
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A CuICoII cryptate for the visible light-driven reduction of CO2†
Julia
Jökel
a,
Esma Birsen
Boydas
b,
Joël
Wellauer
c,
Oliver S.
Wenger
 c,
Marc
Robert
de,
Michael
Römelt
*b and
Ulf-Peter
Apfel
*af
c,
Marc
Robert
de,
Michael
Römelt
*b and
Ulf-Peter
Apfel
*af
aFraunhofer UMSICHT, Osterfelder Str. 3, 46047 Oberhausen, Germany. E-mail: ulf-peter.apfel@umsicht.fraunhofer.de; ulf.apfel@rub.de
bInstitute of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor Str. 2, 12489 Berlin, Germany
cDepartment of Chemistry, Universität Basel, St. Johanns-Ring 19, 4056 Basel, Switzerland
dUniversité Paris Cité, Laboratoire d'Electrochimie Moléculaire, CNRS, F-75013 Paris, France
eInstitut Universitaire de France (IUF), F-76006 Paris, France
fInorganic Chemistry I, Ruhr-Universität Bochum, Universitätsstr. 150, 44801 Bochum, Germany
First published on 27th October 2023
Abstract
Among the rare bimetallic complexes known for the reduction of CO2, CoIICoII and ZnIICoII hexamine cryptates are described as efficient photocatalysts. In close relation to the active sites of natural, CO2-reducing enzymes, we recently reported the asymmetric cryptand {NSNN}m ({NSNN}m = N[(CH2)2SCH2(m-C6H4)CH2NH(CH2)2]3N) comprising distinct sulphur- and nitrogen-rich binding sites and the corresponding CuIMII (MII = CoII, NiII, CuII) complexes. To gain insight into the effect of metals in different oxidation states and sulphur-incorporation on the photocatalytic activity, we herein investigate the CuICoII complex of {NSNN}m as catalyst for the visible light-driven reduction of CO2. After 24 h irradiation with LED light of 450 nm, CuICoII-{NSNN}m shows a high efficiency for the photocatalytic CO2-to-CO conversion with 9.22 μmol corresponding to a turnover number of 2305 and a high selectivity of 98% over the competing H2 production despite working in an acetonitrile/water (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture. Experiments with mononuclear counterparts and computational studies show that the high activity can be attributed to synergistic catalysis between Cu and Co. Furthermore, it was shown that an increase of the metal distance results in the loss of synergistic effects and rather single-sited Co catalysis is observed.
:1) mixture. Experiments with mononuclear counterparts and computational studies show that the high activity can be attributed to synergistic catalysis between Cu and Co. Furthermore, it was shown that an increase of the metal distance results in the loss of synergistic effects and rather single-sited Co catalysis is observed.
Introduction
Artificial photosynthesis is a promising alternative to meet the increasing energy demand which has been, until now, mainly covered by the combustion of fossil fuels.1–3 With the use of abundant resources like sunlight and water, the reduction of CO2 into renewable fuels like CO, CH4, formic acid or methanol, is a viable approach for completing the carbon cycle.4,5 Although tremendous efforts have been devoted to the development of heterogeneous catalysts for the photocatalytic CO2 reduction, low yields together with a missing profound insight into reaction mechanisms make an industrial application not yet feasible.6–8To get a fundamental understanding of important structural features or mechanistic details of an efficient photocatalytic CO2 reduction, homogeneous catalysis is a helpful tool.9 Moreover, the manifold spectroscopic techniques available for homogeneous catalysis allow for a specific fine-tuning of the catalyst's structure on a molecular level.9–12
Typically, homogeneous photocatalytic systems comprise three components: a photosensitiser for harvesting and converting light into electrochemical potential, a sacrificial reductant providing the required electrons and a molecular catalyst for accumulating and transferring them to the substrate.13,14 Due to the cruciality of the last step, many studies focus on the design of molecular catalysts.11,15–17
In contrast to the required harsh conditions and low selectivity associated with the artificial reduction of CO2, the naturally occurring enzymes CO dehydrogenases (CODHs) can convert CO2 selectively into CO under mild conditions.18 They commonly have heterobimetallic active sites comprised of frustrated Lewis pairs (e.g. Ni0FeII, CuIMoVI) embedded in a protecting, sulphur-rich ligand environment.18–21
With CODHs being a role model, numerous homogeneous catalysts for the photocatalytic CO2 reduction have been developed in the past, though, most of them utilizing only one 3d metal within a macrocyclic chelating ligand.15,16,22–25 In addition, among the rare bimetallic catalysts like Co21 or Ni21 (Scheme 1), geometric constraints do often suppress synergistic catalysis between metal centres.26,27 One recent example for actual cooperative effects during CO2 reduction was described by Robert et al.28 Under visible light irradiation, the Co2 bi-quaterpyridine complex Co22 (Scheme 1) selectively (max. 97%) reduces CO2 to formate in basic acetonitrile solution with a turnover frequency (TOF) of 0.23 min−1. Addition of a weak acid such as phenol shifts the reaction to CO with 99% selectivity and an increased catalytic rate of 13.8 min−1. Notably, the product outcome is dependent on the metal-bound η2 isomer of CO2, η2O,O or η2C,O, stabilised under the respective conditions, leading to either formate or CO. However, in both cases, CO2 is bound between the Co centres, highlighting the potential of bimetallic complexes.
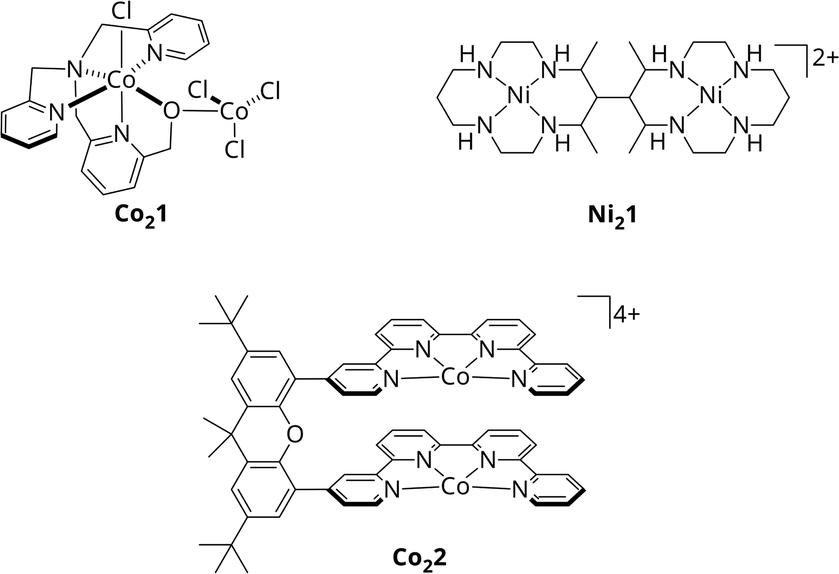 | ||
| Scheme 1 Molecular structures of Co21, Ni21 and Co22.26–28 | ||
In the past, we amongst others reported on the ability of dinuclear azacryptates for the uptake of atmospheric CO2 as bicarbonate between the metal centres including Co2,29 Ni2,30 Cu231 or Zn232 complexes. Beyond these reports, Lu et al. reported on a dinuclear Co2 azacryptate (CoIICoII-{NNNN}m, Scheme 2) as photocatalyst for the visible light-driven CO2 conversion in a water-containing system with [Ru(phen)3](PF6)2 as photosensitiser (PS) and triethanolamine (TEOA) as sacrificial electron donor (SED). Due to synergistic effects between the CoII centres, the cryptate was reported to reach a high catalytic rate of 0.47 s−1 compared to its mononuclear analogue with 0.04 s−1, while exhibiting a high selectivity of 98% for CO evolution even in the presence of water.33 The observed cooperativity was even more pronounced, when one CoII was exchanged by ZnII to yield ZnIICoII-{NNNN}m, resulting in an enhanced activity for CO2-to-CO conversion with 1.80 s−1 and a maintained high selectivity. Due to the stronger binding affinity of ZnII over CoII to OH− in the critical C–O cleaving step, C–O bond cleavage is greatly promoted, highlighting the importance of understanding the role of metals in dual CO2 activation.34
