
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLength-tunable Pd2Sn@Pt core–shell nanorods for enhanced ethanol electrooxidation with concurrent hydrogen production†
Tong
Li
,
Qiuxia
Wang
,
Wenjie
Zhang
 ,
Huaming
Li
,
Yong
Wang
* and
Junfeng
Liu
*
,
Huaming
Li
,
Yong
Wang
* and
Junfeng
Liu
*
Institute for Energy Research, School of Chemistry and Chemical Engineering, Jiangsu University, Zhenjiang 212013, China. E-mail: wangyong@ujs.edu.cn; jliu@ujs.edu.cn
First published on 16th August 2023
Abstract
The electrooxidation of ethanol as an alternative to the oxygen evolution reaction presents a promising approach for low-cost hydrogen production. However, the design and synthesis of efficient ethanol oxidation electrocatalysts remain key challenges. Here, a colloidal procedure is developed to prepare Pd2Sn@Pt core–shell nanorods with an expanded Pt lattice and tunable length. The obtained Pd2Sn@Pt catalysts exhibit superior activity and stability for ethanol electrooxidation compared to Pd2Sn and commercial Pt/C catalysts. By tuning the length of the Pd2Sn@Pt nanorods, remarkable mass activity of up to 4.75 A mgPd+Pt−1 and specific activity of 20.14 mA cm−2 are achieved for the short nanorods owing to their large specific surface area. A hybrid electrolysis system for ethanol oxidation and hydrogen evolution is constructed using Pd2Sn@Pt as the anodic catalyst and Pt mesh as the cathode. The system requires a low cell voltage of 0.59 V for the simultaneous production of acetic acid and hydrogen at a current density of 10 mA cm−2. Density functional theory calculations further reveal that the strained Pt shell reduces energy barriers in the ethanol electrooxidation pathway, facilitating the conversion of ethanol to acetic acid. This work provides valuable guidance for developing highly efficient ethanol electrooxidation catalysts for integrated hydrogen production systems.
Introduction
Hydrogen has garnered significant attention as a promising renewable energy source due to its high energy density, non-toxic nature, and environmental sustainability.1,2 Electrocatalytic water splitting is considered an efficient method for generating high-purity hydrogen fuel. The process of water electrolysis involves two half reactions: the cathodic hydrogen evolution reaction (HER) and the anodic oxygen evolution reaction (OER). The sluggish kinetics of the OER pose a significant challenge in water electrolysis, requiring high electrical input and hindering its widespread implementation.3–6 Despite extensive research efforts to accelerate anodic oxidation, substantial voltages are still necessary, considering the high theoretical equilibrium potential of 1.23 V for OER.7–10To address the challenges associated with the OER in water electrolysis, the electrooxidation of thermodynamically favorable small molecules as an alternative anodic process have been proposed in recent years.11–20 Integrating the oxidation of molecules with the cathodic HER not only reduces the operating voltage of the water electrolysis system but also eliminates the risk of H2/O2 mixture explosions.21,22 Among various potential substrates, ethanol has emerged as a highly attractive candidate due to its low toxicity, high boiling point, and the possibility of deriving it from biomass fermentation.23–26 The electrooxidation of ethanol to produce acetic acid, which holds significant economic value, represents a crucial pathway for synthesizing acetic acid under moderate reaction conditions.27,28 Consequently, there is a strong imperative to develop hybrid water electrolysis systems that combine the ethanol oxidation reaction (EOR) with HER, enabling the simultaneous production of acetic acid and high-purity hydrogen while minimizing energy consumption.
Pt and Pt-based catalysts have been widely recognized as highly efficient electrocatalysts for the EOR.29–32 However, their practical application is severely hindered by the exorbitant cost of Pt. Additionally, these catalysts often suffer from rapid degradation during the EOR process due to the accumulation of carbonaceous intermediates on their surfaces.33–35 Thus, the design and synthesis of high-performance Pt-based electrocatalysts with a low Pt-loading amount are worthwhile endeavors. One common approach to enhancing Pt utilization efficiency is the construction of catalysts in nanoscale dimensions, allowing for control over phase, size and morphology.36–39 Another effective strategy is the engineering of core@Pt-shell nanostructures by depositing Pt atoms onto the surface of a secondary material. This approach offers the advantage of reducing Pt usage while simultaneously enhancing EOR performance through the modulation of electronic interactions between the Pt surface and the core materials.40–42 Strain engineering is an additional crucial strategy for modifying the electronic structure of Pt and enhancing catalytic performance.43–46 Such as He et al. developed methodologies involving the insertion/extraction of phosphorus to induce lattice strain on the Pt surface, leading to strained Pt catalysts with enhanced activity for alcohol oxidation.47 While various strategies have been developed to enhance the performance of Pt-based catalysts, only a limited number of studies have integrated all of the aforementioned strategies into a single catalyst.
Herein, we present a facile electrocatalyst denoted as Pd2Sn@Pt, which consists of intermetallic Pd2Sn nanorod (NR) cores with Pt shells. The synthesis of Pd2Sn NRs grown on the [010] direction was achieved using a novel colloidal method, followed by the growth of Pt on the surface of the NRs. The lattice mismatch between the surface Pt and the Pd2Sn cores induced a significant tensile strain in the Pt shells. This strain plays a crucial role in modulating the binding affinity of reactive species on the catalysts, thereby enhancing the EOR activity. Additionally, the length of the Pd2Sn@Pt NRs was further tuned by adjusting the Pd2Sn core size to maximize their performance. The Pd2Sn@Pt catalysts exhibit exceptional EOR activity in alkaline conditions, surpassing both the performance of Pd2Sn and unstrained Pt/C counterparts. This remarkable performance can be attributed to the controlled morphology, core–shell structure, and surface strain effects. To gain further insights, density functional theory (DFT) calculations were employed to elucidate the influence of tensile strain on the adsorption ability of reaction intermediates along the EOR pathway. Furthermore, we constructed a coupled electrolyzer utilizing Pd2Sn@Pt as the anodic catalyst for EOR and Pt mesh as the cathodic catalyst for HER. This system demonstrated a remarkably low cell voltage of 0.59 V to run the system at a current density of 10 mA cm−2.
