
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceResponsive calcium-derived nanoassemblies induce mitochondrial disorder to promote tumor calcification†
Yan
Zhao
a,
Xinquan
Yu
b,
Weiheng
Kong
a,
Rong-Mei
Kong
 a,
Ensheng
Zhang
a,
Lian
Xia
a,
Jing
Zhang
c,
Fengli
Qu
*ab and
Weihong
Tan
b
a,
Ensheng
Zhang
a,
Lian
Xia
a,
Jing
Zhang
c,
Fengli
Qu
*ab and
Weihong
Tan
b
aCollege of Chemistry and Chemical Engineering, Qufu Normal University, Qufu, Shandong 273165, China. E-mail: fengliquhn@hotmail.com
bDepartment of Pathology, Cancer Hospital of Zhejiang Province, Hangzhou Institute of Medicine, Chinese Academy of Sciences, Hangzhou, Zhejiang 310022, China
cSchool of Chemistry and Chemical Engineering, University of Jinan, Jinan, Shandong 250022, China
First published on 23rd August 2023
Abstract
Physiological calcification of the treated tumor area is considered to be a predictor of good prognosis. Promoting tumor calcification by inducing mitochondrial metabolic disorder and destroying calcium equilibrium has a potential inhibitory effect on tumor proliferation. Here, by promoting calcification by inducing mitochondrial dysfunction combined with triggering a surge of reactive oxygen species, we construct a bioresponsive calcification initiator, termed CaP-AA, using CaHPO4 covalently doped L-ascorbic acid. CaHPO4 releases Ca2+ within the cytoplasm of tumor cells to trigger calcium overload. Meanwhile, exogenous L-ascorbic acid indirectly enhances metabolic balance disruption via pro-oxidant effects. Such Ca2+ overload increases the likelihood of tumor calcification in vivo for tumor inhibition by perturbing mitochondrial homeostasis. The introduction of responsive calcium sources that would, in turn, trigger intratumoral calcification mediated by perturbing mitochondrial homeostasis would be an effective regulatory strategy for tumor therapy.
Introduction
In clinical oncology treatment, some cancer patients would develop internal tumor calcification after radiotherapy, accompanied by a better treatment outcome over time.1,2 Calcification suppresses the proliferation and invasive migration ability of tumor cells, which has been reported as a predictor of prognosis.3 To trigger intratumoral calcification, an exogenous calcium stimulation strategy has been introduced by causing an intracellular Ca2+ increase,4–6 such as calcium carbonate (CaCO3) and calcium peroxide (CaO2).7,8 Increased intracytoplasmic Ca2+ can provoke tumor cells to assume a state of calcium overload.9 However, simply inducing calcium overload may not have the desired optimal effect on tumor injury because steady-state calcium equilibrium can still be sustained through a regulatory mitochondrial calcium signaling network.10,11 That is, through calcium transport channels, the Ca2+ concentration can remain at a normal level, resulting in an inferior anticancer effect.12 To address this dilemma, an in-depth understanding of calcium overload induction and the synergism that originates from mitochondrial regulation in promoting calcium overload can lead to new avenues toward inducing a more desirable calcification outcome in the treatment of tumors.Cellular mitochondria, as key organelles for cellular communication and signal transduction,13,14 play a central regulatory role in integral calcium-related metabolic balance.15 Disruption of mitochondrial metabolism has been validated as an assisted tumor therapy in a number of clinical trials.16,17 In previous studies, transition metal complexes,18 guanidinium salts19 and specific peptides20 could act directly or indirectly on mitochondria by inducing the proliferation of reactive oxygen species (ROS) within the tumor microenvironment (TME). The resultant oxidative stress could induce mitochondrial dysfunction in tumors, thereby interfering with mitochondrial regulation and causing, in turn, effects like stimulating ATP production, inhibiting autophagy, rectifying intracellular physiological functions, and, finally, regulating cell death.21 Disruption of mitochondrial metabolic balance, as a breakthrough in the adjuvant/synergistic enhancement of tumor calcification therapeutic efficacy, is a key determinant of designable therapeutic strategies.
Here, a dual-path effect Ca-derived nanopharmaceutical is established, working as an inducer of mitochondrial disorder to promote tumor calcification by employing tumor acidic-responsive CaHPO4 covalently doped with L-ascorbic acid (L-AA), abbreviated as CaP-AA nanoassemblies. Bioresponsive CaP-AA could release Ca2+ and L-AA within the TME (Scheme 1). (i) Exogenous L-AA is different from the previous cognition of an in vitro antioxidant; in tumor extracellular fluid, pharmacological concentrations of L-AA have been proved to promote in vivo hydrogen peroxide by inducing a pro-oxidation effect, which can cause changes in cellular metabolic activity after its spread into the cells, and these findings have been confirmed repeatedly in many studies.22–24 First, the above effects of L-AA can indirectly change cellular functions and have an indirect effect on the mitochondrial homeostasis disruption (path 1). Second, it augments the disruption of Ca2+ metabolic balance, an effect shown in path 2. (ii) Ca2+ released from CaHPO4 triggers calcium overload, promoting mitochondrial dysfunction in tumor cells, which contributes to excessive ROS production,25,26 accompanied by membrane lipid peroxidation27 and breakdown of the cell membrane structure,28,29 finally resulting in nucleus DNA degradation.30,31 In addition, mitochondrial disorder further increases calcium overflow (path 2) with mitochondrial permeability increasing and intracellular molecules non-selectively spreading between cytosol and mitochondria, leading to destruction of the membrane potential, capacity unit ATP extravasation and mitochondrial swelling.32 With such dual-path effect, this metabolic destruction-assisted calcification therapeutic strategy could induce apoptosis and prompt calcification of tumor cells, showing potential utility in clinical application as a complement for conventional clinical oncology treatment.
 | ||
| Scheme 1 Bioresponsive CaP-AA acts as a calcifying pharmacological agent by inducing mitochondrial disorder and calcium overloading. Path 1: induced metabolic imbalance to promote mitochondrial damage; path 2: mitochondrial disorder to increase calcium overload. | ||
Results
Characterization of acid-responsive CaP-AA
The construction of acid-responsive nanoassemblies with excellent performance hypothesizes that the material should have significant pre- and post-response variability, maintaining a stable before response and disintegrating rapidly during the response. Here, CaP-AA nanoassemblies were constructed by adding PO43− to calcium ascorbate. The strong coordination between PO43− and Ca2+ could replace part of L-AA, thereby forming CaP-AA self-assembled nanoparticles with CaHPO4 as the framework. As shown in Fig. 1A and S1A,† the self-assembled CaP-AA showed nanoscale agglomerates in the transmission electronic microscopy (TEM) image with a hydrodynamic diameter of ∼100 nm, as determined by dynamic light scattering (DLS). It could retain its particle stability for a long time in complex neutral environments such as serum (Fig. S1B†). The X-ray diffraction (XRD) pattern of CaP-AA largely matched the crystal phase of CaHPO4·2H2O (JCPDS No. 72-1240) (Fig. 1B),33 indicating the phase purity of CaP-AA. In addition, the Fourier transform infrared spectroscopy (FT-IR) spectrum showed that CaP-AA had characteristic absorption peaks of CaHPO4 (1300–600 cm−1) and CaAA (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OH: 3600–3100 cm−1 and CC: 2600–1600 cm−1), respectively (Fig. 1C),34,35 suggesting the successful assembly of CaP-AA.
OH: 3600–3100 cm−1 and CC: 2600–1600 cm−1), respectively (Fig. 1C),34,35 suggesting the successful assembly of CaP-AA.