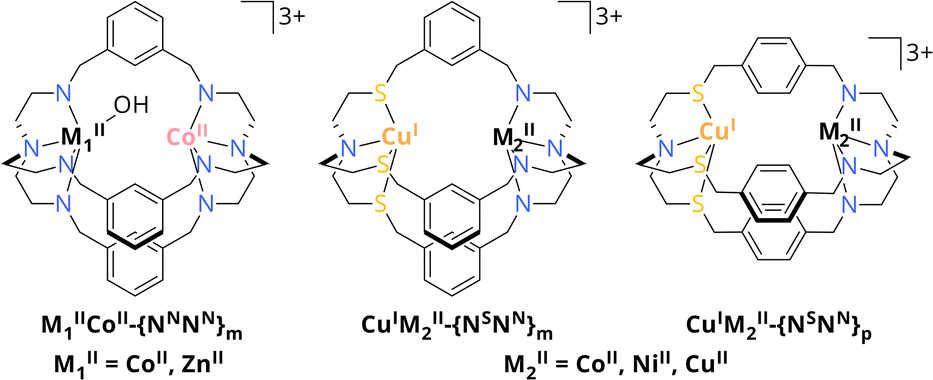 | ||
| Scheme 2 Schematic overview of CoIIM1II-{NNNN}m with M1II = CoII, ZnII and CuIM2II-{NSNN}m/p with M2II = CoII, NiII, CuII. Hydrogen atoms are omitted for clarity.32,33 | ||
These findings pointed our interest towards photocatalysts comprising azacryptates with not only metal centres of different kind but also oxidation states. Especially since the presence of a frustrated Lewis pair is crucial for the reduction of CO2 within the CODHs, we got interested in cooperative metal–metal effects for CO2 reduction.18 Along this line, we recently reported on the synthesis of the asymmetric cryptand {NSNN}m which allows for the site-specific coordination of CuI and either CoII, NiII or CuII through sulphur incorporation within the ligand framework (Scheme 2). Furthermore, the distance between the metal centres can be increased when using {NSNN}p as ligand.35
Thus, we herein report on the photocatalytic activity of CuICoII-{NSNN}m towards CO2 reduction with additional focus on the influence of the metal distance. The selection of CuICoII-{NSNN}m is based on the following considerations: (1) the CoII-{NN} site was already successfully reported for photocatalytic CO2 conversion33,34 and thus, allows for studying alterations of the other catalytic site. (2) Considering the active site's sulphur-rich environment in CO2-converting enzymes, using a catalyst with sulphur donors was desired. (3) CuI-{NS} was synthetically easily accessible due to the matching donor–acceptor properties of CuI and sulphur and exhibits structural similarity to the active site of the [MoCu]-CODH.
Results and discussion
In order to investigate the influence of mixed-valent metal centres on the overall CO2 reduction reactivity, we herein examined the stable heterobimetallic complex CuICoII-{NSNN}m as potential photocatalyst under otherwise similar conditions as was reported by Lu et al.33,35 Like commonly described for bimetallic azacryptates,31,33,34,36CuICoII-{NSNN}m readily absorbs CO2 which is indicated by a change of the UV/vis/NIR spectrum upon purging a MeCN/H2O (4:1) complex solution with CO2 for 5 min, indeed making it a promising candidate as a CO2 reduction catalyst. In the presence of CO2, the absorbance maxima at 490 nm and 618 nm decrease while the absorbance below 378 nm increases, consequently generating an isosbestic point (Fig. S1, ESI†) and thus, indicating the formation of a new species. The presence of a mass peak at m/z = 931.3 in the ESI-MS spectrum matching the [CuICoII-{NSNN}(HCO3)(ClO4)]+ fragment (Fig. S2B, ESI†) suggests a fixation of CO2 as bicarbonate. Moreover, the intensification of bicarbonate-related vibrations at 1641 cm−1 and 1449 cm−1 in the respective IR spectrum (Fig. S2A, ESI†) support this finding, albeit they coincide with ligand-associated vibrations.31,36,37 Notably, under Ar atmosphere, the UV/vis/NIR spectrum remained unchanged within 24 h and illustrates the high stability of CuICoII-{NSNN}m in solution.
Visible light-driven reduction of CO2 to CO with CuICoII-{NSNN}m as catalyst
The photocatalytic CO2 reduction experiments were performed using 2 μM CuICoII-{NSNN}m as catalyst, 0.4 mM [Ru(phen)3](PF6)2 as photosensitizer and 0.3 M triethanolamine (TEOA) as sacrificial reductant in CO2-saturated MeCN/H2O (4:1) at room temperature. The photosystem was illuminated (450 nm LED, light intensity of 1200 mcd, irradiation area 0.8 cm2) for 56 h with two-hourly GC-BID analysis (except for hours 14–22 and 36–44) of the headspace composition as well as GC-MS analysis of the liquid phase composition after 56 h. All given values were averaged over three experiments with typical uncertainties of 2–8%. CO was observed as sole carbon-based product from CO2 reduction (Fig. S3 and S4, ESI†) with a steady increase of evolution during the first 24 h (Fig. 1). Beyond this point, the CO production ceased and the maximum value with 9.22 μM was reached, corresponding to a turnover number (TON) of 2305 and turnover frequency (TOF) of 1.60 min−1. Only H2 was formed as side product from proton reduction with 0.189 μmol (TON of 47, TOF of 3.28 × 10−3 min−1) after 24 h, leading to a high selectivity of 98% for CO2-to-CO conversion. Analysis of the liquid phase furthermore revealed the formation of acetaldehyde which can be attributed to the degradation of TEOA.38 The quantum yield was determined to be 0.15% using a potassium ferrioxalate actinometer (Fig. S5, ESI†). Contrary, in the absence of either CuICoII-{NSNN}m, [Ru(phen)3](PF6)2, TEOA or in the dark, no CO was detected, thus, showing the necessity of each component for the photocatalytic system (Fig. S6, ESI†). The small activity for H2 production with 8.85 × 10−3 min−1 (0.0510 μmol, TON of 13, 24 h) observed in experiments without CuICoII-{NSNN}m is due to [Ru(phen)3](PF6)2 which can also act simultaneously as photosensitiser and catalyst for hydrogen evolution itself.39,40 Irradiation of an Ar purged solution instead of CO2 also generated no CO, excluding a potential CO production through degradation of any of the components of the photocatalytic system. Moreover, the presence of the proton source plays a pivotal role as well (Fig. S7, ESI†). While in neat MeCN the CO production rate is with 3.84 × 10−2 min−1 low (0.221 μmol, TON of 55, 24 h), addition of water with a ratio of MeCN/H2O 9:1 increased the activity to 1.08 min−1 (6.23 μmol CO, TON of 1558, 24 h). The best performance was achieved with the MeCN/H2O mixture of 4:1 while further increase of the water content to 1:1 limited the CO production to 1.37 × 10−2 min−1 (0.0791 μmol, TON of 20, 24 h). Isotope labelling experiments performed under a 13CO2 atmosphere followed by gas chromatography mass spectrometry analysis of the headspace, revealed the formation of 13CO (m/z = 29) as reduction product confirming that CO originates from CO2 (Fig. S8, ESI†).
