
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoredox-catalyzed stereo- and regioselective vicinal fluorosulfonyl-borylation of unsaturated hydrocarbons†
Heyin
Li
,
Mengjun
Huang
,
Zhenlei
Zou
,
Zhen
Wang
,
Yifan
Li
,
Chao
Sun
,
Wangzhe
Chen
,
Yi
Pan
,
Weigang
Zhang
* and
Yi
Wang
 *
*
State Key Laboratory of Coordination Chemistry, Jiangsu Key Laboratory of Advanced Organic Materials, Collaborative Innovation Center of Advanced Microstructures, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: yiwang@nju.edu.cn
First published on 13th November 2023
Abstract
There has been considerable research on sulfur(VI) fluoride exchange (SuFEx) chemistry, which is considered to be a next-generation click reaction, and relies on the unique balance between reactivity and stability inherent in high valent organosulfur. The synthetic versatility of the bifunctional handles containing the fluorosulfonyl group presents great synthetic value and opportunity for drug discovery. However, the direct photoredox-catalyzed fluorosulfonyl-borylation process remains unexplored and challenging due to its system incompatibility and limited synthetic strategies. Herein, we developed a sequential photocatalytic radical difunctionalization strategy for the highly efficient stereoselective synthesis of vicinal fluorosulfonyl borides (VFSBs) with an integrated redox-active SO2F radical reagent. The VFSBs acted as orthogonal synthons, and were subjected to a range of convenient transformations via the cleavage of the C–B and S(VI)–F bonds, including halogenation, Suzuki coupling, hydrogenation, and the SuFEX click reaction, which demonstrated the great potential of the VFSB moieties for use in skeleton linkage and drug modification.
Introduction
Click chemistry has become an essential tool in the fields of drug discovery, materials science, and bioconjugation due to its practicality, modularity, and ability to attach complex substances for chemoproteomic, pharmacological, and various biomimetic applications.1–5 Extensive research has been conducted on sulfur(VI) fluoride exchange (SuFEx) chemistry as a next-generation click reaction6 that relies on unique chemical properties, including hydrolytic stability, resistance to reduction, and chemoselective reactivity at the sulfur center.7–18 The versatile bifunctional handles can be used as valuable linkers and key intermediates in organic synthesis and drug discovery.19–21 However, the construction of bifunctional connectors containing the fluorosulfonyl group, which depends on multiple functional groups for subsequent derivatization, and allowing for the linkage and modification of different drug molecules, has rarely been reported22,23 (Fig. 1A, bottom).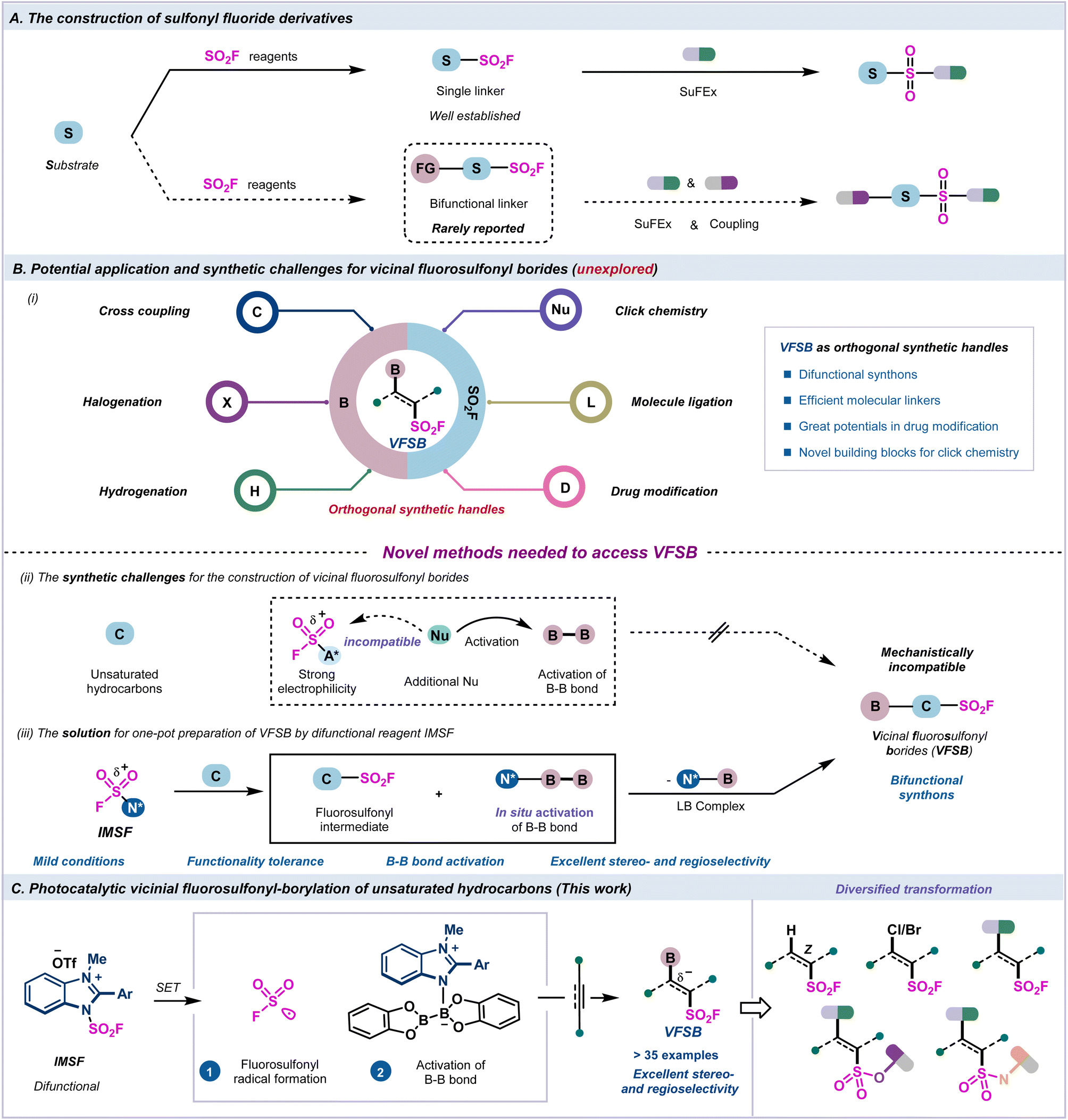 | ||
| Fig. 1 Origin of the reaction design. (A) The construction of sulfonyl fluoride derivatives. (B) Potential application and synthetic challenges for vicinal fluorosulfonyl borides. (C) Photocatalytic vicinal fluorosulfonyl-borylation of unsaturated hydrocarbons. | ||
Recently, indirect methods have been developed for the rapid installation of single fluorosulfonyl groups in organic molecules, including the chloride–fluoride exchange of sulfonyl chlorides,24–26 SO2 insertion/fluorination,27–30 electrophilic fluorination of thiols, and anodic oxidative fluorination.31–34 Additionally, direct functionalizations to access monofunctional sulfonyl fluoride compounds have been realized through emerging fluorosulfonylating reagents35–37 and sulfonyl fluoride building blocks38–43 (Fig. 1A, upper arrow). However, the preparation of bifunctional fluorosulfonyl borides through the direct fluorosulfonyl-borylation of unsaturated hydrocarbons is greatly challenging and thus remains unexplored.