Results and discussion
Pd2Sn@Pt NRs were synthesized through a two-step colloidal procedure. In the first step, Pd2Sn intermetallic NRs were synthesized using oleylamine as the solvent and reducing agent, trioctylphosphine as the surface ligand, and methylamine hydrochloride (MAHC) as the directing agent for NRs growth. The length of Pd2Sn NRs was controlled by adjusting the amount of MAHC in the reaction solvent.48 In the second step, Pt precursor was introduced into the suspension containing Pd2Sn NRs and oleylamine, with glucose acting as the reducing agent (see the ESI† for details). Due to the lower energy barrier for heterogeneous nucleation of Pt on Pd2Sn seeds compared to homogeneous nucleation, Pt grew on the surface of Pd2Sn NRs, forming the Pd2Sn@Pt core–shell structure.49Fig. 1a–c displays the transmission electron microscopy (TEM) micrographs of obtained Pd2Sn@Pt NRs using 50.4, 58.7 and 100 mg of MAHC in the Pd2Sn synthesis process. All the synthesized NRs exhibited highly monodisperse and uniform morphology. With increasing amounts of MAHC, the NRs exhibited different lengths: a small length of 8 × 18 nm (referred to as Pd2Sn-s@Pt), a medium length of 10 × 38 nm (referred to as Pd2Sn-m@Pt), and a long length of 14 × 200 nm (referred to as Pd2Sn-l@Pt), as shown in Fig. S1.† The X-ray diffraction (XRD) patterns matched well with those of Pd2Sn (JCPDS 89-2057) and Pt (JCPDS 87-0636), confirming the presence of intermetallic Pd2Sn cores and Pt shells (Fig. 1d). As the length of the NRs increased, the full width at half maximum of the peak corresponding to the (020) facet decreased significantly, indicating the growth of Pd2Sn NRs along the (010) facet. Despite the variations in length, all the Pd2Sn@Pt NRs exhibited similar Pt ratios based on energy-dispersive X-ray spectroscopy (EDS) analysis (Fig. S2†).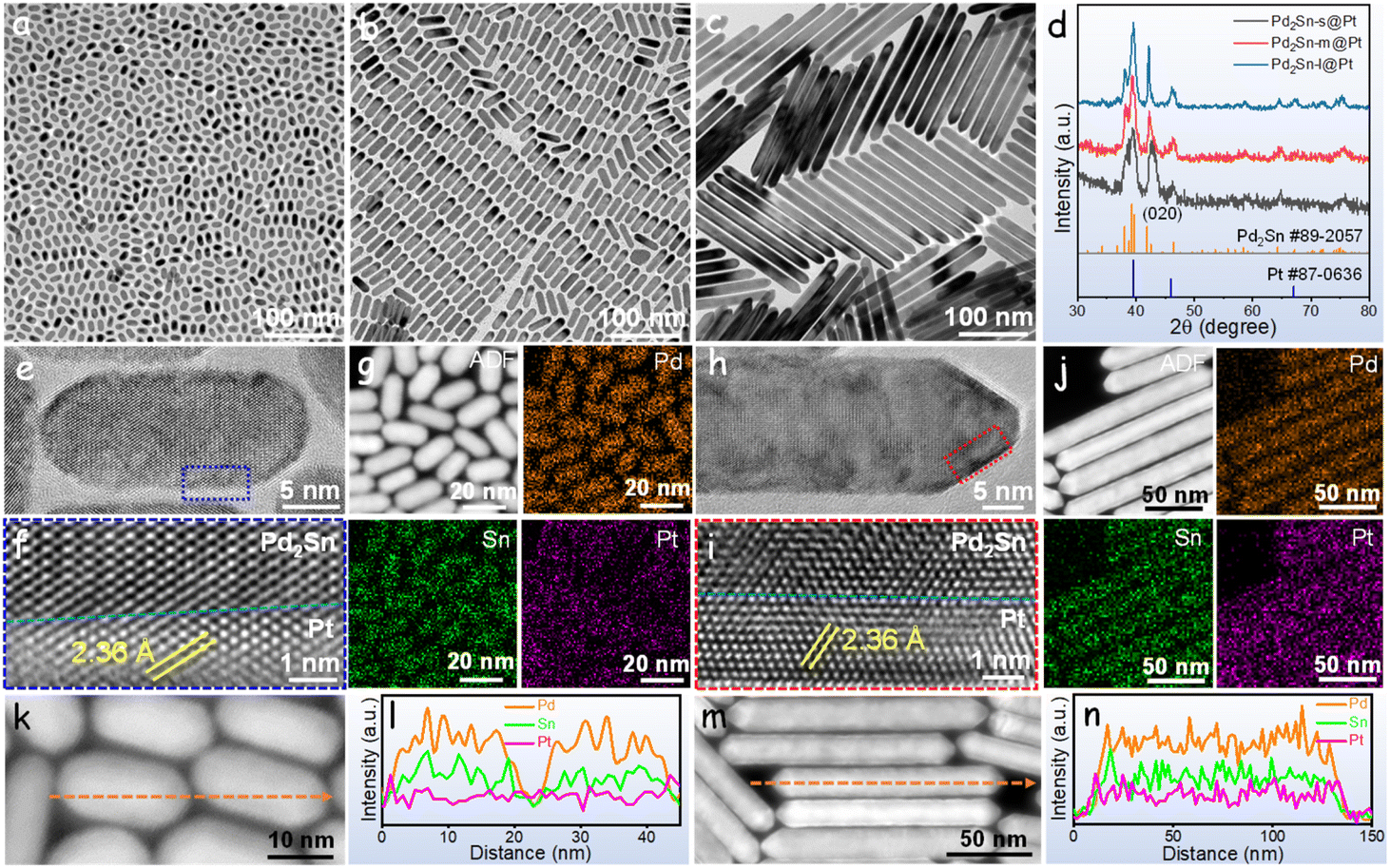 | ||
| Fig. 1 (a–c) TEM micrographs of (a) Pd2Sn-s@Pt, (b) Pd2Sn-m@Pt, (c) Pd2Sn-l@Pt and (d) their XRD patterns. (e) HRTEM image of Pd2Sn-s@Pt and (f) integrated pixel intensity of the crystal phase taken from the dotted rectangle in (e) panel. (g) STEM image and corresponding EDS elemental mappings of Pd2Sn-s@Pt. (h) HRTEM image of Pd2Sn-l@Pt and (i) integrated pixel intensity of the crystal phase taken from the dotted rectangle in (h) panel. (j) STEM image and corresponding EDS elemental mappings of Pd2Sn-l@Pt. (k and l) EDS line scan across the Pd2Sn-s@Pt (l) as indicated by the dashed line in STEM image shown in (k). (m and n) EDS line scan across the Pd2Sn-l@Pt (n) as indicated by the dashed line in STEM image shown in (m). | ||
High-resolution TEM (HRTEM) and scanning transmission electron microscopy EDS (STEM-EDS) mappings were employed to characterize Pd2Sn-s@Pt and Pd2Sn-l@Pt NRs. HRTEM analysis in Fig. 1e and f for Pd2Sn-s@Pt and Fig. 1h and i for Pd2Sn-l@Pt confirmed the presence of orthorhombic Pd2Sn cores and cubic Pt shells on the NRs. The lattice parameter of the Pt shells, indexed to the (111) planes, was measured to be 0.236 nm, which was larger than that of commercial Pt/C (0.228 nm, Fig. S3†). Comparing the lattice parameters of the Pt shell in Pd2Sn@Pt and conventional Pt, a 3.5% tensile strain in the Pt shell was observed. We attribute this lattice strain to the epitaxial growth of the Pt shell on the Pd2Sn core (Fig. S4†). Importantly, the Pt (111) facet was found consistently across the entire surface of the NRs, ranging from 6 atomic layers in short NRs to 10 atomic layers in long NRs (Fig. S5†). Fig. 1g and j revealed that the Pd and Sn elements were located in the interior of the NRs, while Pt was located in the exterior, confirming the core–shell structure with the Pt shell enclosing the Pd2Sn core. This observation was further supported by the EDS line-scan plot (Fig. 1k–n), which clearly demonstrated the accumulation of Pt in the near-surface region.
