
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThieno[3,4-d]pyrimidin-4(3H)-thione: an effective, oxygenation independent, heavy-atom-free photosensitizer for cancer cells†
Luis A.
Ortiz-Rodríguez
 a,
Ye-Guang
Fang
b,
Germain
Niogret
c,
Kaivin
Hadidi
c,
Sean J.
Hoehn
a,
Heather J.
Folkwein
a,
Steffen
Jockusch
d,
Yitzhak
Tor
*c,
Ganglong
Cui
*b,
Liraz
Levi
*ef and
Carlos E.
Crespo-Hernández
*a
a,
Ye-Guang
Fang
b,
Germain
Niogret
c,
Kaivin
Hadidi
c,
Sean J.
Hoehn
a,
Heather J.
Folkwein
a,
Steffen
Jockusch
d,
Yitzhak
Tor
*c,
Ganglong
Cui
*b,
Liraz
Levi
*ef and
Carlos E.
Crespo-Hernández
*a
aDepartment of Chemistry, Case Western Reserve University, Cleveland, OH 44106, USA. E-mail: cxc302@case.edu
bKey Lab of Theoretical and Computational Photochemistry, Ministry of Education, Chemistry College, Beijing Normal University, Beijing, 100875, China
cDepartment of Chemistry and Biochemistry, University of California San Diego, La Jolla, CA 92093, USA
dCenter for Photochemical Sciences, Bowling Green State University, Bowling Green, Ohio 43403, USA
eCelloram Inc, Cleveland, OH 44106, USA
fDepartment of Pediatrics, Case Western Reserve University School of Medicine, Cleveland, Ohio 44106, USA
First published on 25th July 2023
Abstract
All-organic, heavy-atom-free photosensitizers based on thionation of nucleobases are receiving increased attention because they are easy to make, noncytotoxic, work both in the presence and absence of molecular oxygen, and can be readily incorporated into DNA and RNA. In this contribution, the DNA and RNA fluorescent probe, thieno[3,4-d]pyrimidin-4(1H)-one, has been thionated to develop thieno[3,4-d]pyrimidin-4(3H)-thione, which is nonfluorescent and absorbs near-visible radiation with about 60% higher efficiency. Steady-state absorption and emission spectra are combined with transient absorption spectroscopy and CASPT2 calculations to delineate the electronic relaxation mechanisms of both pyrimidine derivatives in aqueous and acetonitrile solutions. It is demonstrated that thieno[3,4-d]pyrimidin-4(3H)-thione efficiently populates the long-lived and reactive triplet state generating singlet oxygen with a quantum yield of about 80% independent of solvent. It is further shown that thieno[3,4-d]pyrimidin-4(3H)-thione exhibits high photodynamic efficacy against monolayer melanoma cells and cervical cancer cells both under normoxic and hypoxic conditions. Our combined spectroscopic, computational, and in vitro data demonstrate the excellent potential of thieno[3,4-d]pyrimidin-4(1H)-thione as a heavy-atom-free PDT agent and paves the way for further development of photosensitizers based on the thionation of thieno[3,4-d]pyrimidine derivatives. Collectively, the experimental and computational results demonstrate that thieno[3,4-d]pyrimidine-4(3H)-thione stands out as the most promising thiobase photosensitizer developed to this date.
Introduction
Photodynamic therapy (PDT) has grown in popularity, primarily in dermatology, because of its simplicity and effortless light exposure accessibility of the skin.1–3 PDT uses a photoactivable drug (a.k.a., photosensitizer) to generate singlet oxygen and other reactive oxygen species, offering a high degree of spatiotemporal selectivity in tumor destruction, noninvasiveness, and reduced side effects.4,5 The development of highly effective photosensitizers has been, however, a major challenge due to difficulties in predicting a priori their intersystem crossing mechanisms, the yield of triplet state population, and the singlet oxygen generation efficacy. Furthermore, the efficacy of a photosensitizer depends not only on the type of target cells and their oxygenation status, but also its ability to penetrate the targeted diseased tissue selectively, and the wavelength of light activation.6,7Transition metals and other heavy atoms have been introduced into organic chromophores as an effective strategy to yield photosensitizers for PDT.8–11 However, the dark cytotoxicity of the heavy-atom-containing photosensitizers continues to be a major concern. In this context, heavy-atom-free photosensitizers (HAFPs) are emerging as an innovative class of photosensitizers exhibiting great potential for preclinical and clinical studies for the photodynamic treatment of cancers. Furthermore, HAFPs are receiving increased attention not only for PDT,12–27 but also for photovoltaic,28 and photocatalytic29,30 applications, as well as for the development of innovative photonic technologies.31,32 Some of the advantages of HAFPs over traditional photosensitizers include the generation of long-lived excited triplet states in high yield, efficient generation of reactive oxygen species, low dark toxicity, good photostability and biocompatibility, straightforward synthetic modification, and low cost.16,19
Most HAFPs developed for PDT so far, however, do not only exhibit low water solubility and poor tumor selectivity, but also aggregate in aqueous media due to their hydrophobic and rigid structures.19,33,34 More broadly, the efficacy of most photosensitizers in preclinical and clinical use depends strictly on the molecular oxygen concentration available in solid tumors, which varies widely by location due to the aggressive proliferation of cancer cells and insufficient blood supply.35,36 Indeed, the interior of certain tumors can exhibit molecular oxygen concentrations of less than 4%, decreasing to 0% locally, which severely limits the efficacy of PDT against in vivo hypoxic tumors. In addition, because the photophysical process itself consumes molecular oxygen, thereby effecting hypoxia, the treatment itself can increase the tumor's resistance to PDT.36,37 There is, therefore, a clinical need to develop water-soluble and nonaggregating photosensitizer alternatives that offer improved target cell selectivity and the ability to photosensitize damage through more than one mechanism, in addition to reactive oxygen species generation.
In this context, site-selected sulfur-substituted nucleobases (a.k.a., thiobases) have emerged as a promising class of heavy-atom-free organic biomolecules for preclinical and clinical PDT applications.13,14,16,38–42 The unique structural, biochemical, and photochemical properties of thiobases offer an attractive strategy for developing highly effective and highly targeted phototherapeutic compounds,15,43 which can be site-selectively incorporated into DNA and RNA sequences, working both in the absence and in the presence of molecular oxygen.13,15,42,44 A single-atom-substitution from oxygen to sulfur, converts DNA and RNA nucleobases into effective UVA chromophores (ε ≥ 104 M−1 cm−1) that exhibit red-shifted absorption maxima from ca. 320 to 380 nm, and with absorption bands extending all the way to the near visible region. These compounds have also been demonstrated to exhibit near-unity triplet yields and high yields of singlet oxygen,15,43 with one of most attractive applications being for the treatment of skin malignancies that are readily accessible to near-visible irradiation.13,39–41,43
Similarly, thieno[3,4-d]pyrimidine derivatives can be easily incorporated into DNA,45–47 and thionation of their carbonyl group would be expected to redshift their absorption spectra and to increase their triplet state population yields,18,20 thus generating reactive oxygen species including singlet oxygen and increasing the probability of phototoxic reactions. To test this hypothesis, we have thionated the fluorescent thieno[3,4-d]pyrimidin-4(3H)-one (ThiaHX) at the C4 position (equivalent to the C6 position in the parent purine derivatives; see comment regarding the nomenclature used in Scheme 1), to generate thieno[3,4-d]pyrimidine-4(3H)-thione (ThiathioHX).
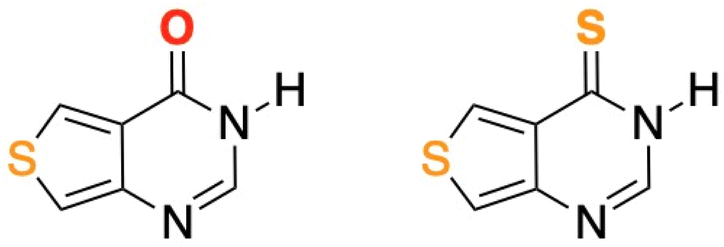 | ||
| Scheme 1 Molecular structures of ThiaHX (left) and ThiathioHX (right). Note, while this heterocyclic system is numbered differently than the native purines, we will use the latter nomenclature for familiarity and clarity throughout this study. Thus, we will consider the carbonyl and thiocarbonyl positions as C6. See ESI† for synthesis, characterization data, and crystal structures. | ||
In this contribution, the photodynamic efficacy of ThiathioHX against monolayer melanoma cells and cervical cancer cells is demonstrated in normoxic and hypoxic conditions. Furthermore, we report a comprehensive characterization of the photophysical, photochemical, and electronic–structure properties of ThiaHX and ThiathioHX both in phosphate buffer solution at pH 7.4 and in acetonitrile studied by steady-state absorption and emission spectroscopies, femtosecond-broadband transient absorption measurements, nanosecond transient absorption, and luminescence measurements. The experimental results are enhanced with complete active space second-order perturbation theory (CASPT2) calculations. Detailed excited state deactivation mechanisms are presented for both molecules. Collectively, the experimental and computational results reported herein reveal the excellent potential of ThiathioHX as a heavy-atom-free PDT agent, paving the way for further development of photosensitizers based on the thionation of thieno[3,4-d]pyrimidine derivatives.