The vicinal fluorosulfonyl borides (VFSBs) are novel and practical bifunctional molecular connectors with great application potential because they can be readily converted to a myriad of diversified and valuable compounds through the transformation of the C–B bond and S(VI)–F bond via halogenation,44 Suzuki coupling,45–49 hydrogenation,50 and elaboration of different drug molecules51–55 (Fig. 1B, i). Thus, there is a robust demand for the development of efficient synthetic strategies for the stereo- and regioselective introduction of fluorosulfonyl and boronated functionalities into unsaturated hydrocarbons.
Typically, a radical borylation process often employs nucleophilic Lewis basic solvents/mediators for the activation of diboron reagents via homolytic cleavage of B–B bonds.56–62 However, exogenous Lewis base-activated diboron species inevitably deplete highly active SO2F reagents, and are unable to engage in the desired fluorosulfonyl-borylation sequence (Fig. 1B, ii). Several issues need to be addressed due to the complexity of the radical process incorporating C–S and C–B bond formation, including the efficiency of B–B bond activation, the reactivities of sulfur- and boron-centered radicals, and the stereo- and regioselectivity of the radical additions to asymmetrical alkynes.
Recently, we developed an air-stable redox-active imidazolium fluorosulfonate (IMSF) reagent that was successfully employed for the stereoselective synthesis of alkenyl sulfonyl fluorides and functional alkyl sulfonyl fluorides.35 Inspired by the in situ activation strategy of radical borylation,44,63 we envisioned that the bench-stable radical fluorosulfonylating reagent IMSF would be ideal for use as an integrated difunctional reagent for the fluorosulfonyl-borylation of unsaturated hydrocarbons. This highly active cationic salt underwent the SET process to generate the SO2F radical. The endogenous imidazole residue subsequently activated the B–B bond for further borylation to realise the fluorosulfonyl-borylation process, and the use of an external Lewis base that may be detrimental to the SO2F moiety is unnecessary (Fig. 1B, iii).
Herein, we successfully prepared a series of VFSB derivatives with excellent stereo- and regioselectivity through a photocatalyzed radical fluorosulfonyl-borylation process (Fig. 1C).
Results and discussion
To showcase the protocol, we selected 3-cyclohexylpropyne (1a) as a pilot substrate to test the fluorosulfonyl-borylation (Table 1). After extensive efforts to identify the optimal conditions, we found that when using 2.5 equivalents of benzimidazolium sulfonate reagent (IMSF, 2a), 2 mol% of 4CzIPN, and 2.5 equivalents of 2,2′-bis-1,3,2-benzodioxaborole (B2Cat2,3) in EtOAc (2 mL) under the irradiation of 60 W blue LEDs, fluorosulfonyl boride 4a was obtained in 23% yield with >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Z/E ratio. Because the catechol boronate product is not stable, the crude reaction mixture was converted to the corresponding 1,1,2,2-tetraethylethylene glycol borate ester (B(EPin)), which is more stable than pinacol borate on silica gel.64
:1 Z/E ratio. Because the catechol boronate product is not stable, the crude reaction mixture was converted to the corresponding 1,1,2,2-tetraethylethylene glycol borate ester (B(EPin)), which is more stable than pinacol borate on silica gel.64
|
|
|||
|---|---|---|---|
| Entry | Variation from the above condition | Yielda |
Z:Eb |
|
a Yield determined by gas chromography (GC) using dodecane as an internal standard.
b The Z:E ratio was determined by 1H NMR and GC.
c IA = Isopropyl acetate.
d Isolated yield.
|
|||
| 1 | None | 23 | >20:1 |
| 2 | 2b instead of 2a | 8 | >20:1 |
| 3 | 2c instead of 2a | 9 | >20:1 |
| 4 | 2d instead of 2a | 5 | >20:1 |
| 5 | fac-Ir(ppy)3 instead of 4CzIPN | 11 | >20:1 |
| 6 | 90 W blue LEDs | 46 | >20:1 |
| 7 | IAc (1 mL), 90 W blue LEDs | 60 | >20:1 |
| 8 | IAc (0.6 mL), 90 W blue LEDs | 95 (70)d |
>20:1 |
| 9 | w/o 4CzIPN | 0 | — |
| 10 | In the darkness | 0 | — |
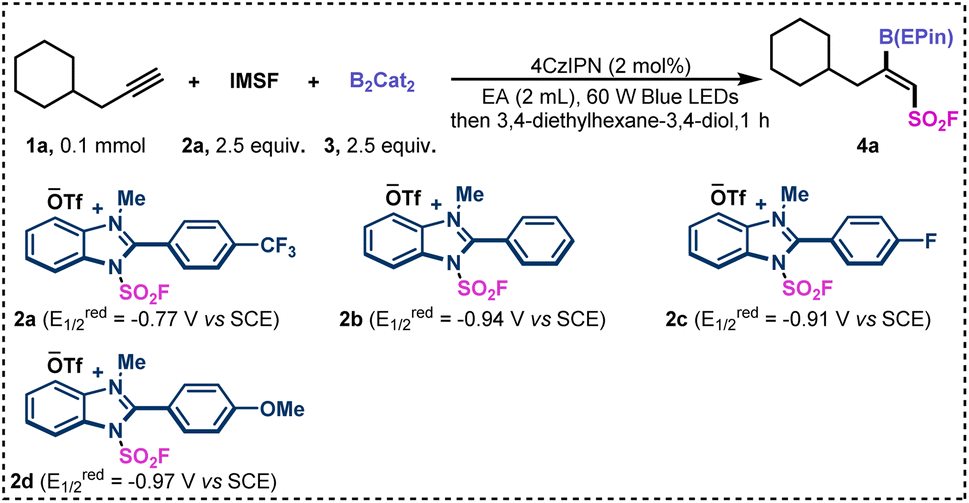
Next, the various benzimidazolium sulfonate reagents were examined (Table 1). When cationic reagents 2b–2d were used, the desired fluorosulfonyl boride 4a was obtained in much lower yields, which may due to the relatively high negative reduction potential (entries 2–4). The yield of 4a decreased when fac-Ir(ppy)3 was used as the photocatalyst instead of 4CzIPN (entry 5). When the reaction proceeded with 90 W blue LEDs, the borylated product was afforded in 46% yield (entry 6).