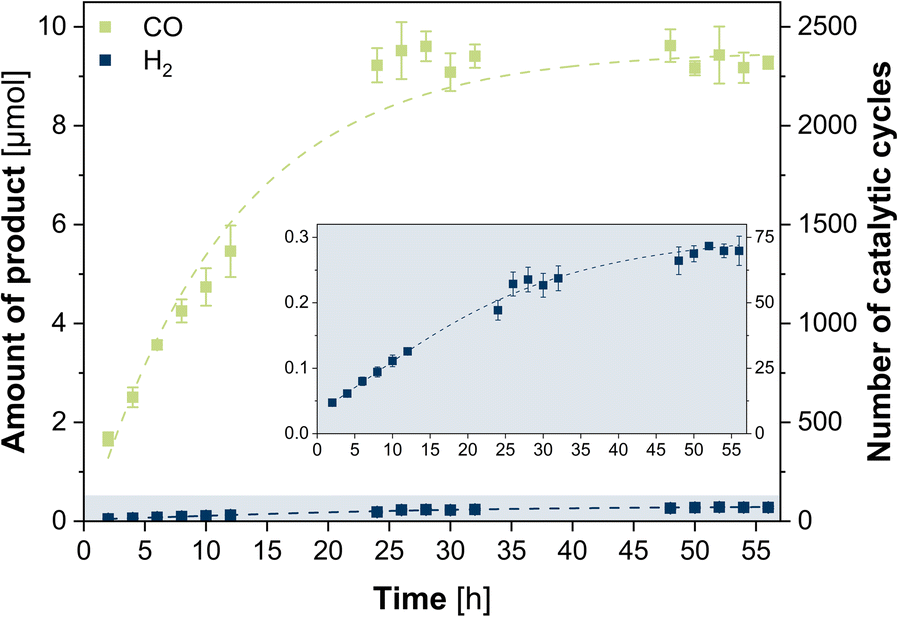 | ||
| Fig. 1 Photocatalytic evolution of CO (green) and H2 (blue) during 56 h catalysed by CuICoII-{NSNN}m (2 μM) in a CO2-saturated MeCN/H2O (4:1) solution containing 0.4 mM [Ru(phen)3](PF6)2 and 0.3 M TEOA under irradiation with blue LED light (λ = 450 nm, 1200 mcd, irradiation area 0.8 cm2). Dashed lines represent the best fit of data. | ||
Since the UV/vis/NIR spectrum of a MeCN/H2O (4:1) solution of 0.6 mM CuICoII-{NSNN}m did not alter during 24 h irradiation, stability of the catalyst under irradiation can be anticipated (Fig. S1, ESI†) which is supported by the continuous production of CO during this time. HPLC analysis of the photocatalytic solution after 24 h irradiation followed by subsequent ESI-MS of the catalyst-containing fraction revealed a signal at m/z = 478.6 in the negative mode corresponding to [CuICoII-{NSNN}(BF4)(ClO4)(-3H)]2− (Fig. S9, ESI†) and thus, further confirming the integrity of CuICoII-{NSNN}m. The durability of the photosystem, however, is limited to 24 h as shown by the ceasing CO production beyond this hour (Fig. 1). Since TEOA is present in large excess and the UV/vis/NIR spectrum of [Ru(phen)3](PF6)2 (Fig. S10, ESI†) shows a steady decrease of the absorbance maximum at 446 nm until 24 h, this finding can be attributed to the photodegradation of [Ru(phen)3](PF6)2. Furthermore, experiments in the presence of mercury revealed when considering the margins a similar CO production rate with 1.27 min−1 (7.34 μmol, TON of 1835, 24 h) and increased H2 production with 3.72 × 10−2 min−1 (0.214 μmol, TON of 54, 24 h) revealing that the catalytic reaction is predominantly homogeneous and does not result from elemental Cu or Co due to catalyst degradation. The absence of any particles in the photocatalytic solution after irradiation for 24 h is further confirmed by particle size analysis using laser diffraction (Fig. S11, ESI†). Consequently, reactivation experiments were performed in which fresh [Ru(phen)3](PF6)2 was added to the catalytic system after 26 h and 52 h (Fig. S12, ESI†). Notably, after addition of fresh photosensitiser, the CO production was increased again, achieving similar catalytic rates like after 24 h, with 1.70 min−1 (11.4 μmol, TON of 2850) and 1.48 min−1 (19.2 μmol, TON of 4800) after 2 h of the first and second re-addition, respectively, ultimately highlighting that [Ru(phen)3](PF6)2 is the main limiting factor herein.
Synergistic catalysis and influence of metal distance on CO2 reduction
To explore the ongoing photocatalytic reaction in detail, investigations on the CO evolution kinetics were performed in a first step. Concentration-dependent experiments with 0.5, 1.0, 1.5 and 2 μM catalyst show at every hour a linear dependence of the concentration versus the amount of CO generated (Fig. S13, ESI†), suggesting a first-order reaction for the CO2-to-CO conversion. To consequently gain a more profound insight into the catalytic activity of the individual metals within CuICoII-{NSNN}m, we also employed the mononuclear counterparts CuI-{NSNN}m and CoII-{NSNN}m as comparative catalyst systems (Fig. 2B).35 While the CO and H2 production with CuI-{NSNN}m was neglectable (CO: 0.01 min−1, 0.0464 μmol, TON of 12, H2: 4.86 × 10−3 min−1, 0.0279 μmol, TON of 7, 24 h), complex CoII-{NSNN}m achieved a catalytic rate of 0.99 min−1 (5.70 μmol, TON 1425, 24 h) for CO and 2.34 × 10−3 min−1 (0.135 μmol, TON 34, 24 h) for H2, corresponding to a 98% selectivity of CO over H2 production. Thus, the nearly absent activity with Cu and the decreased activity with Co suggest that probably only the Co centre is the redox-active site during catalysis within CuICoII-{NSNN}m but synergistically benefiting from the presence of the Cu centre.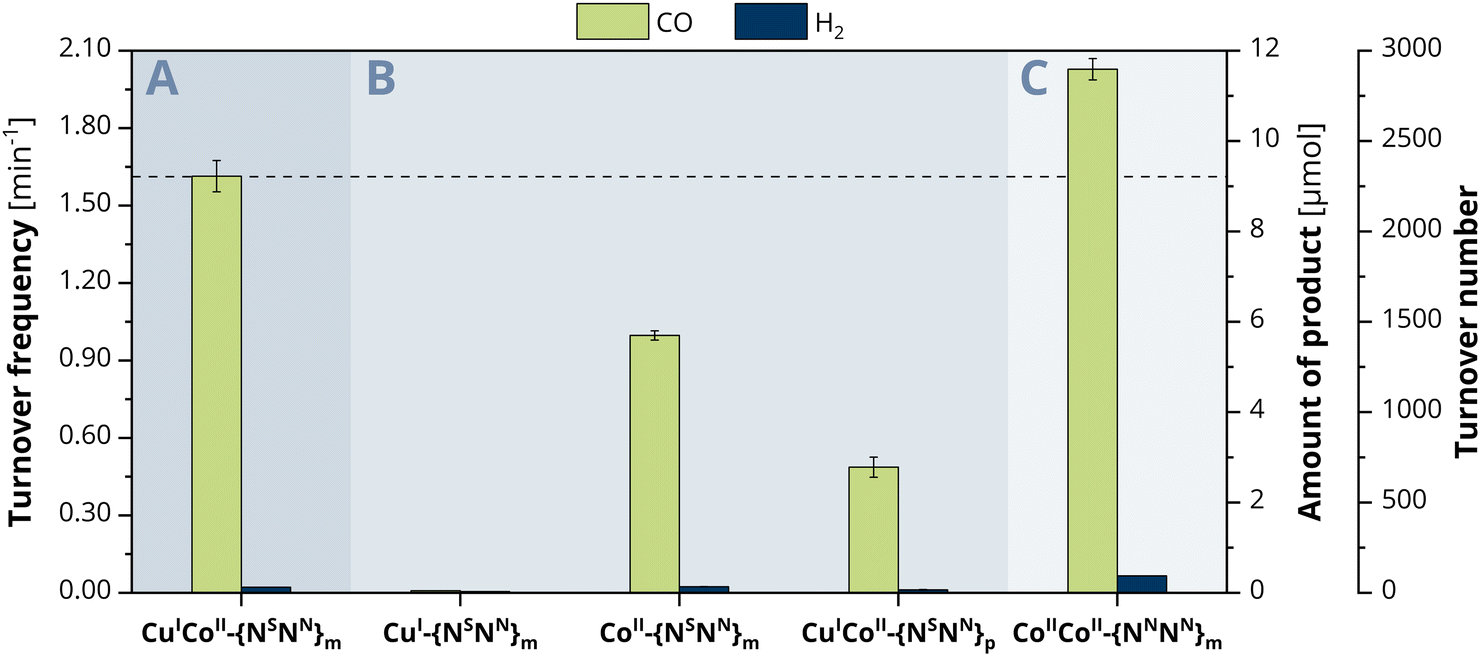 | ||
| Fig. 2 Photocatalytic evolution of CO (green) and H2 (blue) after 24 h irradiation with blue LED light (λ = 450 nm, 1200 mcd, irradiation area 0.8 cm2) of a CO2-saturated MeCN/H2O (4:1) solution containing 0.4 mM [Ru(phen)3](PF6)2 and 0.3 M TEOA with (A) CuICoII-{NSNN}m, (B) CuI-{NSNN}m, CoII-{NSNN}m, and CuICoII-{NSNN}p or (C) CoIICoII-{NNNN}m as catalyst. | ||
We therefore became curious about whether the CO generation can be influenced by a variation of the metal distance between CuI and CoII. We previously described that the distance between the binding sites of the para-substituted analogue of the ligand, {NSNN}p, is roughly 1 Å larger than in {NSNN}m, giving a metal–metal distance of 6.578 Å within the related CuICuII cryptate.35 Due to the lack of a crystal structure of a corresponding meta cryptate, we consequently refer to the distance between the binding sites in the empty ligand and used CuICoII-{NSNN}p as catalyst for comparison. Under otherwise identical conditions, CuICoII-{NSNN}p achieved a CO production rate of 0.48 min−1 (2.78 μmol, TON of 695, 24 h) and a H2 production of 1.17 × 10−2 min−1 (0.0671 μmol, TON of 17, 24 h) (Fig. 2B). These values are significantly smaller compared to those observed with CuICoII-{NSNN}m showing that the metal distance within CuICoII-{NSNN}p is too large for synergistic catalysis to occur; therefore, it is possible that the CoII site is solely responsible for the observed activity. Interestingly, CuICoII-{NSNN}p has a smaller activity for CO production than the mononuclear catalyst CoII-{NSNN}m. We preliminarily attribute this not only to the altered metal distance but also lowered flexibility of the macrocycle caused by the para-substituted spacers bridging the two binding sites.