Results
Steady-state absorption and emission
Fig. 1 shows the absorption spectra of ThiaHX and ThiathioHX (Scheme 1) in phosphate buffer pH 7.4 and in MeCN. As observed for the canonical nucleobases,15,16,43 substitution of the oxygen by a sulfur atom in the carbonyl group at the C6 position (Scheme 1) results in a red shift of the lowest-energy absorption band from a maximum at 308 nm in ThiaHX in phosphate buffer to 367 nm in ThiathioHX (58 nm, 0.64 eV, or 5145 cm−1). An absorption tail that extends to ca. 430 nm is also observed in ThiathioHX. Remarkably, the molar absorption coefficient of the lowest-energy band maximum increases by 62% when going from ThiaHX to ThiathioHX (i.e., 4400 ± 200 and 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 ± 300 M−1 cm−1 for ThiaHX and ThiathioHX, respectively). As observed in phosphate buffer, the absorption maximum of the lowest-energy absorption band of ThiaHX, now at 305 nm in MeCN, redshifts to 369 nm in ThiothiaHX and an absorption tail is also observed extending to ca. 450 nm. The absorption tail shifts to the red in going from phosphate buffer to MeCN, suggesting that the lowest-energy singlet state of ThiathioHX has nπ* character and is close in energy with the lowest-energy ππ* state. Similar than in phosphate buffer, the absorption coefficient of the lowest-energy band maximum in MeCN increases significantly (i.e., 62%) when going from ThiaHX to ThiathioHX (i.e., 4400 ± 200 and 11600 ± 400 M−1 cm−1) ThiaHX and ThiathioHX, respectively. Therefore, thionation of ThiaHX simultaneously redshifts its lowest-energy absorption maxima by ca. 60 nm (5145 cm−1) and increases the absorption coefficient by 62% independent of the solvent, making ThiathioHX a significantly better absorber of near-visible radiation. Using a typical absorption cross section cutoff of 1000 M−1 cm−1,13 ThiathioHX is expected to allow 18% deeper tissue penetration (up to ca. 100 μm) than 2,6-dithiopurine, one of the most promising thiobase PDT agents reported to date.42,48
500 ± 300 M−1 cm−1 for ThiaHX and ThiathioHX, respectively). As observed in phosphate buffer, the absorption maximum of the lowest-energy absorption band of ThiaHX, now at 305 nm in MeCN, redshifts to 369 nm in ThiothiaHX and an absorption tail is also observed extending to ca. 450 nm. The absorption tail shifts to the red in going from phosphate buffer to MeCN, suggesting that the lowest-energy singlet state of ThiathioHX has nπ* character and is close in energy with the lowest-energy ππ* state. Similar than in phosphate buffer, the absorption coefficient of the lowest-energy band maximum in MeCN increases significantly (i.e., 62%) when going from ThiaHX to ThiathioHX (i.e., 4400 ± 200 and 11600 ± 400 M−1 cm−1) ThiaHX and ThiathioHX, respectively. Therefore, thionation of ThiaHX simultaneously redshifts its lowest-energy absorption maxima by ca. 60 nm (5145 cm−1) and increases the absorption coefficient by 62% independent of the solvent, making ThiathioHX a significantly better absorber of near-visible radiation. Using a typical absorption cross section cutoff of 1000 M−1 cm−1,13 ThiathioHX is expected to allow 18% deeper tissue penetration (up to ca. 100 μm) than 2,6-dithiopurine, one of the most promising thiobase PDT agents reported to date.42,48
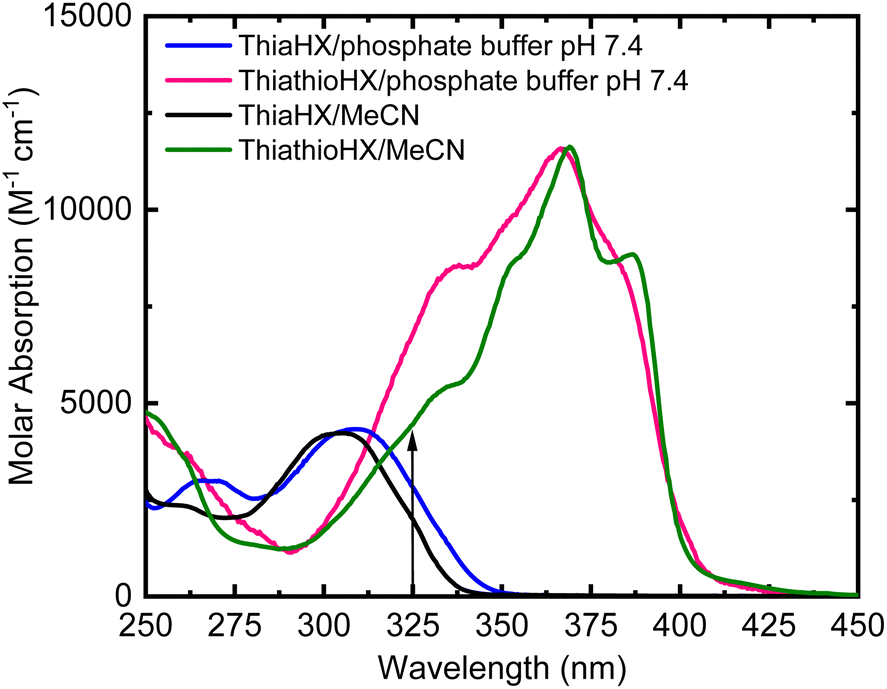 | ||
| Fig. 1 Absorption spectra of ThiaHX and ThiothiaHX in phosphate buffer (pH 7.4) and MeCN. The vertical arrow shows the excitation wavelength used in the femtosecond transient absorption experiments of 325 nm. | ||
The emission spectra of ThiaHX in phosphate buffer (pH 7.4) and in MeCN are presented in Fig. S1.† ThiaHX exhibits a relatively intense emission band with maximum at 386 nm in phosphate buffer and at 371 nm in MeCN. A fluorescence quantum yield of 0.31 ± 0.06 is measured in phosphate buffer, which is an order of magnitude higher than the one obtained in MeCN (0.028 ± 0.008). The blueshift of the emission band in going from phosphate buffer to MeCN is characteristic of an electronic transition with ππ* character. In contrast, ThiathioHX does not exhibit any detectable emission in either solvent, demonstrating that most of the excited state population in ThiathioHX decays nonradiatively to the ground state or to a non-emissive excited state.
Theoretical results
To provide detailed and crucial electronic and nuclear information regarding the deactivation mechanisms of ThiaHX and ThiathioHX, quantum chemical calculations were performed. Based on the molecular structures of both thieno[3,4-d]pyrimidine derivatives, two possible tautomers, N1(H) and N3(H), may be present in solution (Fig. S2 and S3†). To assess their relative stability, single-point calculation (CASPT2(12,10)/aug-cc-pVTZ/PCM) were performed on ground-state optimized structures (CASPT2(12,10)/cc-pVDZ), where explicit solvent interactions were modelled by adding an explicit MeCN molecule or two explicit water molecules. According to these calculations, the N1(H) tautomer is >4 and >5 kcal mol−1 more stable than the N3(H) tautomer in ThiaHX and ThiathioHX, independently of the solvent. Therefore, we conclude that only the N1(H) tautomer of both ThiaHX and ThiathioHX is available in solution. Hence, hereinafter, all the calculations presented were performed only for the N1(H) tautomer of both molecules.CASPT2 calculations were done to optimize the relevant ground- and excited-state structures, as shown in Fig. 2, S4, and S5.† Structurally, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond is significantly longer than its CO counterpart in the S0 state, e.g., 1.7 vs. 1.2 Å. Based on the optimized S0 structures, the excited-state properties of ThiaHX and ThiathioHX at the Franck–Condon region have been explored. The first excited singlet state of ThiaHX has ππ* character, as judged by the relevant molecular orbitals and oscillator strength (Fig. S6 and Table S1†). In contrast, the S1 state of ThiathioHX is a spectroscopically dark state with nπ* character (oscillator strength of ca. 10−5), as shown in Fig. S7 and Table S2.† The S2 state of ThiathioHX is an optically allowed ππ* state and its excitation energy is predicted to be 3.10 eV (Fig. S7 and Table S2†). The predicted value is also close to the experimental values of 3.38 and 3.36 eV in phosphate buffer and in MeCN, respectively.
