
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen spillover and substrate–support hydrogen bonding mediate hydrogenation of phenol catalyzed by palladium on reducible metal oxides†
Yeongseo
An‡
a,
Puranjan
Chatterjee‡
ab,
Pranjali
Naik
 ab,
Sayak
Banerjee
a,
Wenyu
Huang
ab,
Igor I.
Slowing
ab and
Vincenzo
Venditti
*ac
ab,
Sayak
Banerjee
a,
Wenyu
Huang
ab,
Igor I.
Slowing
ab and
Vincenzo
Venditti
*ac
aDepartment of Chemistry, Iowa State University, Hach Hall, 2438 Pammel Drive, Ames, Iowa 50011, USA. E-mail: venditti@iastate.edu; Fax: +1-515-294-7550; Tel: +1-515-294-1044
bDepartment of Energy, Ames Laboratory, Ames, Iowa 50011, USA
cRoy J. Carver Department of Biochemistry, Biophysics and Molecular Biology, Iowa State University, Ames, Iowa 50011, USA
First published on 24th November 2023
Abstract
Substrate–support interactions play an important role in the catalytic hydrogenation of phenolic compounds by ceria-supported palladium (Pd/CeO2). Here, we combine surface contrast solution NMR methods and reaction kinetic assays to investigate the role of substrate–support interactions in phenol (PhOH) hydrogenation catalyzed by titania-supported palladium (Pd/TiO2). We show that PhOH adsorbs on the catalyst via a weak hydrogen-bonding interaction between the –OH group of the substrate and one oxygen atom on the support. Interestingly, we observe that the addition of 20 mM inorganic phosphate results in a ∼2-fold destabilization of the PhOH–support interaction and a corresponding ∼2-fold inhibition of the catalytic reaction, suggesting an active role of the PhOH–TiO2 hydrogen bond in catalysis. A comparison of the data measured here with the results previously reported for a Pd/CeO2 catalyst indicates that the efficiency of the Pd-supported catalysts is correlated to the amount of PhOH hydrogen bonded to the metal oxide support. Since CeO2 and TiO2 have similar ability to uptake activated hydrogen from a noble metal site, these data suggest that hydrogen spillover is the main mechanism by which Pd-activated hydrogens are shuttled to the PhOH adsorbed on the surface of the support. Consistent with this hypothesis, Pd supported on a non-reducible metal oxide (silica) displays negligible hydrogenation activity. Therefore, we conclude that basic and reducible metal oxides are active supports for the efficient hydrogenation of phenolic compounds due to their ability to hydrogen bond to the substrate and mediate the addition of the activated hydrogens to the adsorbed aromatic ring.
Introduction
Phenolic compounds are high-volume petrochemicals currently employed in a myriad of industrial processes, from the production of polymer resins to the synthesis of pharmaceuticals.1 In the past years, much effort has been made toward the development of strategies for the fundamental elaboration and diversification of phenols to unlock new routes for their conversion into higher-value chemicals.1 In one of these applications, hydrogenation of phenol (PhOH) catalyzed by noble metals is used as an efficient route for large-scale production of cyclohexanone and cyclohexanol, which are important precursors for the synthesis of nylons and polyamide resins.2 Recently, we have shown that Pd supported on ceria (Pd/CeO2) can catalyze PhOH hydrogenation to cyclohexanone at room temperature and atmospheric pressure with unprecedented efficiency and selectivity.3 In this catalyst, the supported Pd is required for H2 adsorption and activation, while ceria mediates the adsorption of the small molecule into the catalytically competent substrate–catalyst adduct.3–6 Although it is demonstrated that the hydrogen-bonding interaction between PhOH and ceria has a direct participation in catalysis,4,5 it is unclear if the ability of ceria to mediate catalysis originates from its ability to shuttle the activated hydrogen to the substrate via hydrogen spillover, or to its capacity to facilitate constructive encounters between PhOH and Pd by optimizing the dispersion of the noble metal on the catalyst (Fig. 1). Similarly, it is not understood if other metal oxide supports can assist the hydrogenation reaction via analogous mechanisms.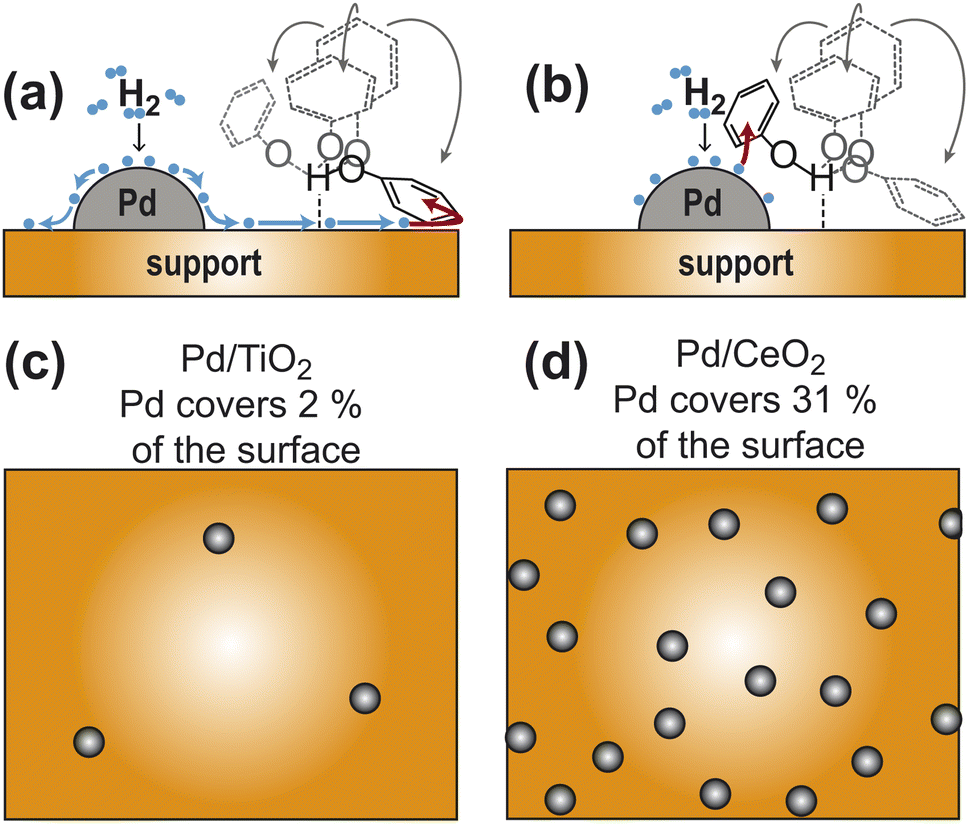 | ||
| Fig. 1 Models for the participation of support in catalysis. (a) Pd-activated hydrogen spills over the support and reduces the aromatic ring of PhOH hydrogen bonded to the support. (b) Hydrogen bonding to the support facilitates the establishment of close contacts between PhOH and a nearby Pd site. Panels (c) and (d) are cartoon representations of a catalyst with a sparse and dense distribution of Pd sites, respectively. The ratio between metallic and support surface area has been chosen to correspond to the Pd/TiO2 and Pd/CeO2 catalysts in (c) and (d), respectively (see Table 1). In all panels the Pd sites are gray spheres and the support surface is an orange box. In panels (a) and (b) the activated hydrogens are blue spheres and four possible orientations of PhOH hydrogen bonded to the support are shown as formula structures. The PhOH orientation that receives hydrogen from the catalyst is colored black. The other three PhOH orientations are gray. Spillover is indicated by blue arrows. The addition of activated hydrogen to the aromatic ring is indicated by red arrows. | ||
Here we use surface contrast NMR (scNMR)5,7 to investigate how the kinetics and thermodynamics of adsorption modulate the PhOH hydrogenation catalyzed by Pd supported on titania (Pd/TiO2). Pd/TiO2 has been previously shown to catalyze selective PhOH hydrogenation to cyclohexanone at mild temperature and pressure.8,9 In this work, we use the Pd/TiO2 catalyst to investigate the contribution of support–substrate interactions to catalysis. We show that the adsorption of PhOH on Pd/TiO2 results in the formation of a weakly bound adduct in which the small molecule is likely hydrogen bonded to the support. In analogy to what we have previously observed for the Pd/CeO2 catalyst,5 we notice that the addition of 20 mM phosphate (at constant pH) results in a ∼2-fold destabilization of the PhOH–titania interaction and in a corresponding ∼2-fold reduction in catalytic conversion, which suggests that the weak substrate–support interaction is competent for the hydrogenation reaction catalyzed by Pd/TiO2. Finally, a comparison of the data measured for the Pd/TiO2 and Pd/CeO2 catalysts indicates that, irrespective of the metal oxide, the kinetics of hydrogenation are correlated to the concentration of substrate hydrogen-bonded to the support. Since the ceria- and titania-based catalysts have drastically different Pd dispersion (Fig. 1) but a comparable ability to uptake activated hydrogen on the support surface,10 our results indicate that hydrogen spillover is the main mechanism mediating catalytic conversion of PhOH hydrogen-bonded to the metal oxide support. Consistent with this conclusion, we show that silica-supported Pd (Pd/SiO2) is incapable to mediate hydrogen spillover on the support and provides an inefficient catalyst for PhOH hydrogenation at mild conditions.
