubMed; (e) E. M. D'Amato, J. Börgel and T. Ritter, Chem. Sci., 2019, 10, 2424 RSC; (f) E. Falk, V. C. M. Gasser and B. Morandi, Org. Lett., 2021, 23, 1422 CrossRef CAS PubMed.
- V. C. M. Gasser, S. Makai and B. Morandi, Chem. Commun., 2022, 58, 9991 RSC.
- For reviews on electrophilic amination, see: (a) L. G. O'Neil and J. F. Bower, Angew. Chem., Int. Ed., 2021, 60, 25640 CrossRef PubMed; (b) D. Jinan, P. P. Mondal, A. V. Nair and B. Sahoo, Chem. Commun., 2021, 57, 13495 RSC; (c) Z. Zhou and L. Kürti, in Methodologies in Amine Synthesis, 2021, p. 1, DOI:10.1002/9783527826186.ch1.
- For examples of arene amination using N–O reagents without iron catalysis, see: (a) M. P. Paudyal, A. M. Adebesin, S. R. Burt, D. H. Ess, Z. Ma, L. Kürti and J. R. Falck, Science, 2016, 353, 1144 CrossRef CAS PubMed; (b) J. J. Farndon, X. Ma and J. F. Bower, J. Am. Chem. Soc., 2017, 139, 14005 CrossRef CAS PubMed; (c) J.-S. Yu, M. Espinosa, H. Noda and M. Shibasaki, J. Am. Chem. Soc., 2019, 141, 10530 CrossRef CAS PubMed; (d) Y. Y. See and M. S. Sanford, Org. Lett., 2020, 22, 2931 CrossRef CAS PubMed; (e) T. Wang, M. Hoffmann, A. Dreuw, E. Hasagić, C. Hu, P. M. Stein, S. Witzel, H. Shi, Y. Yang, M. Rudolph, F. Stuck, F. Rominger, M. Kerscher, P. Comba and A. S. K. Hashmi, Adv. Synth. Catal., 2021, 363, 2783 CrossRef CAS.
- For directed, iron-catalyzed arene functionalization using N–O reagents, see: R. R. Anugu, S. Munnuri and J. R. Falck, J. Am. Chem. Soc., 2020, 142, 5266 CrossRef CAS PubMed.
- J. E. Gillespie, C. Morrill and R. J. Phipps, J. Am. Chem. Soc., 2021, 143, 9355 CrossRef CAS PubMed.
- C. Morrill, J. E. Gillespie and R. J. Phipps, Angew. Chem., Int. Ed., 2022, 61, e202204025 CrossRef CAS PubMed.
- J. E. Gillespie, A. Fanourakis and R. J. Phipps, J. Am. Chem. Soc., 2022, 144, 18195 CrossRef CAS PubMed.
- C. Pratley, S. Fenner and J. A. Murphy, Chem. Rev., 2022, 122, 8181 CrossRef CAS PubMed.
- See ESI† for unsuccessful examples.
- L. Legnani, B. N. Bhawal and B. Morandi, Synthesis, 2017, 49, 776 CAS.
- (a) N. Haga, Y. Endo, K. Kataoka, K. Yamaguchi and K. Shudo, J. Am. Chem. Soc., 1992, 114, 9795 CrossRef CAS; (b) T. J. Snape, in Arene Chemistry, Wiley, 2015, ch. 18, p. 485, DOI:10.1002/9781118754887.ch18.
- In brief, the major product of amination, regardless of where the aminating agent was positioned, was for the sulfonyl product. This includes both when the aminating reagent was exogenously added and when the aminating N–O agent was located on the carboxyl substrate. See ESI† for further details.
- J. J. Farndon, T. A. Young and J. F. Bower, J. Am. Chem. Soc., 2018, 140, 17846 CrossRef CAS PubMed.
- (a) D. P. Curran and A. I. Keller, J. Am. Chem. Soc., 2006, 128, 13706 CrossRef CAS PubMed; (b) E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS PubMed.
- S. Chatterjee, I. Harden, G. Bistoni, R. G. Castillo, S. Chabbra, M. van Gastel, A. Schnegg, E. Bill, J. A. Birrell, B. Morandi, F. Neese and S. DeBeer, J. Am. Chem. Soc., 2022, 144, 2637 CrossRef CAS PubMed.
- (a) E. T. Hennessy, R. Y. Liu, D. A. Iovan, R. A. Duncan and T. A. Betley, Chem. Sci., 2014, 5, 1526 RSC; (b) D.-F. Lu, C.-L. Zhu, Z.-X. Jia and H. Xu, J. Am. Chem. Soc., 2014, 136, 13186 CrossRef CAS PubMed; (c) L. Legnani and B. Morandi, Angew. Chem., Int. Ed., 2016, 55, 2248 CrossRef CAS PubMed; (d) L. Legnani, G. Prina-Cerai, T. Delcaillau, S. Willems and B. Morandi, Science, 2018, 362, 434 CrossRef CAS PubMed; (e) S. Makai, E. Falk and B. Morandi, J. Am. Chem. Soc., 2020, 142, 21548 CrossRef CAS PubMed.
- (a) S. P. de Visser, J. Kaneti, R. Neumann and S. Shaik, J. Org. Chem., 2003, 68, 2903 CrossRef CAS PubMed; (b) A. Tlahuext-Aca, R. A. Garza-Sanchez and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 3708 CrossRef CAS PubMed.
- Single TFA co-ordination is illustratively shown in the proposed mechanism but involvement of more than one cannot be ruled out based on the present information. See ESI† for further discussion.
- Y. Zhou, J. Ni, Z. Lyu, Y. Li, T. Wang and G.-J. Cheng, ACS Catal., 2023, 13, 1863 CrossRef CAS.
- Further competition experiments suggest against carboxylate ligand exchange on the Fe catalyst as being responsible for the observed intermolecular crossover observed in Scheme 3D. See the ESI† for details.
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03293k |
| This journal is © The Royal Society of Chemistry 2023 |