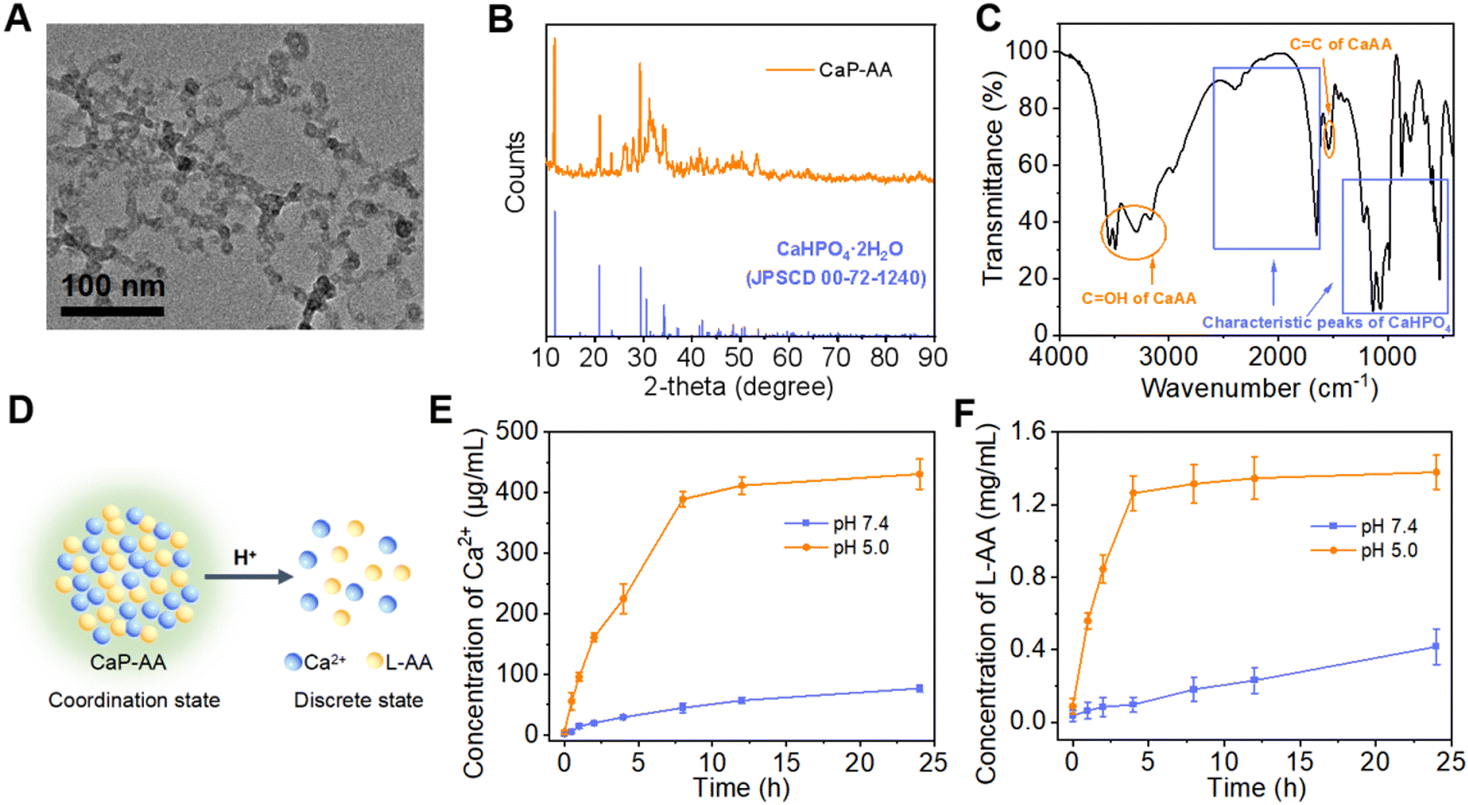 | ||
| Fig. 1 Structure and response properties of CaP-AA. (A) TEM image of CaP-AA. (B) XRD patterns of CaP-AA and the standard CaHPO4·2H2O (JCPDS No. 00-72-1240). (C) FT-IR spectrum of CaP-AA. (D) Schematic of the acid-responsive cleavage of CaP-AA. Plots showing the concentration of (E) Ca2+ and (F) L-AA releasing from CaP-AA after incubation with different pH conditions vs. time. | ||
The potential acid-response properties of CaP-AA (Fig. 1D) were investigated as shown in Fig. S1A;† when CaP-AA was transferred from neutral solution to pH 5 buffer, its hydrodynamic diameter was gradually reduced from ∼100 nm to ∼10 nm, accompanied by a change of zeta potential from negative to positive (Fig. S1C†), indicating that Ca2+ was released during the disintegration of CaP-AA in acidic solution. Moreover, as shown in Fig. 1E and F, at a fixed acidity (pH 5.0), the releasing rates of Ca2+vs.L-AA were positively correlated during the acidolysis of CaP-AA, which was sufficient to verify that CaP-AA was assembled by Ca2+ and L-AA (see Fig. S2† for the detailed quantification method of L-AA).
In vitro calcium overload and mitochondrial disorder
The aforementioned in-solution experiments demonstrated that CaP-AA disintegrated into Ca2+ and L-AA under acidic conditions. At the cellular level, as shown in Fig. 2A, the intracellularly increased Ca2+ could first cause calcium overload, which, in turn, would affect mitochondrial metabolic efficiency by inhibiting mitochondrial respiration. Subsequently, under the premise of metabolic damage, overloaded Ca2+ could increase the likelihood of tumor calcification in vivo for tumor inhibition. This dual-interference strategy could inevitably affect tumor cell proliferation. As shown in Fig. 2B, inhibiting the viability of 4T1 cells was assessed by the MTT cytotoxicity test. CaP-AA exhibited a more pronounced inhibition of proliferation capacity compared to the L-AA or Ca2+ group with the same L-AA/Ca2+ concentration. It was worth mentioning that CaP exhibited a priori toxicity, possibly attributed to the higher concentrations of Ca2+ and PO43− following the acid response, affecting cellular ion homeostasis. Meanwhile, the formation of calcium phosphate precipitates in the cellular environment could also have implications for cell survival. CaP-AA showed lower inhibition against normal 293T cells compared to 4T1 cells (Fig. S3†), mainly owing to the relatively neutral environment of normal cells, accounting for the failed response to acid-responsive CaP-AA. It was worth mentioning that L-AA inhibited the proliferation of tumor cells at high doses, which was also consistent with the conclusions of previous studies,22,23 confirming that L-AA at specific doses would have irreversible effects on tumor cellular activity. Previous studies have proven that pharmacological concentrations of L-AA did not kill tumor cells directly, but by supplying H2O2 as a precursor drug form. Based on this, we draw a conclusion that the inhibition of tumor cell proliferation was caused by the synergistic action of Ca2+ and L-AA. As shown in Fig. 2C and D, using the ROS indicator DCFH-DA, the green fluorescence of CaP-AA-treated groups was caused by calcium-induced mitochondrial imbalance, which could further promote the increase of intracellular oxidative stress. The L-AA group did not cause obvious green fluorescence, which we hypothesized was because the extracellular media were different from the extracellular matrix of tumors in vivo, and thus the pro-oxidation effect of L-AA at this relatively low current dosage was not large enough to be detected by fluorescence imaging, which was not contrary to the inhibition of cell proliferation caused by high doses of L-AA.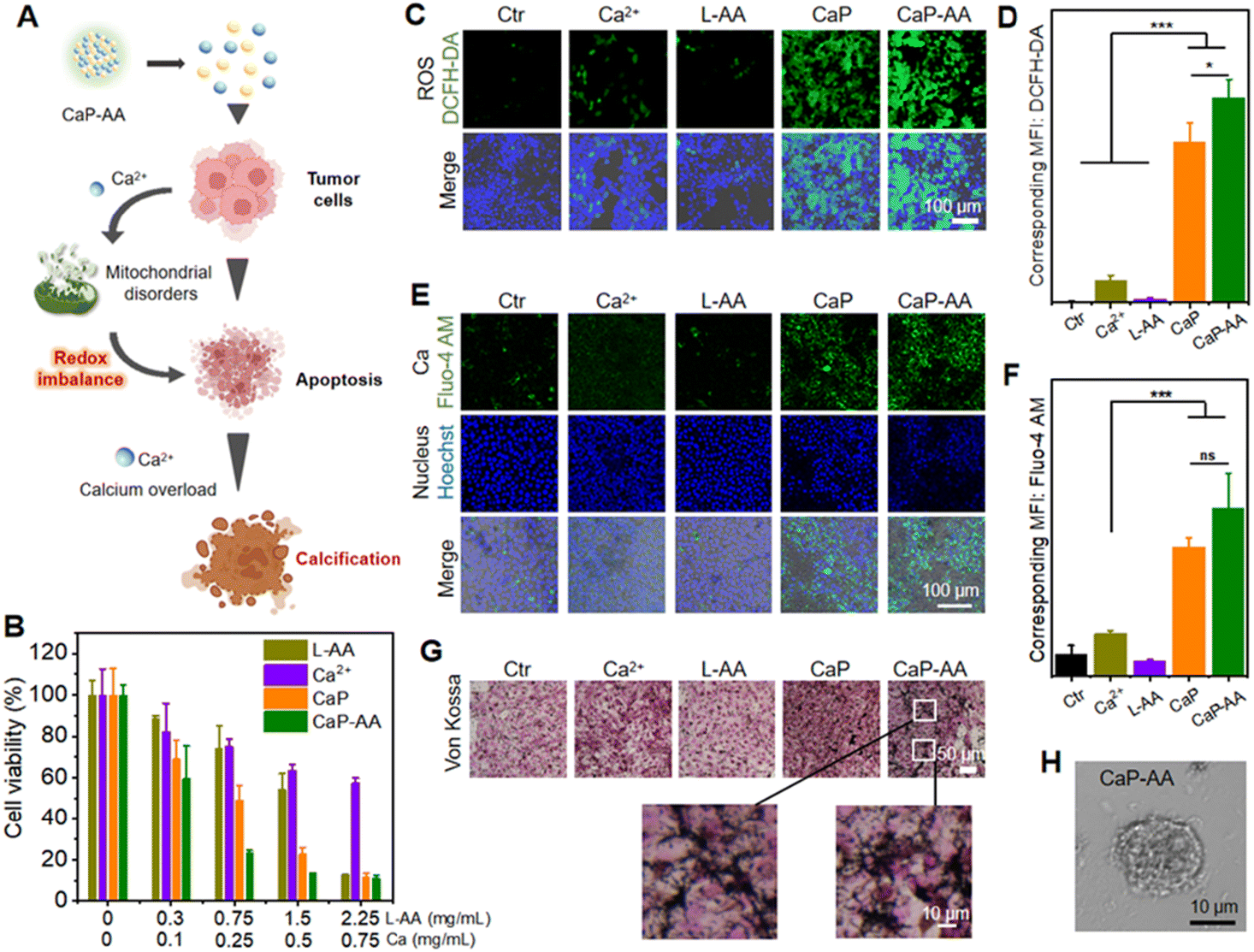 | ||