This result indicated that the light intensity was critical to the fluorosulfonyl-borylation of unsaturated hydrocarbons. When isopropyl acetate (IA) was used as the solvent for the reaction, the yield of product 4a was slightly increased (entry 7). The concentration of the reaction mixture was found to be significant for this transformation, as evidenced by the excellent yield (95% GC yield) obtained when the amount of solvent was decreased (entry 8). In addition, no product was detected in the absence of photocatalyst or light, demonstrating the essential role of all these components.
After we determined the optimized reaction conditions, we next examined the generality of this transformation with different alkynes (Scheme 1). First, we expanded the substrate scope for alkyl-substituted alkynes under standard conditions. Terminal alkynes bearing halides, aromatics, and aliphatics were able to uniformly furnish the target products (4b–4e) in moderate yields (52–62%) with high stereoselectivity. Subsequently, we extended the substrate scope to various alkynes containing saturated carbocyclic structures. Strained cyclopropyl- and cyclobutyl-derivatized substrates, especially fluorinated cyclopropyl- and cyclobutyl-derivatized alkynes, provided the corresponding vicinal fluorosulfonyl borides (4f–4h, 4j) in moderate to satisfactory yields. The reaction can also be applied to saturated five-membered carbocycle-derivatized alkynes to afford (Z)-selective fluorosulfonyl boronate 4i in 64% yield.
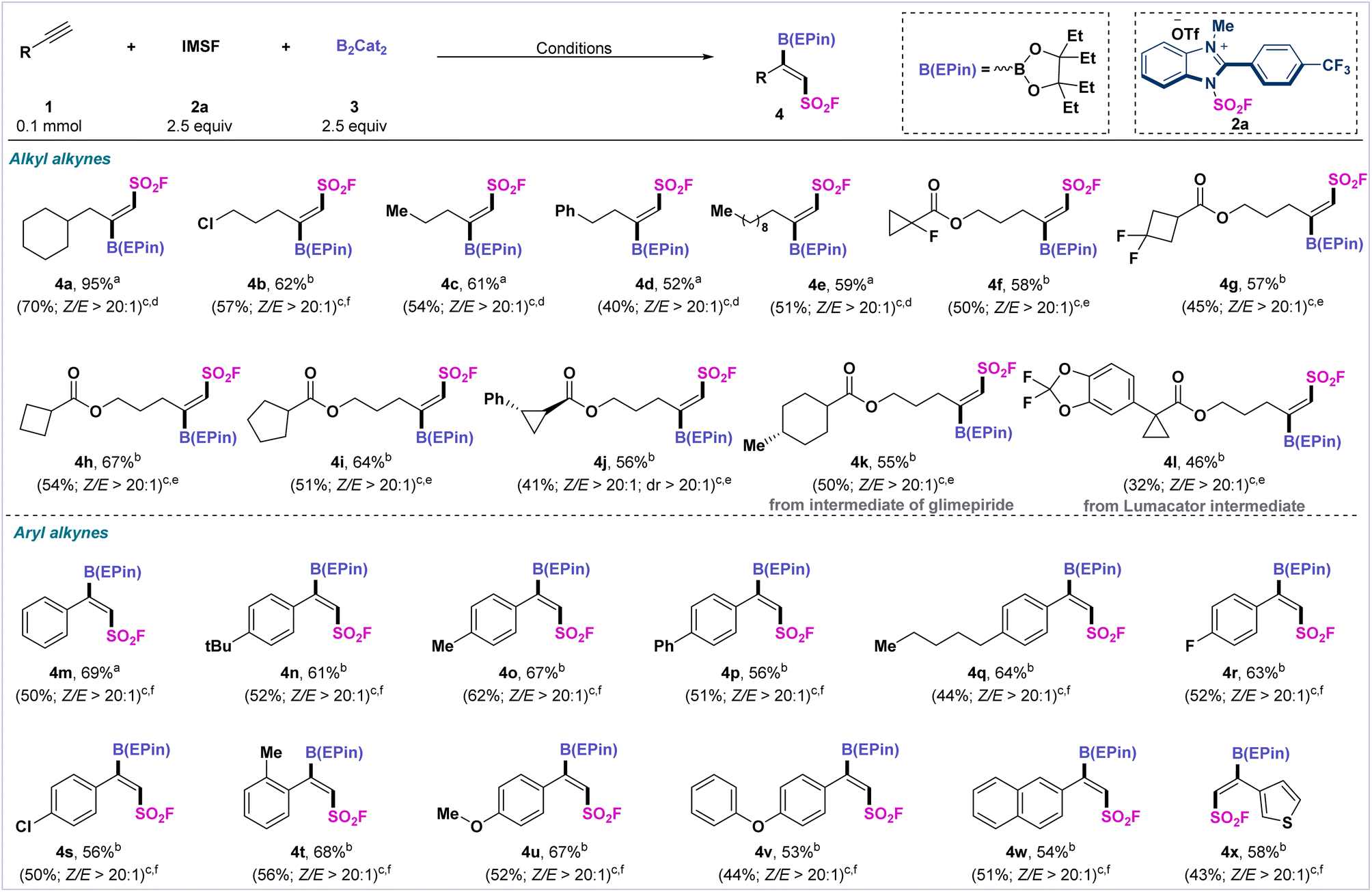 | ||
| Scheme 1 Substrate scope of the alkynes. a Crude yields determined by GC using dodecane as an internal standard. b Crude yields determined by 19F NMR spectroscopy using phenylmethylsulfonyl fluoride as an internal standard. c Values in parentheses are of isolated yields. d Condition A: all reactions were carried out with alkynes 1 (0.10 mmol), 2a (2.5 equiv.), B2Cat2 (3.0 equiv.), and 4CzIPN (2 mol%) in isopropyl acetate (0.6 mL) under Ar and 90 W blue LEDs for 40 min–12 h. Then, 3,4-diethyl-3,4-hexanediol was added to the reaction system, which was stirred for 1 h. e Condition B: isopropyl acetate: ethyl acetate = 2:1 (0.8 mL) was used. f Condition C: 13 h was used. | ||
We also selected a number of alkynes containing natural drug molecules to test the fluorosulfonyl-borylation process. Glimepiride- and lumacator-derivatized terminal alkynes afforded fluorosulfonyl borylated products (4k–4l) in moderate yields with high stereoselectivity. We also extended this fluorosulfonyl-borylation protocol to a range of aryl alkynes. Aryl alkynes with electro-withdrawing or electro-donating substituents produced the desired products 4m–4v in moderate to satisfactory yields with high stereo- and regioselectivity (Z:E > 20:1). Naphthyl- and thienyl-substituted alkynes were also readily converted into the (Z)-fluorosulfonyl products 4w and 4x in moderate yields.