S bond is significantly longer than its CO counterpart in the S0 state, e.g., 1.7 vs. 1.2 Å. Based on the optimized S0 structures, the excited-state properties of ThiaHX and ThiathioHX at the Franck–Condon region have been explored. The first excited singlet state of ThiaHX has ππ* character, as judged by the relevant molecular orbitals and oscillator strength (Fig. S6 and Table S1†). In contrast, the S1 state of ThiathioHX is a spectroscopically dark state with nπ* character (oscillator strength of ca. 10−5), as shown in Fig. S7 and Table S2.† The S2 state of ThiathioHX is an optically allowed ππ* state and its excitation energy is predicted to be 3.10 eV (Fig. S7 and Table S2†). The predicted value is also close to the experimental values of 3.38 and 3.36 eV in phosphate buffer and in MeCN, respectively.
 | ||
| Fig. 2 Ground-state optimized structures of ThiathioHX in water (top, left) and MeCN (top, right) and ThiaHX in water (bottom, left) and in MeCN (bottom, right) obtained at the CASPT2/cc-pVDZ level of theory. | ||
The emission properties were also explored at the CASPT2 level by optimizing the S1 structures. For ThiaHX, the S1 → S0 vertical emission is estimated to be 417 nm, with a reasonably large oscillator strength of 0.12. This theoretical estimation also agrees well with the experimental measurements of emission at 386 and 391 nm in phosphate buffer and in MeCN, respectively. For ThiathioHX, on the other hand, the S1 state has nπ* character and the S1 → S0 vertical emission is significantly weaker due to a negligible oscillator strength of ca. 10−5. Furthermore, the adiabatic excitation energy of the 1ππ* state (i.e., the E00) in water is estimated to be 28470 cm−1 (3.53 eV) for ThiaHX and 20006 cm−1 (2.48 eV) for ThiathioHX, while in MeCN the E00 is estimated to be 28750 cm−1 (3.56 eV) for ThiaHX and 19900 cm−1 (2.47 eV) for ThiathioHX. The predicted values are in good agreement with those obtained experimentally for ThiaHX, 29200 cm−1 (3.61 eV) and 30000 cm−1 (3.72 eV) in phosphate buffer and in MeCN, respectively (Fig. S1†).
Fig. 3 shows the CASPT2(12,10)/aug-cc-pVTZ/PCM//CASPT2(12,10)/cc-pVDZ calculated order of the four lower energy electronic states for ThiaHX and the six lower energy electronic states for ThiathioHX. These electronic states assist in understanding the excited-state relaxation pathways of ThiaHX and ThiathioHX in aqueous solution and MeCN (see Section S2.5 in the ESI†). All relevant minima structures and linear interpolation of internal coordinates (LIIC) corresponding of the electronic states of ThiaHX are reported in Fig. S4, S8, and S9,† while those of ThiathioHX are reported in Fig. S5, S10, and S11,† respectively.
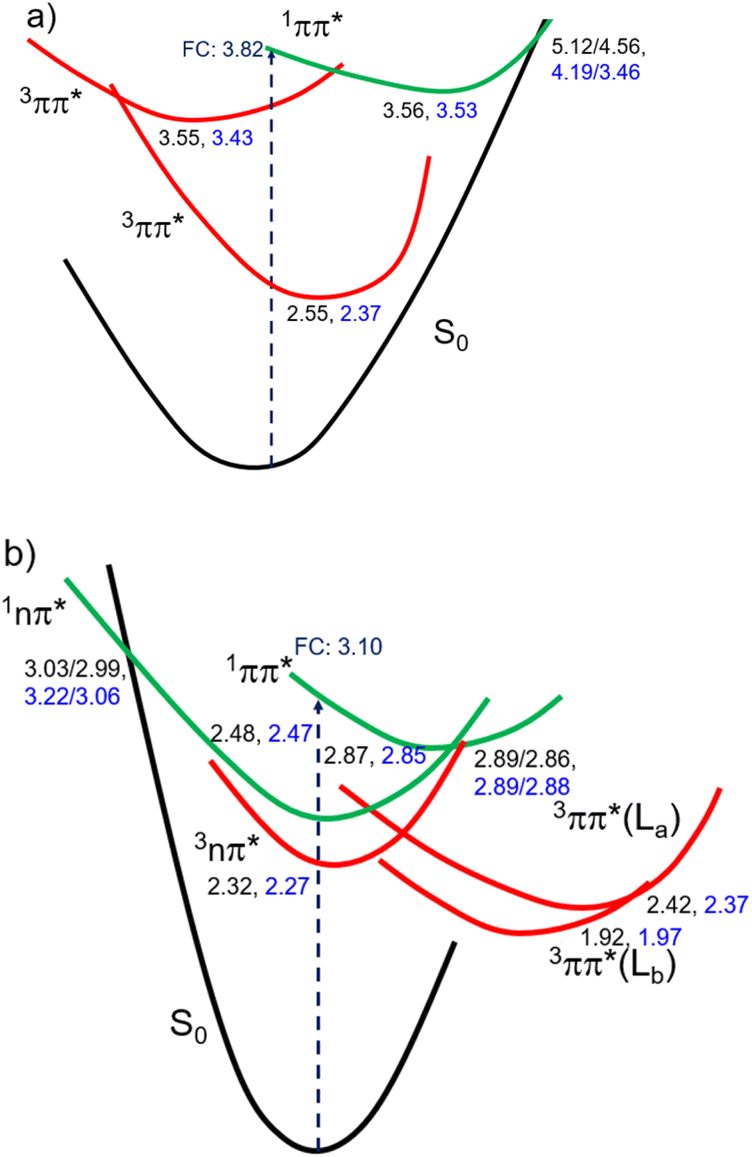 | ||
| Fig. 3 Proposed order of the four lower energy electronic states for ThiaHX (a) and the five lower energy states of ThiathioHX (b) in MeCN (values shown in black) and water (values shown in blue) based on the calculations reported in this work. Values correspond to relative energies (in eV) of key stationary points, conical intersections, and crossing points. | ||
Finally, to assist in the assignment of the transient absorption species (see below), the excited state absorption spectra of the 1ππ* and 3ππ*(T1) minima of ThiaHX and the 1ππ*, 1nπ*, 3ππ*-La, 3nπ* and 3ππ*-Lb minima of ThiathioHX were calculated at the CASPT2(12,10)/aug-cc-pVTZ level of theory in MeCN (Fig. S12†) using the structures optimized at the CASPT2/cc-pVDZ level of theory.