Mechanistic studies of the CO2 reduction
:1), CuICoII-{NSNN}m exhibits one primary reduction wave at −1.05 V vs. Fc+/0 and at −1.59 V vs. Fc+/0. Overlay with the CVs of the monometallic counterparts CuI-{NSNN}m and CoII-{NSNN}m clarifies that the former wave can be assigned to the reduction of CoII to CoI and the latter one corresponds to the reduction of CuI (Fig. S14B, ESI†). The corresponding oxidations for Cu0 to CuI and CoI to CoII appear at −0.78 and −0.62 V vs. Fc+/0, respectively. Additionally, the herein measured potential window displays further the oxidation of CuI to CuII at −0.25 V followed by reduction at −0.76 V vs. Fc+/0. Upon saturation with CO2, the CoII/I redox couple remains nearly unchanged in terms of current but with a minor anodic shift to −1.01 V. Furthermore, an additional diffusion-limited signal can be observed, suggesting an interaction with CO2, followed by a catalytic response at higher potentials (Fig. S14C and S15A, ESI†). Contrary, a pre-catalyst transformation was not observed in the linear sweep voltammogram (LSV) of CoII-{NSNN}m in the presence of CO2 with a catalytic current increase from −1.44 V vs. Fc+/0, suggesting that it follows a different mechanism than CuICoII-{NSNN}m. This is also the case for CuI-{NSNN}m which additionally does not display a catalytic response at considerable potential, supporting its inactivity towards the photocatalytic CO2 reduction. Since the LSVs of CuICoII-{NSNN}m and CuICoII-{NSNN}p show a similar behaviour upon addition of CO2, no reason for the difference in reactivity within the photocatalytic CO evolution can be drawn from the electrochemical studies (Fig. S15A, ESI†).
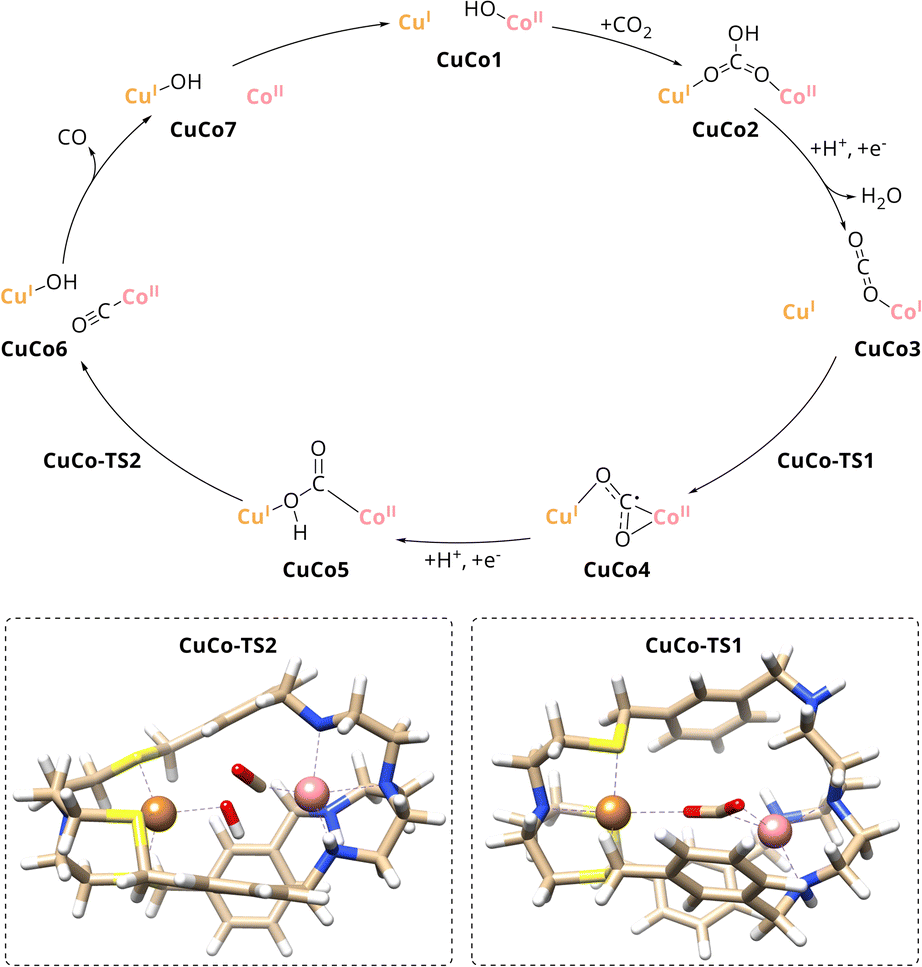 | ||
| Scheme 3 Proposed mechanism for CO2 reduction to CO with CuICoII-{NSNN}m as catalyst. (Inset) Transition state structures for one electron CO2 reduction and C–OH bond cleavage, CuCo-TS1 and CuCo-TS2, respectively. | ||
A comparison of the catalytic cycle with the experimental results of the mononuclear analogues reveals a good agreement of the predictions of the role of the individual metal within CuICoII-{NSNN}m. The nearly absent activity of CuI-{NSNN}m towards the reduction of CO2 is in line with Cu taking over an assistant role in binding during catalysis. In contrast, Co was experimentally proposed as redox-active site due to the activity of CoII-{NSNN}m towards CO2-to-CO conversion which matches the role of Co during the catalytic cycle, too. To further strengthen these findings, smaller computed scaffold models comprising the specific tripodal ligand backbone with an open coordination site in axial position (Fig. S20, ESI†) were considered.33 While one-electron reduction of the CuI-{NS} site alone entails a complex decomposition, addition of an electron to CuI-{NS} and CO2 leads to the formation of the CO2˙− that is only weakly bound to the catalyst (d = 2.83 Å). Hence, in accordance with the experimental results for CuI-{NSNN}m, a pathway for the reaction between CuI-{NS} and CO2 could not be established. Contrary to CuI-{NS} and thus, in good agreement with the experimental results for CoII-{NSNN}m, a reaction profile for the CoII-{NN} site with CO2 was proposed by Lu et al.33 Therein, a simultaneous two-electron transfer step from the Co centre to CO2 yields a η1C CoIII–CO22− intermediate which is transformed in a PCET into CoII-COOH−. Subsequently, C–OH bond cleavage results in CO and OH−, both bound to Co, followed by release of the former. While the simultaneous two-electron reduction of CO2 in CoII-{NN} proceeds with an energy barrier of 19.5 kcal mol−1,33 the one for the stepwise reduction in CuICoII-{NSNN}m with 10.4 kcal mol−1 is significantly lower which is which most likely due to the favourable interaction of Cu stabilising the CO2˙− adduct in CuCo4. Furthermore, the energy barrier of the following C–OH bond cleavage is considerably lower for CuICoII-{NSNN}m (8.9 kcal mol−1) compared to the reported value for the mononuclear case (18.1 kcal mol−1).33 Consequently, benefitting from the synergistic effect between the two metals, with CoII as active site and CuI taking over an assistant role in binding, CO2 reduction to CO is more favoured in the dinuclear cryptate CuICoII-{NSNN}m.