The adjacent fluorosulfonyl alkylborides are also useful bifunctional linkers in relevant fields of drug design and bioconjugation. We applied this radical fluorosulfonyl-borylation protocol for the construction of vicinal fluorosulfonyl alkylborides. By slight variation of the standard conditions, we extended this radical fluorosulfonyl-borylation strategy to a series of unactivated alkenes (Scheme 2). The aryl-substituted butene derivatives afforded the target products 6a–6e in moderate to satisfactory yields (62–86%). Additionally, 1-vinylcyclohexane, allylbenzene, and dodecene were compatible with the reaction system and generated the desired products (6f, 6i–6j) in moderate to satisfactory yields (39–81%). Alkenes bearing boronate were also tolerated and transformed into desired product 6k. It is noteworthy that mono-substituted alkenes derived from liquid crystal building blocks, such as trans, trans-4-butyl-4′-vinylbicyclohexyl, and 4-p-tolyl-4′-vinylbi(cyclohexane), afforded β-fluorosulfonylborides (6l and 6m) in moderate yields. In addition, terminal alkenes bearing ester functionality were also tolerated, and the desired product was smoothly obtained (6n).
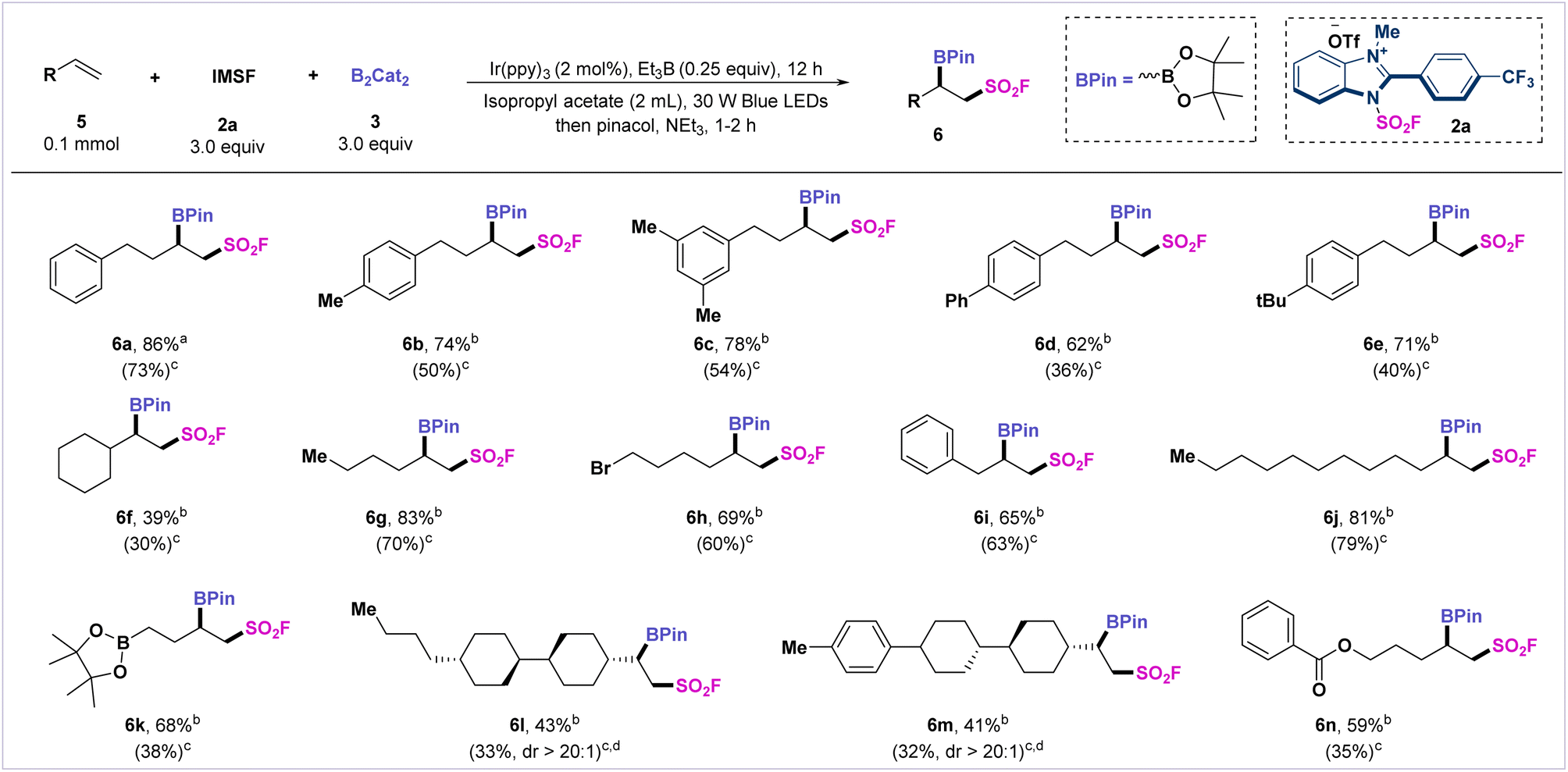 | ||
| Scheme 2 Substrate scope of the olefins. a Crude yields determined by GC using dodecane as an internal standard. b Crude yields determined by 19F NMR spectroscopy using phenylmethylsulfonyl fluoride as an internal standard. c Values in parentheses are of isolated yields. d The diastereomeric ratio was determined by 1H NMR and 19F NMR. | ||
To demonstrate the synthetic value of the VFSB moiety, derivatization of the fluorosulfonyl borides was carried out (Scheme 3). First, 4m was treated with copper bromide or copper chloride to afford β-bromo alkenylsulfonyl fluoride 7 or β-chloro alkenylsulfonyl fluoride 8. The palladium-catalyzed hydrogenation of 4m furnished the desired product 9 with a high Z/E ratio (>20:1). Palladium-catalyzed Suzuki–Miyaura cross-coupling of 4m with tetrahydro-4-pyrone-derived triflate 10 and bromobenzene 12 afforded the corresponding trisubstituted alkenes 11 (60%) and 13 (61%).