Results
Preparation of the Pd/TiO2 catalyst
Titanium(IV) oxide, anatase nanopowder was purchased from Sigma-Aldrich and was used for catalyst preparation without further purification. In anatase TiO2, the predominant crystallographic plane is (101), which is confirmed by the intense peak at 2θ ∼ 25° in the X-ray diffraction (XRD) pattern11,12 (Fig. 2d). Pd nanoparticles were deposited on TiO2 starting from the palladium(II) nitrate precursor via the incipient wetness impregnation method targeting 1 wt% Pd loading (see ESI Methods†). The XRD pattern of Pd/TiO2 did not show the peak at 2θ ∼ 40° that is usually assigned to the (111) facet of the Pd nanoparticle13 (Fig. 2d). This observation indicates that the deposited Pd particle size is below 3 nm. According to the transmission electron micrographs, the TiO2 and Pd/TiO2 particles are spherical in morphology with some irregularities (Fig. 2a and b).14 The average size of 100 Pd/TiO2 particles counted was 33 ± 8 nm (Fig. 2c), which is consistent with the claimed size of the commercial TiO2 nanoparticles (∼25 nm). N2 sorption analysis reported a surface area of 76.0 m2 g−1 for TiO2 and 73.0 m2 g−1 for Pd/TiO2. The dispersion of the Pd nanoparticle was determined to be 38.5% by CO pulse chemisorption. The corresponding Pd particle size was ∼2.7 nm. Given the metallic surface area of 1.6 m2 g−1 obtained by CO pulse chemisorption, the actual TiO2 surface area on the Pd/TiO2 catalyst is calculated as 71.4 m2 g−1 (73.0–1.6 m2 g−1) (Table 1).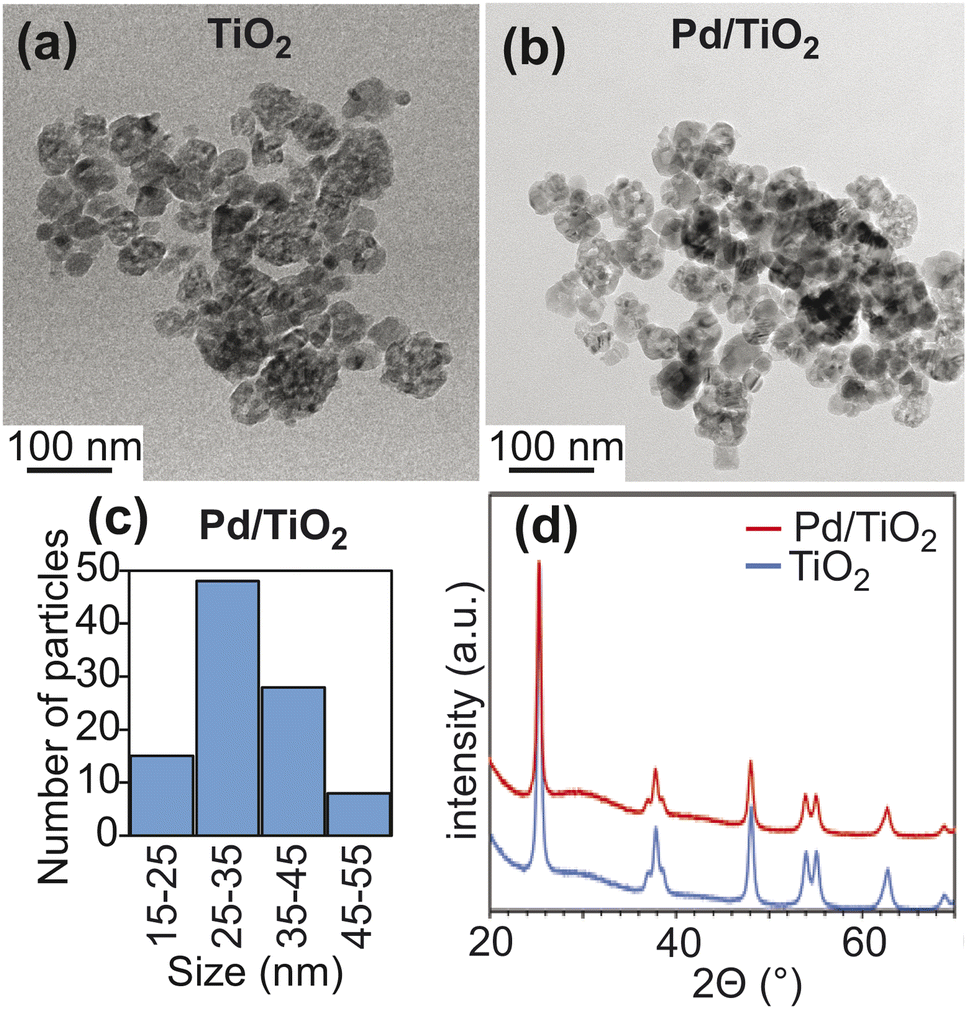 | ||
| Fig. 2 Properties of TiO2 and Pd/TiO2. Panels (a) and (b) show TEM images for TiO2 and Pd/TiO2. Panel (c) shows a histogram of particle size constructed by counting 100 Pd/TiO2 particles (average particle size 33 ± 8 nm). Panel (d) shows wide-angle PXRD patterns for TiO2 (blue) and Pd/TiO2 (red). | ||
| TiO2 | Pd/TiO2 | Pd/CeO2a | Pd/SiO2 | |
|---|---|---|---|---|
| a Data on the Pd/CeO2 catalyst were reported by Egner et al. (2020).5 b 13C ΔR2 values averaged over the ortho, meta, and para positions. | ||||
| Morphology | Spherical | Spherical | Cubic | Spherical |
| Diameter (nm) | 33 ± 8 | 33 ± 8 | 25 ± 8 | 22 ± 5 |
| BET surface area (m2 g−1) | 76.0 | 73.0 | 9.4 | 103.0 |
| Metallic surface area (m2 g−1) | — | 1.6 | 2.9 | 1.4 |
| Support surface area (m2 g−1) | 76.0 | 71.4 | 6.5 | 101.6 |
| Cubic crystallite size (nm) | — | 2.7 | 2.0 | 2.4 |
| % dispersion | — | 38 | 65 | 38 |
| k (μM s−1) @ 0 mM Pi | — | 22.0 ± 0.2 | 30.5 ± 0.2 | 0 |
| k (μM s−1) @ 20 mM Pi | — | 11.5 ± 0.2 | 15.7 ± 0.4 | 0 |
| Aver. 13C ΔR2 (s−1) @ 0 mM Pib | 0.5 ± 0.1 | 0.7 ± 0.1 | 1.6 ± 0.1 | 4.2 ± 0.1 |
| Aver. 13C ΔR2 (s−1) @ 20 mM Pib | 0.2 ± 0.1 | 0.2 ± 0.1 | 0.9 ± 0.1 | 2.6 ± 0.1 |
Effect of TiO2 and Pd/TiO2 on PhOH 13C transverse relaxation
In solution NMR, the interaction between an NMR-visible small molecule and an NMR-invisible nanoparticle can be conveniently detected by measuring the increase in 13C transverse relaxation (R2) of the small molecule upon addition of the nanoparticle (ΔR2).4,7,15 In the particular case of PhOH adsorption on a metal oxide particle, analysis of a plot of ΔR2versus the angle (θ) formed by the relevant 13C–1H bond and the para C–H bond provides thermodynamic and structural information on the adsorption process (Fig. 3). Indeed, we have previously shown that the formation of a hydrogen bond between the PhOH and the metal oxide surface generates a homogeneous increase in 13C R2 for the ortho, meta, and para positions that is the result of a homogeneous restriction of motion across the C–H bonds upon binding (Fig. 3a).4,5 On the other hand, the interaction of PhOH with an oxygen vacancy on the nanoparticle selectively restricts the motion of the para C–H bond (Fig. 3b and d) and causes the ΔR2 for the para carbon to be larger than the ΔR2 values measured for the ortho and meta positions.4,5,16 The data presented in Fig. 3e clearly indicate that the addition of 1 wt% TiO2 to a 10 mM PhOH solution generates ΔR2 values that are independent on θ (average ΔR2 = 0.5 ± 0.1 s−1). This observation indicates the absence (within the detection limit) of PhOH bound to oxygen vacancies and is consistent with the fact the titania is known to produce nanoparticles with a small number of defects.17 Addition of 20 mM inorganic phosphate (Pi) to the sample at pH 7 (that matches the pH of the sample at 0 mM Pi, see ESI Methods†) results in a ∼2-fold decrease in ΔR2 (average ΔR2 = 0.2 ± 0.1 s−1) (Fig. 3e). Since Pi interacts strongly with metal oxide nanoparticles,18 the observed reduction in ΔR2 is a direct consequence of the competition between Pi and PhOH for the absorption sites and corroborates that the measured increase in R2 reports on the PhOH–TiO2 hydrogen-bonding interaction.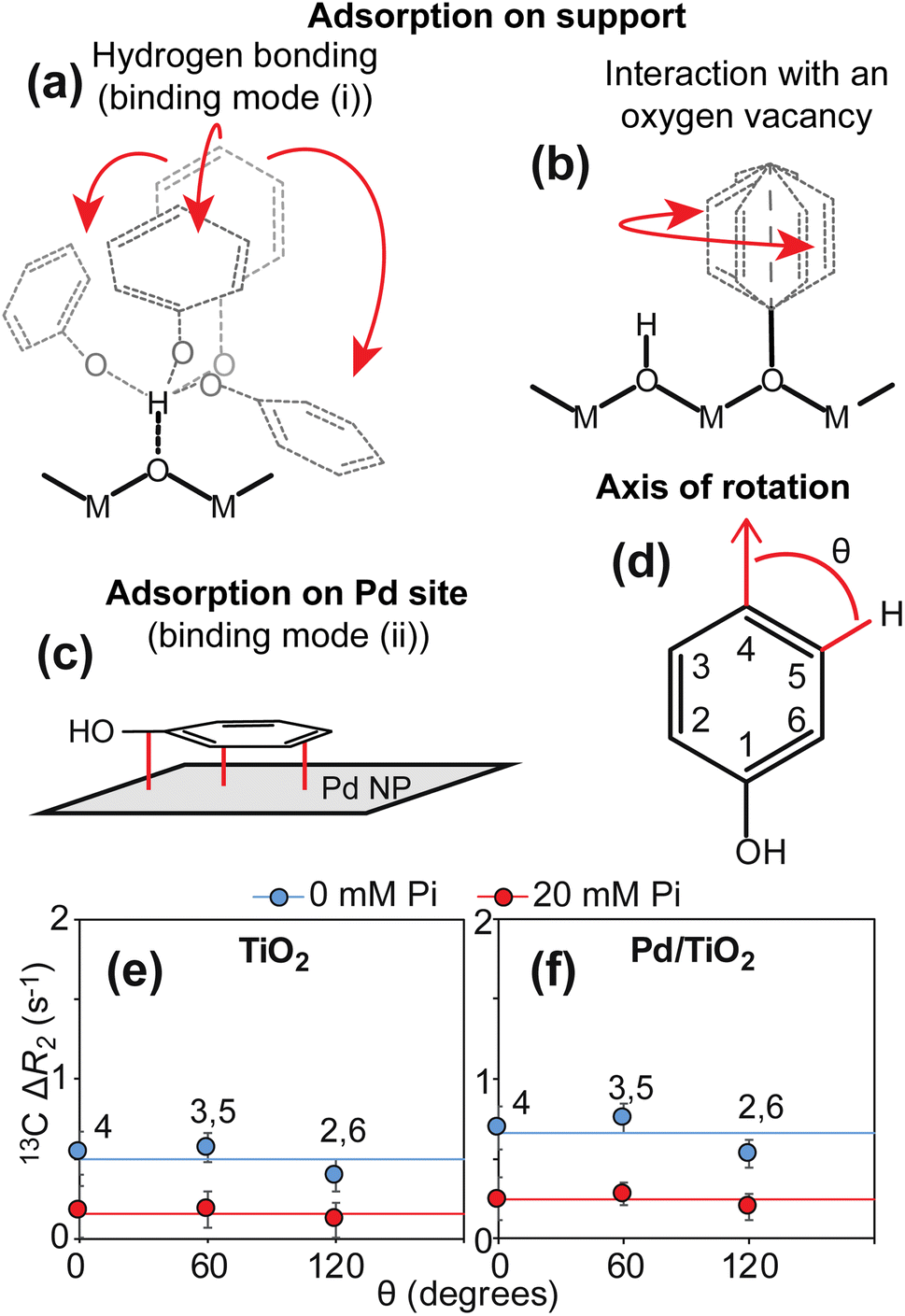 | ||