Femtosecond transient absorption
Femtosecond transient absorption spectroscopy (fs-TAS) was used to study the excited-state dynamics of ThiaHX and ThiathioHX. The setup is described in detail in previous publications.49–51 The transient absorption spectra for ThiaHX and ThiathioHX in phosphate buffer pH 7.4 and in MeCN upon excitation at 325 nm (3.81 eV) are reported in Fig. S13 and S14,† respectively, while a description of the observed results is included in the supporting results section in the ESI.†Table 1 collects the lifetimes extracted from the global and target analysis using a two-component sequential kinetic model, in which the second lifetime models the slow decay of the transient data in a time scale longer than 3 ns. Representative decay traces, best fits, and evolution-associated difference spectra (EADS) in both solvents are shown in Fig. 4 and 5 for ThiaHX and ThiathioHX, respectively.| Lifetime | Phosphate buffer | MeCN |
|---|---|---|
| ThiaHX (τ1) | 0.95 ± 0.04 ps | 375 ± 20 ps |
| ThiaHX (τ2) | >3 ns | >3 ns |
| ThiathioHX (τ1) | 1.2 ± 0.5 ps | 9 ± 1 ps |
| ThiathioHX (τ2) | >3 ns | >3 ns |
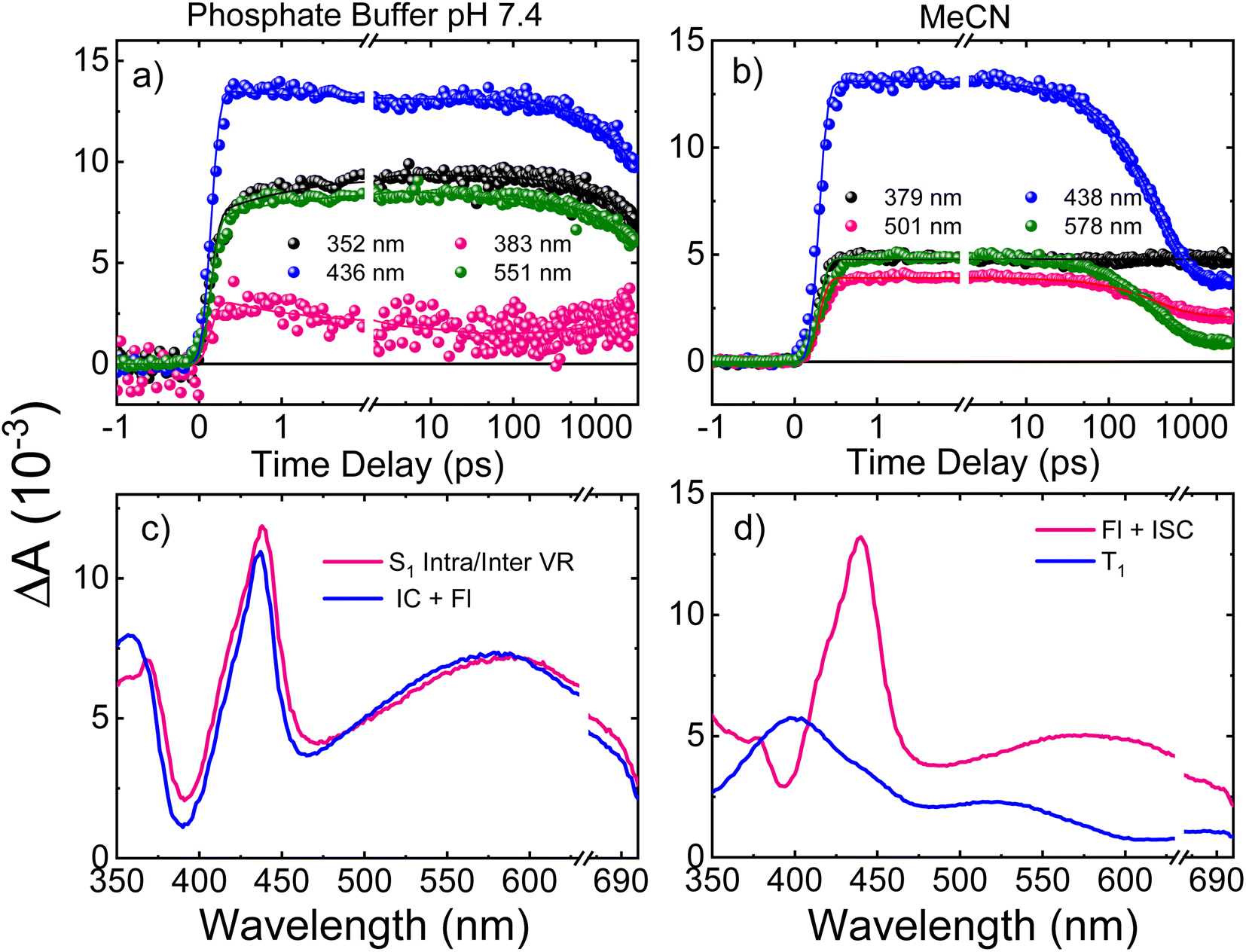 | ||
| Fig. 4 Representative decay traces of ThiaHX in (a) phosphate buffer and in (b) MeCN upon excitation with 325 nm. Evolution-associated difference spectra (EADS) of ThiaHX in (c) phosphate buffer and in (d) MeCN, extracted from the global and target analyses. VR, Fl, IC and ISC stand for vibrational redistribution, fluorescence, internal conversion, and intersystem crossing, respectively. The breaks are covering the overtone of the pump beam at 650 nm. | ||
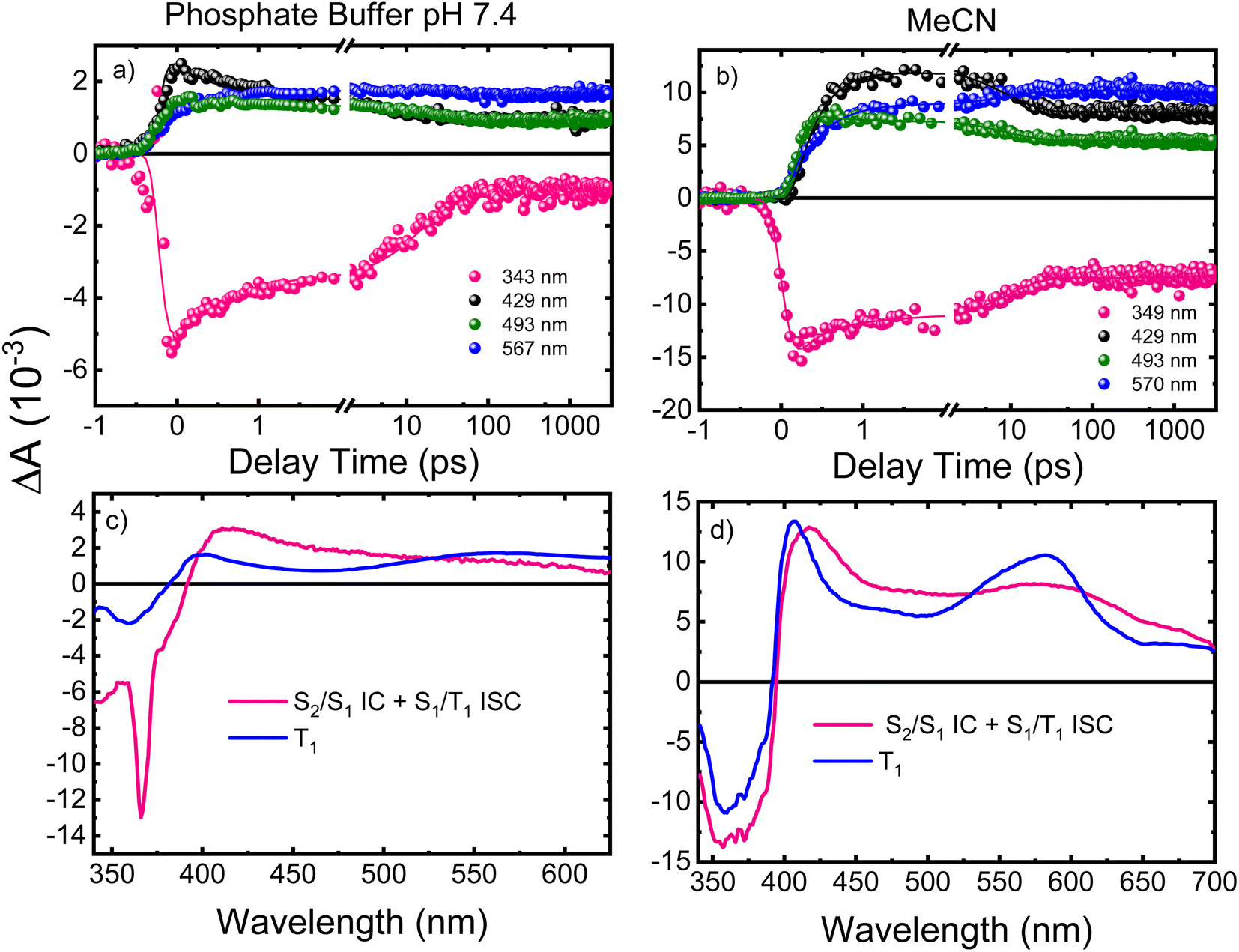 | ||
| Fig. 5 Representative decay traces of ThiathioHX (a) in phosphate buffer and (b) in MeCN upon excitation with 325 nm. EADS extracted from the global and target analyses (c) in phosphate buffer and (d) in MeCN. IC and ISC stand for internal conversion and intersystem crossing, respectively. The negative spike at 367 nm corresponds to stimulated Raman emission from the solvent. | ||
Nanosecond laser flash photolysis and singlet oxygen formation yields
To decipher the photophysical processes of ThiaHX and ThiathioHX that occur at timescales longer than 3 ns, we employed nanosecond laser flash photolysis. As shown in Fig. S15† and Table 2, both ThiaHX and ThiathioHX exhibit population of a long-lived triplet state. The determination of the bimolecular quenching rate constants (kqO2) for the quenching of ThiaHX and ThiathioHX triplet states by molecular oxygen (Fig. S16† and Table 2) yields a kqO2 of 4.4 (±0.1) ×109 and 2.3 (±0.1) ×109 M−1 s−1 for ThiaHX and ThiathioHX in MeCN, respectively.| ThiaHX | ThiathioHX | |||
|---|---|---|---|---|
| Aqueous buffer | MeCN | Aqueous buffer | MeCN | |
| a D2O/Tris, pH 7.5. b Phosphate buffer pH 7.4. | ||||
| ϕ Δ (O2-saturated) | 0.35 ± 0.02a | 0.50 ± 0.02 | 0.85 ± 0.02a | 0.82 ± 0.02 |
| ϕ Δ (air-saturated) | 0.34 ± 0.02a | 0.50 ± 0.02 | 0.75 ± 0.02a | 0.77 ± 0.02 |
| τ T (μs) (deoxygenated) | 10 μsb | 15 μs | 7.3 μsb | 4.0 μs |
| k qO2 (109 M−1 s−1) | — | 4.4 ± 0.1 | — | 2.3 ± 0.1 |
In addition, nanosecond near-infrared emission spectroscopy was used to quantify the singlet oxygen yields of ThiaHX and ThiathioHX. Fig. S17† shows representative singlet oxygen emission decay traces for ThiaHX, ThiathioHX and the phenalenone standard, while Table 2 collects the determined quantum yields. Singlet oxygen quantum yields of 0.35 and 0.50 were determined for ThiaHX under O2-saturated conditions in aqueous buffer and in MeCN, respectively. Under air-saturated conditions, singlet oxygen yields of 0.34 and 0.50 were determined. In the case of ThiathioHX, singlet oxygen quantum yields of 0.85 and 0.82 were determined under O2-saturated conditions in aqueous buffer and in MeCN, respectively. Under air-saturated conditions, singlet oxygen quantum yields of 0.75 and 0.77 were determined in aqueous buffer and in MeCN, respectively.