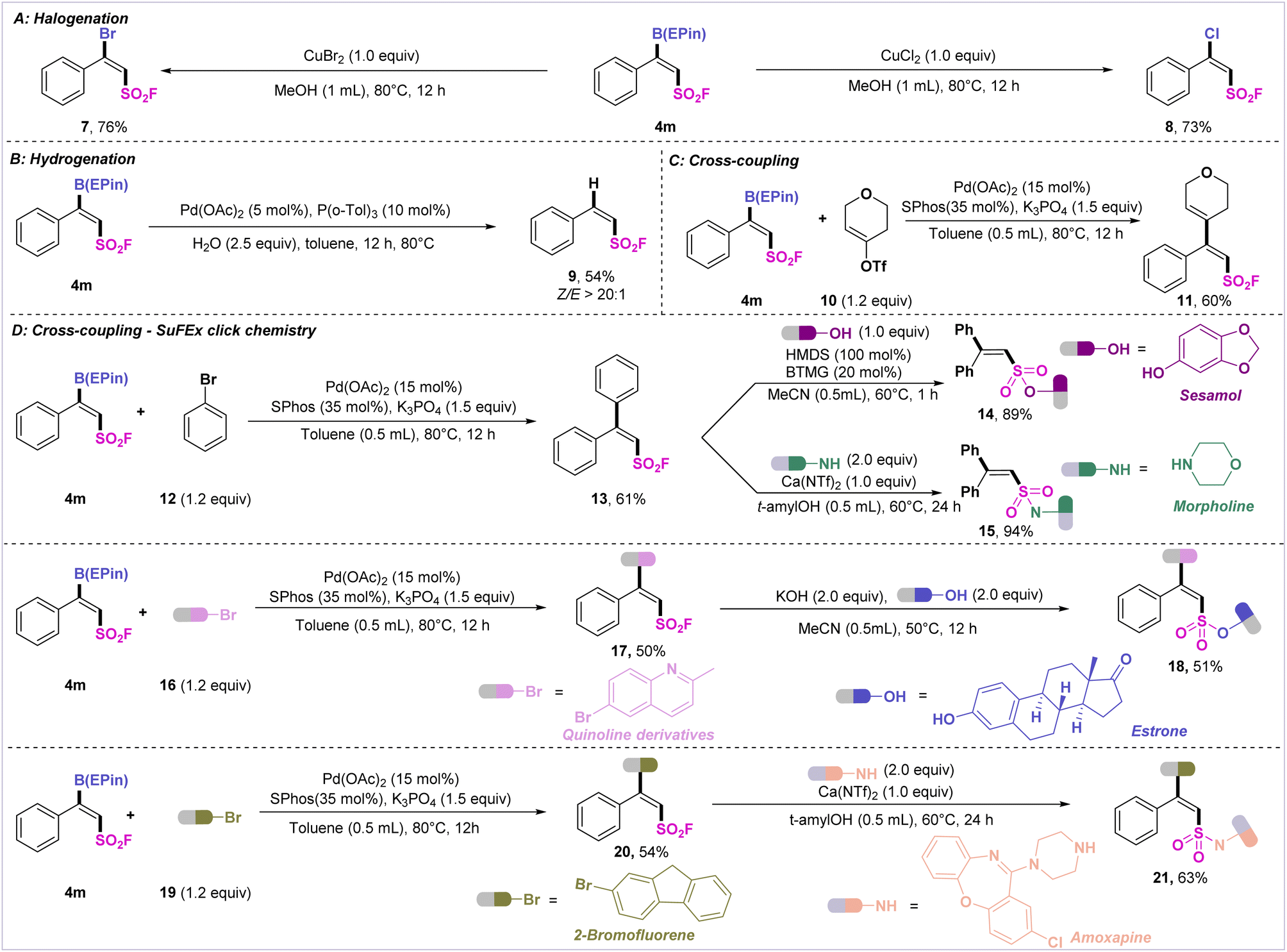 | ||
| Scheme 3 Synthetic applications. (A) Halogenation of fluorosulfonyl boride 4m. (B) Hydrogenation of borylated product 4m. (C) Suzuki–Miyaura cross-coupling of 4m with alkenyl sulfonate 10. (D) Sequential Suzuki–Miyaura cross-coupling and SuFEx click reaction for drug molecule elaboration. | ||
It is well known that sulfonyl fluoride species readily undergo various SuFEx reactions to connect other molecules. Thus, we tentatively attempted to ligate sulfonyl fluoride 13 with sesamol and morpholine, which afforded the desired sulfonated lipid product 14 and sulfonamide 15 in excellent yield. Finally, to verify the continuous functionalization of the VFSBs, 4m was coupled with bromoquinoline and estrone to access the difunctionalized 18. Also, the sequential linkage of 4m with 2-bromofluorene and amoxapine furnished 21 in high efficiency.
To gain insight into this reaction, several mechanistic experiments were carried out (Scheme 4). First, the luminescence quenching experiment indicated that an excited state photocatalyst (PC*) can be quenched by IMSF-2a, which involved an oxidative quenching catalytic cycle (Scheme 4A). Then, the 11B NMR experiments indicated that only in situ-generated imidazole residue can activate the diboron reagent, as evidenced by the newly generated peak of 19.31 ppm (Scheme 4B; for details, see ESI Supplementary Fig. 10–13†). Finally, the radical trapping experiment using 3 equivalents of TEMPO resulted in inhibition of the radical addition. The addition of radical scavenger 1,1-diphenylethylene under the standard conditions yielded only trace amount of the product 4a. The radical captive product 13 was isolated in 66% yield (Scheme 4C).