The computed binding mode of the CO2 radical anion for the meta and para analogue of the CuICoII complex (Fig. 3 and S21, ESI†) also supports the significantly lowered activity of CuICoII-{NSNN}p. While the distance between the binding sites in {NSNN}m is inherently smaller,35 it exhibits a higher flexibility and the corresponding metal complex can consequently adapt an even smaller bite length in the presence of various substrates (Table S3, ESI†). For example, owing to the flexible nature of CuICoII-{NSNN}m, the Cu–Co distance reduces from 6.20 to 4.60 Å for the accommodation of the CO2 radical anion, affording a dual μ2,η2C,O binding mode. In contrast, the lowered flexibility of CuICoII-{NSNN}p only allows for a deviation of the metal distance by 0.19 Å, binding the CO2˙−via its two oxo sites in a μ2,η2O,O fashion, consequently preventing synergistic catalysis.
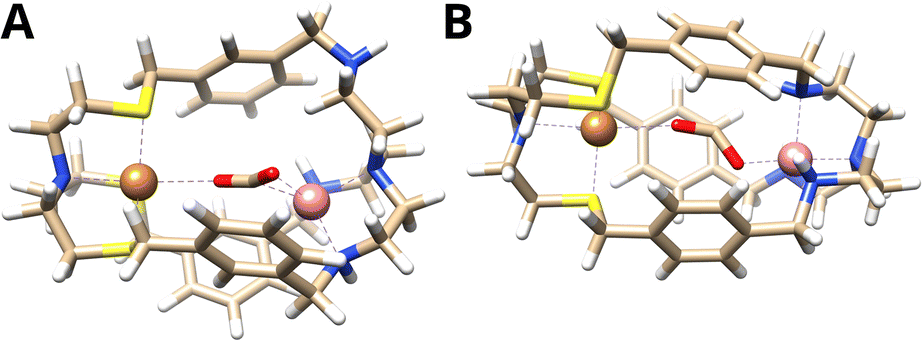 | ||
| Fig. 3 Binding mode of the CO2 radical anion within (A) CuICoII-{NSNN}m and (B) CuICoII-{NSNN}p. | ||
Comparison with the octa-amino, homodinuclear cryptate CoIICoII-{NNNN}
To classify the activity of CuICoII-{NSNN}m and compare it with CoIICoII-{NNNN}m described by Lu et al.,34 we prepared the full-nitrogen system according to the described procedure and tested it as photocatalyst for the CO2 reduction under the herein stated conditions with 2 μM catalyst concentration. Small deviations within the photosystem set-up compared to the one used in literature might lead to a great difference in product outcome and make a direct comparison of the results difficult. In the herein employed photosystem, CoIICoII-{NNNN}m achieved a production rate of 2.01 min−1 (11.6 μmol, TON of 2900, 24 h) and 6.51 × 10−2 min−1 (0.375 μmol, TON of 94, 24 h) for CO and H2, respectively, corresponding to a 97% selectivity for CO2-to-CO conversion (Fig. 2C). Notably, the activity of CuICoII-{NSNN}m towards CO2-to-CO conversion is with 1.60 min−1 smaller with a maintained selectivity over H2 production as with CoIICoII-{NNNN}m. Although CuICoII-{NSNN}m allows for the reduction of CoII to CoI at more anodic potential (−1.01 V vs. Fc+/0) compared to CoIICoII-{NNNN}m (−1.15 V vs. Fc+0, one electron reduction to CoICoII33), the LSV of the latter shows a drastic increase of current upon CO2 saturation after the CoII reduction wave (Fig. S11B, ESI†). For CuICoII-{NSNN}m, a similar current is only observed at roughly 0.4 V more cathodic potentials, providing one reason for the lower activity compared to CoIICoII-{NNNN}m. Although a comparison of the proposed reaction mechanisms for the CO2-to-CO conversion with CuICoII-{NSNN}m and the previously described CoIICoII-{NNNN}m reveal that they follow different pathways for the actual reduction of CO2, the critical C–OH bond cleaving step is similar for both.33 Here, the assisting binding site, CuI-{NS} or CoII-{NN} in case of CuICoII-{NSNN}m and CoIICoII-{NNNN}m, respectively, plays an important role. Since CuI-{NS} exhibits a higher electron density compared to CoII-{NN}, the binding affinity to the negatively charged OH− is less pronounced. Thus, bond cleavage is promoted to a lower extent, consequently resulting in the lower activity of CuICoII-{NSNN}m. Furthermore, next to the synergistic pathway in CoIICoII-{NNNN}m, single-sited catalysis at both Co centres individually is conceivable as well, considerably enhancing the chance for CO2 reduction to occur. Due to the inactivity of the single CuI-{NS} site, this is not the case for CuICoII-{NSNN}m.At this point, we became curious on investigating the effect of incorporating two CoII into {NSNN}m on the photocatalytic CO2 reduction. Experiments with CoIICoII-{NSNN}m however, revealed lower catalytic rates for CO2-to-CO conversion with 1.11 min−1 (6.39 μmol, TON of 1598, 24 h) compared to both, CoIICoII-{NNNN}m and CuICoII-{NSNN}m. Same was for ZnIICoII-{NSNN}m with 1.08 min−1 (6.21 μmol, TON of 1553, 24 h), despite the significantly improved catalysis observed for the corresponding ZnIICoII complex of the full-nitrogen system {NNNN}m.34 Interestingly, the second metal seemingly does not promote the catalysis at all, since the observed rates are in the similar range as for monometallic CoII-{NSNN}m (0.99 min −1). The decreased catalytic rates and formation of a precipitate during irradiation together with a few reports on +II metal complexes revealing decreased activity for CO2-to-CO conversion or tendency to demetallation upon directs N/S exchange in the ligand under reductive conditions,41–43 let us to the conclusion that the lowered activity is due to decomposition of the {NS} site and consequently emphasises the importance of matching donor–acceptor properties of ligand and metal.