| Fig. 3 13C ΔR2 analysis. Panels (a–c) show the expected binding modes for PhOH on the surface of a nanoparticle formed by Pd supported on a metal oxide. In (a), PhOH is hydrogen bonded to the support. The motion of the three aromatic C–H bonds is restricted to a similar extent. In (b), PhOH is bound to an oxygen vacancy on the support. Only one rotatable bond separates the support from the aromatic ring. Therefore, the para C–H group appears static with respect to the axis of rotation. In (c), PhOH is rigidly adsorbed on the Pd site. All the aromatic C–H bonds tumble with the same correlation time as the nanoparticle (i.e. S2 ∼ 1).5 M and O indicate the metal and oxygen atoms of the metal oxide, respectively. The Pd site is shown as a gray surface. Panel (d) shows the angle θ formed by the aromatic C–H bonds with the axis of rotation of a PhOH molecule bound to an oxygen vacancy. Panels (e) and (f) show a plot of the 13C ΔR2 as a function of θ measured for PhOH in the presence of 1 wt% TiO2 and Pd/TiO2, respectively. The ortho (2,6) and meta (3,5) positions generate equivalent NMR peaks and one single ΔR2 value for these peaks is reported. Data measured in the presence of 0 and 20 mM Pi are shown as blue and red spheres, respectively. Solid blue and red lines are the corresponding average ΔR2 values. | ||
The ΔR2 values measured upon addition of Pd/TiO2 in the absence and in the presence of Pi (average ΔR2 = 0.7 ± 0.1 and 0.2 ± 0.1 s−1, respectively) are identical (within error) to the ones measured upon addition of TiO2 (Fig. 3f), suggesting that deposition of Pd does not substantially affect the interaction between PhOH and the nanoparticle surface. This observation is consistent with the fact that deposition of Pd on the titania reduces the surface of the support slightly (from 76.0 to 71.4 m2 g−1) and that the metallic surface area (1.6 m2 g−1) is negligible compared to the support surface area in Pd/TiO2 (Table 1). Finally, it is interesting to note that the ΔR2 induced on PhOH by addition of 1 wt% Pd/TiO2 is considerably smaller than the ΔR2 previously measured in the presence of 1 wt% Pd/CeO2 (average ΔR2 = 1.6 ± 0.1 and 0.9 ± 0.1 s−1 in the absence and in the presence of 20 mM Pi, respectively) (Table 1),5 which suggests that titania has lower affinity for PhOH than ceria.
scNMR characterization of PhOH adsorption on Pd/TiO2
Further characterization of PhOH adsorption on Pd/TiO2 was obtained by scNMR.5,7 Three scNMR experiments were employed: dark-state saturation transfer (DEST), Carr–Purcell–Meiboom–Gill (CPMG) relaxation dispersion (RD), and longitudinal relaxation (R1). The combined analysis of 13C DEST, RD, and R1 returns a comprehensive characterization of the thermodynamics and kinetics of adsorption processes occurring on the μs–s timescale, and provides structural information on the adsorbed species.5,7The addition of 1 wt% TiO2 or Pd/TiO2 to a 10 mM solution of PhOH induces a homogeneous broadening of the DEST profiles and a shift to higher R2 values of the RD curves measured for the ortho, meta, and para carbons of PhOH (ESI Fig. 1†), indicating the formation of a high molecular weight PhOH–nanoparticle adduct in chemical exchange with the free, NMR-visible small molecule.7 The quantitative modeling of the experimental DEST, RD, and R1 data was done using an adsorption/desorption exchange model in which all experimental observables are described by solutions to the McConnell equation (ESI Methods†). Analysis of the reduced χ2 values obtained from modeling of the scNMR experiments indicates that all data can be accounted for by a two-site exchange kinetic model in which PhOH is in equilibrium between a desorbed (D) and an adsorbed (A) state (Fig. 4). The modeled parameters are shown in Fig. 5 and listed in ESI Tables S1 and S2.† Inspection of these data reveals that the interaction between PhOH and TiO2 is weak (kAD ∼ 7700 s−1 and population of adsorbed PhOH, pA, ∼1.0%) and confirms that dispersion of Pd on the nanoparticle surface does not significantly perturb the PhOH–nanoparticle interaction (kAD ∼ 7400 s−1 and pA ∼ 1.2%) (Fig. 5a and b). On the other hand, the addition of 20 mM Pi to the solution results in a detectable destabilization of the interaction, which results in a ∼2-fold reduction of the population of PhOH adsorbed to TiO2 or Pd/TiO2 (pA ∼ 0.4 and 0.6%, respectively). These results agree well with the ∼2-fold decrease in 13C ΔR2 observed upon the addition of Pi (Fig. 3) and confirm that the detected PhOH–Pd/TiO2 adduct involves interactions between the small molecule and titania that can be destabilized by the addition of Pi (note that Pi does not compete with PhOH for absorption on Pd).5 This conclusion is further sported by the model-free (MF) analysis of the 13C R1 and R2 rates obtained for the adsorbed state (see ESI Methods†). Indeed the MF analysis returns an order parameter (S2) < 0.01 for the aromatic C–H groups of PhOH (Fig. 5), which is consistent with the order parameter expected for a PhOH molecule hydrogen bonded to a metal oxide nanoparticle,5,16 and two orders of magnitude smaller than the S2 expected for a PhOH molecule bound to the Pd site of Pd/TiO2.5 Finally, it is important to highlight that, while the scNMR data reported here do not provide evidence of a PhOH–Pd interaction in Pd/TiO2, a prior scNMR study detected and characterized PhOH adsorption on Pd particles of ∼2 nm size deposited on ceria nanocubes.5 This apparent discrepancy is most likely the result of the lower metallic surface area in Pd/TiO2 compared to Pd/CeO2 (1.6 and 2.9 m2 g−1, respectively) (Table 1). Indeed, since the PhOH–Pd interaction detected on Pd/CeO2 by DEST (population ∼ 0.1%) was just above the detection limit of the experiment,5 we expect the ∼2-fold smaller metallic surface area in Pd/TiO2 to further reduce the amount of PhOH adsorbed on the noble metal site and, therefore, push the PhOH–Pd adduct beyond the limit of detection.