| Fig. 2 Intracellular ROS and calcium overload. (A) Schematic of CaP-AA enhancing calcium deposition by inducing oxidative stress. (B) Cell viability of 4T1 cells incubated with different concentrations of indicated NPs (quantified by Ca and L-AA, respectively). (C) DCFH-DA-stained 4T1 cells after different treatments (green: ROS generation; blue: nucleus). (D) Corresponding intracellular ROS content after various treatments. (E) Calcium generation in 4T1 cells after 6 h incubation of different treatments (red: intracellular Ca2+ content; blue: nucleus). (F) Corresponding intracellular Ca content after various treatments (G) Von Kossa-stained 4T1 cells after different treatments. (H) Morphology of 4T1 cells after co-incubation with CaP-AA for 24 h. The significance of the difference was determined through one-way analysis of variance (ANOVA) by *p < 0.05, **p < 0.01, ***p < 0.001, (ns) p ≥ 0.05. | ||
Further determination of calcium release from CaP-AA was accomplished by using the calcium probe Fluo-4 AM as a calcium indicator (Fig. 2E, F and S4†). As compared to L-AA, the addition of CaP or CaP-AA could induce an obvious intracellular green fluorescence increase, indicating that acid-responsive CaHPO4 possessed the potential to increase cellular calcium. In addition, as shown in Fig. 2G, with the increased cellular calcium content of CaP and CaP-AA groups, a large amount of black calcium sedimentation was observed in Von Kossa calcium staining, indicating that excess intracellular calcium could induce calcium sedimentation. This might also contribute to the formation of calcified foci for tumor treatment. In addition, as shown in Fig. 2H, CaP-AA could affect the cell morphology where CaP-AA induced nucleus protrusion, cytoplasmic crinkling, cytosolic damage and lysis accompanied by numerous cell fragments scattered about the typical features of calcified cells.
As mentioned before, the CaP-AA-induced intracellular calcium overload production and redox imbalance could induce mitochondrial imbalance, achieved by influencing the several pathways related to mitochondrial metabolism, e.g., mitochondrial morphology, mitochondrial ATP production ability and mitochondrial membrane potential (Fig. 3A). A mitochondrial localization test was performed, and CaP-AA co-incubated cells possessed a condensed nucleus with weak mitochondrial staining, accompanied by a decrease in ATP production, verifying mitochondrial inactivation (Fig. 3B–D). JC-1 staining was used to monitor mitochondrial integrity in 4T1 cells. As shown in Fig. 3E and F, the fluorescence shift of JC-1 from red to green (CaP-AA vs. Ctr) implied a decrease in mitochondrial membrane potential, which was a sign of early apoptosis, reflecting that CaP-AA could cause mitochondrial dysfunction during antitumor treatment. In summary, at the cellular level, acid-responsive CaP-AA could contribute to mitochondrial imbalance and promote apoptosis by producing a combined effect of calcium overload and redox imbalance.
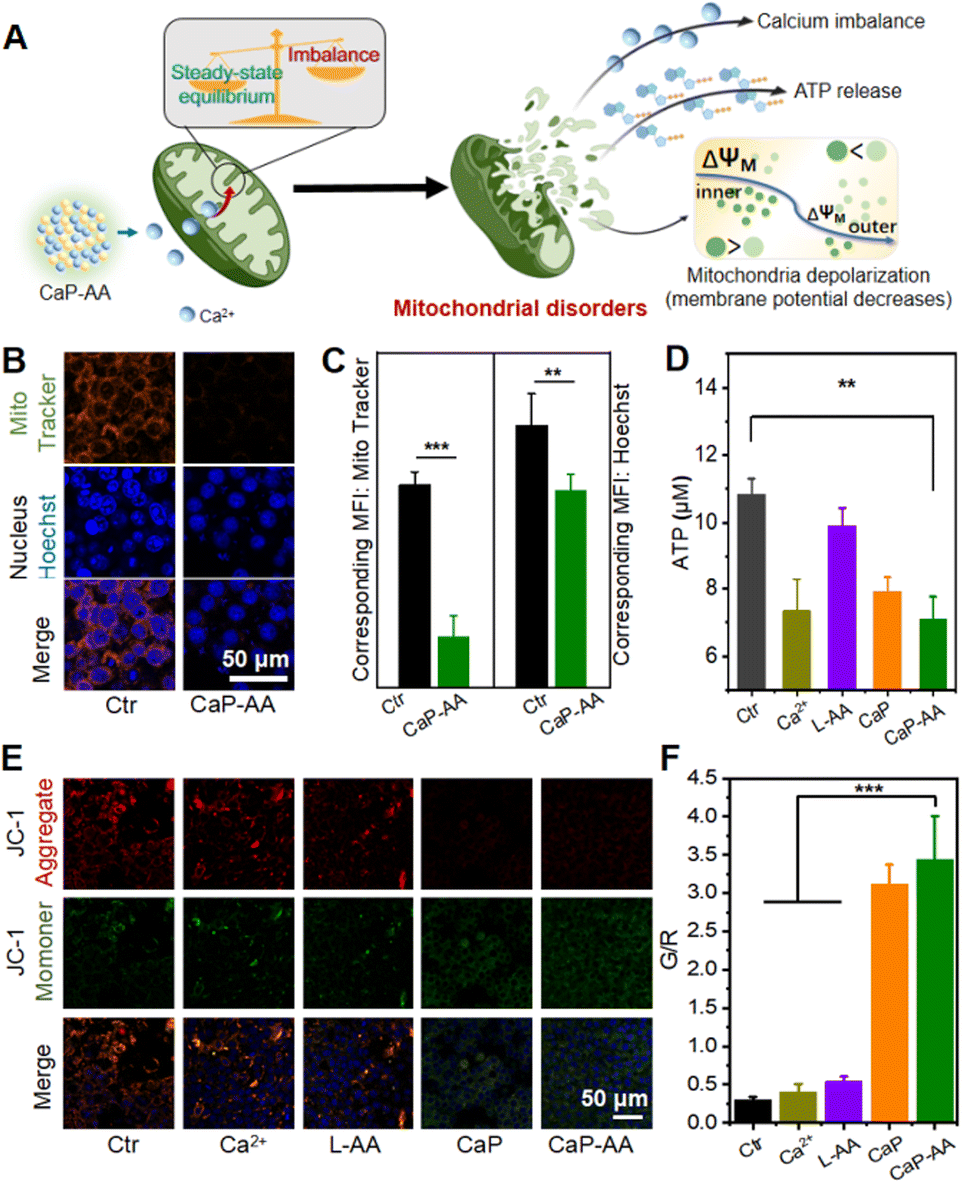 | ||
| Fig. 3 Calcium overload and ROS generation induce mitochondrial disorder. (A) Schematic of CaP-AA-induced mitochondrial imbalance. (B) Changes of the nucleus and mitochondrial membrane potential before and after CaP-AA treatment (red: mitochondrial; blue: nucleus). (C) Corresponding intracellular fluorescence intensity of the mitochondria and nucleus. (D) Changes in ATP amount in 4T1 cells after different treatments. (E) Confocal images of JC-1 staining after different treatments (green: monomer; red: J-aggregates; blue: nucleus). (F) Corresponding quantification of green/red intensity after various treatments. The significance of the difference was determined through one-way analysis of variance (ANOVA) by *p < 0.05, **p < 0.01, ***p < 0.001, (ns) p ≥ 0.05. | ||
Transcriptome alterations caused by CaP-AA