Experimental
General techniques
{NSNN}m and {NSNN}p as well as the corresponding metal complexes CuI-{NSNN}m, CoII-{NSNN}m, CuICoII-{NSNN}m and CuICoII-{NSNN}p were synthesized according to a procedure developed previously within our group.35 For additional characterisation of the beforehand mentioned complexes, CHN experiments were measured with an Elementar vario MICRO cube. CoIICoII-{NNNN}m was synthesised according to a literature-known procedure.33 Mass spectra were measured on a Advion expressionL instrument. IR measurements were performed on a Shimadzu IRTracer-100 attached with a Pike Miracle ATR unit and are reported in [cm−1]. UV/Vis/NIR spectra were recorded with a Shimadzu UV 1900i at 25 °C and are reported in [nm]. X-Band EPR spectra were recorded on a Bruker EMXplus X-band EPR spectrometer. Samples were frozen using a liquid helium recirculating cooling system provided by ColdEdge and were measured at 13 K. The solution in the tube was frozen in liquid nitrogen and kept frozen until measured. 1H NMR spectra for application of Evans' method were obtained on a Bruker DRX 400 MHz NMR spectrometer at room temperature in deuterated acetonitrile (MeCN-d3), followed by evaluation according to a literature-known procedure. The calculated spin-only effective moment was using the following equation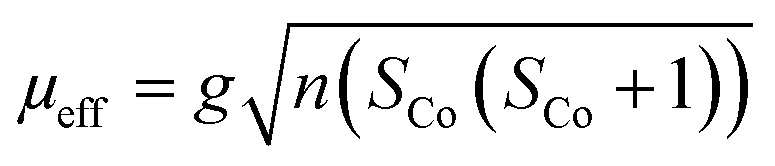 | (1) |
CHN analysis
Synthesis of CoIICoII-{NSNN}m
{NSNN}m (34.9 mg, 53.7 μmol) was dissolved in minimum amount of DCM. MeCN/MeOH (4:1) was added (2.5 mL), followed by the addition of Co(ClO4)2·6H2O (39.3 mg, 107 μmol) dissolved in 0.5 mL MeCN/MeOH (4:1) whereupon a direct colour change to brown was observed. The solution was stirred at room temperature for 24 h. Subsequently, the solvent was removed, the residue taken up in MeCN and filtered through a short silica column. After removal of the solvent, [CoIICoII{NSNN}m](ClO4)4 (CoIICoII-{NSNN}m) was obtained as a brown solid in 85% (53.2 mg, 45.6 μmol). ESI-MS: calcd for [CoIICoII-{NSNN}m(-3H)(MeCN)(OH)(Na)]+ m/z = 845.2; found: 844.9. Anal. calcd for [C36H51Cl4Co2N5O16S3] + 0.8 MeCN + 8H2O + 2 DCM: C 31.45, H 4.89, N 5.37; found: C 31.67, H 4.70, N 5.43. IR (ATR): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3458, 3254, 2943, 2318, 2291, 1638, 1450, 1367, 1101, 930, 802 cm−1. UV/vis/NIR (MeCN): λmax (ε in L mol−1 cm−1) = 384 (202 ± 2), 480 (114 ± 3), 587 (54 ± 1) nm. EPR (MeCN, 13 K): g = 4.29 (hs CoII).47 Evans method (MeCN-d3, 298 K): calcd for two hs CoII μeff = 5.48 μB; exp.: 5.64 μB.
= 3458, 3254, 2943, 2318, 2291, 1638, 1450, 1367, 1101, 930, 802 cm−1. UV/vis/NIR (MeCN): λmax (ε in L mol−1 cm−1) = 384 (202 ± 2), 480 (114 ± 3), 587 (54 ± 1) nm. EPR (MeCN, 13 K): g = 4.29 (hs CoII).47 Evans method (MeCN-d3, 298 K): calcd for two hs CoII μeff = 5.48 μB; exp.: 5.64 μB.
Synthesis of ZnIICoII-{NSNN}m
{NSNN}m (35.0 mg, 53.8 μmol) was dissolved in minimum amount of DCM. MeCN/MeOH (4:1) was added (2.5 mL), followed by the addition of Co(ClO4)2·6H2O (19.7 mg, 53.8 μmol) dissolved in 0.5 mL MeCN/MeOH (4:1) whereupon a direct colour change to light blue was observed. The solution was stirred at room temperature for 24 h. Subsequently, Zn(ClO4)2·6H2O (20.1 mg, 53.8 μmol) dissolved in 0.5 mL MeCN/MeOH (4:1) was added. Upon addition, the colour changed to beige and it was stirred at room temperature for 24 h. The solvent was removed, the residue taken up in MeCN and filtered through a short silica column. After removal of the solvent, [ZnIICoII{NSNN}m](ClO4)4 (ZnIICoII-{NSNN}m) was obtained as a beige solid in 76% (47.9 mg, 40.9 μmol). ESI-MS: calcd for [ZnIICoII-{NSNN}m(ClO4) (-2H)(MeCN)(Na)]+ m/z = 933.1; found: 933.0; [ZnIICoII-{NSNN}m(ClO4)(-H)(MeCN)(Na)(Li)]+ m/z = 941.1; found: 940.5; [ZnIICoII-{NSNN}m(2ClO4)(-2H)(Li)]+ m/z = 975.1; found: 975.9; calcd for [ZnIICoII-{NSNN}m(4ClO4)(H2O)]+ m/z = 1089.5; found: 1088.8. Anal. calcd for [C36H51Cl4CoN5O16S3Zn] + 1.5 MeCN + 6H2O + 2 DCM: C 32.58, H 4.77, N 6.02; found: C 32.25, H 4.54, N 6.11. IR (ATR): = 3460, 3246, 2941, 2317, 2290, 1637, 1450, 1365, 1098, 930, 802 cm−1. UV/vis/NIR (MeCN): λmax (ε in L mol−1 cm−1) = 380 (163 ± 6), 480 (57 ± 7), 571 (25 ± 6) nm. EPR (MeCN, 13 K): g = 4.37 (hs CoII).47 Evans method (MeCN-d3, 298 K): calcd for one hs CoII μeff = 3.87 μB; exp.: 3.17 μB.
Photochemistry
The photocatalytic reduction of CO2 was performed at 25 °C in a 12 mL reactor sealed with a rubber septum containing 2 mL of the catalyst (0.5–2 μM), 0.4 mM [Ru(phen)3](PF6)2 as photosensitizer and 0.3 M TEOA as sacrificial electron donor in degassed MeCN or MeCN/H2O (9:1, 4:1 or 1:1 mixtures). The solution was saturated with either Ar, 12CO2 or 13CO2 and was illuminated with blue LED light (λ = 450 nm, 1200 mcd, 0.8 cm2 irradiation area) for minimum 24 h and maximum 56 h. For measurements in the absence of light, the reaction was performed in a black chamber. To identify the composition of the gas phase, 50 μL of the headspace were analysed two-hourly by GC-BID via hand injection. To analyse the generated liquid products after photocatalysis, 1 mL of the solution was acidified with 100 μL conc. H2SO4 and analysed via headspace GC-MS (Fig. S3, ESI†). To verify the absence of formate/formic acid, the liquid phase was further analysed for propyl formate after derivatisation with n-propanol. Therefore, 400 μL sample were treated with 500 μL n-propanol and 100 μL p-toluene sulfonic acid and analysed via headspace GC-MS (Fig. S4, ESI†). Each photocatalytic reaction was repeated at least three times to confirm the reliability of the data. A summary of the determined data can be found in the ESI (Table S4†).