Phototoxicity and reactive oxygen species generation of ThiathioHX in monolayer cancer cells
The phototoxicity of ThiathioHX under normoxic and hypoxic conditions was evaluated by proliferation assays using PrestoBlue. Accordingly, proliferation of the mouse melanoma cells B16F10 and human cervical adenocarcinoma line HeLa cells were assessed by measuring cell viability following photodynamic treatment with a low UVA irradiation dose of 5 J cm−2 under both oxygenation conditions. Treatment with ThiathioHX but omitting the irradiation dose and treatment with the irradiation dose but without ThiathioHX were used to evaluate dark cytotoxicity and UVA damage, respectively. Inhibition of cell proliferation was determined relative to untreated cells (i.e., no ThiathioHX and no UVA dose).Fig. 6a and b show the decrease in B16F10 and HeLa cell proliferation, respectively, after treatment with ThiathioHX in concentrations ranging from 4 to 166 μM with and without UVA irradiation. Dark cytotoxicity is not observed in the concentration range used in this study in both cell lines, regardless of the oxygenation status. Similarly, the low UVA dose of 5 J cm−2 is not cytotoxic regardless of the oxygenation status. Importantly, when ThiathioHX is combined with a low UVA dose of 5 J cm−2, cell proliferation in both cell lines is significantly inhibited. Half-inhibitory concentrations (IC50) of 22 and 4.7 μM were obtained for the B16F10 cells under hypoxia and normoxia, respectively, while IC50 of 25 and 6.5 μM were obtained for HeLa cells under hypoxia and normoxia, respectively. Collectively, the non-dark cytotoxicity and synergistic toxicity exhibited by ThiathioHX when combined with the low UVA dose confirm that ThiathioHX acts as a powerful phototherapeutic agent and not as a chemotherapeutic agent.
 | ||
| Fig. 6 Cell viability of (a) B16F10 and (b) HeLa cells following treatment with ThiathioHX in combination with a low UVA irradiation dose of 5 J cm−2 and without the irradiation dose. * denotes p < 0.05. | ||
To explore the cellular mechanisms by which ThiathioHX in combination with low UVA affect cancer cell viability, markers of early- and late-apoptosis, and necrosis markers were used. B16F10 and HeLa cells were treated for 1.5 hours and subsequently stained with nuclei marker, 4′,6-diamidino-2-phenylindole (DAPI), the early-apoptosis marker, Annexin V, and the necrosis or late-apoptosis marker, propidium iodide (PI). As shown in Fig. 7, and consistent with the viability results reported in Fig. 6, non-treated cells and cells that were only treated with ThiathioHX but without UVA, or with UVA but without ThiathioHX, exhibit no apoptosis or necrosis. However, when ThiathioHX (125 μM) and the low UVA dose are combined, >90% of the cells exhibit green fluorescence (Annexin V), indicating that decrease in cell viability following treatment is due to apoptosis.
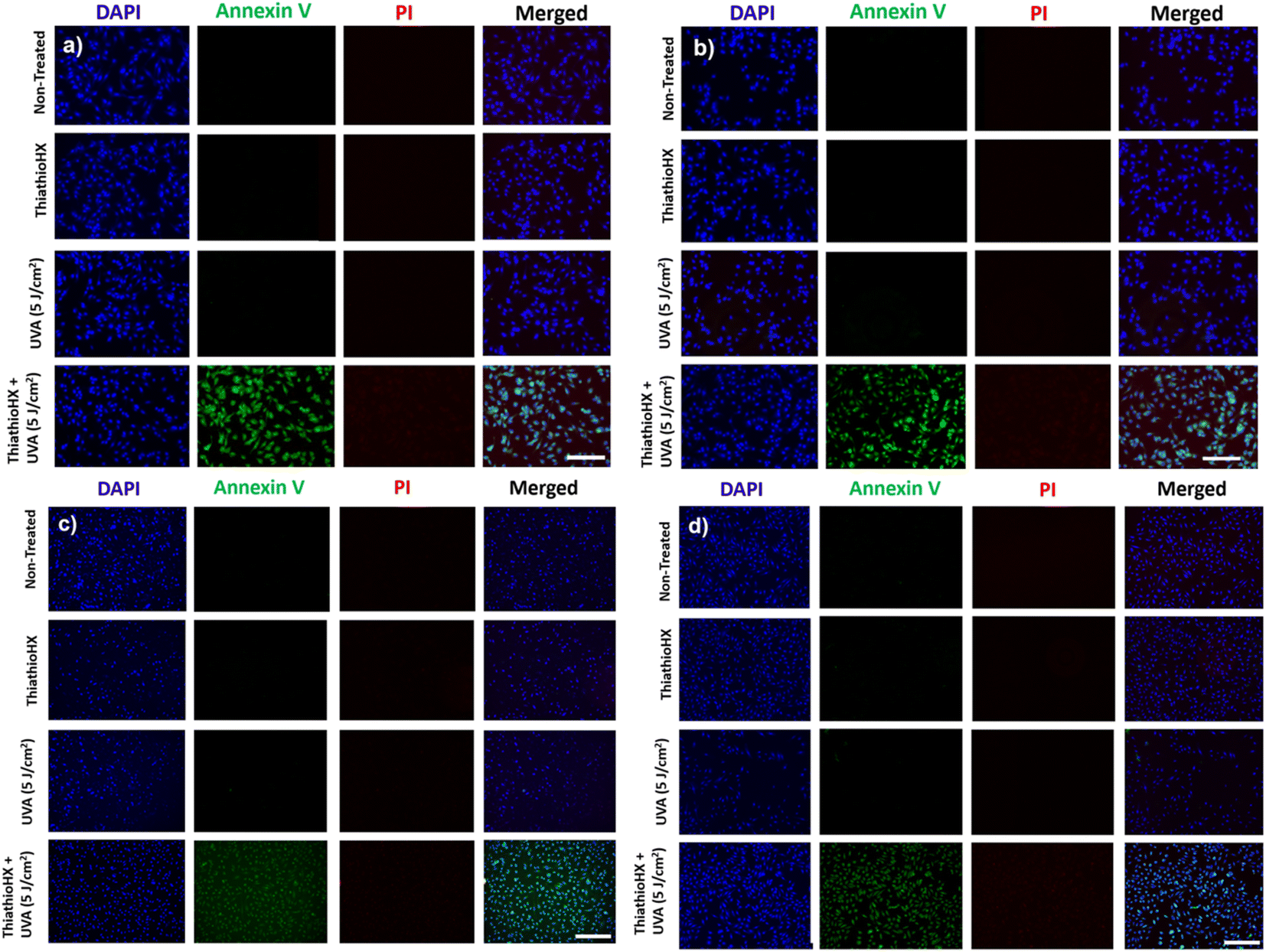 | ||
| Fig. 7 Fluorescence microscopy of B16F10 cells and HeLa cells under hypoxia and normoxia loaded with DAPI (blue), Annexin V (green) and propidium iodide (red). Panels (a) and (b) are the results for B16F10 cells under normoxia and hypoxia, respectively. Panels (c) and (d) are the results for HeLa cells under normoxia and hypoxia, respectively. Concentration of ThiathioHX = 125 μM. Scale bar = 100 μm. | ||
To measure the intracellular reactive oxygen species (ROS) generation in B16F10 and HeLa cells treated with ThiathioHX following UVA photoactivation under normoxia and hypoxia, the fluorescent ROS reporter dye, 2,7-dichlorohydrofluorescein diacetate (DCF-DA) was used (Fig. 8). To establish control conditions, the experiments were also performed without the UVA dose (Fig. S18†). As expected, significantly less ROS are generated under hypoxic conditions (see Fig. 8). Interestingly, small changes in the detection of ROS in the concentration range of 4 to 20 μM in B16F10 and 4 to 40 μM in HeLa cells are observed under normoxia and hypoxia.