To clarify the calcification effects on tumor cells, transcriptomic changes of 4T1 cells were analyzed after incubation with CaP-AA for 24 h. After filtering with the standards of >1.5-fold changes and a false discovery rate <0.05 (Fig. S5†), 558 genes were found in total with differential expression, including 287 upregulated and 271 downregulated genes.Heatmaps of genes with log2(fold change) values greater than 2 were also shown (Fig. 4A). Some of these genes were associated with intracellular calcium metabolism, such as Tnni2 and Mzb1. Tnni2 was an inhibitory subunit of troponin. Its reduction could inhibit calcium-sensitivity to striated muscle actomyosin ATPase, consistent with the abovementioned ATP assay. The reduction of Mzb1 could contribute to the functional diversification of peripheral B cells by downregulating Ca2+ storage, antibody secretion and integrin activation. Some genes were shown to be associated with angiogenesis and immune regulation. As a potent proinflammatory cytokine, interleukin-1β was initially found to be the major endogenous pyrogen, which also played a role in angiogenesis by synergistically inducing the production of the vascular endothelial growth factor with TNF and IL-6. Its reduction implied inhibition of tumor proliferation. Interferon Induced Protein 44 Like (IFI44L) was a precursor to the mouse immunogenic peptide. When it was complexed with MHC, it could generate immune responses upon T cells, stimulating the induction of CD8+ T-cell recognition. These overall analyses suggested that the antitumor effects of CaP-AA-induced calcification acted primarily by affecting cellular metabolism and angiogenesis and might have a synergistic effect with immunotherapy.
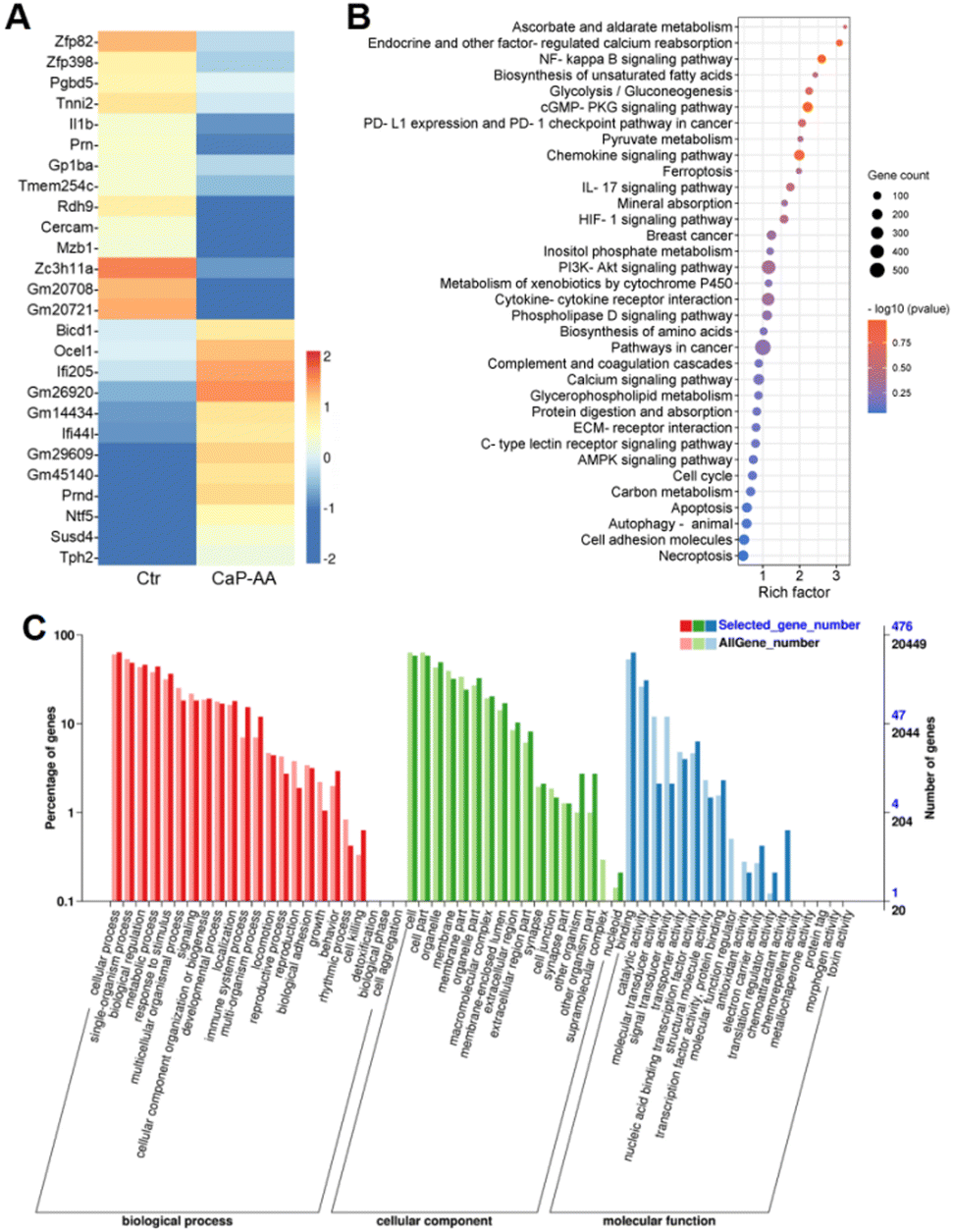 | ||
| Fig. 4 Transcriptome alterations of CaP-AA-treated cells. (A) Heatmap of differentially expressed genes between native and CaP-AA-treated 4T1 cells with log2(fold change) greater than 2. (B) KEGG pathway enrichment analysis of differentially expressed genes between native and CaP-AA-treated 4T1 cells. (C) Gene ontology (GO) annotation classification of differentially expressed genes of 4T1 cells with or without treatment of CaP-AA. GO enrichment map of differentially expressed genes involved in biological processes, cellular components and molecular functions. The horizontal coordinate was the GO classification, and the vertical coordinate on the left was the percentage of the number of genes, while the right was the number of genes. This figure showed the gene enrichment of each secondary function of GO between the differentially expressed gene (selected gene) background and the all-gene background, reflecting the status of each secondary function in both backgrounds. | ||
Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation classification confirmed similar results, i.e., that signal transduction and metabolism were notable (Fig. 4B and S6†). CaP-AA could affect ascorbate and aldehyde metabolism, which, in turn, modulates calcium reabsorption regulated by endocrine and other factors, accompanied by alterations in the hypoxia-inducible factor 1 (HIF-1) and calcium signaling pathways, and all these confirmed the effect of CaP-AA on intracellular calcium balance. Notably, glycolysis, unsaturated fatty acid/amino acid biosynthesis, and protein digestion/absorption pathways were significantly altered, suggesting that CaP-AA-induced calcification could affect multifaceted metabolic processes, primarily by affecting cellular metabolism resulting in cell death. PD-L1 expression and PD-1 checkpoint pathways in tumor were also on the list, implying that further studies should examine the relationship between immune checkpoints and cellular calcification. In addition, cGMP-PKG, IL-17, PI3K-Akt and AMPK signaling pathways were associated with calcification.
