
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn artificial light-harvesting system constructed from a water-soluble metal–organic barrel for photocatalytic aerobic reactions in aqueous media†
Danyang
Li
a,
Linlin
Yang
 b,
Wangjian
Fang
c,
Xinmei
Fu
a,
Hechuan
Li
a,
Jianxu
Li
a,
Xuezhao
Li
a and
Cheng
He
*a
b,
Wangjian
Fang
c,
Xinmei
Fu
a,
Hechuan
Li
a,
Jianxu
Li
a,
Xuezhao
Li
a and
Cheng
He
*a
aState Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian 116012, P. R. China. E-mail: hecheng@dlut.edu.cn
bXinxiang Key Laboratory of Forensic Science Evidence, School of Forensic Medicine, Xinxiang Medical University, Xinxiang 453003, P. R. China
cInstitute for Molecular Design and Synthesis, School of Pharmaceutical Science & Technology, Tianjin University, Tianjin 300072, P. R. China
First published on 31st August 2023
Abstract
An artificial light-harvesting system constructed from a water-soluble host–guest complex can be regarded as a high-level conceptual model of its biological counterpart and can convert solar energy into chemical energy in an aqueous environment. Herein, a water-soluble metal–organic barrel Ga-tpe with twelve sulfonic acid units was obtained by subcomponent self-assembly between Ga3+ ions and tetra-topic ligands with tetraphenylethylene (TPE) cores. By taking advantage of host–guest interactions, cationic dye rhodamine B (RB) was constrained in the pocket of Ga-tpe to promote the Förster resonance energy transfer (FRET) process for efficient photocatalytic aerobic oxidation of sulfides and cross-dehydrogenative coupling (CDC) reaction in aqueous media.
Introduction
The construction of molecular containers has provided a sustainable way to mimic and rival natural organisms and enzymes.1 Among such cage-like hosts, the coordination-driven assembly of metal–organic cages, which can be functionalized with both organic ligands and metal centers, has been considered an excellent candidate to mimic enzymes, due to their precisely controlled, well-defined topology obtained using facile synthesis and stability in common solvents.2,3 With highly selective recognition toward substrates inside their large inner cavity, such discrete coordination constructs have proven useful as molecular devices for substrate encapsulation,4 separation,5 and reaction vessels to catalytically accelerate chemical processes.6,7Specifically, supramolecular assemblies formed by cage hosts and encapsulated guests with essential components being forced into close proximity have also attracted constant attention to developing supramolecular charge/energy donor–acceptor systems, which led to efficiency enhancements in energy, electron, or substance transfers based on short through-space and long through-bond pathways.8,9 For example, inspired by nature, significant advances have been made in using supramolecular donor–acceptor systems to mimic light antenna harvesting systems using the FRET process.10–14 Previously, we reported a highly efficient supramolecular FRET system using a large metal–organic cage as a donor. It was thought that encapsulation of the guest within the inner cavity of the cage would render the interaction between the donor and the acceptor stronger and more robust in close proximity and the rigid coordination cage could provide an isolated microenvironment in which the donors could be fixed at a high local concentration without further aggregation.15
It is nearly universal in natural systems that most host–guest interactions based on biological processes are executed under aqueous conditions.16 Nevertheless, most metal–organic cages have been prepared and studied in organic solvents. It is appealing to create water-soluble metal–organic cages that could be regarded as high-level conceptual models for their biological counterparts.17 Water-soluble metal–organic cages with large inner pockets have found widespread use in the selective storage and transport of a variety of guest molecules, as well as in the promotion of chemical reactions.18–20 To achieve this goal, the design of water-soluble and stable metal–organic cages is highly desired but has been challenging due to the following: first, these discrete coordination structures need to maintain rigidity and stability, which requires the ligands to contain nonpolar aromatic backbones; however, in this case, the aromatic ligand is poorly soluble in aqueous solutions. Second, due to the coordinating ability of water, it can bind to metal centers, compete with ligands, and reduce the formation of metal–organic cages. Third, hydrophobic effects may change the way ligands organize around metal ions, leading to aggregation as opposed to the formation of hollow cages.
Thus, to better imitate natural light-harvesting antenna systems, establishing supramolecular FRET systems in aqueous environments to convert solar energy into chemical energy is of great significance. Herein, an anionic metal–organic barrel Ga-tpe endowed with water solubility was constructed using subcomponent self-assembly for application as an energy donor. Ga-tpe was capable of encapsulating the positively charged guest RB as a suitable energy acceptor. It was envisioned that a FRET process in an aqueous environment could be enabled between the face-capped cage Ga-tpe and the RB dye with a high local donor–acceptor ratio and minimized distance between them. Significantly, the harvested solar energy could be utilized to promote the photooxidation of thioanisole and photocatalyzed aerobic CDC reactions in an aqueous medium. Thus, the RB@Ga-tpe supramolecular system could be regarded as a promising natural counterpart to initiate photocatalytic reactions in a simulated enzymatic environment (Scheme 1).