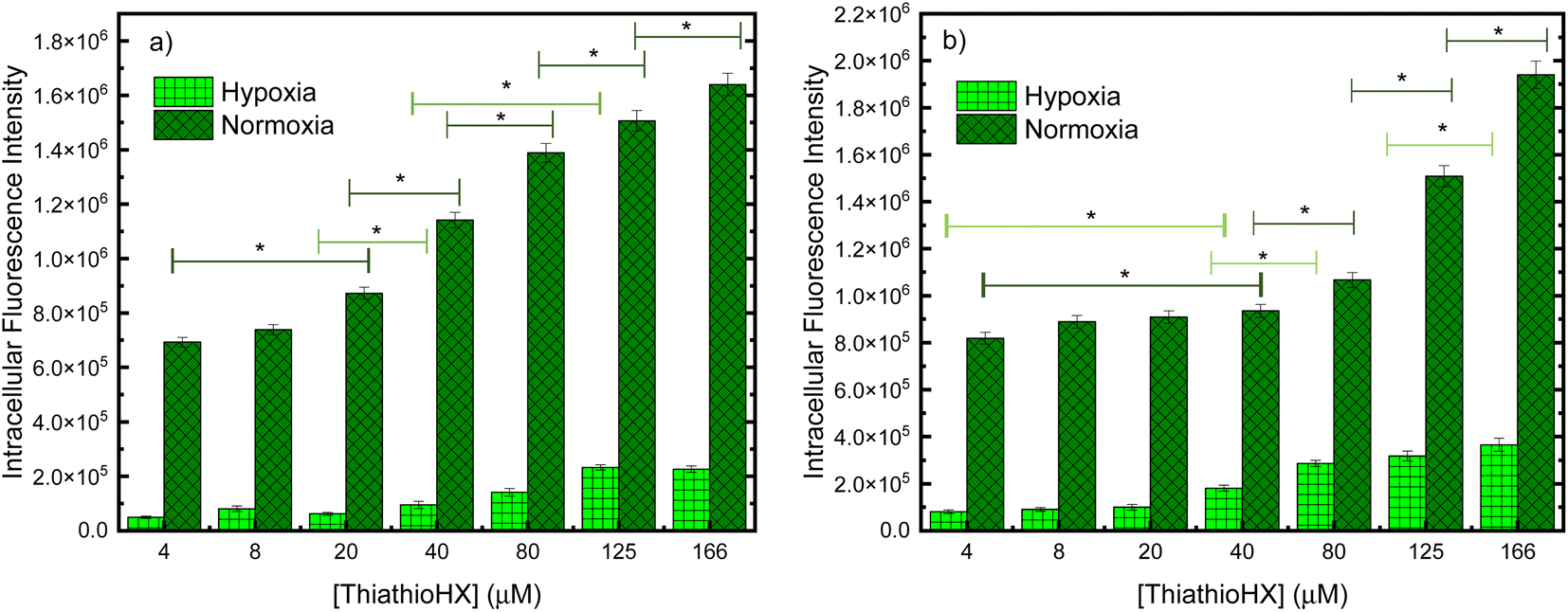 | ||
| Fig. 8 Intracellular fluorescent intensities of ROS sensor DCF-DA in (a) B16F10 cells and (b) HeLa cells. The increase in intracellular ROS generation in cells treated with UVA and ThiathioHX was calculated relative to the control where the cells were treated with 5 J cm−2 of UVA but with no ThiathioHX. * denotes p < 0.05. | ||
Discussion
Electronic relaxation mechanism of ThiathioHX upon UVA photoactivation
Understanding the electronic relaxation mechanisms of prospective PDT agents is essential for comprehending their photochemistry and for establishing design principles for further optimization of PDT properties. The experimental and theoretical results reported in this study allowed us to elucidate the electronic deactivation mechanisms for both thieno[3,4-d]pyrimidine derivatives. A discussion of the electronic relaxation mechanism of ThiathioHX upon UVA photoactivation is reported next, while an analogous discussion of the relaxation mechanism for ThiaHX is presented in the ESI.†Excitation of ThiathioHX at 325 nm populates a transient species that absorbs in the range of 400 to 700 nm in both solvents used (Fig. S14†). Interestingly, prior to time zero, this transient species does not exhibit a clear maximum at wavelengths around 400 to 415 nm. However, right after time zero, well-defined absorption bands appear with maxima at this range form (Fig. S14a and e†). According to the calculated vertical excitation energies for ThiathioHX, excitation at 325 nm populates the 1ππ* state in the Franck–Condon (FC) region, which can relax to its minimum. Near the 1ππ* minimum, there is also an energetically accessible 1ππ*/1nπ* conical intersection that is isoenergetic with the 1ππ* minimum. As shown in Fig. S12b,† the excited state absorption spectrum of the 1ππ* state consists of one band in the spectral range of 400 to 700 nm, while the excited state absorption spectrum of the 1nπ* consists of two bands in the same spectral range. Because only one band is detected prior to time zero and because the 1ππ*/1nπ* conical intersection can be accessed in a barrierless fashion, we propose that the 1ππ*/1nπ* internal conversion occurs at timescales faster than our resolution. We therefore assigned the transient species prior to time zero to excited state absorption of the 1nπ* state.
The calculations also predict that the population reaching the 1nπ* state could intersystem cross to either populate the 3nπ* or the 3ππ* state. We expect the 1nπ* → 3nπ* intersystem crossing to play a minor role according to El-Sayed rules,52,53 while the 1nπ* → 3ππ*-La pathway should play a major role in the intersystem crossing to the triplet manifold in ThiathioHX. Once in the 3ππ*-La potential energy surface, the system is expected to evolve into the energetically lower 3ππ*-Lb state because it can be accessed in a barrierless fashion at the CASPT2(12,10)/aug-cc-pVTZ level of theory (see ESI†). The calculated excited state absorption spectra of both 3ππ*-La and 3ππ*-Lb minima (Fig. S12†) are in good agreement with the transient species observed in the experimental transient absorption data observed after 170 and 210 fs time delays in phosphate buffer and MeCN, respectively.
The transient absorption signal observed at ∼3 ns seems to contain higher contribution of the 3ππ*-Lb state (Fig. S12d†). We speculate, however, that the inter-conversion between both the 3ππ*-La and 3ππ*-Lb minima is likely to occur due to the small energy difference and small associated barriers between these states. The 3ππ* state decays with a lifetime of 7.3 and 4.0 μs in aqueous buffer and MeCN, respectively. Collectively, we propose that the first lifetime (Table 1) corresponds to a combination of the population of the 1nπ* state, following internal conversion from the 1ππ* through the 1ππ*/1nπ* conical intersection, and intersystem crossing to the triplet manifold. Following the population of the 3ππ* state, singlet oxygen generation is sensitized with a quantum yield of ∼80%. The proposed deactivation mechanism is reported in Scheme 2, which takes into consideration the experimental and computational results. Finally, no significant degradation (9%, 210 J cm−2) was observed in both solvents.
 | ||
| Scheme 2 Proposed excited-state relaxation mechanisms for ThiathioHX based on the calculations and experiments reported in this work. | ||
We note that the lifetime associated with the intersystem crossing event is longer than the lifetimes measured for this process in the thiobases and other thionated derivatives, which occur in a sub-picosecond timescale.13,18,44,48,54–59 We proposed that the reason for the slower intersystem crossing is largely due to the weaker spin–orbit coupling (SOC = 83 cm−1, relative to thiobases and other thionated compounds) calculated between the states involved in the intersystem crossing pathways and the energy barrier that must be overcome to access the crossing region. Typically, the SOCs for thiobases and other thionated derivatives are >100 cm−1.18,48,57,59
Photodynamic efficacy of ThiathioHX against monolayer cancer cells
Conventional PDT requires the employment of a photosensitizer that upon photoactivation, populates the reactive triplet state and generates ROS such as singlet oxygen, hydroxyl radical, hydrogen peroxide and super oxide anion, through Type I and/or Type II photosensitization mechanisms.60 These ROS can damage intracellular lipids, proteins and nucleic acids to ultimately cause cell death. UVA photoactivation of ThiathioHX results in efficient population of the long-lived, reactive triplet state and the generation of singlet oxygen in high yield, thus making ThiathioHX a promising photosensitizer for PDT applications. As was demonstrated in the results section, ThiathioHX is highly effective against both tested cell lines under normoxic and hypoxic conditions. Although PDT has been undergoing clinical applications targeted to a wide range of tumors, the high molecular oxygen dependency of the clinical PDT agents limits its development due to the hypoxic nature of solid tumors due to the insufficient blood supply.35,61,62 Hence, the good performance of ThiathioHX under hypoxia further strengthens its candidature as promising PDT agent.As shown in Fig. 8, only small variations in ROS generation in the concentration range of 4 to 20 μM in B16F10, and 4 to 40 μM in HeLa cells, are observed under normoxia and hypoxia. If the phototoxicity induced by ThiathioHX when combined with the low UVA dose was only mediated by ROS generation, then a similar trend should be observed when these results are compared with the cell viability results, which is not the case (i.e., at these concentrations dramatic inhibition of cell proliferation is observed in both cells lines). Therefore, a competing mechanism that does not involve ROS, and, therefore, is independent of molecular oxygen should be playing a role in the phototoxicity mediated by ThiathioHX, particularly in its efficacy under hypoxic conditions. We hypothesized that photocrosslinking or photocycloaddition reactions between the HAFP and biological substrates (e.g., nucleic acids and proteins) such as those observed with psoralen derivatives in psoralen + UVA therapy (PUVA),63–66 and other thiobases44,67 leading to the formation of cytotoxic adducts are prospective candidates for the oxygen-independent phototoxicity observed. This idea is supported by experiments where deoxygenated solutions of ThiathioHX, in presence or absence of calf thymus (CT) DNA, were irradiated (300 J cm−2, λexc = 325 nm). The results showed clear formation of a photoproduct when CT DNA was present in the solution, but not in the absence of CT DNA (Fig. S19†).