Gene ontology (GO) annotation classification statistics showed that the differentially expressed genes were mainly enriched in “cell killing” in the biological process ontology, while “reproduction” and “growth” processes were inhibited, indicating that calcification may affect tumor cell proliferation. In the cellular component ontology, the “supramolecular complex” was mostly suppressed, which might be induced by cell membrane calcification. The terms “molecular transducer activity”, “signal transducer activity” and “molecular function regulator” were reduced, while “chemorepellent activity” was enhanced for genes in the molecular function ontology (Fig. 4C). These genes showed noticeable expression differences exactly corresponding to tumor inhibition.
In vivo tumor growth inhibition by CaP-AA
Inspired by the prominent acid-responsive release of L-AA/Ca2+ which triggered metabolic destruction and calcium mineralization as revealed by in vitro experiments, in vivo antitumor effects of CaP-AA were investigated. The potential biosafety toxicity of CaP-AA during systemic injection needed to be examined. As shown in Fig. S7,† a high dosage of L-AA, CaP or CaP-AA did not cause significant hemolysis, indicating good biocompatibility of these nanoassemblies. As evident in the blood circulation and biodistribution results (Fig. S8 and S9†), CaP-AA was enriched in the liver through rapid blood metabolism. In other words, the depletion of CaP-AA by organs (liver, kidneys, etc.) implied that intravenous injection requires larger doses to achieve effects comparable to intratumoral injections. After 16 days of monitoring, the Ca2+ content in the liver was recovered as normal, indicating that CaP-AA could gradually self-clean from the organism. Meanwhile, with either intratumoral or intravenous injection, hematoxylin-eosin (H&E) histological staining of major organs showed no obvious pathological damage after 16 days of CaP-AA treatment (Fig. S10 and S11†). In addition, blood biochemistry and blood routine of mice after intravenous injection of CaP-AA showed no significant inflammation or organ failure (Tables S1 and S2†), indicating acceptable biocompatibility of CaP-AA for the conduct of animal experiments.The antitumor ability of CaP-AA in vivo was further investigated. After intratumoral injection of CaP-AA, the relative tumor volume and weight curves exhibited a significant tumor suppressive effect owing to the combined effect of calcium overload and redox imbalance to the TME (Fig. 5A, B and S12†). Other treatment groups showed a tumor inhibition effect to some extent. It was worth mentioning that the tumor inhibition effect of L-AA also existed objectively, which verified the auxiliary applicable to select L-AA. During the 16 days of treatment, the bodyweight of mice in each group did not change significantly (Fig. S13†), indicating that these nanoassemblies did not induce significant acute damage to mice.
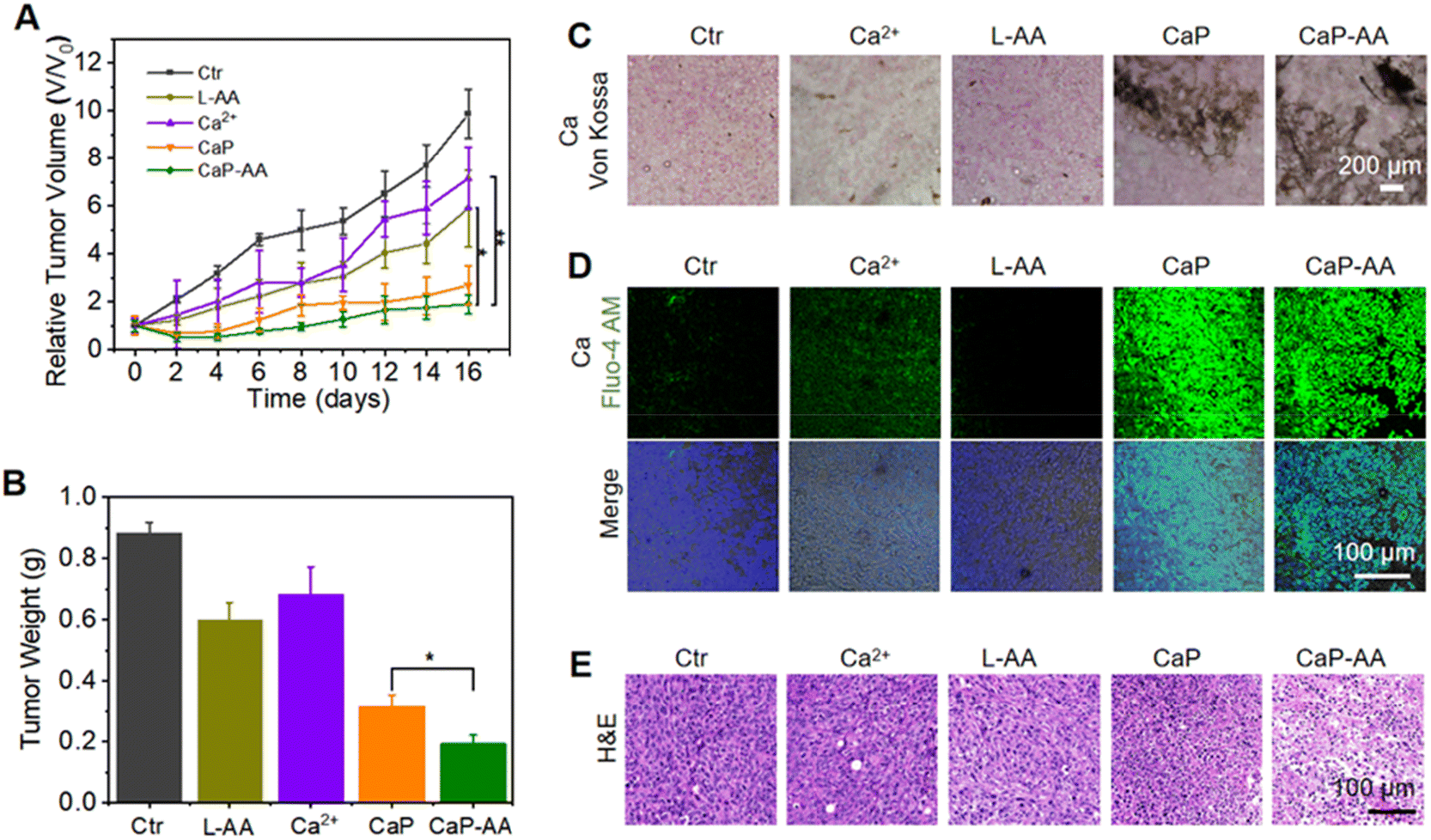 | ||
| Fig. 5 Tumor inhibition properties in vivo. (A) Tumor growth curves after intratumoral injection of different treatment groups. (B) Average tumor weights from different treatment groups at 16 days post-intratumoral injection. (C) Von Kossa-stained tumor slices after different treatments. (D) Calcium- and (E) H&E-stained tumor slices after different treatments. The significance of the difference was determined through one-way analysis of variance (ANOVA) by *p < 0.05, **p < 0.01, ***p < 0.001, (ns) p ≥ 0.05. | ||
Similar to the validation of intracellular calcium sedimentation, variations of calcium content within the tumor region were monitored by Von Kossa staining. CaP and CaP-AA treatment groups showed significant calcium sedimentation, while the CaP-AA-treated group exhibited extensive coverage compared with CaP, mainly by the released L-AA, which could secondarily damage mitochondria by pro-oxidation to maintain oxidative stress in the TME, as shown in Fig. 5C and D. Moreover, an efficient tumor inhibitory effect by CaP-AA could be visualized in pathological sections. As shown in Fig. 5E, after 16 days of CaP-AA-treatment, H&E histological staining showed significant cellular atrophy and necrosis with crinkled nucleus.