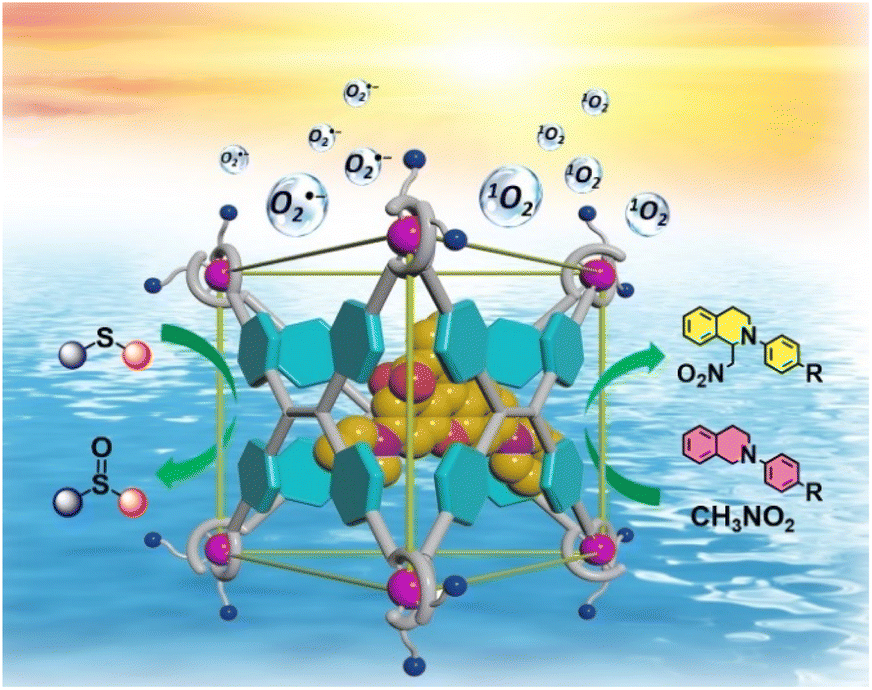 | ||
| Scheme 1 The illustration of the host–guest strategy between Ga-tpe and the encapsulated RB dye in inducing effective FRET processes for highly efficient photocatalytic aerobic reactions. | ||
Results and discussion
Synthesis and characterization of cage Ga-tpe
Ga-tpe was formed by the reaction of 4,4′,4′′,4′′′-(ethene-1,1,2,2-tetrayl)tetra(benzohydrazide) (3 equiv.), 5-sulfosalicylaldehyde sodium salt (12 equiv.), gallium tris(acetylacetonate) (6 equiv.) and NaOH (24 equiv.) in methanol at 60 °C overnight (Fig. 1a). The 1H NMR spectra of Ga-tpe with a single set of ligand environments were recorded in D2O, and the broad signals of Ga-tpe were indicative of the formation of the cage structure (Fig. S7, ESI†). Moreover, the 1H diffusion-ordered (DOSY) NMR spectrum of Ga-tpe showed the formation of a single species with a diffusion coefficient (D) value of 3.47 × 10−11 m2 s−1 (Fig. 1b). Thanks to the twelve sulfonic acid units of the cage Ga-tpe, the water solubility and stability of the cage were realized (Fig. S9, ESI†).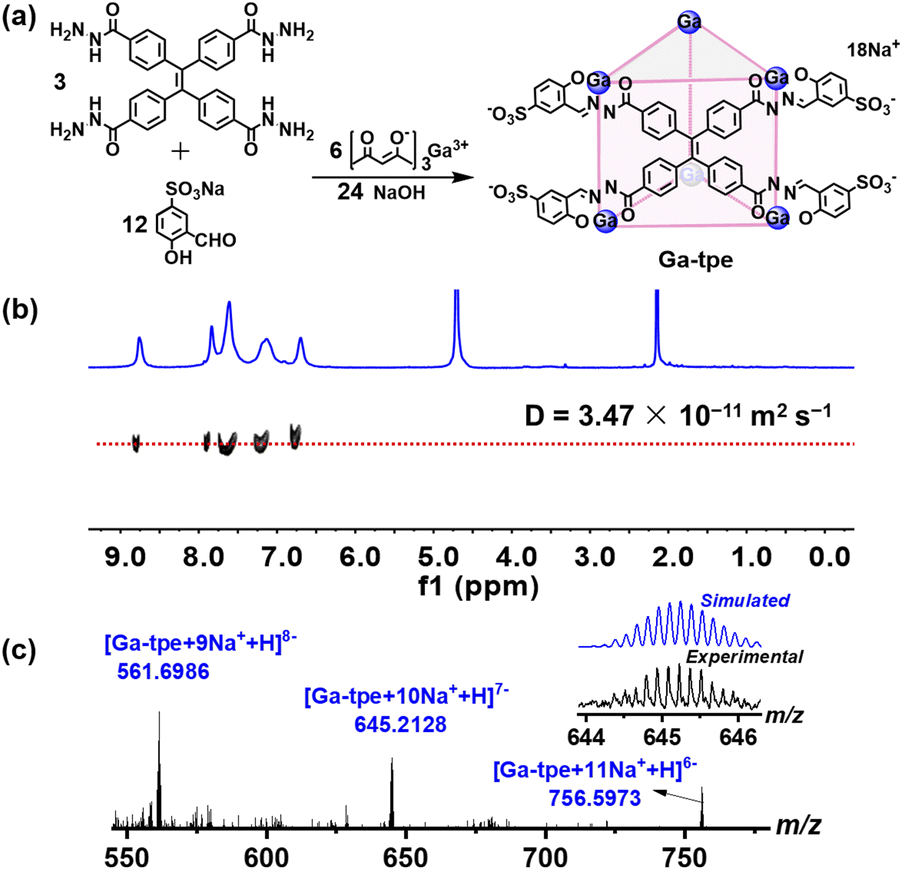 | ||
| Fig. 1 (a) The self-assembly of Ga-tpe; (b) DOSY spectrum of Ga-tpe in D2O; (c) the ESI-MS spectra of Ga-tpe. | ||
Electrospray ionization mass spectrometry (ESI-MS) of Ga-tpe showed a set of signals at m/z = 561.6986, 645.2128, 756.5973, confirming the formation of a Ga6L3 metal–organic barrel (Fig. 1c). A simple comparison with the simulation results based on natural isotopic abundances suggested that the peaks corresponded to a characteristic sequence of signals associated with the loss of different numbers of cations, [Ga6L3·n(Na+)H](17−n)− (n = 9–11).
Unfortunately, several trials were conducted to obtain a single crystal of Ga-tpe but were unsuccessful. The structural optimization calculation was employed,21 providing a clear picture of the structural pattern of Ga-tpe (Fig. 2). The optimized structure of Ga-tpe revealed the formation of a Ga6L3 barrel consisting of three TPE ligands and six gallium ions. There was the possibility of the formation of two isomeric barrels where the ligand panels could be horizontally oriented (Fig. 2a–c) or vertically oriented (Fig. 2e–f). Hence, the six Ga ions were located at the vertices of the barrel, whereas the three ligands served as the three faces of the barrel to form a pocket for the recognition and localization of guests (such as RB or MB) in the host. Moreover, the results suggested that coordination-driven restricted rotation of the TPE-ligand would prohibit nonradiative decay.22
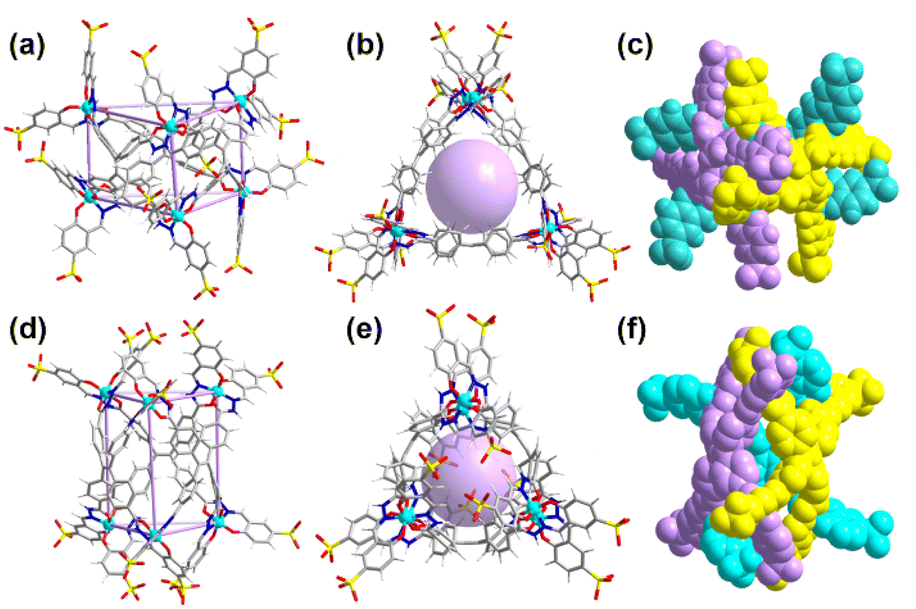 | ||
| Fig. 2 Optimized structure of barrel Ga-tpe. (a) and (b) The different views where ligands were horizontally oriented; (c) the filling pattern of Ga-tpe; (d) and (e) the different views of the other expected geometry where ligands were vertically oriented; (f) the filling pattern of Ga-tpe. Color codes: Ga turquoise, N blue, S yellow, O red, C grey, and H white. | ||
Host–guest complex with encapsulated RB
Driven by possible aromatic interactions, H bond interactions, and electrostatic interactions in the Ga-tpe pocket, the cationic energy acceptor RB with a suitable size was expected to be encapsulated in the Ga-tpe cage as a guest, not only avoiding the aggregation-caused quenching effect of the aggregated dye but also forcing close proximity between the energy donor and acceptor to meet the need for a highly efficient FRET process. The mode of interaction between Ga-tpe and RB was studied by isothermal titration calorimetry assays, and the combined number indicated that the presence of RB triggered the formation of an RB@Ga-tpe 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host–dye complex in water (Fig. S10, ESI†). Curve fitting by computer simulation using an “independent” model showed an association constant of 1.22 × 105 M−1.