Determination of quantum yield
The quantum yields of the photocatalytic CO2 reduction experiments were determined using the equation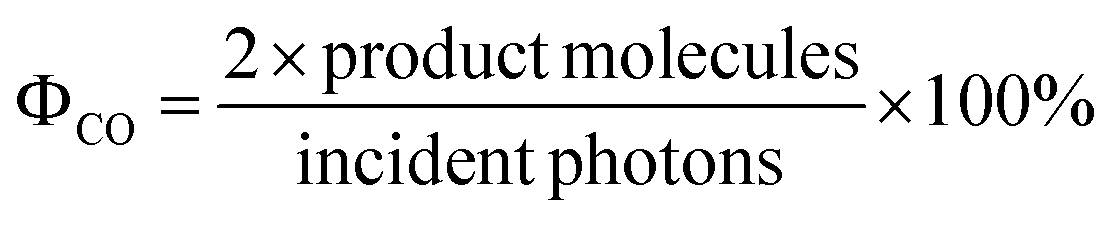 | (2) |
Quantification of CO2RR products
Quantification of the headspace gas composition of the photochemical cell was performed using a Shimadzu GC-2010 Pro equipped with a Shimadzu BID-2010 Plus barrier discharge ionization detector (BID). Gas phase separation was performed via hand injection using a SPL injector and a Carboxen 1010 PLOT fused silica capillary GC column (L × I.D. 30 m × 0.32 mm, average thickness 15 μm). Solvents were cut off previously with a SH-Rxi-1ms fused silica capillary GC column (L × I.D. 30 m × 0.32 mm, average thickness 1.0 μm) and were deflected to a second detector (TCD). Helium was used as carrier gas. The following gaseous products/components were assayed via the GC-BID system: H2, O2, N2, CO, CH4, C2H4, C2H6. Quantification of the liquid phase composition of the photochemical cell was performed using a Shimadzu GCMS-QP2020 system equipped with a MS detector. Phase separation was performed via headspace analysis using a SH-Rtx-200 ms fused silica capillary GC column (L × I.D. 30 m × 0.25 mm, average thickness 1 μm). Helium was used as carrier gas. The following liquid products/components were assayed via the GCMS system: methanol, ethanol, propanol, formate/formic acid, acetate/acetic acid, propionate/propionic acid, acetaldehyde and propionaldehyde. The given values are averaged over three experiments with typical uncertainties of ±2–8%.High-performance liquid chromatography
Analytical HPLC was performed using a Knauer Smartline setup with a four-wavelength detector and a dynamic mixing chamber with reversed-phase chromatography column (Nucleodur C4 gravity, 5 μm, 125 × 4 mm, flow rate 1 mL min−1). MiliQ-water with 0.1% trifluoroacetic acid (buffer A) and MeCN with 0.1% trifluoroacetic acid (buffer B) were used as eluents. For the analysis, a linear gradient from 99% to 1% buffer A was driven over 15 min.Particle size measurement
Particle sizes were measured using a Shimadzu SALD-2300 laser diffraction particle size analyzer equipped with a SALD-BC23 Batch cell. Therefore, 1 mL of the irradiated photocatalytic solution was added to the measurement cell containing 12 mL MeCN/H2O (4:1). The obtained particle sizes were calculated using the Fraunhofer approximation.
Electrochemistry
The electrochemical studies were performed using a PalmSens3 or a PalmSens4 potentiostat in a standard three-electrode setup. A glassy carbon electrode was used as working electrode (WE), an Ag wire as pseudo-reference electrode (PRE) and a Pt wire as counter electrode (CE). The working electrode was prepared by successive polishing with 1.0 and 0.3 μm sandpaper and subsequent sonication in MeCN for 10 min. Tetrabutylammonium hexafluorophosphate ([nBu4N]PF6, 0.1 M) was used as electrolyte in all electrochemical measurements either in anhydrous MeCN or a mixture of MeCN/H2O (4:1). Prior to each experiment, the electrochemical cell was degassed with Ar for 10 min and an Ar or CO2 atmosphere was maintained throughout the measurement. All cyclic voltammograms were recorded at a scan rate of 100 mV s−1 and after every experiment all pseudo-referenced potentials were referenced against the ferrocenium/ferrocene couple (Fc+/0).
Luminescence quenching
Luminescence lifetimes were measured on an Edinburgh Instruments Lifespec II spectrometer with a 472 nm pulsed diode laser (pulse width 75.5 ps).Computational studies
All reported calculations were carried out with ORCA program package in its version 5.0.3.49 The meta-hybrid TPSSh functional50 in conjunction with the ZORA-def2-TZVP(-f) basis set51 was used during geometry optimisations and final energy evaluations. Grimme's semiempirical van der Waals corrections with the Becke-Johnson damping (D3BJ) were employed to describe dispersive and non-covalent interactions. Scalar relativistic effects were approximated through the zeroth order regular approximation (ZORA).52–54 The resolution-of-identity (RI) along with chain-of-spheres algorithms (COSX) were used during the SCF cycles to accelerate the computation of two-electron integrals, while utilising the SARC/J basis set for the former approximation as an auxiliary basis.55–60 Solvent effects were incorporated implicitly within the Conductor-like Polarizable Continuum Model (C-PCM).61,62 All structures were fully optimised without symmetry constraints. Normal mode analysis was performed for all reported structures to identify the nature of the ground state and transition state structures. Free energies were reported in kcal mol−1 at 1 atm and 298 K. An electrode potential was applied by adding −eΦ with a value of Φ = 1.12 V to the electronic energy for each added electron. The Gibbs free energy of a proton in acetonitrile was taken to be −260.2 kcal mol−1.Conclusions
In summary, we herein describe CuICoII-{NSNN}m as efficient catalyst for the visible light-driven reduction of CO2 achieving a CO production rate of 1.60 min−1 with a high selectivity of 98% over H2 production. While CuICoII-{NSNN}m is a stable, homogeneous catalyst, the photosystem durability is limited to 24 h due to photosensitiser degradation, but can be reactivated through addition of new [Ru(phen)3](PF6)2. Experiments with the mononuclear analogues of CuICoII-{NSNN}m clearly reveal that the simultaneous presence of both metals has a beneficial influence on the amount of CO produced. While with CuI-{NSNN}m nearly no CO2 reduction was observed, CoII-{NSNN}m produced CO but exhibits a smaller activity than the dinuclear cryptate (0.99 min−1). Computational studies revealed that in case of CoII-{NSNN}m single CoII-sited catalysis is responsible for CO2 reduction and that the higher activity with CuICoII-{NSNN}m is indeed based on synergistic effects between Cu and Co. Furthermore, experimental and computational studies with CuICoII-{NSNN}p as catalyst show that the increased bite length together with the decreased flexibility of the para-substituted cryptand prevent synergistic CO2 reduction, thus leading to a significantly smaller catalytic rate. Moreover, compared to CoIICoII-{NNNN}m, the exchange of one CoII-{NN} site by CuI-{NS} in CuICoII-{NSNN}m is accompanied by a slight decrease in activity due to the smaller binding affinity of the CuI-{NS} site to OH− which is especially important in the critical C–OH cleaving step. The herein presented results thus provide important insight into the design of bimetallic catalysts and highlight the importance of the interplay of the metal distance as well as binding strength to CO2 for an efficient C–O bond cleavage and synergistic catalysis.Data availability
Supplemental figures, tables and relevant data including Cartesian coordinates of all computationally investigated structures have been provided in the ESI.†Author contributions
J. J. planned and performed synthesis and characterisation of the catalysts, photocatalytic and electrochemical experiments, degradation studies and prepared the original manuscript. E. B. B. carried out all theoretical computations and was involved in writing the manuscript. J. W. performed the luminescence quenching experiments. O. S. W. supervised the luminescence quenching experiments and revised the manuscript. Ma. R. aided in interpreting the results and planning of photocatalytic experiments and revised the manuscript. Mi. R. conceived and supervised all theoretical calculations and was involved in the writing of the manuscript. U.-P. A. conceived the whole research, supervised photocatalytic experiments and finalised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) for financial support under Germany's Excellence Strategy – EXC-2033 390677874 – “RESOLV” & AP242/9-1. This work was further supported by the Fraunhofer Internal Programs under Grant no. Attract 097-602175. J. J. gratefully acknowledges financial support from the Deutsche Bundesstiftung Umwelt. E. B. B. and Mi. R. thank the Deutsche Forschungsgemeinschaft for funding through Emmy-Noether project 5688/1. Partial financial support to Ma. R. from the Institut Universitaire de France (IUF) and from ANR (Agence Nationale de la Recherche, ANR-20-SODR-0003-02) is also gratefully acknowledged.References
- H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS PubMed.
- L. Hammarström, Acc. Chem. Res., 2009, 42, 1859–1860 CrossRef PubMed.