In addition, a high dosage of CaP-AA was selected for intravenous injection to verify the potential clinical application of acid-responsive CaP-AA. As evidenced in tumor volume and weight curves (Fig. S14 and S15†), with increased intravenous injection concentrations, CaP-AA-treated groups exhibited a promising therapeutic effect, caused by the combination of calcium overload and redox imbalance. H&E histological staining showed pronounced cell shrinkage and necrosis with a crinkled nucleus (Fig. S16†). During 16 days of treatment, no significant bodyweight change was noted (Fig. S17†), indicating that CaP-AA possessed admissible bioavailability with potential usage for deep tumor treatment.
Discussion
Inducing micro/nano-material alterations in tumors was gradually becoming an unconventional but promising approach for the treatment of malignant tumors.36–41 In recent years, researchers have begun to investigate calcium-based nanomaterials such as calcium peroxide (CaO2)42 and calcium carbonate (CaCO3),9 as TME-responsive materials for tumor therapeutic effects, and have explored the action mechanism of Ca2+. We here choose CaHPO4, which could be controllable to synthesize and has been widely used in supplements.43,44 As a responsive calcium source, CaHPO4 had inherent biocompatibility, weakly damaging normal tissues. Based on this, we attempted to investigate whether CaHPO4 possessed different effects on the cellular energy unit mitochondria of tumor cells. It has to be acknowledged that calcium-derived nanopharmaceuticals have the disadvantage of being less toxic at low doses compared to Fenton or Fenton-like effects of transition-state metals, so when targeting tumor treatment, calcium-derived agents required relatively high concentrations, which made calcium-derived agents usually selected for combination with other agents to enhance the therapeutic effect.45,46L-AA has been proven as a prodrug for tumor tissue.22,23,47L-AA at pharmacological concentrations could act as a precursor tumor drug and could spontaneously interact with internal metal ions (iron, etc.) within the extracellular microenvironment to produce hydrogen peroxide,22,23 which could diffuse into cells and cause intracellular ATP depletion, reduce glutathione levels, etc., ultimately leading to cell death. These made L-AA an efficient and viable material to be chosen. However, free L-AA itself was easy to be oxidized, which was not conducive to the pro-oxidation effect of L-AA on tumor cells. Here, calcium ascorbate as a coordination compound could stabilize reduced L-AA, so it is advisable to form calcium ascorbate. CaP-AA was synthesized by using calcium ascorbate as an intermediate. The construction of CaP-AA was an optimization that strikes a balance between structure and properties.CaP-AA was co-assembled by Ca2+ and L-AA to explore the many advantages of combined action for tumor therapy. The combination of the two could not only construct responsive structures, but also play a complementary role in the TME. Exogenous calcium sources generated Ca2+ when acids enter the cell, causing calcium surge in tumor cells. Ca2+ could directly act on mitochondria,48–50 affect their normal functions, and stimulate the changes of mitochondrial membrane potential and ATP production. L-AA did not directly act on mitochondria; on the other hand, it indirectly changes the redox equilibrium state in tumor cells through pro-oxidation,22,23,51 which could further result in an imbalance of cell metabolism. In this design, CaP-AA acted on the tumor cells with the combination of direct and indirect effects, by affecting the functional organelle mitochondria, and finally inducing damage to tumor cells. Different from inducing conspicuous oxidative stress, CaP-AA induced oxidative stress in tumor cells through affecting mitochondrial function, damaging the ability of mitochondria to maintain cell homeostasis, which fundamentally disrupts the pathway of cell maintenance function.
The intratumoral injected therapy effect of CaP-AA was far superior to that of the tail vein injection mode, the main reason was the enrichment effect of nanopharmaceuticals in liver, and fewer nanopharmaceutical could enter tumor areas through EPR, which was the common shortcoming of all nanopharmaceuticals. In this study, we recommended the use of intratumoral injection to simulate interventional therapy, which could avoid chronic liver injury caused by nanopharmaceutical accumulation, and could also avoid hypercalcemia caused by blood circulation under high dose injection. It was worth mentioning that CaP-AA was designed to be acid-responsive nanoassembled in the tumor area, which meant that the normal tissue damage could be significantly reduced. In addition, studies have claimed that L-AA could be used as a collagen deposition agent to promote the deposition of collagen fibers,52 which could be used as a calcium source to cause the formation of calcification foci after tumor tissue injury. Whether the calcified tumor structure could affect the spillover and dissociation of residual tumor cells, and whether it could reduce the possibility of systemic metastasis and recurrence would be the follow-up of this work.
Experimental
Synthesis of CaP-AA NPs
20 mg mL−1 of BSA aqueous solution, 30 mg mL−1 of L-AA aqueous solution (ultrasonic assisted dissolution) and 30 mg mL−1 of CaCO3 liquid suspension (use after uniform vibration) were pre-prepared for the following experiments. 200 μL of CaCO3 and 800 μL of L-AA were mixed to form calcium ascorbate (CaAA). 100 μL of BSA and 200 μL of CaAA were dissolved in 800 μL of water, and then 800 μL of ethanol and 200 μL of PB buffer (100 mM, pH 7.4, without NaCl) were added with vigorous stirring for 0.5 h. The resulting CaP-AA was washed several times and then re-dispersed in water. In order to prevent the oxidation and deterioration of L-AA, the as-prepared CaP-AA was stored under nitrogen for a long time. The prepared CaP-AA was mixed with PEG-5000 aqueous solution for 24 h.The Ca content in CaP-AA was quantified by ICP-MS. The amount of L-AA was determined by a mediated colorimetric ABTS strategy: 100 μL of H2O2 (200 mM), 1 μL of HRP (1 nM) and 100 μL of ABTS (2 mM) were mixed with 20 μL of different concentrations of L-AA such that colorimetric ABTS would be reduced with the addition of L-AA. The concentration of L-AA in CaP-AA was detected by this method.
CaP was synthesised by complex decomposition using a similar ingredient proportion. 20 mg mL−1 of BSA aqueous solution and 30 mg mL−1 of CaCl2 solution were pre-prepared for the following experiments. 100 μL of BSA and 40 μL of CaCl2 were dissolved in 1000 μL of water, and then 800 μL of ethanol and 200 μL of PB buffer (100 mM, pH 7.4, without NaCl) were added with vigorous stirring for 0.5 h. pH was adjusted to 5.0–6.0 during the synthesis. The resulting precipitates CaHPO4 (CaP) were washed several times and then re-dispersed in water.
Decomposition of CaP-AA NPs
The release ability of CaP-AA was determined by dialysis. 500 μL of concentrated CaP-AA aqueous solution (Ca: 40 mg mL−1, quantified by ICP, L-AA: 120 mg mL−1) was placed in a dialysis bag with a cut-off molecular weight of 3 kDa. CaP-AA aqueous solution was dialyzed against 50 mL of MES buffer (pH 5.0) or 50 mL of PBS buffer (pH 7.4), respectively. The released Ca2+ was measured by ICP-MS. The release of L-AA was measured with the above procedure.Calcium overload in vitro: cells were incubated under 37.0 °C within 5% CO2 and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin.
To investigate in vitro ROS-generating activity, confocal images of DCFH-DA staining were measured within cancer cells, and 4T1 cells were seeded into 0.3 mL culture dishes (5 × 103 cells per dish) for 24 h. Afterwards, the cells were treated with Ca2+, L-AA, CaP and CaP-AA (10 μL, Ca content: 0.25 mg mL−1, L-AA content: 0.75 mg mL−1), respectively. After another 6 h incubation, DCFH-DA (1 μM) and Hoechst (1 μg mL−1) were added into each well and incubated for another 20 min at 37.0 °C. Fluorescence images were captured by CLSM.
To measure cellular viability, 4T1 cells or 293T cells were cultivated in wells of a 96-well plate and incubated with different concentrations of Ca2+, L-AA, CaP and CaP-AA, respectively. After incubation of 12 h, cellular viability was tested by the MTT assay.
For confocal imaging of living and dead cells, 4T1 cells were seeded into 0.3 mL culture dishes (5 × 103 cells per dish) for 24 h. Then, those cells were treated with Ca2+, L-AA, CaP and CaP-AA (10 μL, Ca content: 0.25 mg mL−1, L-AA content: 0.75 mg mL−1), respectively. After incubation for 12 h, DPBS co-stained with Calcein-AM (2 mM) and PI (4.5 μM) was added into each well and incubated for 20 min at 37.0 °C. Fluorescence images were captured by CLSM.