:1 host–dye complex in water (Fig. S10, ESI†). Curve fitting by computer simulation using an “independent” model showed an association constant of 1.22 × 105 M−1.
The formation of the host–guest complex was further evidenced by the 1H NMR titration experiment between metal–organic barrel Ga-tpe and RB (Fig. 3a). The addition of RB to Ga-tpe showed significant upfield shifts (0.42 ppm and 0.38 ppm) where the methyl (H1 and –CH3) and the methylene (H2 and –CH2–) protons of RB were observed. Furthermore, H–H interactions between the aromatic rings, –NH– of Ga-tpe and methyl (H1) protons of RB could be observed in the nuclear Overhauser effect (NOESY) spectrum of RB@Ga-tpe (Fig. 3b), which showed the guest RB to reside within the pocket of Ga-tpe and the donor host and acceptor guest were close enough in space for efficient energy transfer. The binding constant of RB with Ga-tpe was detected by UV-Vis titration in water and nonlinear fitting of the titration curve with an association constant (Ka = 1.52 × 105 M−1) (Fig. 3c). With a gradually increasing ratio of RB to Ga-tpe, the absorption peak of Ga-tpe at 385 nm gradually decreased, causing significant variation in these bands with sharp isosbestic points at 360 nm and 415 nm, suggesting the formation of a host–guest complex between Ga-tpe and RB.
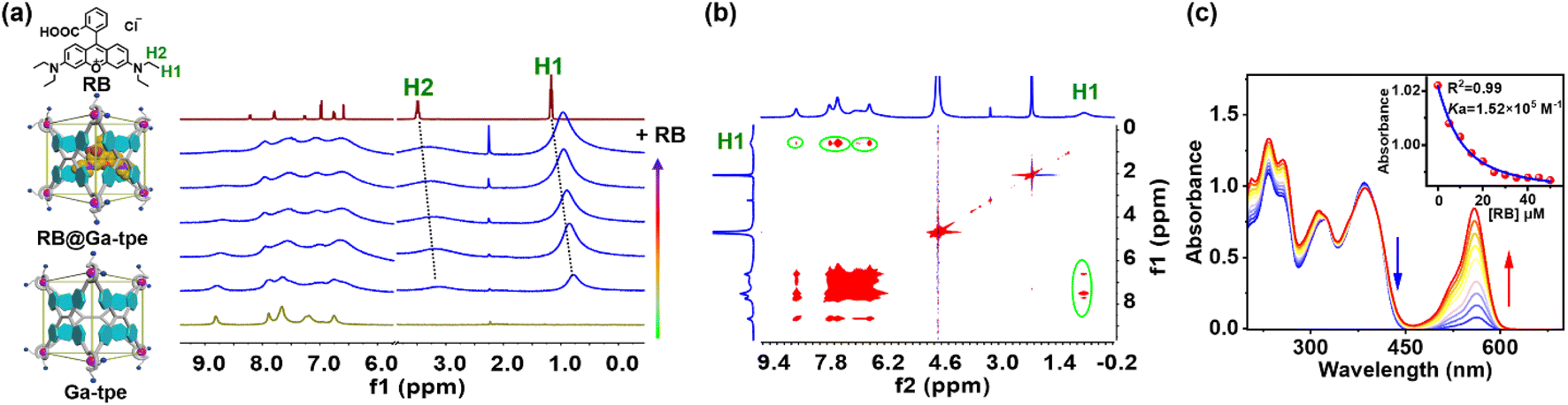 | ||
| Fig. 3 (a) The 1H NMR titration spectra between Ga-tpe ([Ga-tpe] = 2.0 × 10−3 M) and RB (1 equiv. per session) in D2O; (b) the NOESY spectrum of Ga-tpe with RB in D2O; (c) the absorbance spectra of Ga-tpe ([Ga-tpe] = 1.0 × 10−5 M) upon the addition of RB in water. Inset: nonlinear fitting of the titration curve at 385 nm, showing the calculated associate constant Ka = 1.52 × 105 M−1. | ||
Artificial light harvesting by Förster resonance energy transfer
The effective FRET process requires not only an appropriate distance and dipole orientation between different chromophores but also a match in the spectral overlap between the donor emission and acceptor excitation. RB was chosen as an energy acceptor because of the good overlap between the fluorescence band of Ga-tpe and the absorption spectra of RB (Fig. 4a). The FRET process was shown by fluorescence titration of RB with the aqueous solution of Ga-tpe (Fig. 4b). Interestingly, with an increasing concentration of RB in Ga-tpe, the broad emission peak of Ga-tpe at 505 nm was gradually decreased, while a new emission peak of RB@Ga-tpe at 590 nm appeared when excited at 385 nm. Moreover, the CIE chromaticity diagram confirmed the obvious fluorescence color change from blue to pink under UV light (Fig. 4c). Alternatively, the addition of Ga-tpe to the solution of RB showed that the intensity of Ga-tpe and RB gradually increased when excited at 385 nm, revealing the highly efficient energy transfer from Ga-tpe to RB (Fig. 4d).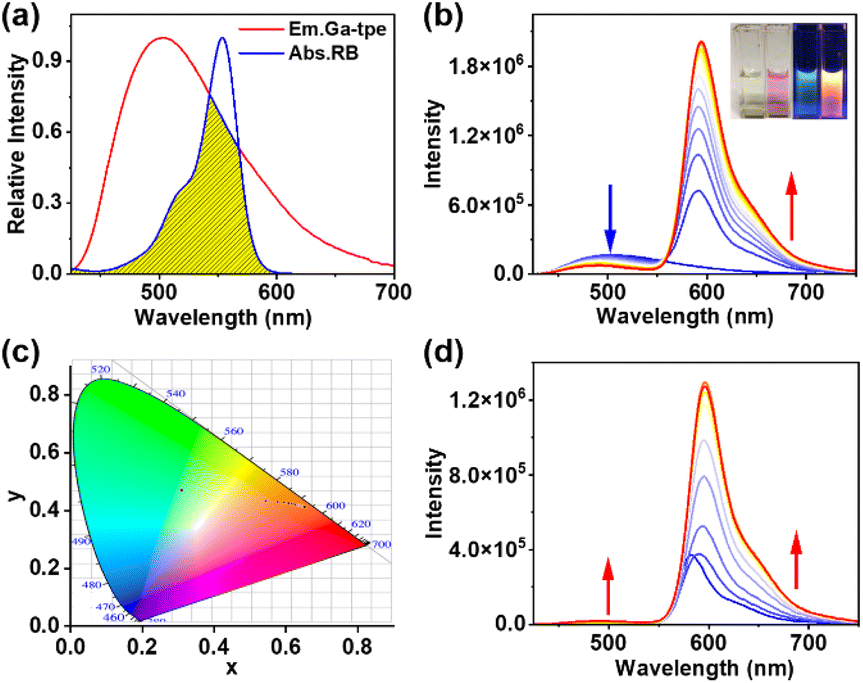 | ||
| Fig. 4 (a) Normalized fluorescence spectrum of Ga-tpe and the absorbance spectrum of RB; (b) fluorescence spectrum of Ga-tpe ([Ga-tpe] = 1.0 × 10−5 M) in water with different concentrations of RB (0.05 equiv. per session); (c) the CIE chromaticity diagram changes of Ga-tpe upon titration with RB; (d) fluorescence spectrum of RB ([RB] = 1.0 × 10−5 M) in water with different concentrations of Ga-tpe. | ||