- J. Barber, Solar Fuels and Photosynthesis, Royal Society of Chemistry, London, 2012 Search PubMed.
- Z. Wang, H. Song, H. Liu and J. Ye, Angew. Chem., Int. Ed., 2020, 59, 8016–8035 CrossRef CAS PubMed.
- J. Strunk, Heterogeneous Photocatalysis – From Fundamentals to Applications in Energy Conversion and Depollution, Wiley-VCH, Weinheim, Germany, 2021 Search PubMed.
- H. Shen, T. Peppel, J. Strunk and Z. Sun, Sol. RRL, 2020, 4, 1900546 CrossRef CAS.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- M. Dilla, R. Schlögl and J. Strunk, ChemCatChem, 2017, 9, 696–704 CrossRef CAS.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS PubMed.
- F. Möller, S. Piontek, R. G. Miller and U.-P. Apfel, Chem.–Eur. J., 2018, 24, 1471–1493 CrossRef PubMed.
- J. Bonin, A. Maurin and M. Robert, Coord. Chem. Rev., 2017, 334, 184–198 CrossRef CAS.
- K. J. Lee, N. Elgrishi, B. Kandemir and J. L. Dempsey, Nat. Rev. Chem, 2017, 1, 0039 CrossRef CAS.
- S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283–295 CrossRef CAS.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- E. Boutin, L. Merakeb, B. Ma, B. Boudy, M. Wang, J. Bonin, E. Anxolabéhère-Mallart and M. Robert, Chem. Soc. Rev., 2020, 49, 5772–5809 RSC.
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2017, 7, 70–88 CrossRef CAS.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- H. Dobbek, Science, 2001, 293, 1281–1285 CrossRef CAS PubMed.
- H. Dobbek, L. Gremer, R. Kiefersauer, R. Huber and O. Meyer, Proc. Natl. Acad. Sci. U.S.A., 2002, 99, 15971–15976 CrossRef CAS PubMed.
- J.-H. Jeoung and H. Dobbek, Science, 2007, 318, 1461–1464 CrossRef CAS PubMed.
- M. Beley, J.-P. Collin, R. Ruppert and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1984, 1315 RSC.
- M. Hammouche, D. Lexa, M. Momenteau and J. M. Saveant, J. Am. Chem. Soc., 1991, 113, 8455–8466 CrossRef CAS.
- Z. Guo, F. Yu, Y. Yang, C.-F. Leung, S.-M. Ng, C.-C. Ko, C. Cometto, T.-C. Lau and M. Robert, ChemSusChem, 2017, 10, 4009–4013 CrossRef CAS PubMed.
- H. Rao, C.-H. Lim, J. Bonin, G. M. Miyake and M. Robert, J. Am. Chem. Soc., 2018, 140, 17830–17834 CrossRef CAS PubMed.
- C.-Y. Zhu, Y.-Q. Zhang, R.-Z. Liao, W. Xia, J.-C. Hu, J. Wu, H. Liu and F. Wang, Dalton Trans., 2018, 47, 13142–13150 RSC.
- K. Mochizuki, S. Manaka, I. Takeda and T. Kondo, Inorg. Chem., 1996, 35, 5132–5136 CrossRef CAS.
- Z. Guo, C. Cometto, L. Chen, B. Ma, H. Fan, T. Groizard, W.-L. Man, S.-M. Yiu, K.-C. Lau, M. Robert and T.-C. Lau, Nat. Catal., 2019, 2, 801–808 CrossRef CAS.
- J. Caballero-Jiménez, F. Habib, D. Ramírez-Rosales, R. Grande-Aztatzi, G. Merino, I. Korobkov, M. K. Singh, G. Rajaraman, Y. Reyes-Ortega and M. Murugesu, Dalton Trans., 2015, 44, 8649–8659 RSC.
- F. Möller, K. Merz, C. Herrmann and U.-P. Apfel, Dalton Trans., 2016, 45, 904–907 RSC.
- J.-M. Chen, W. Wei, X.-L. Feng and T.-B. Lu, Chem.–Asian J., 2007, 2, 710–719 CrossRef PubMed.
- M. M. El-Hendawy, N. J. English and D. A. Mooney, Inorg. Chem., 2012, 51, 5282–5288 CrossRef CAS PubMed.
- T. Ouyang, H.-H. Huang, J.-W. Wang, D.-C. Zhong and T.-B. Lu, Angew. Chem., Int. Ed., 2017, 56, 738–743 CrossRef CAS PubMed.
- T. Ouyang, H.-J. Wang, H.-H. Huang, J.-W. Wang, S. Guo, W.-J. Liu, D.-C. Zhong and T.-B. Lu, Angew. Chem., Int. Ed., 2018, 57, 16480–16485 CrossRef CAS PubMed.
- J. Jökel, F. Nyßen, D. Siegmund and U.-P. Apfel, Dalton Trans., 2021, 50, 14602–14610 RSC.
- F. Möller, K. Merz, C. Herrmann and U.-P. Apfel, Dalton Trans., 2016, 45, 904–907 RSC.
- F. Möller, L. Castañeda-Losada, J. R. C. Junqueira, R. G. Miller, M. L. Reback, B. Mallick, M. van Gastel and U.-P. Apfel, Dalton Trans., 2017, 46, 5680–5688 RSC.
- L. K. Weavers, I. Hua and M. R. Hoffmann, Water Environ. Res., 1997, 69, 1112–1119 CrossRef CAS.
- J.-M. Lehn and R. Ziessel, Proc. Natl. Acad. Sci. U.S.A., 1982, 79, 701–704 CrossRef CAS PubMed.
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Proc. Natl. Acad. Sci. U.S.A., 2012, 109, 15673–15678 CrossRef CAS PubMed.
- P. Gerschel, K. Warm, E. R. Farquhar, U. Englert, M. L. Reback, D. Siegmund, K. Ray and U.-P. Apfel, Dalton Trans., 2019, 48, 5923–5932 RSC.
- L. Iffland, D. Siegmund and U.-P. Apfel, Z. Anorg. Allg. Chem., 2020, 646, 1–9 CrossRef.
- M. Obermeier, F. Beckmann, R. S. Schaer, O. S. Wenger and M. Schwalbe, Front. Chem., 2021, 9, 751716 CrossRef CAS PubMed.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- R. S. Drago, Physical Methods for Chemists, Saunders College Pub., Ft. Worth, 1992 Search PubMed.
- G. L. Miessler, P. J. Fischer and D. A. Tarr, Inorganic Chemistry, Pearson, Boston, 2014, Fifth edition Search PubMed.
- L. Banci, A. Bencini, C. Benelli, D. Gatteschi and C. Zanchini, in Structures versus Special Properties, Springer Berlin Heidelberg, Berlin, Heidelberg, 1982, vol. 52, pp. 37–86 Search PubMed.
- P. G. Alsabeh, A. Rosas-Hernández, E. Barsch, H. Junge, R. Ludwig and M. Beller, Catal. Sci. Technol., 2016, 6, 3623–3630 RSC.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS.
- C. van Wüllen, J. Chem. Phys., 1998, 109, 392–399 CrossRef.
- B. I. Dunlap, J. W. D. Connolly and J. R. Sabin, J. Chem. Phys., 1979, 71, 3396–3402 CrossRef CAS.
- O. Vahtras, J. Almlöf and M. W. Feyereisen, Chem. Phys. Lett., 1993, 213, 514–518 CrossRef CAS.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- R. Izsák and F. Neese, J. Chem. Phys., 2011, 135, 144105 CrossRef PubMed.
- R. Izsák, F. Neese and W. Klopper, J. Chem. Phys., 2013, 139, 094111 CrossRef PubMed.
- D. A. Pantazis, X.-Y. Chen, C. R. Landis and F. Neese, J. Chem. Theory Comput., 2008, 4, 908–919 CrossRef CAS PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Garcia-Ratés and F. Neese, J. Comput. Chem., 2020, 41, 922–939 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02679e |
| This journal is © The Royal Society of Chemistry 2023 |