To investigate the calcium content in vitro, 4T1 cells were seeded into 0.3 mL culture dishes (5 × 103 cells per dish) for 24 h. Afterwards, the cells were treated with Ca2+, L-AA, CaP and CaP-AA (10 μL, Ca content: 0.25 mg mL−1, L-AA content: 0.75 mg mL−1), respectively. After different incubation times, Fluo-4 AM was added into each well and incubated for another 300 min at 37.0 °C. Fluorescence images were captured by CLSM. Meanwhile, intracellular calcium was subjected to Von Kossa staining.
Mitochondrial homeostasis assessment
To investigate the in vitro mitochondrial potential, confocal images of JC-1 staining were measured within cancer cells, and 4T1 cells were seeded into 0.3 mL culture dishes (5 × 103 cells per dish) for 24 h. Afterwards, the cells were treated with Ca2+, L-AA, CaP and CaP-AA (10 μL, Ca content: 0.25 mg mL−1, L-AA content: 0.75 mg mL−1), respectively. After another 6 h incubation, the cells were stained with JC-1 (10 μg mL−1) and Hoechst (1 μg mL−1) for another 20 min at 37.0 °C. Fluorescent images were captured by CLSM.To investigate in vitro ATP changes, 4T1 cells were cultivated in wells of a 6-well culture plate (1 × 105 cells per dish) for 24 h. Afterwards, the cells were treated with Ca2+, L-AA, CaP and CaP-AA (10 μL, Ca content: 0.25 mg mL−1, L-AA content: 0.75 mg mL−1) within another 6 h incubation. The content of ATP of cell lysate was quantified using an ATP assay kit.
Transcriptomic assessment
To investigate the transcriptomic changes of 4T1 cells in vitro, 4T1 cells were seeded into 10 cm dishes (1 × 108 cells per dish) for 24 h. Afterwards, the cells were treated with CaP-AA (1 mL, Ca content: 2 mg mL−1, L-AA content: 6 mg mL−1). After another 12 h incubation, total RNA was extracted using Trizol reagent. Samples were delivered to Baimaike Co., Ltd (Beijing) to perform transcriptomic analysis. Differential genes were screened according to fold changes greater than, or equal to, 1.5 and a false discovery rate of less than 0.05. The raw reads were further processed with a bioinformatics pipeline tool, BMKCloud (https://www.biocloud.net) online platform.Antitumor effect in vivo
All animal protocols and all injection procedures and post-operative care were supervised and approved by the Animal Care and Use Committee of Hunan University (SYXK 2018-0006). Balb/c mice received subcutaneous injection of 4T1 cells at the right (5 × 105 cells) flanks.Mice were divided into 5 groups randomly (6 mice for each group). After tumor sizes reached ∼100 mm3, different formulations, (1) DPBS, (2) Ca2+, (3) L-AA, (4) CaP and (5) CaP-AA, were intratumorally injected into tumors at the dosage of 50 μL (Ca content: 0.25 mg mL−1, L-AA content: 0.75 mg mL−1). Meanwhile, another 2 groups (5 mice for each group) were selected for intravenous injection of 200 μL of CaP-AA at concentrations of 2 mg mL−1 and 4 mg mL−1, respectively. From day 0, the tumor sizes and bodyweights of each mouse were monitored every 2 days. A caliper was used to measure tumor volumes, and tumor sizes were calculated by the following formula: “volume = length × width2/2”.
On day 16 post-injection, tumor cryo-sections were collected from different groups for hematoxylin–eosin (H&E) and calcium staining.
In addition, the circulation time and biodistribution of CaP-AA were investigated. For the blood circulation test, mice were intravenously injected with CaP-AA (Ca: 2 mg mL−1, 200 μL), and then 20 μL of blood was collected from each mouse at the indicated time points, weighed, and chemically digested. The Ca content in blood was determined by ICP. At the same time, mouse organs were collected at different times to determine the enrichment biodistribution.
Conclusions
To achieve controllable calcification, a tumor-responsive calcium nanopharmaceutical agent, CaP-AA (CaHPO4 covalently doped L-ascorbic acid) nanoassembly, as an initiator for tumor calcium-mineralization, was successfully synthesized. Bioresponsive CaHPO4 could release Ca2+ within the TME, which could then trigger intracellular calcium overload, promoting mitochondrial disorder. The released Ca2+ in the interstitial tumor cells could result in calcium precipitates. Among them, exogenous L-AA could, secondarily, enhance the effect of CaHPO4 on metabolic balance by pro-oxidant effects, finally indirectly increasing the likelihood of tumor calcification to inhibit tumor growth. The introduction of this dual-path exogenous acid-responsive calcium source through mitochondrial metabolic destruction for stimulated tumor calcification could be an innovative tool for tumor treatment.Data availability
Data are available on request to the authors.Author contributions
This manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Y. Z., X. Y., and W.·K.: conceptualization and data curation; Y. Z., X. Y., R.-M. K. and F. Q.: formal analysis; Y. Z. and E. Z.: investigation; Y. Z., J. Z. and F. Q.: resources; W. T.: supervision and validation; Y. Z. and W.·K.: roles/writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Pioneer R&D Program of Zhejiang (2022C03031), the National Key Research and Development Program of China (2021YFA0910103), the National Natural Science Foundation of China (22274141, 22074080, 22007038, 22207062, and 22174086), the Natural Science Foundation of Shandong Province (ZR2022ZD28, ZR2020KB020, ZR2022QB223, ZR2022QB209, and ZR2020MB056), the Changjiang Scholar Program of the Ministry of Education of China (Q2019258), the Taishan Scholar Program of Shandong Province (tsqn201909106), the Qingchuang Science and Technology Project of Universities in Shandong Province, China (2021KJ070) and the Open Project of Chemistry Department of Qingdao University of Science and Technology (QUSTHX202008). Cartoons in Scheme 1, Fig. 2A and 3A were created with https://BioRender.com.Notes and references
- T. W. McLennan and R. A. Castellino, Radiology, 1975, 115, 87–89 CrossRef CAS PubMed
.