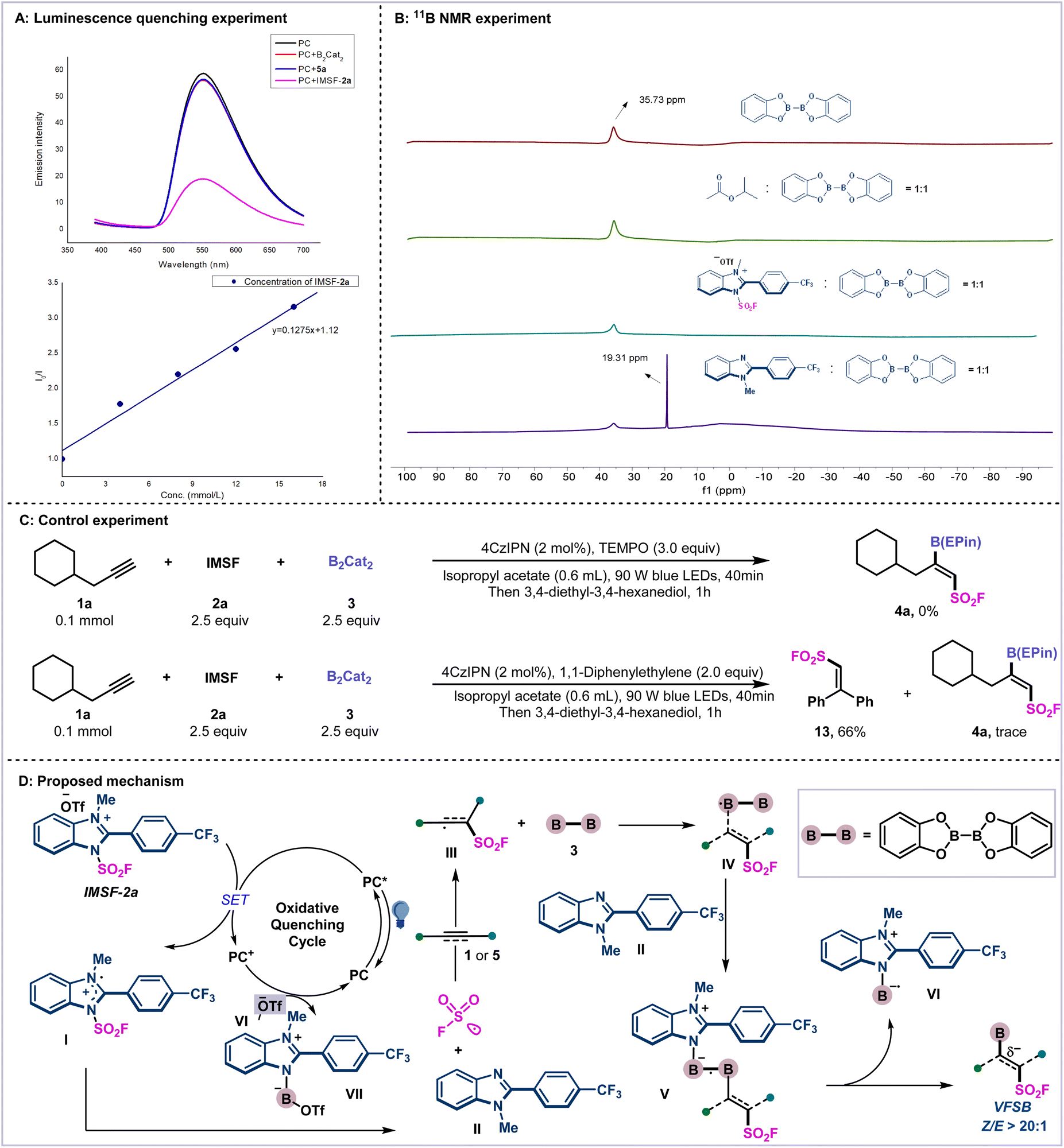 | ||
| Scheme 4 Mechanistic studies and proposed mechanism. (A) Luminescence quenching experiment. (B) 11B NMR experiments. (C) Control experiments. (D) Proposed mechanism. | ||
From the above mechanistic experiments, we speculate on the possible mechanism of the reaction in Scheme 4D. First, under irradiation, cationic reagent 2a can be reduced by PC* to generate radical intermediate I, and it releases the SO2F radical and imidazole residue II. Then, the addition of the SO2F radical to the alkynes regioselectively furnishes vinylic radical intermediate III. Subsequent addition of vinyl radical III to B2cat2 afforded Z-vinyl diboron radical species IV. The control of stereoselectivity is governed by steric repulsion between the fluorosulfonyl group and the boronates.44 Then, the activation of diboron reagent by in situ-generated imidazole residue II forms a highly reactive B–N heteroleptic intermediate V, which leads to the desired bifunctional products 4 or 6 and imidazole-stabilized boryl radical species VI. Finally, photo-oxidation of VI followed by coupling with −OTf affords boryl imidazolium salt VII and regenerates PC.
Experimental
General procedure for the synthesis of products 4a and 4c–4e. Condition A
Under argon, corresponding alkynes 1a and 1c–1e (0.1 mmol) were added to a solution of 4CzIPN (2 mol%), 2,2′-bis-1,3,2-benzodioxaborole 3 (0.25 mmol, 2.5 equiv.), and IMSF reagent 2a (0.2 mmol, 2.0 equiv.) in dried isopropyl acetate (0.6 mL) at room temperature. The tube was then exposed to a 90 W blue LED for approximately 40 min–12 h, after which 3,4-diethyl-3,4-hexanediol was added to the reaction system, which was stirred for 1 h. The reaction was monitored by TLC analysis to determine completion, and the reaction mixture was evaporated in vacuo. The crude products were directly purified by flash chromatography on silica gel to yield the desired products.General procedure for the synthesis of products 4b and 4f–4l. Condition B
Under argon, corresponding alkynes 1b and 1f–1l (0.1 mmol) were added to a solution of 4CzIPN (2 mol%), 2,2′-bis-1,3,2-benzodioxaborole 3 (0.25 mmol, 2.5 equiv.), and IMSF reagent 2a (0.2 mmol, 2.0 equiv.) in dried isopropyl acetate:ethyl acetate = 2:1 (0.8 mL) at room temperature. The tube was then exposed to a 90 W blue LED for approximately 40 min–12 h, after which 3,4-diethyl-3,4-hexanediol was added to the reaction system, which was stirred for 1 h. The reaction was monitored by TLC analysis for completion, and the reaction mixture was evaporated in vacuo. The crude products were directly purified by flash chromatography on silica gel to yield the desired products.
General procedure for the synthesis of products 4m–4x. Condition C
Under argon, corresponding alkynes 1m–1x (0.1 mmol) were added to a solution of 4CzIPN (2 mol%), 2,2′-bis-1,3,2-benzodioxaborole 3 (0.25 mmol, 2.5 equiv.), and IMSF reagent 2a (0.2 mmol, 2.0 equiv.) in dried isopropyl acetate (0.6 mL) at room temperature. The tube was then exposed to a 90 W blue LED for approximately 13 h, after which 3,4-diethyl-3,4-hexanediol was added to the reaction system, which was stirred for 1 h. The reaction was monitored by TLC analysis for completion, and the reaction mixture was evaporated in vacuo. The crude products were directly purified by flash chromatography on silica gel to yield the desired products.General procedure for the synthesis of product 6
Under argon, corresponding olefin 5 (0.1 mmol) was added to a solution of fac-Ir(ppy)3 (2 mol%), BEt3 (0.025 mmol, 0.25 equiv.), 2,2′-bis-1,3,2-benzodioxaborole 3 (0.3 mmol, 3.0 equiv.), and IMSF reagent 2a (0.3 mmol, 3.0 equiv.) in dried isopropyl acetate (2 mL) at room temperature. The tube was then exposed to a 30 W blue LED for approximately 12 h, after which pinacol and triethylamine was added to the reaction system, which was stirred for 1–2 h. The reaction was monitored by TLC analysis for completion, and the reaction mixture was evaporated in vacuo. The crude products were directly purified by flash chromatography on silica gel to yield the desired products.Conclusion
The first fluorosulfonyl-borylation of unsaturated hydrocarbons using inexpensive and scalable benzimidazolium sulfonate as the difunctional reagent was successfully established under mild conditions. The highly active cationic reagent IMSF generated the SO2F radical through the SET process and simultaneously in situ-generated imidazole residue to enable activation of the B–B bond. The vicinal fluorosulfonyl borides (VFSBs) are a combination of sulfonyl fluorides with boronates, and they provided a powerful bifunctional linker for selective assembly of different drug molecules. Based on the varieties of the VFSB scaffold, derivatizations of the borides and the sulfonyl fluorides can be readily realized, and they exhibit superior compatibility and extensive application of VFSB in the development of synthetic, bioconjugation, and medicinal tools.Data availability
The data that support the findings of this study are available in the ESI† or upon request from the corresponding author.Author contributions
H. L. conducted all experiments and characterized the novel compounds. Y. W., W. Z., and H. L. designed the experiments. W. Z., H. L., and Y. W. wrote the manuscript. Y. P. was responsible for the funding application. M. H., Z. Z., Z. W., Y. L., C. S., and W. C. contributed to the analysis and interpretation of the data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21971107, 2201101, and 22271147) and the China Postdoctoral Science Foundation (2021T140309 and 2021M691511).Notes and references
- P. Wu, M. Malkoch, J. N. Hunt, R. Vestberg, E. Kaltgrad, M. G. Finn, V. V. Fokin, K. B. Sharpless and C. J. Hawker, Chem. Commun., 2005, 2005, 5775–5777 RSC.