Fig. 2 illustrates the X-ray photoelectron spectroscopy (XPS) spectra of Pd 3d, Sn 3d, and Pt 4f for both Pd2Sn-s@Pt and Pd2Sn-s NRs. The Pd 3d spectra of both NRs exhibited two doublets. For Pd2Sn-s@Pt, the Pd0 species was found at a higher binding energy of 335.8 eV (Pd 3d5/2) compared to Pd2Sn-s NRs.50 The slight blueshift in the binding energy can be attributed to the presence of more electronegative Pt, which induces electron displacement toward Pt. The second peak observed at binding energy of 336.3 eV (Pd 3d5/2) corresponds to Pd2+ species, likely formed due to surface oxidation during the synthesis of Pd2Sn NRs. Similarly, two doublets were identified in the Sn 3d spectra, corresponding to Sn0 (485.3 eV) and Sn2+ (486.9 eV) chemical states. The Pt 4f region of Pd2Sn-s@Pt also exhibited two doublets.50 The main Pt component, assigned to a Pt0 chemical environment, was observed at a binding energy of 71.2 eV (Pt 4f7/2), accounting for 83% of the total detected Pt.50 Another component corresponding to Pt2+ at 72.7 eV (Pt 4f5/2) was also identified, resulting from slight surface oxidation of Pd2Sn-s@Pt NRs when exposed to air. The atomic ratio of Pd, Sn and Pt at the surface of the NRs, as determined by XPS, was found to be Pd/Sn/Pt = 51/31/18. It is noteworthy that the measured Pt content is significantly higher than the value detected by EDS analysis. Considering that the detected thickness by XPS typically corresponds to the outermost 2–3 nm of the surface, it can be concluded that Pd2Sn-s@Pt NRs possess a Pt-rich surface, further confirming the formation of Pd2Sn core and Pt shell architectures.
 | ||
| Fig. 2 XPS spectrum of Pd2Sn-s@Pt and Pd2Sn-s in the (a) Pd 3d, (b) Sn 3d and (c) Pt 4f regions. | ||
The Fourier transform infrared (FT-IR) showed the presence of oleylamine and trioctylphosphine as ligands on the NRs surface (Fig. S6†), to ensure unobstructed access to the catalytic active sites, these long-chain ligands were exchanged with a short, air-stable, and environmentally friendly inorganic molecule, ammonium thiocyanate.51 Subsequently, the NRs, along with commercial carbon support, were evaluated for their electrocatalytic performance in an alkaline electrolyte. Fig. 3a shows the cyclic voltammetry (CV) curves of Pd2Sn@Pt and commercial Pt/C electrocatalysts in 1 M KOH aqueous solution at a scan rate of 50 mV s−1. The peaks observed in the range of 0 to 0.5 V can be attributed to the adsorption (during the cathodic scan) and desorption (during the anodic scan) of hydrogen on the surface of Pt (Fig. S7†). Based on the coulombic charges in this region, the electrochemically active surface area (ECSA) of the surface Pt was estimated.52 As shown in Fig. S8,† the Pd2Sn@Pt-based electrocatalysts exhibited larger ECSA values compared to the Pt/C catalyst. Notably, the Pd2Sn-s@Pt catalyst demonstrated the highest ECSA among the Pd2Sn@Pt catalysts, which is ascribed to the high specific area originating from its short length. Additionally, the ECSA normalized by both the Pd and Pt content of the catalysts was also calculated through CO stripping experiments, which were performed by electrochemical oxidation of a pre-adsorbed saturated CO adlayer. As shown in Fig. S9,† Pd2Sn-s@Pt exhibited higher ECSA values than the longer length of Pd2Sn@Pt catalysts. Consequently, the combination of the Pt shell structure and the tunable length of the Pd2Sn@Pt catalysts enables high Pt utilization efficiency and an increased number of catalytically active sites, indicating the potential of Pd2Sn-s@Pt catalyst as an efficient electrocatalyst towards EOR.