Compared to that of Ga-tpe (1.37%), the quantum yield of RB@Ga-tpe increased to 15.76%, which further confirmed the occurrence of the FRET process (Fig. S12, Table S1 ESI†). In addition, the fluorescence lifetime of Ga-tpe followed a double exponential decay, which showed τint = 1.04 ns when emitting at 405 nm (Fig. S13, Table S2 ESI†). In contrast, the fluorescence lifetime of RB@Ga-tpe was found to be shorter in the presence of RB (τint = 0.98 ns), which suggested that energy could be transferred from the donor Ga-tpe to the acceptor RB. As a result, the ΦET of RB@Ga-tpe reached 56.1% (ESI, Table S3†).
In addition, the importance of host–guest interaction in the FRET system was identified by preventing the FRET process by binding an inhibition guest in the pocket of Ga-tpe. Methylene blue 2B (MB) is a cationic dye similar to RB, which is smaller than RB. MB was chosen as a competitive guest introduced into the host–guest system. The fluorescence and UV-Vis absorption titration indicated that MB was a suitable competitive guest for binding into the pocket of Ga-tpe to replace RB (Fig. S14, ESI†). Notably, upon the gradual addition of MB to the mature RB@Ga-tpe FRET system, the emission peak of the system at 590 nm gradually decreased (Fig. S15, ESI†). This showed that MB competed with RB for binding into Ga-tpe to inhibit the FRET process, resulting in a decrease in the emission intensity of RB@Ga-tpe. The effect of the host–guest interaction on the FRET processes was also identified using the fluorescence titration of RB with a solution of ligand L. Even though the absorption of RB overlapped well with the emission of L, the broad emission peak of ligand L at 520 nm slightly decreased upon the gradual addition of RB to a solution of ligand L (Fig. S16, ESI†).
Research on reactive oxygen species (ROS)
To better display the versatility of RB@Ga-tpe, we explored its use in photocatalytic oxidation reactions. Subsequently, we investigated the capacity of RB@Ga-tpe to generate ROS using 3,3′,5,5′-tetramethylbenzidine (TMB) as a probe molecule. Upon adding RB@Ga-tpe into a methanol solution containing TMB, the characteristic absorption peak of the oxidized TMB at around 670 nm increased significantly with Xe light irradiation time in an air atmosphere (Fig. 5a), suggesting the formation of ROS. For comparison, the absorption became lower under similar conditions when performed with Ga-tpe or RB only (Fig. 5b and S18, ESI†), respectively, indicating that the artificial light-harvesting system benefitted oxygen activation.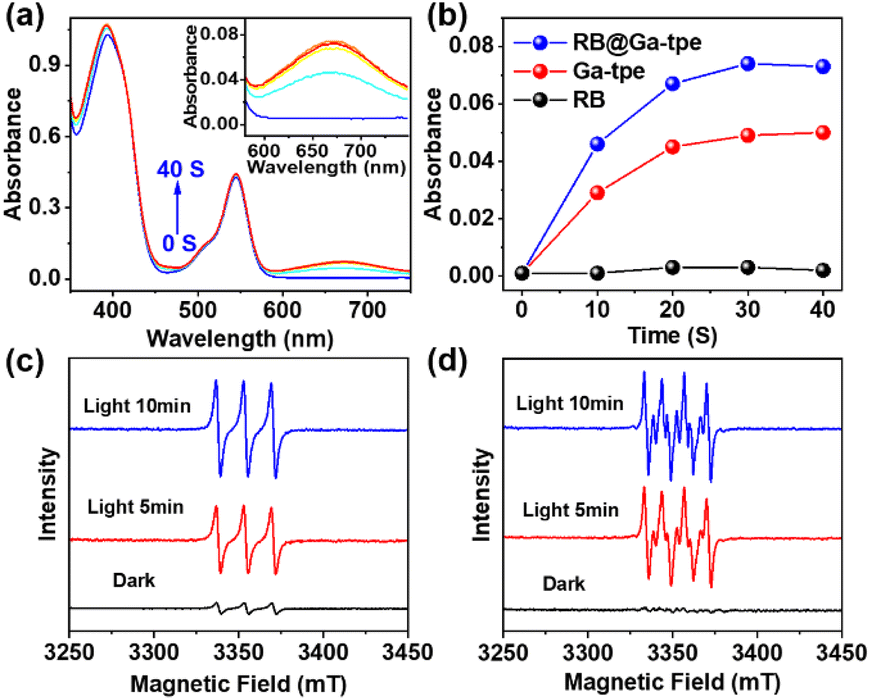 | ||
| Fig. 5 (a) Evolution of the UV-Vis spectra during the continuous irradiation of RB@Ga-tpe with Xe light in the presence of TMB in an air atmosphere; (b) time-dependent corresponding absorption of TMB at 670 nm; (c) the detection of 1O2 in a mixture of TEMP and RB@Ga-tpe in methanol solution under xenon lamp irradiation using EPR spectra in an air atmosphere; (d) the detection of O2˙− in a mixture of DMPO and RB@Ga-tpe in methanol solution under xenon lamp irradiation using EPR spectra in an air atmosphere. | ||
To clarify the various types of ROS that were generated when using RB@Ga-tpe, the electron paramagnetic resonance (EPR) spectra using 2,2,6,6-tetramethylpiperidine (TEMP) and 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as ROS trapping agents were recorded under xenon lamp irradiation and in an air-saturated CH3OH solution (Fig. 5c and d). The obvious characteristics of singlet oxygen (1O2) and superoxide (O2˙−) were trapped. The results showed that the active oxygen species 1O2 and O2˙− could be produced by using RB@Ga-tpe as a photocatalyst, generated by an energy transfer process and single electron transfer.