- L. Zhang, X. Chen, P. Xue, H. Y. Sun, I. D. Williams, K. B. Sharpless, V. V. Fokin and G. Jia, J. Am. Chem. Soc., 2005, 127, 15998–15999 CrossRef CAS PubMed.
- E. J. Yoo, M. Ahlquist, S. H. Kim, I. Bae, V. V. Fokin, K. B. Sharpless and S. Chang, Angew. Chem., Int. Ed., 2007, 46, 1730–1733 CrossRef CAS PubMed.
- M. Whiting, J. C. Tripp, Y.-C. Lin, W. Lindstrom, A. J. Olson, J. H. Elder, K. B. Sharpless and V. V. Fokin, J. Med. Chem., 2006, 49, 7697–7710 CrossRef CAS PubMed.
- A. Krasinski, V. V. Fokin and K. B. Sharpless, Org. Lett., 2004, 6, 1237–1240 CrossRef CAS PubMed.
- J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed.
- B. Gao, S. Li, P. Wu, J. E. Moses and K. B. Sharpless, Angew. Chem., Int. Ed., 2018, 57, 1939–1943 CrossRef CAS PubMed.
- S. Li, P. Wu, J. E. Moses and K. B. Sharpless, Angew. Chem., Int. Ed., 2017, 56, 2903–2908 CrossRef CAS PubMed.
- C. J. Smedley, Q. Zheng, B. Gao, S. Li, A. Molino, H. M. Duivenvoorden, B. S. Parker, D. J. D. Wilson, K. B. Sharpless and J. E. Moses, Angew. Chem., Int. Ed., 2019, 58, 4552–4556 CrossRef CAS PubMed.
- G. J. Brighty, R. C. Botham, S. Li, L. Nelson, D. E. Mortenson, G. Li, C. Morisseau, H. Wang, B. D. Hammock, K. B. Sharpless and J. W. Kelly, Nat. Chem., 2020, 12, 906–913 CrossRef CAS PubMed.
- S. Sun, B. Gao, J. Chen, K. B. Sharpless and J. Dong, Angew. Chem., Int. Ed., 2021, 60, 21195–21199 CrossRef CAS PubMed.
- G. Meng, T. Guo, T. Ma, J. Zhang, Y. Shen, K. B. Sharpless and J. Dong, Nature, 2019, 574, 86–89 CrossRef CAS PubMed.
- H.-L. Qin, Q. Zheng, G. A. L. Bare, P. Wu and K. B. Sharpless, Angew. Chem., Int. Ed., 2016, 55, 14155–14158 CrossRef CAS PubMed.
- J. Thomas and V. V. Fokin, Org. Lett., 2018, 20, 3749–3752 CrossRef CAS PubMed.
- P. Chatelain, C. Muller, A. Sau, D. Brykczynska, M. Bahadori, C. N. Rowley and J. Moran, Angew. Chem., Int. Ed., 2021, 60, 25307–25312 CrossRef CAS PubMed.
- Q. Zheng, H. Xu, H. Wang, W.-G. H. Du, N. Wang, H. Xiong, Y. Gu, L. Noodleman, K. B. Sharpless, G. Yang and P. Wu, J. Am. Chem. Soc., 2021, 143, 3753–3763 CrossRef CAS PubMed.
- F. Liu, H. Wang, S. Li, G. A. L. Bare, X. Chen, C. Wang, J. E. Moses, P. Wu and K. B. Sharpless, Angew. Chem., Int. Ed., 2019, 58, 8029–8033 CrossRef CAS PubMed.
- H. Zhou, P. Mukherjee, R. Liu, E. Evrard, D. Wang, J. M. Humphrey, T. W. Butler, L. R. Hoth, J. B. Sperry, S. K. Sakata, C. J. Helal and C. W. am Ende, Org. Lett., 2018, 20, 812–815 CrossRef CAS PubMed.
- H. Wang, J. Wu, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2022, 61, e202202061 CrossRef CAS PubMed.
- S. Jin, K. Liu, S. Wang and Q. Song, J. Am. Chem. Soc., 2021, 143, 13124–13134 CrossRef CAS PubMed.
- G. Gao, J. Yan, K. Yang, F. Chen and Q. Song, Green Chem., 2017, 19, 3997–4001 RSC.
- X. Nie, T. Xu, Y. Hong, H. Zhang, C. Mao and S. Liao, Angew. Chem., Int. Ed., 2021, 60, 22035–22042 CrossRef CAS PubMed.
- N. L. Frye, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2022, 61, e202115593 CrossRef CAS PubMed.
- K. O. Stepannikova, B. V. Vashchenko, O. O. Grygorenko, M. V. Gorichko, A. Y. Cherepakha, Y. S. Moroz, Y. M. Volovenko and S. Zhersh, Eur. J. Org Chem., 2021, 2021, 6530–6540 CrossRef CAS PubMed.
- T. A. Bianchi and L. A. Cate, J. Org. Chem., 1977, 42, 2031–2032 CrossRef CAS.
- S. R. Dubbaka and P. Vogel, Tetrahedron, 2005, 61, 1523–1530 CrossRef CAS.
- A. T. Davies, J. M. Curto, S. W. Bagley and M. C. Willis, Chem. Sci., 2017, 8, 1233–1237 RSC.
- T. S.-B. Lou, S. W. Bagley and M. C. Willis, Angew. Chem., Int. Ed., 2019, 58, 18859–18863 CrossRef CAS PubMed.