 | ||
| Fig. 3 (a and b) CV curves of catalysts in a 1 M KOH solution (a) and in a 1 M KOH + 1 M ethanol solution (b). (c) Comparison of specific and mass activities of the catalysts. (d) CA profile of catalysts in a 1 M KOH + 1 M ethanol solution. (e) CA curves of catalysts with CV reactivation every 1000 s. (f) 1H NMR analysis of the electrolytes before and after CA measurements for Pd2Sn-s@Pt catalyst. | ||
Fig. S10† shows the LSV curves carried out in a 1 M KOH aqueous solution with and without 1 M ethanol. The absence of ethanol resulted in the occurrence of OER at a high potential above 1.5 V. However, in the presence of ethanol, the OER was significantly suppressed due to the dominant oxidation of ethanol on the catalyst surface. When Pd2Sn-s@Pt was used as the catalyst for EOR, a remarkably low potential of only 0.54 V was required to achieve a current density of 10 mA cm−2, which is notably lower than the OER potential by 1.29 V. To investigate the electrocatalytic activity of different length of Pd2Sn@Pt and commercial Pt/C catalysts for EOR, CV measurements were conducted in a solution containing 1 M KOH and 1 M ethanol. As shown in Fig. 3b, all catalysts exhibited two distinct peaks associated with the oxidation of ethanol in alkaline conditions. The forward peak corresponds to the oxidation of ethanol molecules, while the backward peak is attributed to the elimination of reaction intermediates.53 During the forward scan, a significantly lower onset potential was required for ethanol oxidation on the Pd2Sn@Pt catalysts compared to the commercial Pt/C catalyst (Fig. S11†), indicating lower activation energy and superior reaction activity of Pd2Sn@Pt toward ethanol oxidation. The Pd2Sn@Pt catalysts exhibited (Pd + Pt) mass-normalized current densities of 4.75, 3.72, and 2.91 A mgPd+Pt−1 for Pd2Sn-s@Pt, Pd2Sn-m@Pt, and Pd2Sn-l@Pt, respectively. These activities are much higher than that 0.60 A mgPt−1 for commercial Pt/C catalyst. The activities of the catalysts, normalized to the geometric area of the electrode, exhibited a consistent trend, with the Pd2Sn-s@Pt catalyst demonstrating the highest activity (Fig. S12†). To evaluate their intrinsic catalytic activities, the specific activities of these catalysts were calculated by normalizing their currents to the corresponding ECSA (Fig. S13†). The specific activity of all the Pd2Sn@Pt catalysts is higher than that of the Pt/C catalyst, particularly the Pd2Sn-s@Pt catalyst, which is calculated to be as high as 20.14 mA cm−2 (Fig. 3c). This is 11 times higher than the specific activity of the commercial Pt/C catalyst (1.81 mA cm−2). To the best of our knowledge, the mass and specific activity values for Pd2Sn-s@Pt are the highest among the published Pd- and Pt-based electrocatalysts (Table S1†). The Tafel plots obtained from the CV curves in the potential range between 0.4 and 0.5 V are shown in Fig. S14.† Among the catalysts, the Pd2Sn-s@Pt catalyst exhibits the smallest Tafel slope of 141 mV dec−1, indicating faster charge-transfer kinetics for EOR in alkaline medium. Furthermore, the charge transfer at the electrolyte/catalyst interface was evaluated by electrochemical impedance spectroscopy (Fig. S15†). In the Nyquist plots, the semicircles observed at high frequency range are associated with the charge transfer resistance. The Pd2Sn-s@Pt catalyst exhibited the smallest semicircle among the catalysts, demonstrating its superior charge-transfer capability during the EOR process.
To further understand the effect of the Pt shell in Pd2Sn@Pt catalysts on their catalytic performance, the mass and specific activities were normalized to the amount of Pt and their corresponding ECSA. As shown in Fig. S16 and S17,† Pd2Sn-s@Pt exhibits the highest mass activity of 20.0 A mgPt−1 and specific activity of 15.2 mA cm−2, which are 34 and 10 times higher than that of Pt/C respectively. This indicates that the presence of the strained Pt shell plays a crucial role in enhancing the EOR activity. Furthermore, Pd2Sn-s NRs without Pt layers and spherical Pd2Sn@Pt nanoparticles were also synthesized, and their EOR activity was measured under identical conditions. As shown in Fig. S18–S20,† the presence of a Pt shell on the Pd2Sn-s@Pt catalyst significantly enhanced the activity in comparison to the Pd2Sn-s catalyst alone. Additionally, despite the larger surface area of spherical Pd2Sn@Pt nanoparticles compared to Pd2Sn-s@Pt, their EOR performance was relatively inferior, indicating the crucial role of nanorod morphology in enhancing EOR activity (Fig. S21–S23†). Specifically, the superior EOR performance of Pd2Sn-s@Pt is attributed to the synergistic effects of the core–shell structures, the tensile strain present on the surface of Pt, and the tunable nanorod morphology.