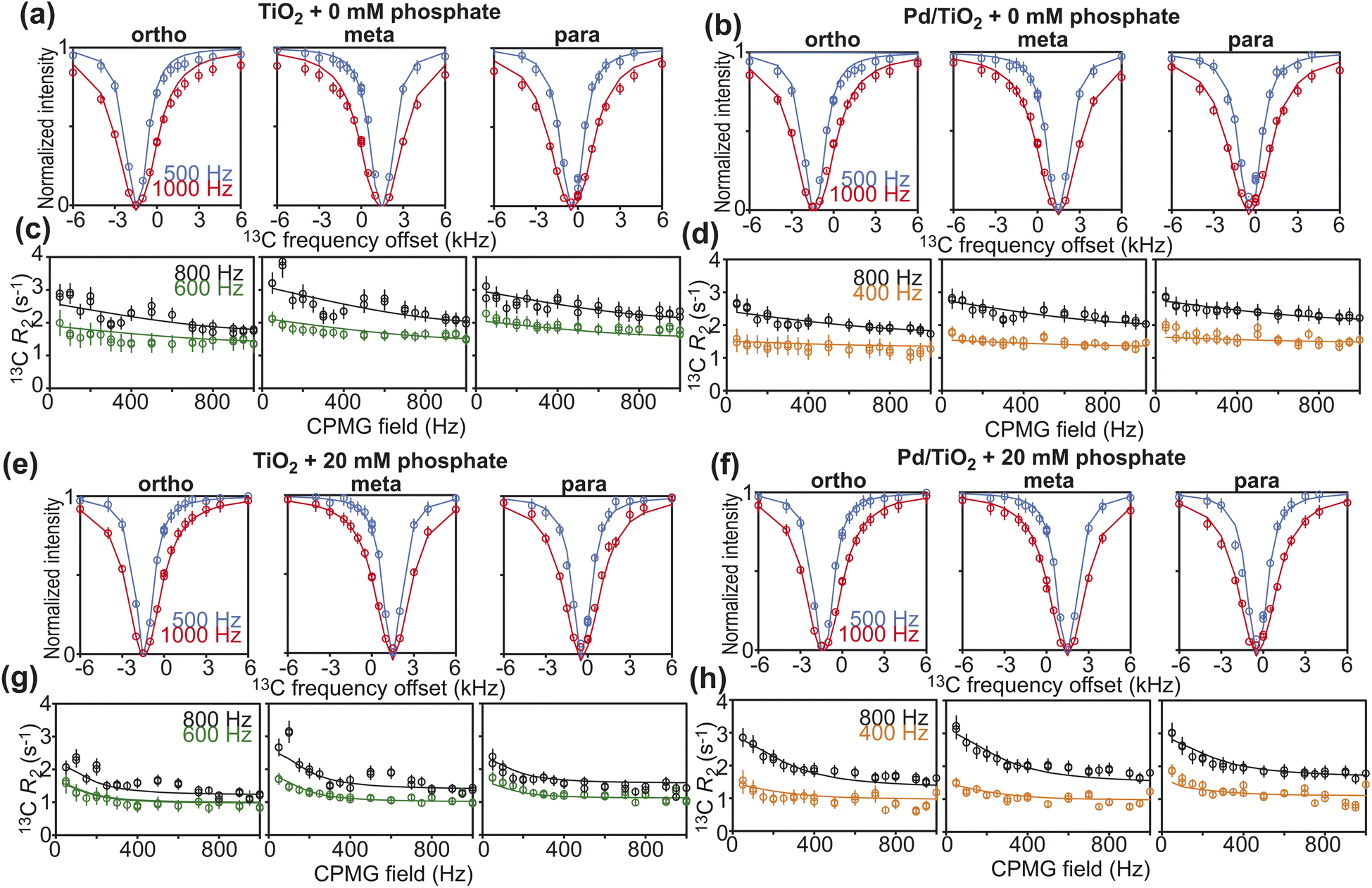 | ||
| Fig. 4 scNMR analysis. 13C DEST (top) and 13C RD (bottom) profiles measured for the ortho (left), meta (center), and para (right) 13C spins of PhOH in the presence of (a and c) 1 wt% TiO2, (e and g) 1 wt% TiO2 and 20 mM Pi, (b and d) 1 wt% Pd/TiO2, and (f and h) 1 wt% Pd/TiO2 and 20 mM Pi. 13C DEST profiles were measured at saturation fields of 500 (blue) and 1000 (red) Hz. The R2 rates for the RD profiles were acquired at 600 (green) and 800 (black) MHz for the TiO2 data and 400 (orange) and 800 (black) MHz for the Pd/TiO2 data. Experimental data are shown in circles, and the best-fit to a two-site exchange model are shown as solid lines. The agreement between experimental and back-calculated 13C R1 data is shown in ESI Fig. S2.† The fits return reduced χ2 values of 1.4, 0.6, 0.6, and 1.0 for the TiO2 and 0 mM Pi, TiO2 and 20 mM Pi, Pd/TiO2 and 0 mM Pi, and Pd/TiO2 and 20 mM Pi, respectively. | ||
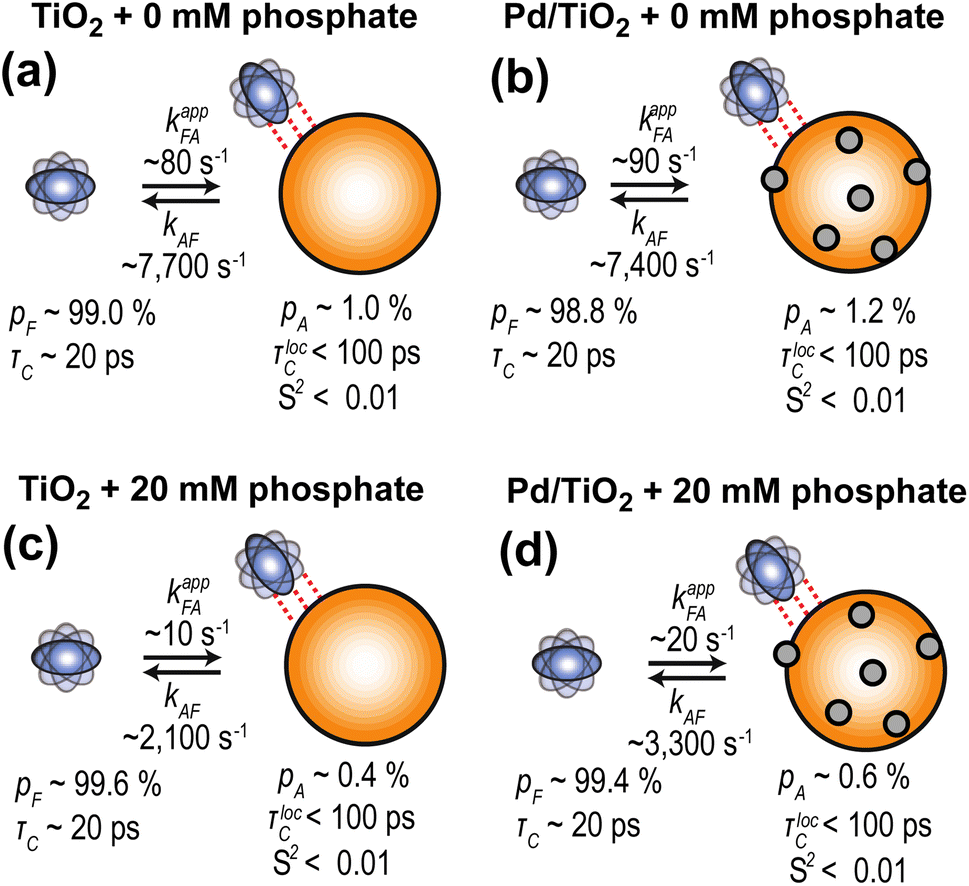 | ||
| Fig. 5 Kinetic model for PhOH absorption on TiO2 and Pd/TiO2. Results of the simultaneous quantitative modeling of the 13C DEST, RD, and R1 data measured on 10 mM PhOH in the presence of (a) 1 wt% TiO2, (b) 1 wt% Pd/TiO2, (c) 1 wt% TiO2, and 20 mM phosphate, and (d) 1 wt% Pd/TiO2 and 20 mM phosphate. All data were interpreted using a two-site exchange model. TiO2 is an orange sphere, and Pd NPs are gray spheres. PhOH is a blue rotating oblate. Dashed red lines indicate weak PhOH–nanoparticle interactions. Rate constants (kxy, where x and y are the states in thermodynamic equilibrium; kappxy represents the apparent rate constant), populations of the two states (pF and pA for free and adsorbed states, respectively), the order parameter of the adsorbed state (S2), and the rotational correlation time for local motion (τlocC) were fit as global parameters. Chemical shift changes were fit as peak-specific parameters (see ESI Methods†). Best fit parameters are shown. A complete report of the fitted parameters is available in the ESI Tables S1 and S2.† | ||
Kinetics of PhOH hydrogenation by Pd/TiO2
When multiple catalyst–support interaction modes are possible, the observed rate of a catalytic reaction is given by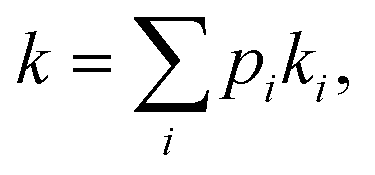 where pi and ki are the fractional abundance and turnover number of interaction mode i, respectively.19–21 In the case of the PhOH hydrogenation reaction catalyzed by Pd/TiO2, the two main binding modes at play are (i) PhOH hydrogen bonded to titania (detected by our scNMR experiments) and (ii) PhOH tightly adsorbed to the Pd site (below the detection limit of our scNMR experiments). To determine which binding mode is chiefly responsible for the catalytic conversion of PhOH, we have measured the rate of PhOH hydrogenation to cyclohexanone catalyzed by 1 wt% Pd/TiO2 at pH 7.0, 35 °C, and 1 bar H2 in the absence and the presence of 20 mM Pi. Since the addition of 20 mM Pi resulted in a ∼2-fold reduction in binding mode (i) (Fig. 5) without affecting binding mode (ii),5 we expect Pi to reduce catalytic conversion by ∼50% if the reaction proceeds by binding mode (i) and to induce a negligible effect on k if the reaction proceeds by binding mode (ii) (Fig. 6a). Results presented in Fig. 6b indicate that the addition of 20 mM Pi to the reaction mixture reduces the rate of PhOH hydrogenation by ∼2-fold (from 22.0 to 11.5 μM s−1) (Table 1), clearly indicating that hydrogen bonding of PhOH to titania is chiefly responsible for the addition of the Pd-activated hydrogen on the aromatic molecule.