Finally, Fig. 8 shows the fluorescence microscopy images of B16F10 and HeLa cells that were fixed after 1.5 hours of treatment and that were subsequently stained with nuclei marker, DAPI, the early-apoptosis marker, Annexin V, and the necrosis or late-apoptosis marker, PI. Evidently, apoptosis is the likely pathway in both cell lines and under both oxygenation conditions because >90% of the cells exhibited bright green fluorescence, which is characteristic of Annexin V. Unlike necrosis, which is known as a passive cell death caused by high levels of cell damage,68 apoptosis is preferred in PDT, as it is a controlled and organized process of autonomous cellular dismantling that does not result in inflammation and activation of the immune response.69 Furthermore, the employment of apoptosis as the primary cell death mechanism in ThiathioHX highlights the efficacy of lower doses of the photosensitizer, facilitating the desired cell death while providing a reliable indication of the optimal treatment dosage range.68 Collectively, these results, together with the spectroscopic and computational results, demonstrate that ThiathioHX stands out as the most promising thiobase photosensitizer developed to this date.
Conclusion
The investigation of the photophysics of thieno[3,4-d]pyrimidin-4(3H)-one and of its thionated derivative in aqueous and acetonitrile solutions shows that sulfur substitution redshifts its lowest-energy absorption band by about 60 nm (5145 cm−1), while simultaneously quenching its fluorescence and increasing its absorption coefficient by 62% independent of solvent. Thionation effectively alters the electronic relaxation mechanism, with a large fraction of the excited-state population reaching a long-lived and reactive triplet state. The intersystem crossing to the triplet state is independent of solvent environment, which is important because intracellular environments are diverse.ThiathioHX is a highly effective HAFP against monolayer cancer cells under both normoxic and hypoxic conditions. Our observations reveal the potential of ThiathioHX as a HAF PDT agent, and pave the way for further development of photosensitizers based on the thionation of thieno[3,4-d]pyrimidine derivatives. Notably, irradiation of ThiathioHX at 400 or 410 nm allows about 20% deeper tissue penetration (up to ca. 100 μm) than 2,6-dithiopurine or 4-thiothymidine, two of the most promising thiobase PDT agents reported in the literature.16,42,44,48 We conclude that thieno[3,4-d]pyrimidine-4(3H)-thione stands out as the most promising thiobase photosensitizer reported to this date.
Data availability
All data associated with the research in this manuscript are available from the corresponding authors on reasonable request.Author contributions
L. A. O.-R.: investigation, formal analysis, writing-original draft, writing review, and editing. Y. G. F.: investigation, formal analysis, writing-original draft, writing review, and editing. G. N.: investigation, writing review, and editing. K. H.: investigation, writing review, and editing. S. J. H.: investigation, writing review, and editing. H. J. F.: investigation, writing review, and editing. S. J.: investigation, writing review, and editing. Y. T.: funding acquisition, project administration, resources, supervision, visualization, validation, writing review, and editing. G. C.: funding acquisition, project administration, resources, supervision, visualization, validation, writing review, and editing. L. L.: funding acquisition, project administration, resources, supervision, visualization, validation, writing review, and editing. C. E. C.-H.: conceived the study, funding acquisition, project administration, resources, supervision, visualization, validation, writing-review, and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. A. O.-R., and C. E. C.-H. acknowledge the National Science Foundation (Grants No. CHE-2246805 & CHE-1800052). L. A. O.-R. also acknowledge the NSF-AGEP and the NOA-AGEP program for support. Y. T. thanks the National Institutes of Health for generous support (through grant GM 139407) and the Chemistry & Biochemistry MS and X-ray Facilities. G. C. thanks the National Natural Science Foundation of China (21688102). This work made use of the High-Performance Computing Resource in the Core Facility for Advanced Research Computing at CWRU.References
- C. A. Morton, R.-M. Szeimies, A. Sidoroff and L. R. Braathen, J. Eur. Acad. Dermatol. Venereol., 2013, 27, 536–544 CrossRef CAS.
- C. A. Morton, R.-M. Szeimies, N. Basset-Séguin, P. G. Calzavara-Pinton, Y. Gilaberte, M. Haedersdal, G. F. L. Hofbauer, R. E. Hunger, S. Karrer, S. Piaserico and others, J. Eur. Acad. Dermatol. Venereol., 2020, 34, 17–29 CrossRef CAS.
- C. A. Morton, R.-M. Szeimies, N. Basset-Seguin, P. Calzavara-Pinton, Y. Gilaberte, M. Haedersdal, G. F. L. Hofbauer, R. E. Hunger, S. Karrer, S. Piaserico and others, J. Eur. Acad. Dermatol. Venereol., 2019, 33, 2225–2238 CrossRef CAS PubMed.
- H. Abrahamse and M. R. Hamblin, Biochem. J., 2016, 473, 347–364 CrossRef CAS.
- R. R. Allison and C. H. Sibata, Photodiagn. Photodyn. Ther., 2010, 7, 61–75 CrossRef CAS PubMed.
- M. Verhille, P. Couleaud, R. Vanderess, D. Brault, M. Barberi-Heyob and C. Frochot, Curr. Med. Chem., 2010, 17, 3925–3943 CrossRef CAS PubMed.
- S. K. Attili, R. Dawe and S. Ibbotson, Photodiagn. Photodyn. Ther., 2011, 8, 53–57 CrossRef.
- P. C. A. Bruijnincx and P. J. Sadler, Adv. Inorg. Chem., 2009, 61, 1–62 CrossRef CAS.
- N. A. Smith and P. J. Sadler, Philos. Trans. R. Soc., A, 2013, 371, 20120519 CrossRef PubMed.
- F. Schmitt, P. Govindaswamy, G. Süss-Fink, W. H. Ang, P. J. Dyson, L. Juillerat-Jeanneret and B. Therrien, J. Med. Chem., 2008, 51, 1811–1816 CrossRef CAS.
- S. Monro, K. L. Colón, H. Yin, J. Roque III, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2018, 119, 797–828 CrossRef PubMed.
- R. Brem, M. Guven and P. Karran, Free Radical Biol. Med., 2017, 107, 101–109 CrossRef CAS PubMed.
- M. Pollum, S. Jockusch and C. E. Crespo-Hernández, J. Am. Chem. Soc., 2014, 136, 17930–17933 CrossRef CAS PubMed.
- M. Pollum, B. Ashwood and M. Lam, J. Am. Chem. Soc., 2016, 138, 1457–11460 CrossRef PubMed.
- B. Ashwood, M. Pollum and C. E. Crespo-Hernández, Photochem. Photobiol., 2019, 95, 33–58 CrossRef CAS PubMed.
- L. A. Ortiz-Rodríguez and C. E. Crespo-Hernández, Chem. Sci., 2020, 11, 11113–11123 RSC.
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88 RSC.
- L. A. Ortiz-Rodríguez, S. J. Hoehn, A. Loredo, L. Wang, H. Xiao and C. E. Crespo-Hernández, J. Am. Chem. Soc., 2021, 143, 2676–2681 CrossRef PubMed.
- V. N. Nguyen, Y. Yan, J. Zhao and J. Yoon, Acc. Chem. Res., 2020, 54, 207–220 CrossRef PubMed.
- J. Tang, L. Wang, A. Loredo, C. Cole and H. Xiao, Chem. Sci., 2020, 11, 6701–6708 RSC.
- G. Turkoglu, G. K. Koygun, M. N. Z. Yurt, N. Demirok and S. Erbas-Cakmak, Org. Biomol. Chem., 2020, 18, 9433–9437 RSC.