The durability of the catalysts was evaluated through chronoamperometry (CA) testing at a constant potential of 0.724 V vs. RHE. As shown in Fig. 3d, all the catalysts initially experienced a noticeable decay in activities. However, the Pd2Sn-s@Pt catalyst demonstrated remarkable durability, retaining a high mass activity of 54 mA mgPd+Pt−1 even after 12 hours of operation. In contrast, the commercial Pt/C catalyst showed negligible activities, indicating its inferior durability compared to the Pd2Sn-s@Pt catalyst. To investigate the initial activity decay during the measurements, CA curves were recorded in consecutive periods of 1000 s. Between these periods, 5 CV cycles in the range from −0.924 to 0.4 V vs. Hg/HgO at a rate of 50 mV s−1 were applied. As shown in Fig. 3e, the activity of the Pd2Sn-s@Pt catalyst was almost fully recovered after each CV activation, suggesting that the decrease in current density during stability measurements was primarily due to the blocking of active/adsorption sites by reaction intermediates rather than structural degradation of the catalyst. In contrast, the Pt/C and Pd2Sn-s catalysts only exhibited partial recovery of activity after reactivation, indicating the enhanced anti-poisoning and desorption capabilities of reaction intermediates on the Pd2Sn@Pt surface (Fig. S24†). Fig. S25† shows the CV curves of the catalysts after 12 h of stability measurement and subsequent reactivation by CV cycles. As compared with the initial activity, Pd2Sn-s@Pt catalyst exhibits a negligible decrease of only 2.0% in mass activity, demonstrating its superior stability. Furthermore, TEM analysis after the stability test showed little variation in size and geometry, confirming the excellent structural stability of the Pd2Sn-s@Pt catalyst (Fig. S26†). These results highlight the significant impact of the Pt tensile strain and the core–shell structure on the catalytic stability during alkaline EOR processes.
To gain insights into the ethanol oxidation pathway on the Pd2Sn@Pt catalyst, 1H nuclear magnetic resonance (1H NMR) analysis was conducted on the electrolyte before and after EOR measurements. As presented in Fig. 3f, a new peak appeared at 1.81 ppm, associated with the ionized acetate, was detected in the electrolyte after EOR measurements, indicating the production of acetic acid during the EOR process. Therefore, acetic acid was the unique C2 product resulting from the ethanol oxidation reaction. To further investigate the potential occurrence of the C1 pathway in the EOR process, the electrolyte following the CA measurement was subjected to high-performance liquid chromatography (HPLC) analysis. Fig. S27† displays the standard curve depicting the integral area of the HPLC peak corresponding to acetic acid concentration. By utilizing this standard curve, the faradaic efficiency of the produced acetic acid was estimated to be approximately 98%. Thus, only a minor portion of ethanol undergoes the C1 pathway, primarily due to the substantial energy consumption necessary for breaking the C–C bond within ethanol. Considering that acetate was the primary detected oxidation product, it can be concluded that the ethanol oxidation on the Pd2Sn-s@Pt catalyst in an alkaline solution predominantly follows the reactive-intermediate pathway. This pathway involves the initial oxidation of ethanol to acetaldehyde and further oxidized to acetic acid.
DFT calculations were conducted to clarify the origin of enhanced EOR performance of the strained Pt surface in Pd2Sn@Pt catalyst. In the reactive-intermediate pathway, the ethanol and different intermediate species adsorbed on the Pt (111) and strained Pt (111) surfaces, ultimately leading to the formation of the final C2 product, CH3COOH, as schematically shown in Fig. 4a and b. Fig. 4c and d shows the calculated free energy diagrams for the oxidation of ethanol to acetic acid on both the Pt (111) and lattice strained Pt (111) surfaces. The results indicate that the rate-determining step (RDS) for both Pt surfaces is the oxidation of (CH3CO)ads to (CH3COOH)ads. In the RDS, the strained Pt (111) surface exhibits a lower energy barrier of 1.64 eV compared to the unstrained Pt (111) surface, which has a higher energy barrier of 1.81 eV. This indicates that the lattice expansion of the strained Pt surface further increases the reaction kinetics for EOR. Additionally, the adsorption free energies of OH were calculated to be 0.67 and 0.91 eV for the strained and unstrained Pt (111) surfaces, respectively. The higher adsorption ability of OH on the strained Pt facilitates the conversion of (CH3CO)ads to (CH3COOH)ads in the RDS and promotes the removal of carbonaceous species bonded to the catalyst surface, thereby improving the EOR activity and stability.46 These DFT calculation results are consistent with the experimental observations. Therefore, besides the tunable length of NRs, the lattice expansion of the Pt shell in Pd2Sn@Pt also contributes to the excellent catalytic performance of EOR.
 | ||
| Fig. 4 (a and b) Schematic illustration for the ethanol oxidation pathway on (a) Pt (111) and (b) strained Pt (111) surfaces. (c and d) Free energy diagrams for electrochemical EOR on the (c) Pt (111) and (d) strained Pt (111) surfaces. | ||
The excellent electrocatalytic performance of Pd2Sn-s@Pt catalyst for ethanol oxidation has inspired the design of a hybrid electrolyzer for anodic EOR with concurrent hydrogen production at the cathode, as shown in Fig. 5a. In this system, a glassy carbon electrode modified with Pd2Sn-s@Pt serves as the anode, while a platinum (Pt) mesh acts as the cathode. Fig. 5b shows the LSV curves of the two-electrode system measured in a 1 M KOH with 1 M ethanol electrolyte. The Pd2Sn-s@Pt catalyst exhibited high activity in the hybrid electrolyzer system, achieving current densities of 10 mA cm−2 at a remarkably low cell voltage of 0.59 V (Table S2†). In contrast, a conventional alkaline electrolysis cell employing commercial Pt/C as the cathode and commercial IrO2 as the anode, with 1 M KOH as the electrolyte, required a significantly higher cell voltage of 1.73 V to achieve the same current densities for water splitting (Fig. S28†). The low voltage requirement of the hybrid electrocatalytic system indicates its low energy consumption, making it an energy-efficient approach for hydrogen production. Furthermore, similar hybrid electrolyzers utilizing Pd2Sn-s or commercial Pt/C as the anodic catalyst were also assembled and tested. While all the hybrid systems required lower cell voltages compared to the conventional water splitting system, the Pd2Sn-s@Pt‖Pt system exhibited the best performance, attributed to its high activity toward EOR. Fig. 5c shows the durability of the hybrid electrolyzer evaluated using CA at a fixed voltage of 0.8 V. The Pd2Sn-s@Pt‖Pt system demonstrated better durability compared to other catalyst-based systems. Importantly, after the long-term stability measurement and subsequent reactivation of the anodic catalysts using 5 CV cycles, the activity was almost fully recovered with minimal variations compared to the initial activity, as shown in Fig. 5d. However, Pd2Sn-s and Pt/C based electrolyzer showed significant decrease in current density, demonstrating the better stability of Pd2Sn-s@Pt based anodic electrolyzer system (Fig. S29†). This activity decay in the long-term measurements is inferred to be associated with the adsorption of C2 intermediates on the anode, similar to the behavior observed in the three-electrode system for ethanol oxidation. Overall, the hybrid system utilizing Pd2Sn-s@Pt as the anode not only enhances the economic value of the anodic ethanol oxidation but also demonstrates high energy efficiency for cathodic hydrogen generation.