where pi and ki are the fractional abundance and turnover number of interaction mode i, respectively.19–21 In the case of the PhOH hydrogenation reaction catalyzed by Pd/TiO2, the two main binding modes at play are (i) PhOH hydrogen bonded to titania (detected by our scNMR experiments) and (ii) PhOH tightly adsorbed to the Pd site (below the detection limit of our scNMR experiments). To determine which binding mode is chiefly responsible for the catalytic conversion of PhOH, we have measured the rate of PhOH hydrogenation to cyclohexanone catalyzed by 1 wt% Pd/TiO2 at pH 7.0, 35 °C, and 1 bar H2 in the absence and the presence of 20 mM Pi. Since the addition of 20 mM Pi resulted in a ∼2-fold reduction in binding mode (i) (Fig. 5) without affecting binding mode (ii),5 we expect Pi to reduce catalytic conversion by ∼50% if the reaction proceeds by binding mode (i) and to induce a negligible effect on k if the reaction proceeds by binding mode (ii) (Fig. 6a). Results presented in Fig. 6b indicate that the addition of 20 mM Pi to the reaction mixture reduces the rate of PhOH hydrogenation by ∼2-fold (from 22.0 to 11.5 μM s−1) (Table 1), clearly indicating that hydrogen bonding of PhOH to titania is chiefly responsible for the addition of the Pd-activated hydrogen on the aromatic molecule.
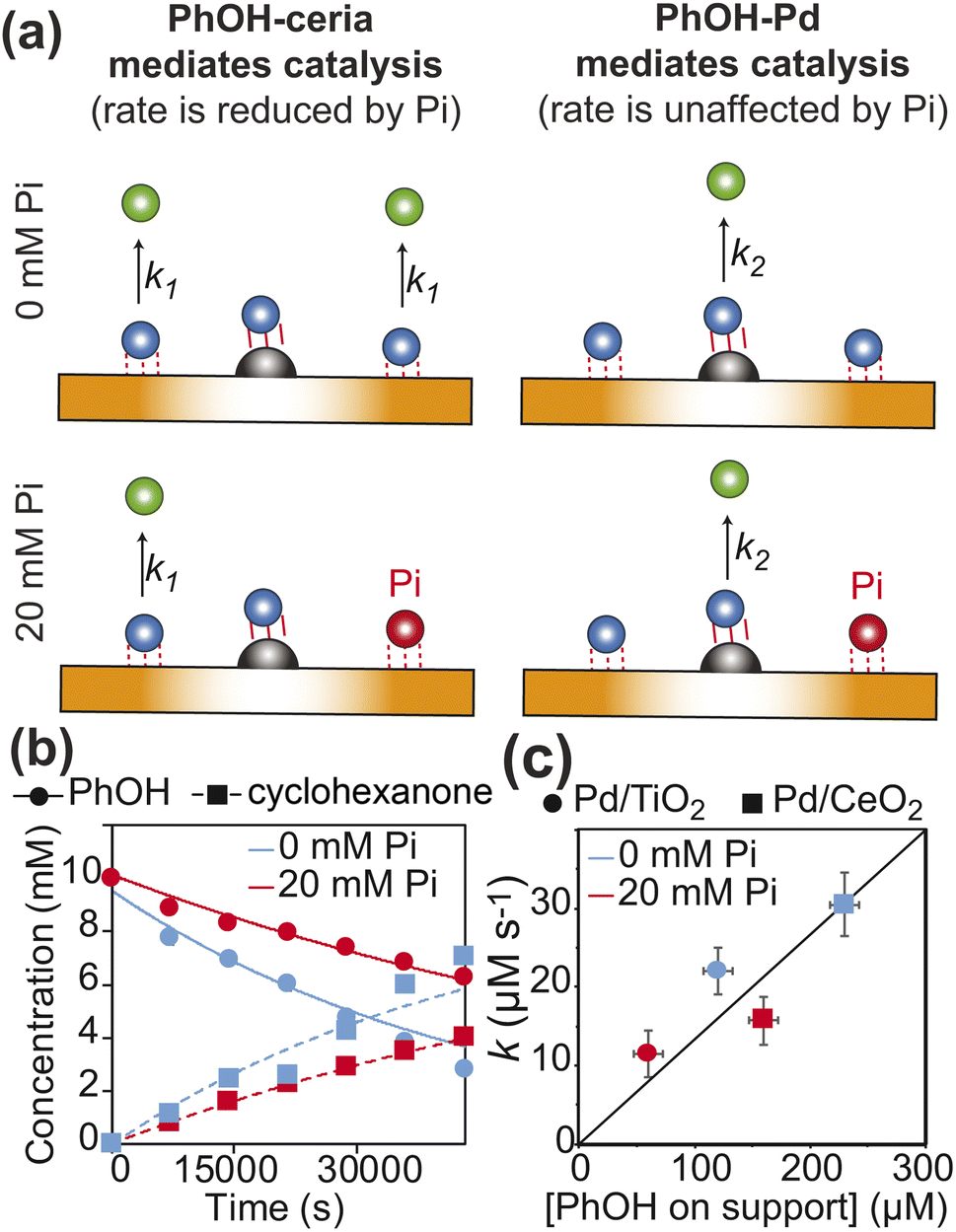 | ||
Fig. 6 Kinetics of PhOH hydrogenation. (a) Models of the dependence of the PhOH hydrogenation reaction from the concentration of inorganic phosphate (Pi). If the hydrogenation of PhOH proceeds from the substrate–support interaction with microscopic rate constant k1 (left panel), a ∼2-fold reduction of the PhOH–support interaction upon addition of 20 mM Pi results in a ∼2-fold inhibition of the observed reaction rate 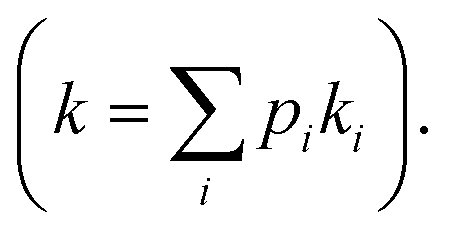 If the reaction proceeds from the substrate–Pd adduct with microscopic rate constant k2 (right panel), the addition of Pi is not expected to affect the observed rate of PhOH hydrogenation. PhOH, cyclohexanone, and Pi are represented as a blue, green, and red spheres, respectively. The Pd site is shown as a gray semi-sphere. The metal oxide support is shown as an orange surface. Red lines indicate the PhOH–catalyst interactions. (b) PhOH hydrogenation catalyzed by Pd/TiO2 in the presence of 0 (blue) and 20 (red) mM Pi is modelled as a pseudo-first-order reaction. The experimental concentration of PhOH and cyclohexanone are shown as circles and squares, respectively. Best-fit lines are shown as solid and dashed curves, respectively. Results of the fit are shown in Table 1. (c) The rate of PhOH hydrogenation (k) is plotted versus the concentration of PhOH bound to the support determined by scNMR. Circles and squares are the data measured for the Pd/TiO2 (this work) and Pd/CeO2 (Egner et al., 2020)5 catalysts, respectively. Data obtained in the presence of 0 and 20 mM Pi are colored blue and red, respectively. If the reaction proceeds from the substrate–Pd adduct with microscopic rate constant k2 (right panel), the addition of Pi is not expected to affect the observed rate of PhOH hydrogenation. PhOH, cyclohexanone, and Pi are represented as a blue, green, and red spheres, respectively. The Pd site is shown as a gray semi-sphere. The metal oxide support is shown as an orange surface. Red lines indicate the PhOH–catalyst interactions. (b) PhOH hydrogenation catalyzed by Pd/TiO2 in the presence of 0 (blue) and 20 (red) mM Pi is modelled as a pseudo-first-order reaction. The experimental concentration of PhOH and cyclohexanone are shown as circles and squares, respectively. Best-fit lines are shown as solid and dashed curves, respectively. Results of the fit are shown in Table 1. (c) The rate of PhOH hydrogenation (k) is plotted versus the concentration of PhOH bound to the support determined by scNMR. Circles and squares are the data measured for the Pd/TiO2 (this work) and Pd/CeO2 (Egner et al., 2020)5 catalysts, respectively. Data obtained in the presence of 0 and 20 mM Pi are colored blue and red, respectively. | ||
Pd/TiO2versus Pd/CeO2
Metal oxides are commonly used as supports for hydrogenation catalysts.10 Therefore, understanding the mechanisms underlying the different performances of various metal oxide supports in hydrogenation reactions is of fundamental importance in catalysis science. We have investigated the performance of Pd/TiO2 (this work) and Pd/CeO2 (Egner et al. 2020)5 as PhOH hydrogenation catalysts under identical experimental conditions (10 mM PhOH, 1 wt% catalysts, pH 7.0, 35 °C, and 1 bar H2), and observed that Pd/CeO2 catalyzes the reaction ∼ 1.5 times faster than Pd/TiO2 (Table 1). Interestingly, by plotting the hydrogenation rate versus the concentration of PhOH weakly adsorbed on the support determined by scNMR experiments acquired in the absence and the presence of 20 mM Pi, it is apparent that the efficiency of the catalyst is correlated to the amount of the PhOH adsorbate, irrespective of the metal oxide used as the support (Fig. 6c). This observation indicates that the more pronounced basic properties of ceria compared to titania17 enhance the affinity of the support for PhOH from 0.2 μmol m−2 in Pd/TiO2 to 3.5 μmol m−2 in Pd/CeO2 (calculated using the support surface areas from Table 1 and the concentrations of PhOH–support adduct in the absence of Pi from Fig. 6c) and make CeO2 a better support for the hydrogenation reaction (note that PhOH acts as the hydrogen bond donor in its interaction with the metal oxide at pH 7.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 and, therefore, it is expected to have a higher affinity for more basic surfaces).