- A. A. Buglak, A. Charisiadis, A. Sheehan, C. J. Kingsbury, M. O. Senge and M. A. Filatov, Chem. – Eur. J., 2021, 27, 9934–9947 CrossRef CAS.
- V. N. Nguyen, S. Qi, S. Kim, N. Kwon, G. Kim, Y. Yim, S. Park and J. Yoon, J. Am. Chem. Soc., 2019, 141, 16243–16248 CrossRef CAS.
- P. P. P. Kumar, P. Yadav, A. Shanavas, S. Thurakkal, J. Joseph and P. P. Neelakandan, Chem. Commun., 2019, 55, 5623–5626 RSC.
- K. M. Farrell, M. M. Brister, M. Pittelkow and T. I. Sølling, J. Am. Chem. Soc., 2018, 140, 11214–11218 CrossRef CAS PubMed.
- X. Luo, Y. Yang and X. Qian, Chin. Chem. Lett., 2020, 31, 2877–2883 CrossRef CAS.
- J. Tang, M. A. Robichaux, K.-L. Wu, J. Pei, N. T. Nguyen, Y. Zhou, T. G. Wensel and H. Xiao, J. Am. Chem. Soc., 2019, 141, 14699–14706 CrossRef CAS PubMed.
- Y. Wu and W. Zhu, Chem. Soc. Rev., 2013, 42, 2039–2058 RSC.
- D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97–113 RSC.
- S. Fukuzumi and K. Ohkubo, Chem. Sci., 2013, 4, 561–574 RSC.
- M. A. Filatov, Org. Biomol. Chem., 2020, 18, 10–27 RSC.
- Y. Dong, B. Dick and J. Zhao, Org. Lett., 2020, 22, 5535–5539 CrossRef CAS PubMed.
- J. M. Brown and W. R. Wilson, Nat. Rev. Cancer, 2004, 4, 437–447 CrossRef CAS PubMed.
- W. R. Wilson and M. P. Hay, Nat. Rev. Cancer, 2011, 11, 393–410 CrossRef CAS PubMed.
- J. D. Chapman, C. C. Stobbe, M. R. Arnfield, R. Santus, J. Lee and M. S. McPhee, Radiat. Res., 1991, 126, 73–79 CrossRef CAS PubMed.
- P. Vaupel, M. Höckel and A. Mayer, Antioxid. Redox Signaling, 2007, 9, 1221–1236 CrossRef CAS PubMed.
- M. Hockel and P. Vaupel, J. Natl. Cancer Inst., 2001, 93, 266–276 CrossRef CAS PubMed.
- A. Massey, Y. Z. Xu and P. Karran, Curr. Biol., 2001, 11, 1142–1146 CrossRef CAS.
- O. Reelfs, P. Karran and A. R. Young, Photochem. Photobiol. Sci., 2012, 11, 148–154 CrossRef CAS PubMed.
- E. Gemenetzidis, O. Shavorskaya O, Y. Z. Xu and G. Trigiante, J. Dermatol. Treat., 2013, 24, 209–214 CrossRef CAS PubMed.
- M. Pollum, L. Guan, S. Ahsanuddin, E. Baron, M. Lam and C. E. Crespo-Hernández, J. Invest. Dermatol., 2016, 136, S105 CrossRef.
- M. Pollum, M. Lam, S. Jockusch and C. E. Crespo-Hernández, ChemMedChem, 2018, 13, 1044–1050 CrossRef CAS PubMed.
- M. Pollum, L. Martínez-Fernández, C. E. Crespo-Hernández, Photoinduced Phenomena in Nucleic Acids I, 2015, vol. 355, pp. 245−327 Search PubMed.
- L. A. Ortiz-Rodríguez, C. Reichardt, S. J. Hoehn, S. Jockusch and C. E. Crespo-Hernández, Nat. Commun., 2020, 11, 1–9 CrossRef PubMed.
- M. Sholokh, R. Sharma, D. Shin, R. Das, O. A. Zaporozhets, Y. Tor and Y. Mély, J. Am. Chem. Soc., 2015, 137, 3185–3188 CrossRef CAS PubMed.
- A. R. Rovira, A. Fin and Y. Tor, Chem. Sci., 2017, 8, 2983–2993 RSC.
- Y. Li, A. Fin, L. McCoy and Y. Tor, Angew. Chem., Int. Ed., 2017, 56, 1303–1307 CrossRef CAS PubMed.
- L. A. Ortiz-Rodríguez, S. J. Hoehn, C. Acquah, N. Abbass, L. Waidmann and C. E. Crespo-Hernández, Phys. Chem. Chem. Phys., 2021, 23, 25048–25055 RSC.
- C. Reichardt, R. A. Vogt and C. E. Crespo-Hernández, J. Chem. Phys., 2009, 131, 224518 CrossRef PubMed.
- C. Reichardt, C. Wen, R. A. Vogt and C. E. Crespo-Hernández, Photochem. Photobiol. Sci., 2013, 12, 1341–1350 CrossRef CAS PubMed.
- M. M. Brister and C. E. Crespo-Hernández, J. Phys. Chem. Lett., 2019, 10, 2156–2161 CrossRef CAS PubMed.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- M. A. El-Sayed, Acc. Chem. Res., 1968, 1, 8–16 CrossRef CAS.
- L. Martínez-Fernández, G. Granucci, M. Pollum, C. E. Crespo-Hernández, M. Persico and I. Corral, Chem. – Eur. J., 2017, 23, 2619–2627 CrossRef PubMed.
- C. Reichardt, C. Guo and C. E. Crespo-Hernández, J. Phys. Chem. B, 2011, 115, 3263–3270 CrossRef CAS PubMed.
- C. Reichardt and C. E. Crespo-Hernández, Chem. Commun., 2010, 46, 5963–5965 RSC.
- S. Mai, M. Pollum, L. Martínez-Fernández, N. Dunn, P. Marquetand, I. Corral, C. E. Crespo-Hernández and L. González, Nat. Commun., 2016, 7, 1–8 Search PubMed.
- R. Borrego-Varillas, D. C. Teles-Ferreira, A. Nenov, I. Conti, L. Ganzer, C. Manzoni, M. Garavelli, A. de Paula and G. Cerullo, J. Am. Chem. Soc., 2018, 140, 16087–16093 CrossRef CAS PubMed.
- D. C. Teles-Ferreira, I. Conti, R. Borrego-Varillas, A. Nenov, I. H. M. Van Stokkum, L. Ganzer, C. Manzoni, A. M. de Paula, G. Cerullo and M. Garavelli, Chem. – Eur. J., 2020, 26, 336–343 CrossRef CAS PubMed.
- M. S. Baptista, J. Cadet, A. Greer and A. H. Thomas, Photochem. Photobiol., 2021, 97, 1456–1483 CrossRef CAS PubMed.
- P. Vaupel and A. Mayer, Cancer Metastasis Rev., 2007, 26, 225–239 CrossRef CAS PubMed.
- Z. Zhou, J. Song, L. Nie and X. Chen, Chem. Soc. Rev., 2016, 45, 6597–6626 RSC.
- G. D. Cimino, H. B. Gamper, S. T. Isaacs and J. E. Hearst, Annu. Rev. Biochem., 1985, 54, 1151–1193 CrossRef CAS PubMed.
- N. Kitamura, S. Kohtani and R. Nakagaki, J. Photochem. Photobiol., C, 2005, 6, 168–185 CrossRef CAS.
- D. Kanne, H. Rapoport and J. E. Hearst, J. Med. Chem., 1984, 27, 531–534 CrossRef CAS PubMed.
- B. R. Vowels, E. K. Yoo and F. P. Gasparro, Photochem. Photobiol., 1996, 63, 572–576 CrossRef CAS PubMed.
- A. Favre, C. Saintomé, J. L. Fourrey, P. Clivio and P. Laugâa, J. Photochem. Photobiol., B, 1998, 42, 109–124 CrossRef CAS.
- N. L. Oleinick, R. L. Morris and I. Belichenko, Photochem. Photobiol. Sci., 2002, 1, 1–21 CrossRef CAS.
- G. Kroemer, B. Dallaporta and M. Resche-Rigon, Annu. Rev. Physiol., 1998, 60, 619–642 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Material and methods, supporting experimental and computational results and discussion, femtosecond and nanosecond transient absorption data, singlet oxygen phosphorescence decay traces, tables and figures with vertical excitation energies and oscillator strengths, computed main electronic configurations, ground-state relative energies, linearly interpolated internal coordinates paths, optimized excited state geometries, calculated excited state absorption spectra, synthesis and analytical data, and Cartesian coordinates of relevant geometries. CCDC 2091571 and 2091575. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02592f |
| This journal is © The Royal Society of Chemistry 2023 |