- J. Wu, Y. Chen, J. Xin, J. Qin, W. Zheng, S. Feng, C. Sun, M. Tian, Z. Lu and B. Wang, Adv. Funct. Mater., 2021, 31, 2101284 CrossRef CAS
- N. Tang, H. Li, L. Zhang, X. Zhang, Y. Chen, H. Shou, S. Feng, X. Chen, Y. Luo, R. Tang and B. Wang, Angew. Chem., Int. Ed., 2021, 60, 6509–6517 CrossRef CAS PubMed
- W. Bao, M. Liu, J. Meng, S. Liu, S. Wang, R. Jia, Y. Wang, G. Ma, W. Wei and Z. Tian, Nat. Commun., 2021, 12, 6399 CrossRef CAS PubMed
- P. Zheng, B. Ding, R. Shi, Z. Jiang, W. Xu, G. Li, J. Ding and X. Chen, Adv. Mater., 2021, 33, 2007426 CrossRef CAS PubMed
- B. Xie, H. Zhao, Y.-F. Ding, Z. Wang, Y. Wang, C. Gao and R. Wang, J. Controlled Release, 2023, 357, 572–579 CrossRef CAS PubMed
- Q. Guan, L.-L. Zhou, F.-H. Lv, W.-Y. Li, Y.-A. Li and Y.-B. Dong, Angew. Chem., Int. Ed., 2020, 59, 18042–18047 CrossRef CAS PubMed
- H. Kong, Q. Chu, C. Fang, G. Cao, G. Han and X. Li, Adv. Sci., 2021, 8, 2100241 CrossRef CAS PubMed
- Y. Li, S. Zhou, H. Song, T. Yu, X. Zheng and Q. Chu, Biomaterials, 2021, 277, 121080 CrossRef CAS PubMed
- J. F. Garbincius and J. W. Elrod, Physiol. Rev., 2021, 102, 893–992 CrossRef PubMed
- D. Chaudhuri, D. J. Artiga, S. A. Abiria and D. E. Clapham, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1872–1880 Search PubMed
- S. Romero-Garcia and H. Prado-Garcia, Int. J. Oncol., 2019, 54, 1155–1167 CAS
- H.-G. Sprenger and T. Langer, Trends Cell Biol., 2019, 29, 888–900 CrossRef CAS PubMed
- C. Chen, R. Zhang, J. Zhang, Y. Zhang, H. Zhang, Z. Wang, X. Huang, S. Chen, T. K. Kwok Ryan, W. Y. Lam Jacky, D. Ding and Z. Tang Ben, CCS Chem., 2021, 4, 2249–2257 CrossRef
- T. Pathak and M. Trebak, Pharmacol. Ther., 2018, 192, 112–123 CrossRef CAS PubMed
- P. Zheng, B. Ding, Z. Jiang, W. Xu, G. Li, J. Ding and X. Chen, Nano Lett., 2021, 21, 2088–2093 CrossRef CAS PubMed
- L. Xu, R. Xu, P. E. Saw, J. Wu, S.-X. Cheng and X. Xu, Nano Lett., 2021, 21, 7569–7578 CrossRef CAS PubMed
- Y. Li, J. Dang, Q. Liang and L. Yin, Biomaterials, 2019, 209, 138–151 CrossRef CAS PubMed
- J. J. Gu, Q. Zhang, C. Mavis, M. S. Czuczman and F. J. Hernandez-Ilizaliturri, Blood, 2015, 126, 4008 CrossRef
- Q. Li, J. Yang, C. Chen, X. Lin, M. Zhou, Z. Zhou and Y. Huang, J. Controlled Release, 2020, 325, 38–51 CrossRef CAS PubMed
- M. d. P. S. Idelchik, U. Begley, T. J. Begley and J. A. Melendez, Semin. Cancer Biol., 2017, 47, 57–66 CrossRef CAS PubMed
- Q. Chen, M. G. Espey, M. C. Krishna, J. B. Mitchell, C. P. Corpe, G. R. Buettner, E. Shacter and M. Levine, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13604–13609 CrossRef CAS PubMed
- Q. Chen, M. G. Espey, A. Y. Sun, J.-H. Lee, M. C. Krishna, E. Shacter, P. L. Choyke, C. Pooput, K. L. Kirk, G. R. Buettner and M. Levine, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8749–8754 CrossRef CAS PubMed
- Y. Ma, J. Chapman, M. Levine, K. Polireddy, J. Drisko and Q. Chen, Sci. Transl. Med., 2014, 6, 222ra218 Search PubMed
- C. Tang, Y. Wei, L. Gu, Q. Zhang, M. Li, G. Yuan, Y. He, L. Huang, Y. Liu and Y. Zhang, Adv. Sci., 2020, 7, 1902536 CrossRef CAS PubMed
- H. Dumbuya, S. Y. Hafez and E. Oancea, FASEB J., 2020, 34, 11605–11623 CrossRef CAS PubMed
- J. Szymański, J. Janikiewicz, B. Michalska, P. Patalas-Krawczyk, M. Perrone, W. Ziółkowski, J. Duszyński, P. Pinton, A. Dobrzyń and M. R. Więckowski, Int. J. Mol. Sci., 2017, 18, 1576 CrossRef PubMed
- T. Pozzan and R. Rizzuto, Nat. Cell Biol., 2000, 2, E25–E27 CrossRef CAS PubMed
- S. Feno, G. Butera, D. Vecellio Reane, R. Rizzuto and A. Raffaello, Oxid. Med. Cell. Longevity, 2019, 2019, 9324018 Search PubMed
- R. Esterberg, T. Linbo, S. B. Pickett, P. Wu, H. C. Ou, E. W. Rubel and D. W. Raible, J. Clin. Invest., 2016, 126, 3556–3566 CrossRef PubMed
- L. Kazak, E. T. Chouchani, I. G. Stavrovskaya, G. Z. Lu, M. P. Jedrychowski, D. F. Egan, M. Kumari, X. Kong, B. K. Erickson, J. Szpyt, E. D. Rosen, M. P. Murphy, B. S. Kristal, S. P. Gygi and B. M. Spiegelman, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 7981–7986 CrossRef CAS PubMed
- A. C. Boese and S. Kang, Redox Biol., 2021, 42, 101870 CrossRef CAS PubMed
- M.-N. de Noirfontaine, E. Garcia-Caurel, D. Funes-Hernando, M. Courtial, S. Tusseau-Nenez, O. Cavani, J. Jdaini, C. Cau-Dit-Coumes, F. Dunstetter and D. Gorse-Pomonti, J. Nucl. Mater., 2021, 545, 152751 CrossRef CAS
- J.-Y. Mevellec, S. Quillard, P. Deniard, O. Mekmene, F. Gaucheron, J.-M. Bouler and J.-P. Buisson, Spectrochim. Acta, Part A, 2013, 111, 7–13 CrossRef CAS PubMed
- N. O. Metreveli, K. K. Jariashvili, L. O. Namicheishvili, D. V. Svintradze, E. N. Chikvaidze, A. Sionkowska and J. Skopinska, Ecotoxicol. Environ. Saf., 2010, 73, 448–455 CrossRef CAS PubMed
- Z. Jiang, Y. Liu, R. Shi, X. Feng, W. Xu, X. Zhuang, J. Ding and X. Chen, Adv. Mater., 2022, 34, 2110094 CrossRef CAS PubMed
- Y. Liu, Z. Jiang, S. Tong, Y. Sun, Y. Zhang, J. Zhang, D. Zhao, Y. Su, J. Ding and X. Chen, Adv. Mater., 2023, 35, 2203291 CrossRef CAS PubMed
- Z. Sun and Y. Hou, BMEMat, 2023, 1, e12012 CrossRef
- X. Xiao, Q. Zong, J. Li and Y. Yuan, CCS Chem., 2022, 4, 3878–3888 CrossRef CAS
- G. Li, Y. Yang, Y. Zhang, P. Huang, J. Yan, Z. Song, Q. Yuan and J. Huang, CCS Chem., 2021, 4, 1654–1670 CrossRef
- Y.-L. Ma, M. Quan, X.-L. Lin, Q. Cheng, H. Yao, X.-R. Yang, M.-S. Li, W.-E. Liu, L.-M. Bai, R. Wang and W. Jiang, CCS Chem., 2020, 3, 1078–1092 CrossRef
- L. Ruan, G. Song, X. Zhang, T. Liu, Y. Sun, J. Zhu, Z. Zeng and G. Jiang, Biomater. Sci., 2021, 9, 6830–6841 RSC
- W. S. Al-Qahtani and P. Virk, Toxicol. Environ. Chem., 2015, 97, 968–988 CrossRef CAS
- J. A. Lapre, H. T. De Vries and R. van der Meer, Am. J. Physiol.: Gastrointest. Liver Physiol., 1991, 261, G907–G912 CrossRef CAS PubMed
- F. Chen, B. Yang, L. Xu, J. Yang and J. Li, ChemMedChem, 2021, 16, 2278–2286 CrossRef CAS PubMed
- C. Ji, Z. Lu, Y. Xu, B. Shen, S. Yu and D. Shi, J. Biomed. Mater. Res., 2018, 106, 2544–2552 CrossRef CAS PubMed
- B. Frei and S. Lawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11037–11038 CrossRef CAS PubMed
- A. L. Chernorudskiy and E. Zito, J. Mol. Biol., 2017, 429, 620–632 CrossRef CAS PubMed
- L. Contreras, I. Drago, E. Zampese and T. Pozzan, Biochim. Biophys. Acta, Bioenerg., 2010, 1797, 607–618 CrossRef CAS PubMed
- Z. Xu, D. Zhang, X. He, Y. Huang and H. Shao, Curr. Genomics, 2016, 17, 215–219 CrossRef CAS PubMed
- C. Shuai, X. Chen, C. He, M. Chen, S. Peng and W. Yang, Colloids Surf., B, 2023, 225, 113251 CrossRef CAS PubMed
- A. Hinek, H. J. Kim, Y. Wang, A. Wang and T. F. Mitts, J. Dermatol. Sci., 2014, 75, 173–182 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary materials and characterization, assisted analysis of data and results. See DOI: https://doi.org/10.1039/d3sc02945j |
| This journal is © The Royal Society of Chemistry 2023 |