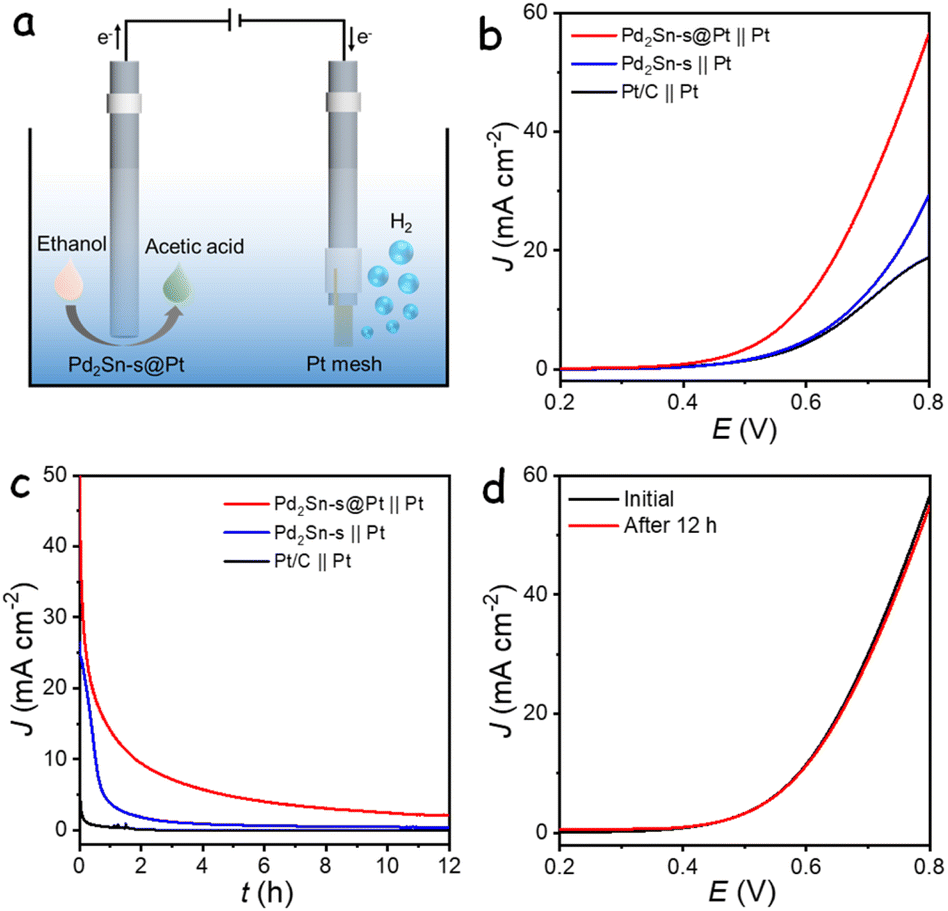 | ||
| Fig. 5 (a) Schematic diagram of the two-electrode electrolyzer. (b) LSV curves and (c) CA curves of the cell in 1 M KOH containing 1 M ethanol electrolyte. (d) LSV curves of Pd2Sn-s@Pt before and after 12 h CA measurement with reactivation. | ||
Conclusions
In summary, we present a facile colloidal strategy to engineer Pd2Sn@Pt core–shell NRs with lattice-expanded Pt shell and tunable nanorod length. The resulting Pd2Sn@Pt catalysts exhibit significantly enhanced electrocatalytic EOR performance under alkaline conditions. The Pd2Sn-s@Pt catalyst, with shorter nanorods, shows the highest mass activity up to 4.75 A mgPd+Pt−1 and a specific activity of 20.14 mA cm−2, making it one of the most efficient electrocatalysts for EOR. DFT calculations provide insights into the improved activity of Pd2Sn@Pt, highlighting the reduced energy barriers for the RDS of ethanol oxidation and the enhanced adsorption capacity of OH on the strained Pt surface. Furthermore, the Pd2Sn@Pt catalyst demonstrates excellent performance as an anodic electrocatalyst in a hybrid electrolyzer, enabling simultaneous acetic acid generation at the anode and hydrogen production at the cathode. The hybrid electrolyzer operates at a remarkably low cell voltage of 0.59 V to attain a current density of 10 mA cm−2. This work provides a promising avenue for the development of advanced core–Pt shell structures with precise morphological control, offering new opportunities for efficient electrocatalytic EOR and low-cost hydrogen production technology.Data availability
The data supporting this study is available within the ESI.†Author contributions
TL designed the experiments, produced all the materials, performed all the electrochemical measurements, and wrote a first draft of the manuscript. QW, WZ, HL and YW joined the characterization data analysis and discussion. JL conceived the project, guided and supervised the work. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 22008091), the funding for scientific research startup of Jiangsu University (No. 19JDG044, 4111510015), the Jiangsu Provincial Program for High-Level Innovative and Entrepreneurial Talents Introduction, and the Jiangsu Distinguished Professors project (No. 1711510024).Notes and references
- H. Yang, Y. Ji, Q. Shao, W. Zhu, M. Fang, M. Ma, F. Liao, H. Huang, Y. Zhang, J. Yang, Z. Fan, Y. Li, Y. Liu, M. Shao and Z. Kang, Energy Environ. Sci., 2023, 16, 574–583 RSC.