4 and, therefore, it is expected to have a higher affinity for more basic surfaces).
In addition to revealing the basis for the outstanding performance of Pd/CeO2 in catalytic hydrogenation of PhOH, our data suggest that hydrogen spillover is the main mechanism mediating the reduction of PhOH bound to the metal oxide support. Indeed, since ceria and titania have comparable abilities to uptake the Pd-activated hydrogen,10 similar amounts of hydrogen atoms are expected to spillover onto the two supports, therefore resulting in the support-independent direct proportionality between catalytic efficiency and amount of PhOH adsorbed on the support observed in Fig. 6c. On the other hand, the correlation in Fig. 6c is inconsistent with the hypothesis that the better overall performance of the ceria-based catalyst is the result of a denser surface distribution of Pd sites that facilitates close encounters between the adsorbed PhOH and Pd on Pd/CeO2 compared to Pd/TiO2. Indeed, the latter hypothesis would suggest that Pd/CeO2 catalyzes the reaction at a higher rate than Pd/TiO2 when the same amount of PhOH is adsorbed on the support (note that a denser distribution of Pd sites increases the probability that the hydrogen bonded PhOH is located nearby Pd, Fig. 1). However, the data presented in Fig. 6c clearly indicate that similar concentrations of PhOH–support adduct result in similar conversion rates irrespective of the metal oxide used as the support (for example, compare the data reported for Pd/TiO2 at 0 mM Pi to the ones measured for Pd/CeO2 at 20 mM Pi in Fig. 6c).
A non-reducible support results in inefficient PhOH hydrogenation
To further test our hypothesis that hydrogen spillover is the main mechanism mediating hydrogenation of PhOH by Pd-supported on metal oxides, we have investigated the ability of Pd supported on Stöber silica (Pd/SiO2) to adsorb and reduce PhOH. Silica (SiO2) is a non-reducible support that is incapable to uptake Pd-activated hydrogen. Therefore, we expect Pd/SiO2 to provide an inefficient catalyst for the PhOH hydrogenation reaction.Pd/SiO2 was prepared as described in methods. TEM images show that Pd/SiO2 presents a spherical morphology with an average particle size of 22 ± 5 nm (Fig. 7a and Table 1). N2 sorption analysis returned a catalyst surface area of 103.0 m2 g−1, and CO pulse chemisorption reported a 38.0% dispersion for Pd. The Pd particle size was calculated to be ∼2.4 nm, and the metallic surface area was 1.4 m2 g−1. Therefore, the actual SiO2 surface area on the catalyst is calculated as 101.6 m2 g−1 (103.0–1.4 m2 g−1) (Table 1).
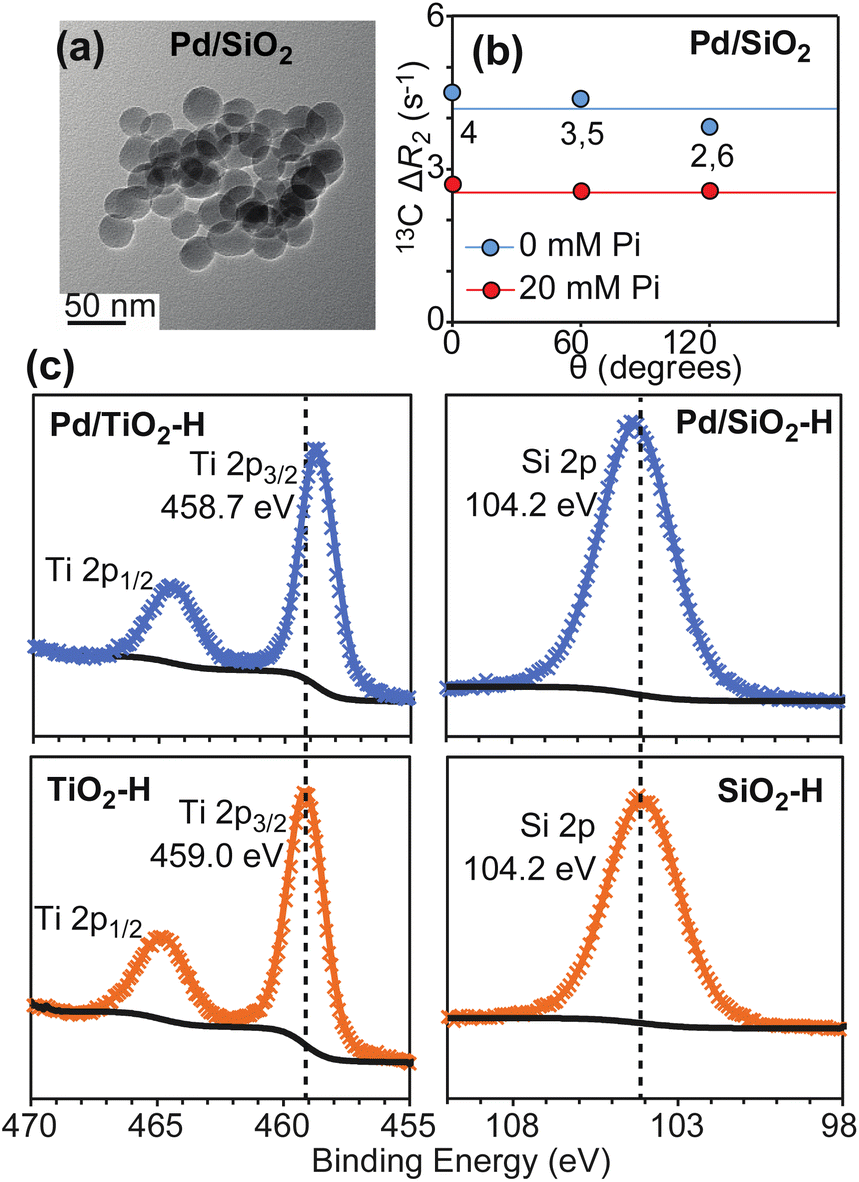 | ||
| Fig. 7 Properties of Pd/SiO2. Panel (a) shows a TEM image for the investigated Pd/SiO2 material. Panel (b) shows a plot of the 13C ΔR2 as a function of θ measured for PhOH in the presence of 1 wt% Pd/SiO2. The ortho (2,6) and meta (3,5) positions generate equivalent NMR peaks and one single ΔR2 value for these peaks is reported. Data measured in the presence of 0 and 20 mM Pi are shown as blue and red spheres, respectively. Solid blue and red lines are the corresponding average ΔR2 values. Panel (c) shows XPS spectra measured for TiO2 (bottom left), Pd/TiO2 (top left), SiO2 (bottom right), and Pd/SiO2 (top right) after hydrogen treatment (see ESI Methods†). The shift of the XPS Ti 2p peaks after hydrogen treatment is consistent with Ti reduction in the presence of supported Pd. | ||
To confirm that hydrogen spillover does not occur in Pd/SiO2, the SiO2 and TiO2 supports as well as the Pd/SiO2 and Pd/TiO2 catalysts were incubated under a flow of H2 gas at 100 °C for 2 hours. The H2 treated samples (SiO2–H, TiO2–H, Pd/SiO2–H, and Pd/TiO2–H, respectively) were then analyzed by X-ray Photoelectron Spectroscopy (XPS) without air exposure. The XPS spectra reported in Fig. 7c show that deposition of Pd on SiO2 does not shift the Si 2p peak after H2 treatment at 100 °C, which indicates that Si has the same oxidation state in SiO2–H and Pd/SiO2–H. On the other hand, comparison of the XPS spectra measured for Pd/TiO2–H and TiO2–H clearly indicates that the presence of Pd causes a negative shift of the Ti 2p binding energy by ∼0.3 eV, which is consistent with spillover of Pd-activated hydrogen on the support and consequent reduction of the Ti sites. In this respect, it is important to notice, that deposition of Pd on TiO2 does affect the oxidation of Ti intrinsically under reduction conditions, as evidenced by the fact that the XPS data measured for Pd/TiO2 before H2 treatment (Pd/TiO2–I) and for TiO2–H display Ti 2p3/2 peaks at the same binding energy (459.0 eV, ESI Fig. S3†).