Photooxidation of sulfides to sulfoxides
To use the output energy to simulate the natural photosynthetic process, the above artificial light-harvesting system based on RB@Ga-tpe was applied as a photocatalyst for the oxidation of thioanisole in water (Table 1). As expected, an excellent yield (90%) and selectivity capability (>99%) on the sulfide being transformed into sulfoxide have been achieved in an aqueous solution and when irradiated with a Xe light (Table 1, entry 1). Exhilaratingly, the yield of 1a increased dramatically upon using the light-harvesting system RB@Ga-tpe compared with using either RB or Ga-tpe only (Table 1, entries 2 and 3). Meanwhile, the control experiments using ligand L or L&RB yielded a trace of the product under the same conditions, confirming that the supramolecular host using the preorganization of an artificial light-harvesting system is essential (Table 1, entries 4 and 5). Furthermore, a series of control experiments were carried out to confirm that the presence of light, photosensitizer, and O2 is essential for the catalytic reaction under similar conditions (Table 1, entries 6–8). Then, 395 nm light (the near absorption of Ga-tpe) triggered the photocatalytic reaction, the slightly high yield (RB@Ga-tpe as the photocatalyst) was 55%, and the lower yield (Ga-tpe or RB as the photocatalyst) was obtained (Table 1, entries 9–11). The results indicated that the photooxidation reaction could also use 395 nm light through the absorption of Ga-tpe, expanding the range of the catalytic light source.|
|
|||||
|---|---|---|---|---|---|
| Entry | Photocatalyst | Light | Conv. (%) | Selectivity (%) | |
| 1a | 1b | ||||
| a Reaction conditions, thioanisole (0.2 mmol, 1 equiv.), catalyst (0.5 mol%), water (2 ml), room temperature, 12 h, air. b Ar. The yields were determined by GC analysis (using 1,3,5-trimethoxybenzene as the internal standard). | |||||
| 1 | RB@Ga-tpe | Xe | 90 | >99 | <1 |
| 2 | RB | Xe | 13 | >99 | <1 |
| 3 | Ga-tpe | Xe | n.d. | 0 | 0 |
| 4 | L | Xe | 10 | >99 | <1 |
| 5 | L&RB | Xe | 30 | >99 | <1 |
| 6 | Blank | Xe | n.d. | 0 | 0 |
| 7b | RB@Ga-tpe | Xe | n.d. | 0 | 0 |
| 8 | RB@Ga-tpe | Dark | n.d. | 0 | 0 |
| 9 | Ga-tpe | 395 nm | 23 | >99 | <1 |
| 10 | RB | 395 nm | n.d. | 0 | 0 |
| 11 | RB@Ga-tpe | 395 nm | 55 | >99 | <1 |
Encouraged by the remarkable photocatalytic performance, the substrate compatibility of RB@Ga-tpe with various sulfides was further explored. As shown in Table S4,†RB@Ga-tpe exhibited excellent activity and selectivity toward other methylphenyl sulfide derivatives with different substituents, such as methoxy (–OMe) and fluorine (–F). Due to the limited solubility of the substrate in aqueous solution, when a small amount of methanol was added to the system, the yield of sulfoxides (1e–i) was significantly improved. In the photooxidation of sulfides, a series of quenching experiments were carried out by introducing various scavengers, such as sodium azide (NaN3, a scavenger of 1O2), 1,4-benzoquinone (BQ, a scavenger of O2˙−), isopropanol (IPA, a scavenger of ˙OH), and AgNO3 (as an electron trapping agent), respectively. The photooxidation reactions were effectively prohibited by adding sodium azide and BQ, whereas other scavengers exerted less influence than them, which revealed that 1O2 and O2˙− should be the dominant reactive species (Fig. S19a and S20, ESI†).23
Photocatalytic aerobic CDC reaction
The RB@Ga-tpe assembly with catalytic efficiency for the oxidation of sulfides was then expanded to other photocatalytic oxidative organic transformations, such as photocatalytic aerobic CDC reactions (Table 2), a highly efficient and powerful strategy for the construction of new carbon–carbon and carbon–heteroatom bonds, which has been considered a waste-minimized and sustainable synthetic alternative to classic coupling procedures.24 In particular, the photocatalytic C–H functionalization of tertiary amines is the most atom-economic and sustainable reaction process. The aerobic CDC reaction of N-phenyl-1,2,3,4-tetrahydroisoquinoline 2a and nitromethane with RB@Ga-tpe as a photocatalyst was selected as a model photocatalytic reaction. The blank reaction was performed under the same conditions, and a moderate yield was shown (50%, Table 2, entry 5). Alone, Ga-tpe, RB, or L, as the photocatalyst, produced relatively low yields in aqueous solution (52–65%, Table 2, entries 2–4). For comparison, the activity of RB@Ga-tpe in the photocatalytic CDC reaction was investigated; an excellent yield of 90% was achieved under the same conditions (Table 2, entry 1). However, a moderate reaction yield was obtained by the assembly resembling L&RB (70%, Table 2, entry 7). These above results revealed that light energy harvested using Ga-tpe could be efficiently transferred to RB within the host–guest encapsulated system, and the RB@Ga-tpe system could be an ideal reaction platform for organic reactions in aqueous solutions.|
|
|||
|---|---|---|---|
| Entry | Photocatalyst | Light | Yield rate (%) |
| a Reaction conditions: N-phenyl-1,2,3,4-tetrahydroisoquinoline (0.1 mmol, 1 equiv.), nitromethane (0.3 ml), catalyst (1.0 mol%), water solution (2 ml), room temperature, 12 h. The yields were determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as an internal standard. | |||
| 1 | RB@Ga-tpe | Xe | 90 |
| 2 | Ga-tpe | Xe | 65 |
| 3 | RB | Xe | 59 |
| 4 | L | Xe | 52 |
| 5 | Blank | Xe | 50 |
| 6 | RB@Ga-tpe | Dark | n.d. |
| 7 | L&RB | Xe | 70 |
| 8 | Ga-tpe | 395 nm | 34 |
| 9 | RB | 395 nm | 25 |
| 10 | RB@Ga-tpe | 395 nm | 51 |

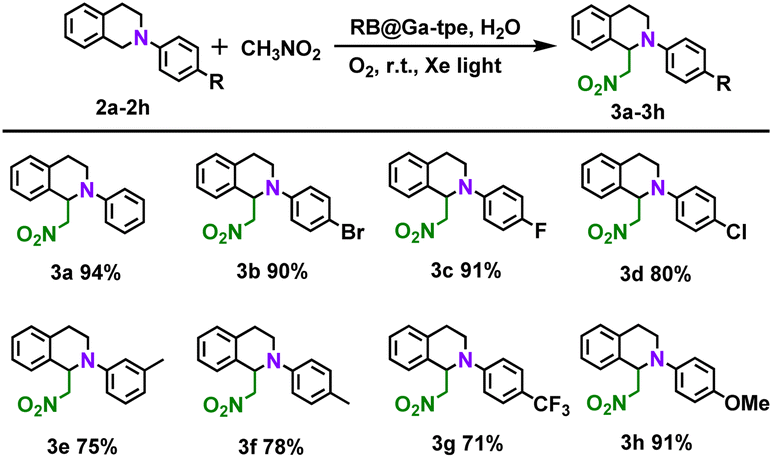
A series of quenching experiments were also carried out by introducing various scavengers into the CDC reaction (Fig. S19b, ESI†). Unsurprisingly, the abovementioned studies demonstrated that the superoxide radical anion is the active intermediate in the photocatalytic CDC reactions. According to the above findings and literature results,25 the photocatalytic CDC reaction mechanism between N-phenyl-1,2,3,4-tetrahydroisoquinoline and nitromethane was proposed (Fig. S21, ESI†). The RB@Ga-tpe assembly was excited using Xe light in an aqueous solution, and the excited state of Ga-tpe transferred the energy to the acceptor dye RB to form the excited state of RB. As shown in Table 3, we further explored the substrate scope; generally, N-phenyl-1,2,3,4-tetrahydroisoquinoline derivatives with different electronic properties could be converted to the desired reaction products 3a–h in excellent yields (71–94%).