- Y. Liu, D. Yu, Y. Guo, J.-C. Xiao, Q.-Y. Chen and C. Liu, Org. Lett., 2020, 22, 2281–2286 CrossRef CAS PubMed.
- T. Zhong, J.-T. Yi, Z.-D. Chen, Q.-C. Zhuang, Y.-Z. Li, G. Lu and J. Weng, Chem. Sci., 2021, 12, 9359–9365 RSC.
- M. Kirihara, S. Naito, Y. Nishimura, Y. Ishizuka, T. Iwai, H. Takeuchi, T. Ogata, H. Hanai, Y. Kinoshita, M. Kishida, K. Yamazaki, T. Noguchi and S. Yamashoji, Tetrahedron, 2014, 70, 2464–2471 CrossRef CAS.
- L. Wang and J. Cornella, Angew. Chem., Int. Ed., 2020, 59, 23510–23515 CrossRef CAS PubMed.
- Y. Cao, B. Adriaenssens, A. de A. Bartolomeu, G. Laudadio, K. T. de Oliveira and T. Noël, J. Flow Chem., 2020, 10, 191–197 CrossRef CAS.
- G. Laudadio, A. de A. Bartolomeu, L. M. H. M. Verwijlen, Y. Cao, K. T. de Oliveira and T. Noël, J. Am. Chem. Soc., 2019, 141, 11832–11836 CrossRef CAS PubMed.
- W. Zhang, H. Li, X. Li, Z. Zou, M. Huang, J. Liu, X. Wang, S. Ni, Y. Pan and Y. Wang, Nat. Commun., 2022, 13, 3515 CrossRef CAS PubMed.
- X. Nie, T. Xu, J. Song, A. Devaraj, B. Zhang, Y. Chen and S. Liao, Angew. Chem., Int. Ed., 2021, 60, 3956–3960 CrossRef CAS PubMed.
- T. Guo, G. Meng, X. Zhan, Q. Yang, T. Ma, L. Xu, K. B. Sharpless and J. Dong, Angew. Chem., Int. Ed., 2018, 57, 2605–2610 CrossRef CAS PubMed.
- C. J. Smedley, G. Li, A. S. Barrow, T. L. Gialelis, M.-C. Giel, A. Ottonello, Y. Cheng, S. Kitamura, D. W. Wolan, K. B. Sharpless and J. E. Moses, Angew. Chem., Int. Ed., 2020, 59, 12460–12469 CrossRef CAS PubMed.
- B. Moku, W.-Y. Fang, J. Leng, E. A. B. Kantchev and H.-L. Qin, ACS Catal., 2019, 9, 10477–10488 CrossRef CAS.
- X. Zhang, W.-Y. Fang and H.-L. Qin, Org. Lett., 2022, 24, 4046–4051 CrossRef CAS PubMed.
- J. Leng, W. Tang, W.-Y. Fang, C. Zhao and H.-L. Qin, Org. Lett., 2020, 22, 4316–4321 CrossRef CAS PubMed.
- W.-Y. Fang, S.-M. Wang, Z.-W. Zhang and H.-L. Qin, Org. Lett., 2020, 22, 8904–8909 CrossRef CAS PubMed.
- A. S. Barrow, C. J. Smedley, Q. Zheng, S. Li, J. Dong and J. E. Moses, Chem. Soc. Rev., 2019, 48, 4731–4758 RSC.
- W. Zhang, Z. Zou, W. Zhao, S. Lu, Z. Wu, M. Huang, X. Wang, Y. Wang, Y. Liang, Y. Zhu, Y. Zheng and Y. Pan, Nat. Commun., 2020, 11, 2572 CrossRef CAS PubMed.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed.
- A. Suzuki, Acc. Chem. Res., 1982, 15, 178–184 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- A. C. Frisch and M. Beller, Angew. Chem., Int. Ed., 2005, 44, 674–688 CrossRef CAS PubMed.
- A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656–2670 CrossRef CAS PubMed.
- C. You, M. Sakai, C. G. Daniliuc, K. Bergander, S. Yamaguchi and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 21697–21701 CrossRef CAS PubMed.
- G.-. Zha, Q. Zheng, J. Leng, P. Wu, H.-L. Qin and K. B. Sharpless, Angew. Chem., Int. Ed., 2017, 56, 4849–4852 CrossRef CAS PubMed.
- Z. Liu, J. Li, S. Li, G. Li, K. B. Sharpless and P. Wu, J. Am. Chem. Soc., 2018, 140, 2919–2925 CrossRef CAS PubMed.
- S. Kitamura, Q. Zheng, J. L. Woehl, A. Solania, E. Chen, N. Dillon, M. V. Hull, M. Kotaniguchi, J. R. Cappiello, S. Kitamura, V. Nizet, K. B. Sharpless and D. W. Wolan, J. Am. Chem. Soc., 2020, 142, 10899–10904 CrossRef CAS PubMed.
- G.-F. Zha, S.-M. Wang, K. P. Rakesh, S. N. A. Bukhari, H. M. Manukumar, H. K. Vivek, N. Mallesha and H.-L. Qin, Eur. J. Med. Chem., 2019, 162, 364–377 CrossRef CAS PubMed.
- A. Marra, J. Dong, T. Ma, S. Giuntini, E. Crescenzo, L. Cerofolini, M. Martinucci, C. Luchinat, M. Fragai, C. Nativi and A. Dondoni, Chem.–Euro. J., 2018, 24, 18981–18987 CrossRef CAS PubMed.
- J. Wu, L. He, A. Noble and V. K. Aggarwal, J. Am. Chem. Soc., 2018, 140, 10700–10704 CrossRef CAS PubMed.
- F. Sandfort, F. S. Kalthoff, F. J. R. Klauck, M. J. James and F. Glorius, Chem.–Euro. J., 2018, 24, 17210–17214 CrossRef CAS PubMed.
- Y. Cheng, C. M. Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2018, 140, 6221–6225 CrossRef CAS PubMed.
- Y. Cheng, C. M. Lichtenfeld and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 16832–16836 CrossRef CAS PubMed.
- A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS PubMed.
- J. Hu, G. Wang, S. Li and Z. Shi, Angew. Chem., Int. Ed., 2018, 57, 15227–15231 CrossRef CAS PubMed.
- L. Candish, M. Teders and F. Glorius, J. Am. Chem. Soc., 2017, 139, 7440–7443 CrossRef CAS PubMed.
- T. M. Monos, R. C. McAtee and C. R. J. Stephenson, Arylsulfonylacetamides as bifunctional reagents for alkene aminoarylation, Science, 2018, 361, 1369–1373 CrossRef CAS PubMed.
- N. Oka, T. Yamada, H. Sajiki, S. Akai and T. Ikawa, Org. Lett., 2022, 24, 3510–3514 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03101b |
| This journal is © The Royal Society of Chemistry 2023 |