Next, we investigated the ability of Pd/SiO2 to adsorb PhOH on the support by 13C ΔR2 experiments. Analysis of the NMR data indicates that PhOH is efficiently adsorbed on Pd/SiO2 (average ΔR2 = 4.2 ± 0.1 s−1, Fig. 7b and Table 1) and that addition of 20 mM Pi results in significant reduction in ΔR2 (average ΔR2 = 2.6 ± 0.1 s−1, Fig. 7b and Table 1). In analogy to what reported above for Pd/TiO2 and previously for Pd/CeO2,4,5 this ΔR2 trend indicates formation of SiO2–PhOH interactions that are destabilized by addition of Pi.
Finally, we established if Pd/SiO2 can function as a catalyst for PhOH hydrogenation under the mild conditions tested for Pd/TiO2 and Pd/CeO2 (1 wt% catalyst, 1 bar of H2, and 35 °C). Interestingly, no signs of substrate degradation or formation of hydrogenated products were detected after 12 hour incubation of Pd/SiO2 with 10 mM PhOH (Table 1), confirming that the latter material is a worse hydrogenation catalyst than Pd/TiO2. Of note, these results cannot be ascribed to different oxidation states of the Pd site in Pd/SiO2 and Pd/TiO2. Indeed, XPS data measured for these materials indicate that palladium deposits as metallic Pd on both metal oxides (ESI Fig. S3†). Therefore, we concluded that the low PhOH hydrogenation rate obtained with Pd/SiO2 originates by its inability to uptake Pd-activated H2via spillover.
Conclusion
The outcome of a catalytic reaction is strictly dependent on the interactions that substrates, intermediates, and products form with the catalyst. Therefore, understanding how the kinetics and thermodynamics of adsorption regulate the efficiency of heterogeneous catalysts is of paramount importance in the design and optimization of catalytic processes. In this work, we have combined scNMR and reaction kinetics experiments to investigate how the structure, dynamics, and thermodynamic partitioning of the surface adsorbed species determine the PhOH hydrogenation reaction catalyzed by Pd/TiO2. We showed that PhOH forms a weak interaction with the TiO2 support that can be destabilized by the addition of 20 mM Pi. Interestingly, we noticed that the rate of PhOH hydrogenation is directly correlated to the amount of substrate adsorbed on titania (i.e., a ∼2-fold reduction in the concentration of adsorbed PhOH resulted in a ∼2-fold reduction in catalytic conversion) (Table 1), indicating direct participation of the substrate–support interaction in catalysis.In analogy with what we have previously observed for the adsorption of phenolic compounds on ceria nanoparticles, the formation of the catalytically competent PhOH–titania interaction likely involves the establishment of a hydrogen bond between the –OH group of the small molecule and one oxygen on the nanoparticle.3–5,22 Consistent with this hypothesis, the S2 modelled for the PhOH–titania adduct based on scNMR data is ≪0.1, which agrees well with the S2 predicted for the C–H group of an aromatic ring connected to a rigid surface by three rotatable bonds.5,16 Interestingly, the high conformational freedom of the adsorbed PhOH allows the substrate to adopt configurations in which the aromatic ring is adjacent to the surface of the heterogeneous catalyst and ideally positioned for the addition of activated hydrogens on the aromatic ring (Fig. 1a and 3a). In this respect, a comparison of the data measured on Pd/TiO2 (this work) and Pd/CeO2 (Egner et al. 2020)5 revealed that the rate of the PhOH hydrogenation reaction is directly correlated to the amount of PhOH hydrogen bonded to the metal oxide support (Fig. 6c). Since titania and ceria have similar abilities to uptake hydrogen activated by a supported noble metal,10 our data suggest that hydrogen spillover on the support is the main mechanism by which the Pd-activated hydrogen is shuttled to the PhOH hydrogen bonded to the metal oxide support. The latter conclusion is consistent with the fact that strong Pd–substrate interactions were observed to inhibit the rate of hydrogenation of 2-hydroxy- and 2-methoxy-PhOH,4 and that using a support that does not mediate hydrogen spillover such as silica results in inefficient hydrogenation catalysis. Overall, the results presented in this work indicate that ceria provides the ideal support for the PhOH hydrogenation reaction thanks to its redox properties that permit spillover of the activated hydrogen and to its basic properties that facilitate the establishment of the catalytically competent substrate–support hydrogen bonding interaction.
Experimental methods
Synthesis of Pd/TiO2
25 mg of palladium(II) nitrate dihydrate from Sigma-Aldrich was dissolved in ca. 500 μL deionized water. 1 g of titanium(IV) oxide nanopowder (anatase, <25 nm particle size) from Aldrich was impregnated with the Pd precursor solution by incipient wetness method targeting 1 wt% Pd loading. The catalyst was then dried under vacuum and reduced under H2 flow (15 cm3 min−1) for 2 h at 100 °C. The reduction temperature was kept under 100 °C to prevent the formation of a TiOx layer on the Pd surface.23Synthesis of Pd/SiO2
Silica nanoparticles were synthesized following the Stöber method. Briefly, in a 250 mL round-bottom flask, 190 mL ethanol was mixed with 10 mL DI water, followed by the addition of ∼3.75 mL concentrated ammonium hydroxide solution. The mixture was stirred at 550 rpm and 40 °C for ∼1 h. Then, ∼2.5 mL of tetraethylorthosilicate (TEOS) was added to the mixture and stirred for 24 h at 40 °C. The material was filtered with thorough washing by ethanol followed by drying at 100 °C overnight to obtain ∼0.5 g of dry silica powder. The Pd/SiO2 catalyst was synthesized following the protocol described above for preparations of Pd/TiO2.Transmission electron microscopy
TiO2 and Pd/TiO2 nanoparticles were imaged in a FEI Tecnai G2 F20 field emission transmission electron microscope (TEM) operating at 200 kV. Samples were prepared by placing 3–4 drops of ethanol suspension (∼0.1 mg mL−1) onto lacey-carbon-coated copper grids and dried in air. Size distribution histograms and average particle size were obtained from TEM images by measuring the diameter of 100 particles using the Digital Micrograph® software.Inductively coupled plasma-optical emission spectroscopy (ICP-OES)
Pd loadings were measured in a PerkinElmer Optima 2100 DV ICP-OES instrument. Samples (10 mg) were sonicated and vortexed for 24 h in 2 mL aqua regia. The solution was diluted to 12.0 mL using deionized water (17.4 Mohm cm). The solution was filtered and the amount of palladium in the filtrate was determined by comparing the ICP-OES signal with the signal obtained from calibration standards.Surface area measurements
Specific surface areas of the materials were calculated from N2 sorption isotherms measured in a Micromeritics Tristar Analyzer at −196 °C. Prior to the measurement, the samples were pretreated at 100 °C for 6 h under N2 flow. The specific surface areas of the materials were calculated using the Brunauer–Emmett–Teller (BET) method.CO pulse chemisorption
Micromeritics Autochem II 2920 Chemisorption Analyzer was used for the CO pulse chemisorption. A glass wool bed was inserted in a quartz U-tube, and 100 mg of Pd/TiO2 was loaded on top of the wool. The catalyst was then reduced under H2 flow (10% in Ar) at a flow rate of 30 cm3 min−1 for 60 min at 100 °C, followed by treatment with a He flow (flow rate: 50 cm3 min−1) for 30 min at 100 °C to remove excess surface bonded H atoms. The temperature was then lowered to 40 °C under the same He flow and held for 30 min. The catalyst was then subjected to CO pulses (10% in He). The palladium dispersion and surface area were calculated as described in ESI Methods.†XPS sample preparation and experiments