- B. Zhou, R. Gao, J. Zou and H. Yang, Small, 2022, 18, 2202336 CrossRef CAS PubMed.
- T. Binninger and M. Doublet, Energy Environ. Sci., 2022, 15, 2519–2528 RSC.
- S. Iqbal, B. Safdar, I. Hussain, K. Zhang and C. Chatzichristodoulou, Adv. Energy Mater., 2023, 13, 2203913 CrossRef CAS.
- S. Yuan, J. Peng, B. Cai, Z. Huang, A. T. Garcia-Esparza, D. Sokaras, Y. Zhang, L. Giordano, K. Akkiraju, Y. G. Zhu, R. Hübner, X. Zou, Y. Román-Leshkov and Y. Shao-Horn, Nat. Mater., 2022, 21, 673–680 CrossRef CAS PubMed.
- H. Sun, X. Xu, H. Kim, W. Jung, W. Zhou and Z. Shao, Energy Environ. Mater., 2022, e12441 Search PubMed.
- J. Yang, Y. Shen, Y. Sun, J. Xian, Y. Long and G. Li, Angew. Chem., Int. Ed., 2023, 62, e202302220 CrossRef CAS PubMed.
- G. Hai, H. Gao, X. Huang, L. Tan, X. Xue, S. Feng and G. Wang, Chem. Sci., 2022, 13, 4397–4405 RSC.
- Q. Kang, D. Lai, W. Tang, Q. Lu and F. Gao, Chem. Sci., 2021, 12, 3818–3835 RSC.
- H. Sun, X. Xu, Y. Song, W. Zhou and Z. Shao, Adv. Funct. Mater., 2021, 31, 2009779 CrossRef CAS.
- C. Wang, W. Gao, X. Wan, B. Yao, W. Mu, J. Gao, Q. Fu and D. Wen, Chem. Sci., 2022, 13, 13956–13965 RSC.
- T. Wang, L. Tao, X. Zhu, C. Chen, W. Chen, S. Du, Y. Zhou, B. Zhou, D. Wang, C. Xie, P. Long, W. Li, Y. Wang, R. Chen, Y. Zou, X. Fu, Y. Li, X. Duan and S. Wang, Nat. Catal., 2021, 5, 66–73 CrossRef.
- F. Sun, J. Qin, Z. Wang, M. Yu, X. Wu, X. Sun and J. Qiu, Nat. Commun., 2021, 12, 4182 CrossRef CAS PubMed.
- G. Montaña-Mora, X. Qi, X. Wang, J. Chacón-Borrero, P. R. Martinez-Alanis, X. Yu, J. Li, Q. Xue, J. Arbiol, M. Ibáñez and A. Cabot, J. Electroanal. Chem., 2023, 936, 117369 CrossRef.
- H. Sun, H. Kim, S. Song and W. Jung, Mater. Rep.: Energy, 2022, 2, 100092 CAS.
- H. Sun, L. Li, Y. Chen, H. kim, X. Xu, D. Guan, Z. Hu, L. Zhang, Z. Shao and W. Jung, Appl. Catal., B, 2023, 325, 122388 CrossRef CAS.
- D. Du, Q. Geng, L. Ma, J. Li, W. Dong, Q. Hua, L. Fan, R. Shao, X. Wang, C. Li and Y. Yamauchi, Chem. Sci., 2022, 13, 3819–3825 RSC.
- T. Wang, X. Cao and L. Jiao, Angew. Chem., Int. Ed., 2022, 61, e202213328 CrossRef CAS PubMed.
- H. Sun, W. Zhang, J. Li, Z. Li, X. Ao, K. Xue, K. K. Ostrikov, J. Tang and C. Wang, Appl. Catal., B, 2021, 284, 119740 CrossRef CAS.
- J. Li, L. Li, X. Ma, X. Han, C. Xing, X. Qi, R. He, J. Arbiol, H. Pan, J. Zhao, J. Deng, Y. Zhang, Y. Yang and A. Cabot, Adv. Sci., 2023, 2300841 CrossRef CAS PubMed.
- Y. Guo, X. Yang, X. Liu, X. Tong and N. Yang, Adv. Funct. Mater., 2023, 33, 2209134 CrossRef CAS.
- Y. Xu, M. Liu, M. Wang, T. Ren, K. Ren, Z. Wang, X. Li, L. Wang and H. Wang, Appl. Catal., B, 2022, 300, 120753 CrossRef CAS.
- J. Liu, Q. Wang, T. Li, Y. Wang, H. Li and A. Cabot, Nano Res., 2023, 16, 2041–2048 CrossRef CAS.
- J. Chang, G. Wang, C. Li, Y. He, Y. Zhu, W. Zhang, M. Sajid, A. Kara, M. Gu and Y. Yang, Joule, 2023, 7, 587–602 CrossRef CAS.
- S. Bai, Y. Xu, K. Cao and X. Huang, Adv. Mater., 2021, 33, 2005767 CrossRef CAS PubMed.
- J. Li, X. Wang, C. Xing, L. Li, S. Mu, X. Han, R. He, Z. Liang, P. Matinez, Y. Yi, Q. Wu, H. Pan, J. Arbiol, C. Cui, Y. Zhang and A. Cabot, Chem. Eng. J., 2022, 440, 135817 CrossRef CAS.
- Q. Wang, T. Li, S. Yan, W. Zhang, G. Lv, H. Xu, H. Li, Y. Wang and J. Liu, Inorg. Chem., 2022, 61, 16211–16219 CrossRef CAS PubMed.
- Q. Wang, J. Liu, T. Li, T. Zhang, J. Arbiol, S. Yan, Y. Wang, H. Li and A. Cabot, Chem. Eng. J., 2022, 446, 136878 CrossRef CAS.
- P. Xu, S. Zhao, T. Wang, W. Ji, Z. Chen and C. Au, J. Mater. Chem. A, 2022, 10, 10150–10161 RSC.
- X. Wang, Z. Zhang, Y. Ge, K. Shen and J. Qian, Small Struct., 2023, 2200390 CrossRef.
- L. Hui, X. Zhang, Y. Xue, X. Chen, Y. Fang, C. Xing, Y. Liu, X. Zheng, Y. Du, C. Zhang, F. He and Y. Li, J. Am. Chem. Soc., 2022, 144, 1921–1928 CrossRef CAS PubMed.
- G. Zhang, D. Cao, S. Guo, Y. Fang, Q. Wang, S. Cheng, W. Zuo, Z. Yang and P. Cui, Small, 2022, 18, 2202587 CrossRef CAS PubMed.
- J. Zhang, M. Yuan, T. Zhao, W. Wang, H. Huang, K. Cui, Z. Liu, S. Li, Z. Li and G. Zhang, J. Mater. Chem. A, 2021, 9, 20676–20684 RSC.
- S. Li, Y. Wang, Y. Li, X. Fang, Y. Liu, M. Li, Z. Wang, Y. Gao, H. Sun, F. Gao, X. Zhang and X. Dai, Nano Res., 2022, 15, 2877–2886 CrossRef CAS.
- Y. Sun, L. Huang, Q. Shan, G. Li, Z. Zheng, Q. Jiang, Y. Jiang and Z. Xie, ACS Appl. Energy Mater., 2022, 5, 10907–10914 CrossRef CAS.
- W. Tong, B. Huang, P. Wang, Q. Shao and X. Huang, Natl. Sci. Rev., 2021, 8, nwaa088 CrossRef CAS PubMed.
- C. Atlan, C. Chatelier, I. Martens, M. Dupraz, A. Viola, N. Li, L. Gao, S. J. Leake, T. U. Schülli, J. Eymery, F. Maillard and M. Richard, Nat. Mater., 2023, 22, 754–761 CrossRef CAS PubMed.
- L. Tao, Z. Xia, Q. Zhang, Y. Sun, M. Li, K. Yin, L. Gu and S. Guo, Sci. Bull., 2021, 66, 44–51 CrossRef CAS PubMed.
- Z. Lyu, X. Zhang, X. Liao, K. Liu, H. Huang, J. Cai, Q. Kuang, Z. Xie and S. Xie, ACS Catal., 2022, 12, 5305–5315 CrossRef CAS.
- Q. Xue, Z. Ge, Z. Yuan, J. Huang, B. He and Y. Chen, Mater. Today Phys., 2023, 31, 100980 CrossRef CAS.
- P. Li, K. Liu, J. Ye, F. Xue, Y. Cheng, Z. Lyu, X. Liao, W. Wang, Q. Zhang, X. Chen, M. Liu and S. Xie, J. Mater. Chem. A, 2019, 7, 17987–17994 RSC.
- Y. Chen, J. Pei, Z. Chen, A. Li, S. Ji, H. Rong, Q. Xu, T. Wang, A. Zhang, H. Tang, J. Zhu, X. Han, Z. Zhuang, G. Zhou and D. Wang, Nano Lett., 2022, 22, 7563–7571 CrossRef CAS PubMed.
- S. Luo, L. Zhang, Y. Liao, L. Li, Q. Yang, X. Wu, X. Wu, D. He, C. He, W. Chen, Q. Wu, M. Li, E. Hensen and Z. Quan, Adv. Mater., 2021, 33, 2008508 CrossRef CAS PubMed.
- L. Bu, N. Zhang, S. Guo, X. Zhang, J. Li, J. Yao, T. Wu, G. Lu, J.-Y. Ma, D. Su and X. Huang, Science, 2016, 354, 1410–1414 CrossRef CAS PubMed.
- A. Alinezhad, L. Gloag, T. M. Benedetti, S. Cheong, R. F. Webster, M. Roelsgaard, B. B. Iversen, W. Schuhmann, J. J. Gooding and R. D. Tilley, J. Am. Chem. Soc., 2019, 141, 16202–16207 CrossRef CAS PubMed.
- G. Liu, W. Zhou, Y. Ji, B. Chen, G. Fu, Q. Yun, S. Chen, Y. Lin, P. F. Yin, X. Cui, J. Liu, F. Meng, Q. Zhang, L. Song, L. Gu and H. Zhang, J. Am. Chem. Soc., 2021, 143, 11262–11270 CrossRef CAS PubMed.
- T. He, W. Wang, F. Shi, X. Yang, X. Li, J. Wu, Y. Yin and M. Jin, Nature, 2021, 598, 76–81 CrossRef CAS PubMed.
- Z. Luo, J. Lu, C. Flox, R. Nafria, A. Genç, J. Arbiol, J. Llorca, M. Ibáñez, J. R. Morante and A. Cabot, J. Mater. Chem. A, 2016, 4, 16706–16713 RSC.
- Y. Xia, K. D. Gilroy, H.-C. Peng and X. Xia, Angew. Chem., Int. Ed., 2017, 56, 60–95 CrossRef CAS PubMed.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-Ray Photoelectron Spectroscopy, Perkin-Elmer Corporation, 1992 Search PubMed.
- A. T. Fafarman, W. Koh, B. T. Diroll, D. K. Kim, D. K. Ko, S. J. Oh, X. Ye, V. Doan-Nguyen, M. R. Crump, D. C. Reifsnyder, C. B. Murray and C. R. Kagan, J. Am. Chem. Soc., 2011, 133, 15753–15761 CrossRef CAS PubMed.
- C. Wei, S. Sun, D. Mandler, X. Wang, S. Z. Qiao and Z. J. Xu, Chem. Soc. Rev., 2019, 48, 2518–2534 RSC.
- Q. Mao, K. Deng, H. Yu, Y. Xu, Z. Wang, X. Li, L. Wang and H. Wang, Adv. Funct. Mater., 2022, 32, 2201081 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02771f |
| This journal is © The Royal Society of Chemistry 2023 |