|
a Reaction conditions: N-phenyl-1,2,3,4-tetrahydroisoquinoline derivative (0.1 mmol, 1 equiv.), nitromethane (0.3 ml). RB@Ga-tpe assembly solution (RB:Ga-tpe = 1:1, 1.0 mol%, 2 ml), Xe light, oxygen ball, room temperature, 18 h. The yields were determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as an internal standard.
|
|---|
|
Overall, these results confirmed that the photocatalytic aerobic reaction was initiated by the supramolecular assembly in the aqueous phase, and dye-loaded capsule RB@Ga-tpe could be regarded as one kind of biological counterpart. The host–guest species RB@Ga-tpe with excellent photocatalytic ability can serve as an efficient light-harvesting system where the output energy was successfully utilized for catalytic chemical transformation for practical application in the aqueous phase. The reason why the dye-loaded capsule RB@Ga-tpe exhibited high catalytic efficiency might be that the host–guest arrangement between the cage Ga-tpe and RB in a confined space avoids unnecessary fluorescence quenching, and the photobleaching effect of RB upon long-time irradiation was also decreased because of the formation of a supramolecular assembly. Thus, we believe that such an efficient host–guest system with an energy transfer process would provide new insight into the fabrication of artificial photosynthetic systems.
Conclusions
In summary, a new negatively charged metal–organic barrel Ga-tpe endowed with water solubility was reported for the construction of a light-harvesting system by host–guest interactions to accelerate light-driven aerobic reactions in aqueous media. The energy acceptor RB was matched to generate the FRET process by rendering the host–guest interaction between the donor and the acceptor stronger and more robust in close proximity. The artificial light-harvesting system RB@Ga-tpe had suitable potential for the photooxidation of sulfides to sulfoxides reaction and photocatalytic CDC reactions in simulating the photosynthesis process. Overall, this work presents an ideal way for the creation of a water-soluble artificial light-harvesting system via host–guest encapsulation to enhance the efficiency of the FRET process, serving as a versatile platform for mimicking natural photosynthesis in a simulated enzymatic environment. The further development of metal–organic host–guest assemblies in aqueous solutions that mimic natural organisms will be facilitated and accelerated in the future.Experimental
Preparation of Ga-tpe
A mixture of 4,4′,4′′,4′′′-(ethene-1,1,2,2-tetrayl)tetra (benzohydrazide) (84.7 mg, 0.15 mmol), 5-sulfosalicylaldehyde sodium salt (134.8 mg, 0.6 mmol), NaOH (48 mg, 1.2 mmol), and gallium tris(acetylacetonate) (110.1 mg, 0.3 mmol) was dissolved in 30 mL CH3OH and then heated at 60 °C for 24 h to give a clear yellow solution. The mixture was repeatedly recrystallized from dichloromethane and acetone to afford a yellow solid with 65.6% yield (based on the solid dried under vacuum). 1H NMR (400 MHz, D2O) δ 8.76 (s, 4H), 7.84 (s, 4H), 7.61 (s, 12H), 7.13 (s, 8H), 6.70 (s, 4H). 13C NMR (100 MHz, MeOD) δ 193.78, 180.44, 170.44, 168.75, 161.46, 156.96, 147.08, 134.59, 133.11, 132.02, 128.35, 122.01, 118.21.General procedure for the RB@Ga-tpe photocatalytic oxidation of thioanisole
Thioanisole (0.2 mmol, 1 equiv.), catalyst (0.5 mol%), and H2O (2 ml) were added to a glass reactor and irradiated with a Xe lamp at room temperature for 12 h in an air atmosphere. At the end of the reaction, it was extracted using dichloromethane. The yields were monitored using gas chromatography (GC) and 1H NMR analysis relative to the internal standard 1,3,5-trimethoxybenzene.General procedure for the RB@Ga-tpe photocatalytic aerobic CDC reaction
N-Phenyl-1,2,3,4-tetrahydroisoquinoline (0.1 mmol, 1 equiv.), nitromethane (0.3 ml), catalyst (1.0 mol%), and H2O (2 ml) were added to a glass reactor and irradiated using a Xe lamp at room temperature for 12 h in an air atmosphere. After the indicated time, it was extracted using ethyl acetate and concentrated under vacuum distillation. The yields were determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as an internal standard.Data availability
All the data supporting this article have been included in the main text and the ESI.†Author contributions
C. He and D. Li designed the research and co-wrote the manuscript. D. Li, H. Li, and J. Li performed all experiments. W. Fang carried out structural optimization calculations. X. Fu performed ESI-MS. L. Yang and X. Li assisted in analyzing the data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China 22171033 and the Fundamental Research Funds for the Central Universities [DUT17RC(3)094].Notes and references
- (a) T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed; (b) T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed; (c) D. Zhang, T. K. Ronson and J. R. Nitschke, Acc. Chem. Res., 2018, 51, 2423–2436 CrossRef CAS PubMed.
- (a) Y. Tamura, H. Takezawa and M. Fujita, J. Am. Chem. Soc., 2020, 142, 5504–5508 CrossRef CAS PubMed; (b) M. D. Ludden, C. G. P. Taylor and M. D. Ward, Chem. Sci., 2021, 12, 12640–12650 RSC; (c) S. Hasegawa, S. L. Meichsner, J. J. Holstein, A. Baksi, M. Kasanmascheff and G. H. Clever, J. Am. Chem. Soc., 2021, 143, 9718–9723 CrossRef CAS PubMed; (d) M. Ueda, N. Kishida, L. Catti and M. Yoshizawa, Chem. Sci., 2022, 13, 8642–8648 RSC; (e) L. R. Holloway, P. M. Bogie, Y. Lyon, C. Ngai, T. F. Miller, R. R. Julian and R. J. Hooley, J. Am. Chem. Soc., 2018, 140, 8078–8081 CrossRef CAS PubMed; (f) R. Banerjee, D. Chakraborty and P. S. Mukherjee, J. Am. Chem. Soc., 2023, 145, 7692–7711 CrossRef CAS PubMed.
- (a) W.-X. Gao, H.-N. Zhang and G.-X. Jin, Coord. Chem. Rev., 2019, 386, 69–84 CrossRef CAS; (b) X.-X. Gou, T. Liu, Y.-Y. Wang and Y.-F. Han, Angew. Chem., Int. Ed., 2020, 59, 16683–16689 CrossRef CAS PubMed; (c) H. Wang, Y. Li, N. Li, A. Filosa and X. Li, Nat. Rev. Mater., 2021, 6, 145–167 CrossRef CAS; (d) Y. Hou, Z. Zhang, S. Lu, J. Yuan, Q. Zhu, W.-P. Chen, S. Ling, X. Li, Y.-Z. Zheng, K. Zhu and M. Zhang, J. Am. Chem. Soc., 2020, 142, 18763–18768 CrossRef CAS PubMed; (e) J. Dong, Y. Liu and Y. Cui, Acc. Chem. Res., 2021, 54, 194–206 CrossRef CAS PubMed.
- (a) M. D. Ward, C. A. Hunter and N. H. Williams, Acc. Chem. Res., 2018, 51, 2073–2082 CrossRef CAS PubMed; (b) A. Galan and P. Ballester, Chem. Soc. Rev., 2016, 45, 1720–1737 RSC; (c) H. Amouri, C. Desmarets and J. Moussa, Chem. Rev., 2012, 112, 2015–2041 CrossRef CAS PubMed.
- (a) D. Zhang, T. K. Ronson, Y.-Q. Zou and J. R. Nitschke, Nat. Rev. Chem, 2021, 5, 168–182 CrossRef CAS PubMed; (b) D. Zhang, T. K. Ronson, R. Lavendomme and J. R. Nitschke, J. Am. Chem. Soc., 2019, 141, 18949–18953 CrossRef CAS PubMed; (c) P. C. Purba, M. Maity, S. Bhattacharyya and P. S. Mukherjee, Angew. Chem., Int. Ed., 2021, 60, 14109–14116 CrossRef CAS PubMed; (d) D. Chakraborty, R. Saha, J. K. Clegg and P. S. Mukherjee, Chem. Sci., 2022, 13, 11764–11771 RSC.
- (a) R. Saha, B. Mondal and P. S. Mukherjee, Chem. Rev., 2022, 122, 12244–12307 CrossRef CAS PubMed; (b) C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed; (c) Y. Xue, X. Hang, J. Ding, B. Li, R. Zhu, H. Pang and Q. Xu, Coord. Chem. Rev., 2021, 430, 213656–221378 CrossRef CAS.
- (a) J.-S. Wang, K. Wu, C. Yin, K. Li, Y. Huang, J. Ruan, X. Feng, P. Hu and C.-Y. Su, Nat. Commun., 2020, 11, 4675–4684 CrossRef CAS PubMed; (b) Q.-Q. Wang, S. Gonell, S. H. A. M. Leenders, M. Duerr, I. Ivanovic-Burmazovic and J. N. H. Reek, Nat. Chem., 2016, 8, 225–230 CrossRef CAS PubMed.
- (a) L. Zhao, X. Jing, X. Li, X. Guo, L. Zeng, C. He and C. Duan, Coord. Chem. Rev., 2019, 378, 151–187 CrossRef CAS; (b) M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910–1921 CrossRef CAS PubMed.
- (a) H. Sunohara, K. Koyamada, H. Takezawa and M. Fujita, Chem. Commun., 2021, 57, 9300–9930 RSC; (b) D. M. Daiton, S. R. Ellis, E. M. Nichols, R. A. Mathies, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2015, 137, 10128–10131 CrossRef PubMed; (c) A. J. Musser, P. P. Neelakandan, J. M. Richter, H. Mori, R. H. Friend and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 12050–12059 CrossRef CAS PubMed.
- (a) Y.-X. Hu, W.-J. Li, P.-P. Jia, X.-Q. Wang, L. Xu and H.-B. Yang, Adv. Opt. Mater., 2020, 8, 2000265 CrossRef CAS; (b) K. Wang, K. Velmurugan, B. Li and X.-Y. Hu, Chem. Commun., 2021, 57, 13641–13654 RSC; (c) X.-M. Chen, X. Chen, X.-F. Hou, S. Zhang, D. Chen and Q. Li, Nanoscale Adv., 2023, 5, 1830–1852 RSC.
- (a) M. Hao, G. Sun, M. Zuo, Z. Xu, Y. Chen, X. Y. Hu and L. Wang, Angew. Chem., Int. Ed., 2020, 59, 10095–10100 CrossRef CAS PubMed; (b) J.-J. Li, H.-Y. Zhang, X.-Y. Dai, Z.-X. Liu and Y. Liu, Chem. Commun., 2020, 56, 5949–5952 RSC; (c) Y. Xia, M. Chen, S. Li, M. Li, X. Li, T. Yi and D. Zhang, J. Mater. Chem. C, 2022, 10, 12332–12337 RSC; (d) Z. Lian, J. He, L. Liu, Y. Fan, X. Chen and H. Jiang, Nat. Commun., 2023, 14, 2752–2762 CrossRef CAS PubMed; (e) P. Sutar, V. M. Suresh, K. Jayaramulu, A. Hazra and T. K. Maji, Nat. Commun., 2018, 9, 3587–3598 CrossRef PubMed.
- (a) K. Acharyya, S. Bhattacharyya, H. Sepehrpour, S. Chakraborty, S. Lu, B. Shi, X. Li, P. S. Mukherjee and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 14565–14569 CrossRef CAS PubMed; (b) Y. Li, Y. Dong, L. Cheng, C. Qin, H. Nian, H. Zhang, Y. Yu and L. Cao, J. Am. Chem. Soc., 2019, 141, 8412–8415 CrossRef CAS PubMed; (c) L. Xu, Z. Wang, R. Wang, L. Wang, X. He, H. Jiang, H. Tang, D. Cao and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9908–9913 CrossRef CAS PubMed.
- (a) W.-J. Li, X.-Q. Wang, D.-Y. Zhang, Y.-X. Hu, W.-T. Xu, L. Xu, W. Wang and H.-B. Yang, Angew. Chem., Int. Ed., 2021, 60, 18761–18768 CrossRef CAS PubMed; (b) Y.-N. Jing, S.-S. Li, M. Su, H. Bao and W.-M. Wan, J. Am. Chem. Soc., 2019, 141, 16839–16848 CrossRef CAS PubMed.
- (a) D. Zhang, W. Yu, S. Li, Y. Xia, X. Li, Y. Li and T. Yi, J. Am. Chem. Soc., 2021, 143, 1313–1317 CrossRef CAS PubMed; (b) Z. Zhang, Z. Zhao, Y. Hou, H. Wang, X. Li, G. He and M. Zhang, Angew. Chem., Int. Ed., 2019, 58, 8862–8866 CrossRef CAS PubMed; (c) J. R. Piper, L. Cletheroe, C. G. P. Taylor, A. J. Metherell, J. A. Weinstein, I. V. Sazanovich and M. D. Ward, Chem. Commun., 2017, 53, 408–411 RSC; (d) A. Kumar, R. Saha and P. S. Mukherjee, Chem. Sci., 2021, 12, 5319–5329 RSC; (e) S. Ahmed, A. Kumar and P. S. Mukherjee, Chem. Mater., 2022, 34, 9656–9665 CrossRef CAS; (f) P.-P. Jia, Y.-X. Hu, Z.-Y. Peng, B. Song, Z.-Y. Zeng, Q.-H. Ling, X. Zhao, L. Xu and H.-B. Yang, Inorg. Chem., 2023, 62, 1950–1957 CrossRef CAS PubMed.
- D. Li, X. Liu, L. Yang, H. Li, G. Guo, X. Li and C. He, Chem. Sci., 2023, 14, 2237–2244 RSC.
- (a) G. V. Oshovsky, D. N. Reinhoudt and W. Verboom, Angew. Chem., Int. Ed., 2007, 46, 2366–2393 CrossRef CAS PubMed; (b) L. L. K. Taylor, I. A. Riddell and M. M. J. Smulders, Angew. Chem., Int. Ed., 2019, 58, 1280–1307 CrossRef CAS PubMed.
- (a) E. G. Percástegui, T. K. Ronson and J. R. Nitschke, Chem. Rev., 2020, 120, 13480–13544 CrossRef PubMed; (b) O. Bistri and O. Reinaud, Org. Biomol. Chem., 2015, 13, 2849–2865 RSC.
- (a) P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697–1699 CrossRef CAS PubMed; (b) G. Wu, Y. Chen, S. Fang, L. Tong, L. Shen, C. Ge, Y. Pan, X. Shi and H. Li, Angew. Chem., Int. Ed., 2021, 60, 16594–16599 CrossRef CAS PubMed; (c) E. G. Percástegui, J. Mosquera, T. K. Ronson, A. J. Plajer, M. Kieffer and J. R. Nitschke, Chem. Sci., 2019, 10, 2006–2018 RSC; (d) P. Howlader, B. Mondal, P. C. Purba, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2018, 140, 7952–7960 CrossRef CAS PubMed.
- (a) H. Takezawa, K. Shitozawa and M. Fujita, Nat. Chem., 2020, 12, 574–578 CrossRef CAS PubMed; (b) D. H. Leung, D. Fiedler, R. G. Bergman and K. N. Raymond, Angew. Chem., Int. Ed., 2004, 43, 963–966 CrossRef CAS PubMed; (c) W. Cullen, M. C. Misuraca, C. A. Hunter, N. H. Williams and M. D. Ward, Nat. Chem., 2016, 8, 231–236 CrossRef CAS PubMed.
- (a) W. Cullen, H. Takezawa and M. Fujita, Angew. Chem., Int. Ed., 2019, 58, 9171–9173 CrossRef CAS PubMed; (b) L.-X. Cai, S.-C. Li, D.-N. Yan, L.-P. Zhou, F. Guo and Q.-F. Sun, J. Am. Chem. Soc., 2018, 140, 4869–4876 CrossRef CAS PubMed; (c) X. Han, C. Guo, C. Xu, L. Shi, B. Liu, Z. Zhang, Q. Bai, B. Song, F. Pan, S. Lu, X. Zhu, H. Wang, X.-Q. Hao, M.-P. Song and X. Li, ACS Nano, 2023, 17, 3723–3736 CrossRef CAS PubMed.
- H. Wang, C. H. Liu, K. Wang, M. Wang, H. Yu, S. Kandapal, R. Brzozowski, B. Xu, M. Wang, P. Eswara, M. P. Nieh, J. Cai and X. Li, J. Am. Chem. Soc., 2019, 141, 16108–16116 CrossRef CAS PubMed.
- (a) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed; (b) X. Yan, T. R. Cook, P. Wang, F. Huang and P. J. Stang, Nat. Chem., 2015, 7, 342–348 CrossRef CAS PubMed; (c) X. Yan, M. Wang, T. R. Cook, M. Zhang, M. L. Saha, Z. Zhou, X. Li, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2016, 138, 4580–4588 CrossRef CAS PubMed; (d) V. M. Suresh, A. De and T. K. Maji, Chem. Commun., 2015, 51, 14678–14681 RSC.
- (a) S.-D. Wang, C.-L. Lai, Y.-X. Zhang, S.-T. Bao, K.-L. Lv and L.-L. Wen, J. Mater. Chem. A, 2022, 10, 20975–20983 RSC; (b) M. Forchetta, F. Sabuzi, L. Stella, V. Conte and P. Galloni, J. Org. Chem., 2022, 87, 14016–14025 CrossRef CAS PubMed.
- (a) Y. Zhi, S. Ma, H. Xia, Y. Zhang, Z. Shi, Y. Mu and X. Liu, Appl. Catal., B, 2019, 244, 36–44 CrossRef CAS; (b) Y. Cheng, W. Ji, X. Wu, X. Ding, X. Liu and B. Han, Appl. Catal., B, 2022, 306, 121110–121122 CrossRef CAS.
- (a) C.-Q. Ma, X.-L. Li, N. Han, Y. Wang, R.-Z. Wang, S. Yu, Y.-B. Wang and L.-B. Xing, J. Mater. Chem. A, 2022, 10, 16390–16395 RSC; (b) Y. Wang, C.-Q. Ma, X.-L. Li, R.-Z. Dong, H. Liu, R.-Z. Wang, S. Yu and L.-B. Xing, J. Mater. Chem. A, 2023, 11, 2627–2633 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02943c |
| This journal is © The Royal Society of Chemistry 2023 |