A clean oven-dried flat-bottomed Schlenk flask (100 mL) was cooled under vacuum and then cycled with UHP hydrogen. Then under a heavy purge of hydrogen, the sample was introduced into the flask, and the vacuum was pulled. The sample was kept in a vacuum at 100 °C for 30 min to ensure complete removal of surface moisture. Then hydrogen was flowed at 15 cm3 min−1 for 2 h at 100 °C. The color of the Pd-impregnated samples darkened over time. At the end of 2 h, the flask was cooled under hydrogen flow, sealed, and introduced into a dry glove box. The reduced samples were stored in a glass-on-glass sample storage tube until further use. The samples after this treatment were named TiO2–H, Pd/TiO2–H, SiO2–H, and Pd/SiO2–H, respectively. All these samples were loaded in the air-free XPS chamber directly from the glovebox to ensure no surface reoxidation takes place during handling. XPS data on the on TiO2, Pd/TiO2, SiO2, and Pd/SiO2 before and after H2 treatment were acquired using a Kratos Amicus XPS system. The XPS data were modelled using the CasaXPS software.Hydrogenation of phenol
10 mM PhOH solutions were prepared in deionized water (17.4 Mohm cm), and 20 mM phosphate buffer, respectively, and the pH was adjusted to 7.0 ± 0.1 using dilute HCl or NaOH. In a typical reaction, the catalyst (20 mg, 5 mol%) and PhOH solution (4 mL, 10 mM) were added to a 10 mL glass tube. The tube was sealed and purged with H2 for 10 min at a rate of 20 cm3 min−1. After 10 min, the pressure relief needle was removed, and the H2 supply was stopped. The reaction tube was then placed in an oil bath preset at 35 °C under constant stirring (800 rpm). After the desired time, the septum was removed, and the catalyst was separated by centrifugation. The solution was extracted 4 times with 1 mL ethyl acetate. A 50 μL aliquot of the extracted solution was taken and analyzed by GC-MS (Agilent 7890A, 5975C) with an HP-5MS column, using 100 μL resorcinol (55 mM) as internal standard. The run started at 60 °C, the temperature was then ramped to 150 °C at 5 °C min−1, then to 300 °C at a ramp of 20 °C min−1 and finally held constant at 300 °C for 3 min. The reaction kinetic data were analyzed assuming a pseudo-first-order reaction. The time dependency of the concentrations of PhOH and cyclohexanone are given by:| [PhOH] = [PhOH]0e−kt | (1) |
| [Cyclohexanone] = [PhOH]0(1 − e−kt) | (2) |
Preparation of the NMR samples
Nanoparticle suspensions were prepared in an agarose gel matrix, as previously described.22 The pH of the phenol solution (10 mM) prepared in D2O was adjusted to 7.0 ± 0.1 using dilute DCl or NaOD. The pH was read directly from the pH meter without correction for isotope effects. Then nanoparticles (1 wt%) and agarose (1 wt%) were added to the solution. For the samples prepared in the presence of phosphate, sodium phosphate was added to a 20 mM final concentration and pH adjusted to 7.0 ± 0.1 using dilute DCl or NaOD. The mixture was then sonicated for 1 min (1:2 on/off duty cycle) at 25% power on a 500 W horn-tipped sonicator, heated to 80 °C for 10 min, transferred to a 5 mm NMR tube, and allowed to cool at room temperature for 10–15 min. The reference samples that did not contain nanoparticles were prepared using the same procedures as above. Phenol and agarose were purchased from Sigma-Aldrich. DCl and NaOD were purchased from Cambridge Isotope Laboratories. All chemicals were used without further purification.
NMR spectroscopy
13C R2 NMR data were collected at 25 °C on Bruker 400 and 800 MHz spectrometer equipped with triple resonance z-gradient cryoprobes. 13C R2 was measured using a double INEPT-based CPMG sequence with proton decoupling during the 13C relaxation period.5 The 13C carrier frequency was set at the center of the three peaks of the aromatic ring in PhOH. The CPMG field strength was set to 1 kHz. 14 different relaxation delays were acquired in an interleaved manner to obtain the 13C R2 relaxation decay curve. The observed relaxation decay was fitted using a monoexponential decay function ae−R2t, where a is the scaling factor, R2 is the transverse relaxation rate, and t is the variable relaxation delay. The experiments were acquired with 32 dummy scans and 128 scans with a recycle delay of 4 s between each scan. NMR spectra were processed and analyzed using the Mnova NMR software (https://mestrelab.com/software/mnova/nmr/).13C DEST, 13C RD, and 13C R1 NMR data were collected at 25 °C on Bruker 400, 600, and 800 MHz spectrometers equipped with triple resonance z-gradient cryoprobes and using the pulse sequences shown in the literature.5 Details on the NMR data acquisition and analysis are provided in ESI Methods.†
Data availability
All data are available upon request.Author contributions
Y. A., P. C., P. N., S. B. and V. V. designed and performed the research; Y. A., P. C., W. H., I. I. S. and V. V. analyzed the data; Y. A., P. C. and V. V. wrote the manuscript; W. H., I. I. S. and V. V. acquired funding for the project.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by funds from the National Institute of General Medical Sciences with grant no. R35GM133488 (to V.V.), and by the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences, through the Ames Laboratory Catalysis Science Program (to I. I. S.). The Ames Laboratory is operated for the U.S. Department of Energy by Iowa State University under contract no. DE-AC02-07CH11358. W. H. acknowledges support from National Science Foundation grant CHE-2108307.References
- M. Weber, M. Weber and M. Kleine-Boymann, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- M. T. Musser, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- N. C. Nelson, J. S. Manzano, A. D. Sadow, S. H. Overbury and I. I. Slowing, ACS Catal., 2015, 5, 2051–2061 CrossRef CAS.
- Y. An, P. Naik, I. I. Slowing and V. Venditti, ACS Appl. Nano Mater., 2020, 3, 11282–11288 CrossRef CAS.
- T. K. Egner, et al. , ChemCatChem, 2020, 12, 4160–4166 CrossRef CAS.
- P. J. Naik, et al. , ACS Catal., 2021, 11, 10553–10564 CrossRef CAS.
- Y. An, S. L. Sedinkin and V. Venditti, Nanoscale Adv., 2022, 4, 2583–2607 RSC.
- D. Zhang, F. Ye, T. Xue, Y. Guan and Y. M. Wang, Catal. Today, 2014, 234, 133–138 CrossRef CAS.
- H. Zhou, et al. , Green Chem., 2017, 19, 3585–3594 RSC.
- A. Beck, et al. , J. Phys. Chem. C, 2022, 126, 17589–17597 CrossRef CAS PubMed.
- N. Pineda-Aguilar, L. L. Garza-Tovar, E. M. Sánchez-Cervantes and M. Sánchez-Domínguez, J. Mater. Sci.: Mater. Electron., 2018, 29, 15464–15479 CrossRef CAS.
- C. Z. Wen, H. B. Jiang, S. Z. Qiao, H. G. Yang and G. Q. Lu, J. Mater. Chem., 2011, 21, 7052–7061 RSC.
- R. Liang, A. Hu, J. Persic and Y. N. Zhou, Nano-Micro Lett., 2013, 5, 202–212 CrossRef CAS.
- L. Wu, D. Buchholz, D. Bresser, L. Gomes Chagas and S. Passerini, J. Power Sources, 2014, 251, 379–385 CrossRef CAS.
- M. Xie and R. Bruschweiler, J. Phys. Chem. Lett., 2020, 11, 10401–10407 CrossRef CAS PubMed.
- J. R. Sachleben, V. Colvin, L. Emsley, E. W. Wooten and A. P. Alivisatos, J. Phys. Chem. B, 1998, 102, 10117–10128 CrossRef CAS.
- E. T. Kho, S. Jantarang, Z. Zheng, J. Scott and R. Amal, Engineering, 2017, 3, 393–401 CrossRef.
- X. Wang, B. Liu and J. Liu, Langmuir, 2018, 34, 15871–15877 CrossRef CAS PubMed.
- R. R. Dotas, et al. , J. Mol. Biol., 2020, 432, 4481–4498 CrossRef CAS PubMed.
- R. Ghose, J. Mol. Biol., 2019, 431, 145–157 CrossRef CAS PubMed.
- H. P. Lu, L. Xun and X. S. Xie, Science, 1998, 282, 1877–1882 CrossRef CAS PubMed.
- T. K. Egner, P. Naik, N. C. Nelson, I. I. Slowing and V. Venditti, Angew. Chem., Int. Ed., 2017, 56, 9802–9806 CrossRef CAS PubMed.
- S. Zhang, et al. , Nano Lett., 2016, 16, 4528–4534 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02913a |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |