
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic asymmetric indolization by a desymmetrizing [3 + 2] annulation strategy†
Changhui
Wu
 a,
Zhiqian
Chang
a,
Chuanyong
Peng
a,
Chen
Bai
a,
Junhao
Xing
a and
Xiaowei
Dou
*ab
a,
Zhiqian
Chang
a,
Chuanyong
Peng
a,
Chen
Bai
a,
Junhao
Xing
a and
Xiaowei
Dou
*ab
aDepartment of Chemistry, School of Science, China Pharmaceutical University, Nanjing, 211198, P. R. China. E-mail: dxw@cpu.edu.cn
bState Key Laboratory of Coordination Chemistry, Nanjing University, Nanjing, 210023, P. R. China
First published on 29th June 2023
Abstract
A new catalytic asymmetric indolization reaction by a desymmetrizing [3 + 2] annulation strategy is developed. The reaction proceeds via a rhodium-catalyzed enantioposition-selective addition/5-exo-trig cyclization/dehydration cascade between ortho-amino arylboronic acids and 2,2-disubstituted cyclopentene-1,3-diones to produce N-unprotected cyclopenta[b]indoles bearing an all-carbon quaternary stereocenter in high yields with good enantioselectivities. A quantitative structure–selectivity relationship (QSSR) model was established to identify the optimal chiral ligand, which effectively controlled the formation of the stereocenter away from the reaction site. Density functional theory (DFT) calculations, non-covalent interaction analysis, and Eyring analysis were performed to understand the key reaction step and the function of the ligand.
Introduction
Chiral indoles are ubiquitous in natural products, biologically active compounds, and functional molecules.1,2 Continuous efforts have been devoted to developing efficient methods to access chiral indoles, and remarkable successes have been achieved in catalytic asymmetric transformations of indolyl substrates over the past decades.3 Compared to asymmetric functionalization of indoles, asymmetric indolization represents a fundamentally different approach to chiral indoles by de novo construction of the indole ring. Therefore, it offers an irreplaceable tool to create novel chiral indole structures and to access otherwise difficult to prepare chiral indoles.4 However, the lack of stereocenter in the indole ring can pose huge challenges for catalytic asymmetric indolization. In recent years, with the elegant design of substrate and reaction cascade, several catalytic asymmetric indolization reactions have been developed, which successfully addressed the challenges in asymmetric construction of axially chiral indoles.5–8 In contrast, only a handful of examples have been disclosed for the synthesis of centrally chiral indoles via asymmetric indolization. One strategy is the asymmetric Fischer indolization developed by List group9a and Mukherjee group9d (Scheme 1a). Another strategy is the exquisitely designed asymmetric cascade reaction of 2-alkynyl aniline derivatives reported by Lu group10a and Zhu group10b among others10 (Scheme 1b). Despite these elegant works, developing new strategies for asymmetric indolization to produce valuable chiral indoles is still highly desirable.11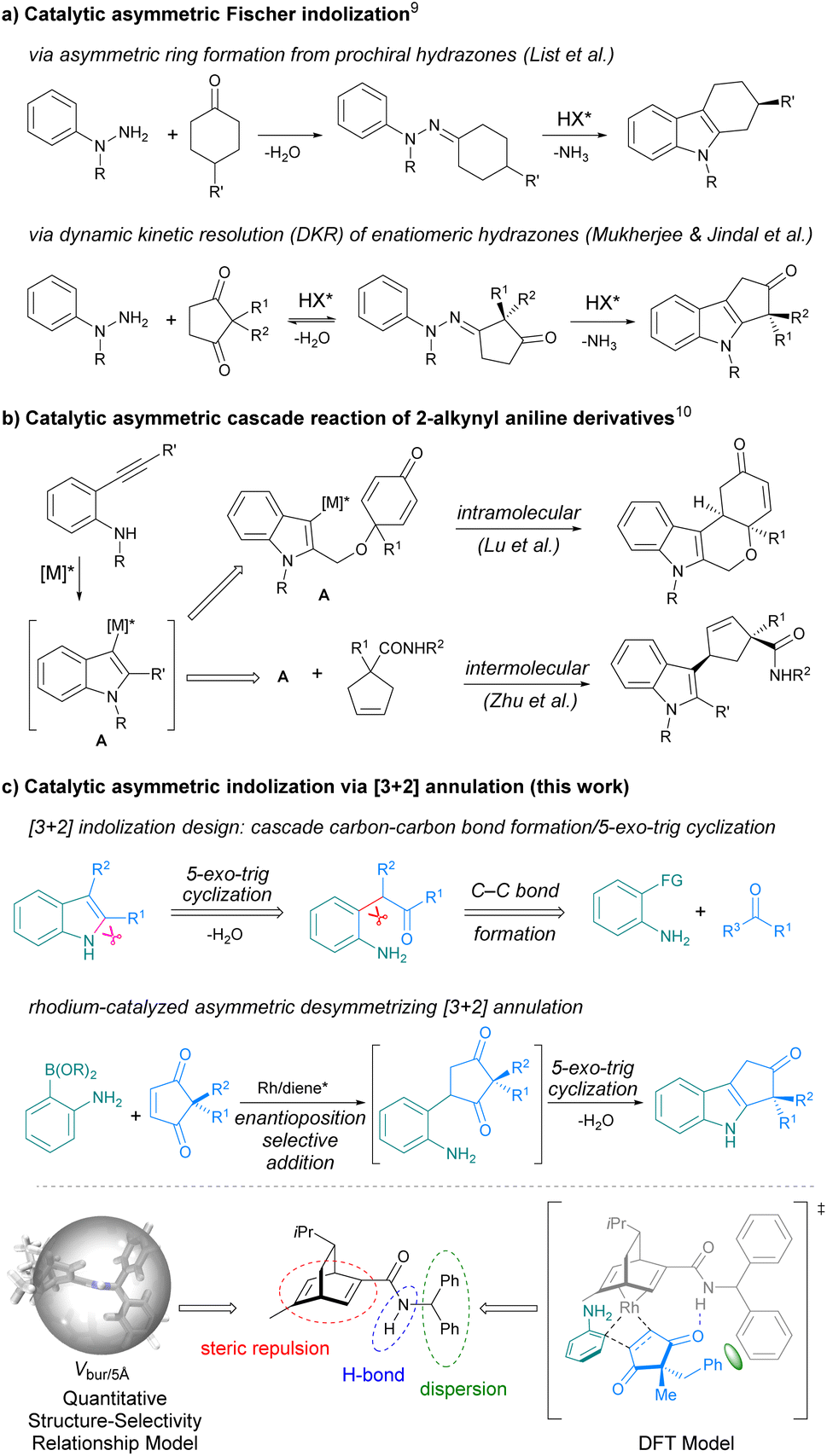 | ||
| Scheme 1 Construction of centrally chiral indoles by catalytic asymmetric indolization. | ||
Inspired by the Reissert indole synthesis,12 which involves intramolecular dehydrating cyclization of the in situ generated β-aniline-substituted α-keto esters as the key step, we envisaged that an intermolecular [3 + 2] indolization reaction could be realized through cascade carbon–carbon bond formation/5-exo-trig cyclization/dehydration reaction between functionalized anilines and appropriate carbonyl substrates (Scheme 1c-top). When a prochiral carbonyl substrate was employed, the asymmetric [3 + 2] indolization shall be feasible. In light of our experience in asymmetric synthesis by rhodium-catalyzed conjugate addition of arylborons to α,β-unsaturated carbonyls,13 we proposed to use ortho-amino arylborons under rhodium catalysis to construct the C–C bond,14,15 and 2,2-disubstituted cyclopentene-1,3-diones16 were selected as the prochiral carbonyl substrates. Ideally, the reactions between ortho-aminoarylborons and 2,2-disubstituted cyclopentene-1,3-diones could proceed via a conjugate addition/5-exo-trig cyclization/dehydration cascade to produce cyclopenta[b]indoles bearing an all-carbon quaternary stereocenter (Scheme 1c-middle).17 In addition, the new strategy may overcome some limitations of the existing methods, such as the lengthy reaction time9 and the tedious preparation of substrates.10 However, there is a major challenge to be considered: the quaternary carbon stereocenter is away from the reaction site while the enantioselectivity is determined at the initial enantioposition-selective conjugate addition step (Scheme 1c-middle). Thus, the identification of a suitable catalyst that can effectively recognize the remote quaternary carbon stereocenter is crucial for the success of the asymmetric indolization.
Herein, we report a rhodium-catalyzed asymmetric indolization reaction via enantioposition-selective addition/5-exo-trig cyclization/dehydration cascade between ortho-aminoarylboronic acids and 2,2-disubstituted cyclopentene-1,3-diones, delivering N-unprotected cyclopenta[b]indoles bearing an all-carbon quaternary stereocenter in high yields with good to excellent enantioselectivities. The optimal chiral ligand that can effectively controls the remote quaternary carbon stereocenter was identified through a quantitative structure–selectivity relationship (QSSR) model, and the steric repulsion as well as the hydrogen bonding and dispersion interactions between the ligand and the substrate were found to collectively contribute to the high enantioselectivity (Scheme 1c-bottom).
Results and discussion
We initiated our investigation with 2-benzyl-2-methyl cyclopentene-1,3-dione 1a and 2-aminophenylboronic acid 2a as model substrates. After preliminary screening of the reaction conditions, it was found the reaction proceeded well in EtOH to give the indole product, and the chiral diene18-ligated rhodium catalysts gave promising results. As shown in Scheme 2, when using ester–diene ligands (L1, L2, and L3), the reaction exhibited promising enantioselectivities. Interestingly, the enantioselectivity of the reaction was improved when the corresponding amide–diene ligand was used (L1 vs. L4). Several other amide–diene19 ligands were then tested. Intuitively, the enantioselectivity of the reaction increases with the size of the R group in the amide–diene ligands (R = Me, 89.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10.5 er; R = tBu, 93:7 er; R = 2,6-iPr2C6H3, 95:5 er). Thus, a quantitative structure–selectivity relationship model was developed to understand this system (see ESI-Section 5† for details).20 There is a linear correlation between the percent buried volume Vbur/5 Å with a radius of 5 Å centered on the hydrogen atom of the amide in ligand and the measured Gibbs energy difference ΔΔG‡ between two competing diastereomeric transition states (Scheme 2, training set). Leave-one-out (LOO) cross-validation and 3-fold cross-validation were implemented to avoid overfitting. Guided by this model, we synthesized ligands featuring different Vbur/5 Å (L6, L11, L12, L13, L14, L15) and tested their performances (Scheme 2, validation set). The newly synthesized ligands fitted well with the established model. Notably, ligand L15 featuring the largest percent buried volume exhibited the best enantiocontrol, however, it was not used for further study due to the lower reactivity. Pleasingly, ligands L14 and L18 proved to be optimal when considering both reactivity and enantioselectivity, and ligand L14 was selected for further investigation due to its easier preparation.
:10.5 er; R = tBu, 93:7 er; R = 2,6-iPr2C6H3, 95:5 er). Thus, a quantitative structure–selectivity relationship model was developed to understand this system (see ESI-Section 5† for details).20 There is a linear correlation between the percent buried volume Vbur/5 Å with a radius of 5 Å centered on the hydrogen atom of the amide in ligand and the measured Gibbs energy difference ΔΔG‡ between two competing diastereomeric transition states (Scheme 2, training set). Leave-one-out (LOO) cross-validation and 3-fold cross-validation were implemented to avoid overfitting. Guided by this model, we synthesized ligands featuring different Vbur/5 Å (L6, L11, L12, L13, L14, L15) and tested their performances (Scheme 2, validation set). The newly synthesized ligands fitted well with the established model. Notably, ligand L15 featuring the largest percent buried volume exhibited the best enantiocontrol, however, it was not used for further study due to the lower reactivity. Pleasingly, ligands L14 and L18 proved to be optimal when considering both reactivity and enantioselectivity, and ligand L14 was selected for further investigation due to its easier preparation.
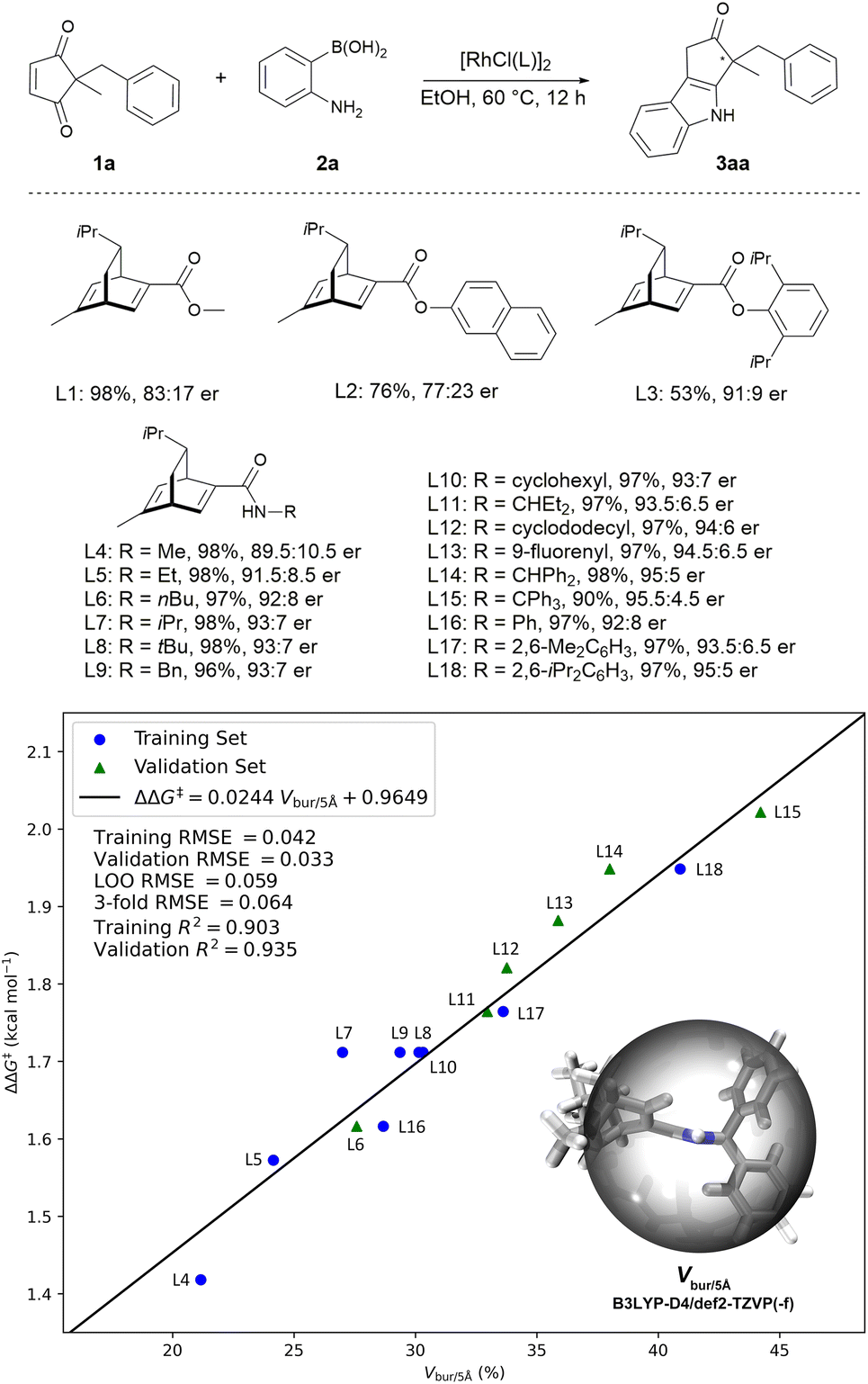 | ||
| Scheme 2 Ligand optimization for the model asymmetric indolization reaction between cyclopentene-1,3-dione 1a and 2-aminophenylboronic acid 2a. Reaction conditions: 1a (0.10 mmol), 2a (0.20 mmol), and Rh catalyst (3 mol% Rh), in EtOH (1 mL) at 60 °C for 12 h. Yield was the isolated yield of 3aa, and the er was determined by HPLC analysis on a chiral stationary phase. Measured ΔΔG‡ were calculated using the formula ΔΔG‡ = −RT ln(er), where R is the ideal gas constant, and T is temperature. | ||
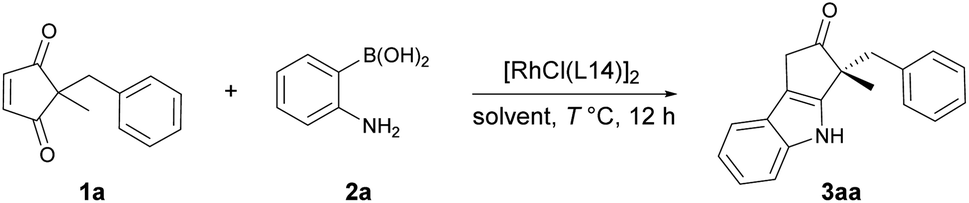
With ligand L14 in hand, further optimization was conducted to establish the optimal reaction conditions (Table 1). Similar yield and enantioselectivity were obtained when iPrOH was used instead of EtOH (entries 1 and 2). tBuOH led to a decreased yield (entry 3) while MeOH led to a diminished enantioselectivity (entry 4). Reducing the amount of catalyst to 1 mol% Rh slightly affected the yield and enantioselectivity (entry 5). When the reaction temperature was lowered, the conjugate addition reaction still proceeded well, but the subsequent intramolecular cyclization was not complete. Thus, the reaction was carried out successively at 30 °C and 60 °C to complete the conjugate addition and cyclization respectively, which further improved the enantioselectivity (entry 6, 96:4 er).
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | T | Yieldb | erc |
| a Reaction conditions: 1a (0.10 mmol), 2a (0.20 mmol), and Rh catalyst (3 mol% Rh), in the corresponding solvent (1 mL) at T °C for 12 h. b Isolated yield of 3aa. c Determined by HPLC analysis on a chiral stationary phase. d Rh catalyst (1 mol% Rh) was used. e 1a (0.10 mmol), 2a (0.15 mmol), and Rh catalyst (3 mol% Rh), in EtOH (1 mL) at 30 °C for 12 h, then at 60 °C for 5 h. | ||||
| 1 | EtOH | 60 | 98 | 95:5 |
| 2 | iPrOH | 60 | 98 | 95:5 |
| 3 | tBuOH | 60 | 91 | 95:5 |
| 4 | MeOH | 60 | 98 | 92:8 |
| 5d | EtOH | 60 | 81 | 93.5:6.5 |
| 6 | EtOH | 30/60 | 98 | 96:4 |
With the optimal reaction conditions in hand, the scope of 2,2-disubstituted cyclopentene-1,3-diones for asymmetric indolization was explored (Scheme 3). Cyclopentene-1,3-diones substituted with methyl and various benzylic groups, including those with electron-donating and electron-withdrawing groups at para, meta, and ortho positions were all suitable (3aa to 3ka). Replacement of the phenyl moiety with a naphthyl (3la, 3ma) or a heteroaryl unit (3na) did not affect the reaction outcome. Cyclopentene-1,3-diones bearing allylic substituent (3oa) and different types of alkyl substituents (3pa to 3ra) also exhibited good yields and high enantioselectivities. The ester and amide groups were tolerated (3ra, 3ta), and the enantioselectivity for the bulky amide-containing substrate was much higher (85:15 er versus 97:3 er). The 2-ethyl-2-benzyl substituted substrate exhibited lower enantioselectivity (3ua) compared to its methyl congener (3aa), probably due to the diminished steric difference between the two substituents at 2-position. The spirocyclic substrates were also suitable for this reaction (3va, 3wa). Notably, the 7-substituted spiroindane substrate gave the highest enantioselectivity (3wa, 99:1 er), which might be partially attributed to better enantio-face recognization induced by the rigid spiro structure and the substituent. The absolute configuration of 3aa was determined by X-ray crystallography analysis, and the absolute configuration of other products was assigned by analogy.
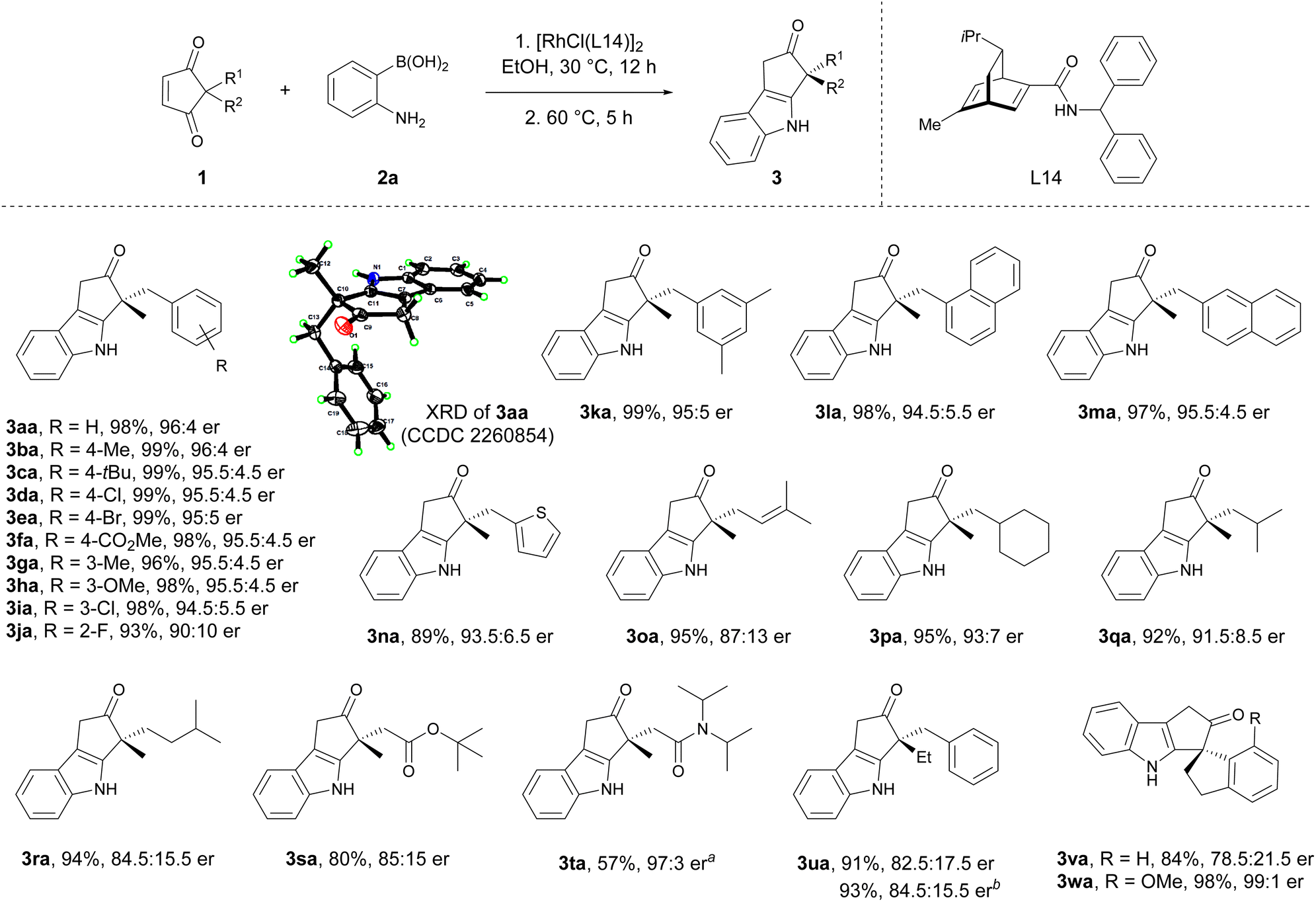 | ||
| Scheme 3 The scope of 2,2-disubstituted cyclopentene-1,3-diones. Reaction conditions: 1 (0.20 mmol), 2a (0.30 mmol), and Rh catalyst (3 mol% Rh), in EtOH (2 mL) at 30 °C for 12 h, then heating at 60 °C for 5 h. a Rh catalyst (8 mol% Rh) was used. b [RhCl(L11)]2 was used as the catalyst. | ||
The scope of 2-aminoarylborons was also explored. Although 2-aminophenylboronic acid is commercially available, other 2-aminoarylboronic acids are not readily accessible. Given the availability and well-established preparation of 2-aminoarylboronic acid pinacol esters,21 they were employed for the scope study. It was found the pinacol esters exhibited lower reactivity at 30 °C, therefore the previously established conditions were slightly modified, and the reaction was conducted constantly at 60 °C. As shown in Scheme 4, the reaction using 2-aminophenylboronic acid pinacol ester proceeded smoothly to give the indolization product in 98% yield with 94.5:5.5 er (3aa), which is comparable to the optimal result from 2-aminophenylboronic acid. The N-methyl 2-aminophenylboronic acid pinacol ester was also tested under the standard conditions, and the N-methyl product was obtained in a moderate yield (43%) with a high enantioselectivity (95:5 er). The lower reactivity of the N-methyl reagent might be attributed to the lower reactivity of the corresponding bulkier arylrhodium species. All the other 2-aminoarylboronic acid pinacol esters bearing either electron-donating or electron-withdrawing substituents at different positions worked well to produce the substituted chiral indoles in high yields with good enantioselectivities (3ab to 3ai). It is worth noting that the electron-withdrawing groups such as –CF3 (3ac), –CO2Me (3ae), and –CN (3ai) are generally not well tolerated in the Fischer indolization, as they would result in diminished reactivity of the corresponding arylhydrazine substrates.9
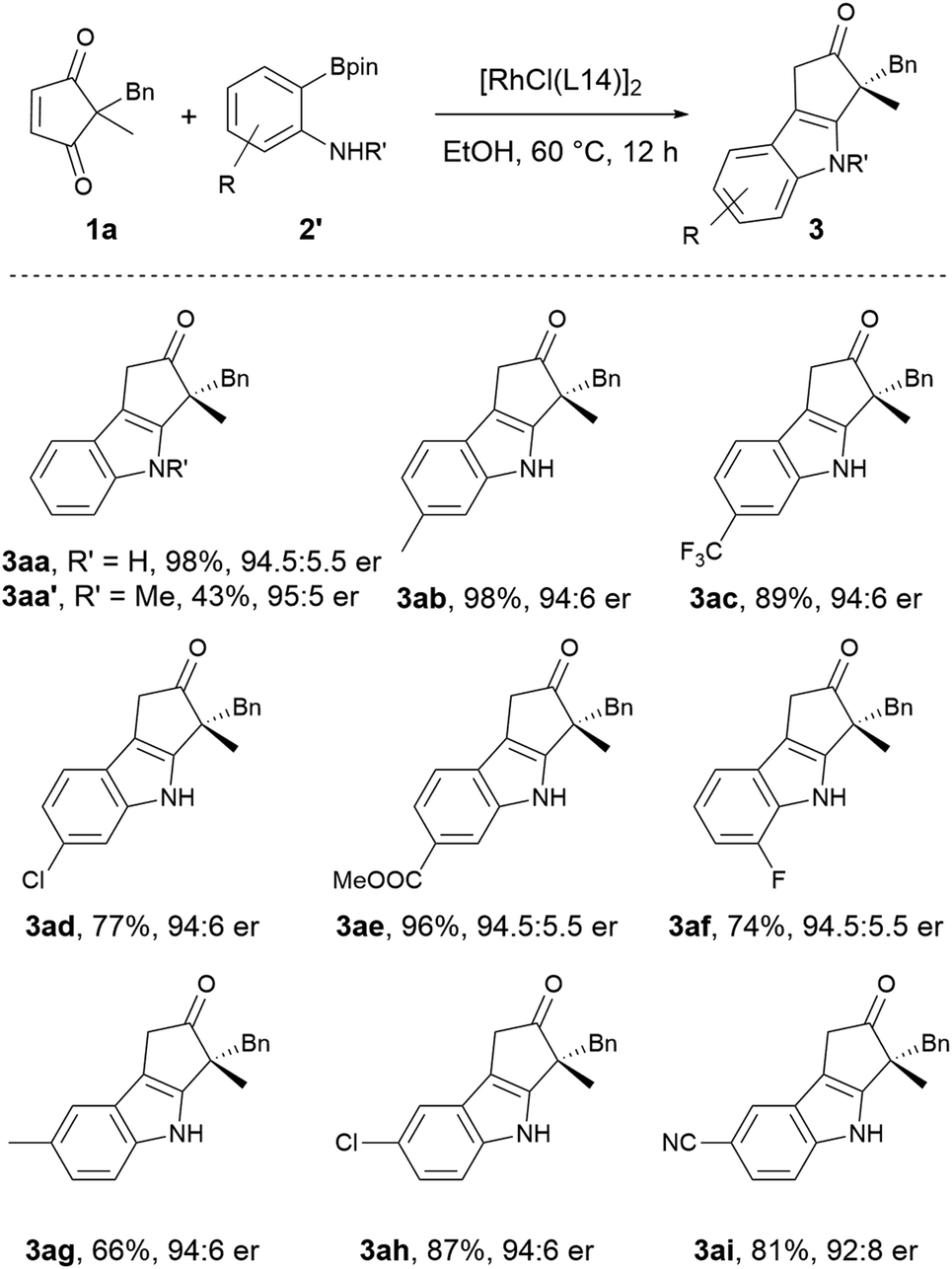 | ||
| Scheme 4 The scope of 2-aminoarylboronic acid pinacol esters. Reaction conditions: 1a (0.20 mmol), 2′ (0.30 mmol), and Rh catalyst (3 mol% Rh), in EtOH (2 mL) at 60 °C for 12 h. | ||
A 2 mmol scale synthesis of product 3aa was conducted to demonstrate the scalability of the asymmetric indolization reaction, and 3aa could still be obtained in 98% yield with 95:5 er under standard conditions. Synthetic transformations of 3aa to other indole derivatives were illustrated in Scheme 5. Oxidation of 3aa with DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone) under mild conditions produced 4, a chiral analogue of natural product bruceolline E featuring the cyclopenta[b]indole-1,2-dione core.22 Reduction of the ketone moiety in 3aa with NaBH4 was highly effective, giving the chiral indole 5 bearing continuous quaternary–tertiary stereocenters in a highly diastereoselective manner. Protection of the free NH moiety with a Boc group could be performed under conventional conditions (6). In addition, conversion of the ketone to vinyl-OTf was viable (7), which could undergo Pd-catalyzed Kumada coupling to construct new carbon–carbon bond on the cyclopenta[b]indole ring (8).
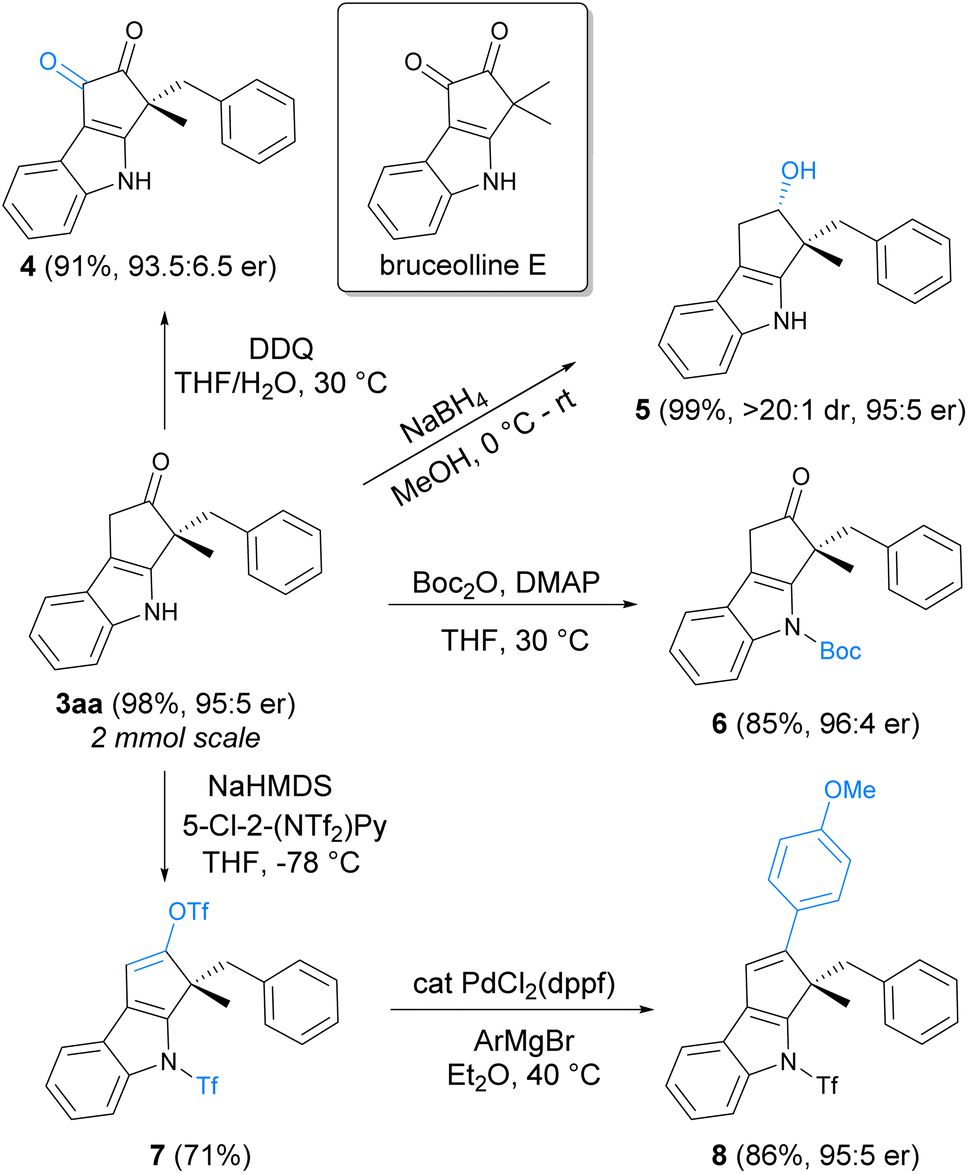 | ||
| Scheme 5 Synthetic transformations of the chiral indole product. | ||
To further explain the origin of enantioselectivity, we performed DFT calculations on the enantiocontrol step of the reaction (Fig. 1). We first identified the transition states for the enantioposition-selective alkene insertion step (with L8-ligated catalyst as a model), which was found to follow Hayashi's stereochemical model.24 These transition states are stabilized by hydrogen bonding interactions between the N–H of the ligand and the carbonyl group of the substrate (see ESI-Section 6† for details). The surface distance projection maps of ligand–rhodium–substrate complex reflexed the relative spatial relationships between the substrate and the ligand.25 For the transition state (TSDisf) leading to the unfavorable product, the benzyl group is closer to the ligand, and its rotation is hindered by the tert-butyl and methyl groups of the ligand. For the transition state (TSFav) leading to the favorable product, the rotation of the benzyl group is not affected, which has a more favorable entropy effect. The ΔΔGTS is 1.63 kcal mol−1, and the calculated er is in good agreement with the experimental result (ercalc = 92:8 versus erexp = 93:7).
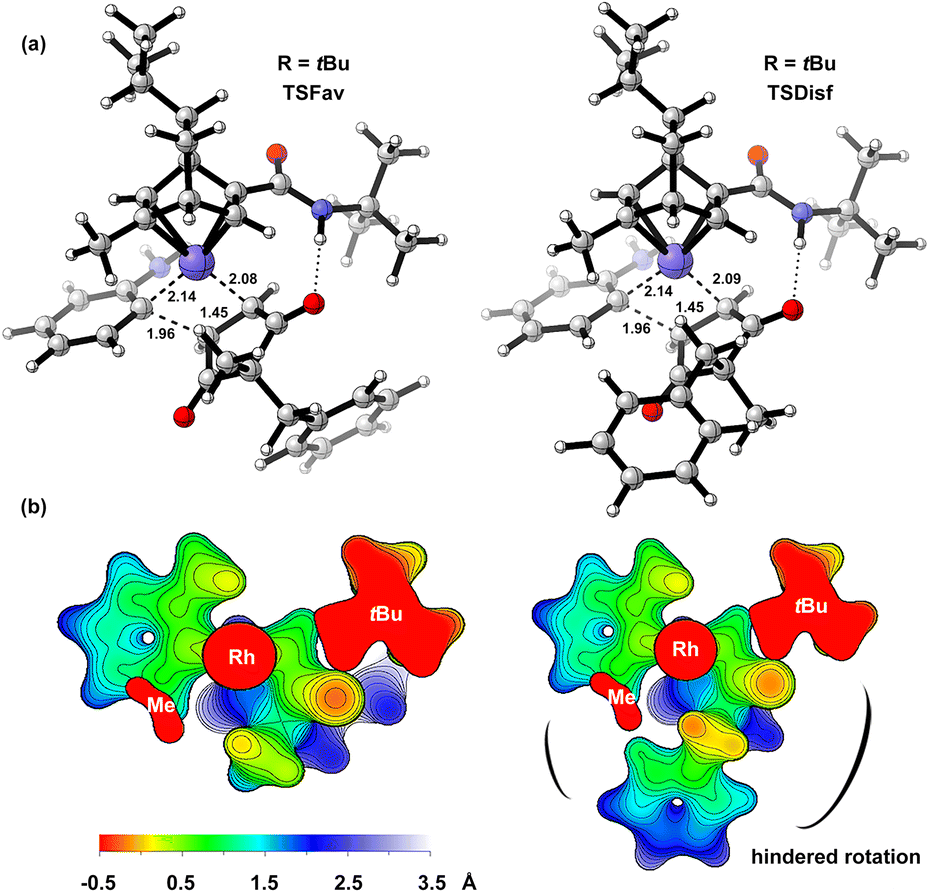 | ||
| Fig. 1 The DFT model of transition states for the alkene insertion with L8-ligated catalyst (L8, R = tBu) established at the SMD(ethanol)//PWPB95-D4/def2-TZVPP//C-PCM(ethanol)/PBE0-D4/def2-TZVP(for Rh)/def2-SVP (for other atoms) level.23 (a) The optimized structures of transition states with ΔΔGTS = 1.63 kcal mol−1 and ercalc = 92:8 (erexp = 93:7). (b) The surface distance projection maps displayed from −0.5 Å to 3.5 Å centered on the rhodium atom. | ||
The non-covalent interaction analysis was also applied to transition states to understand the effect of the size of ligand substituent on enantioselectivity (Fig. 2).26 In the favorable transition state of ligand L8 (R = tBu), there is a CH⋯π interaction between tBu and benzyl group. In the favorable transition state of ligand L15 (R = CPh3), there are some π⋯π interactions between CPh3 and benzyl group. The electron density ρ(r), the Laplacian of electron density ∇2ρ(r), and the energy density H(r) of critical points also indicate these non-covalent interactions are van der Waals interactions which are usually dominated by dispersion effects.27 These interactions are absent in the favorable transition state of ligand L4 (R = Me) as well as in those unfavorable transition states, thought to contribute greatly to the enantioselectivity (see ESI-Section 6† for details). As the size of the substituent on the ligand increases, its interactions with the benzyl group increase, leading to improved enantioselectivity.
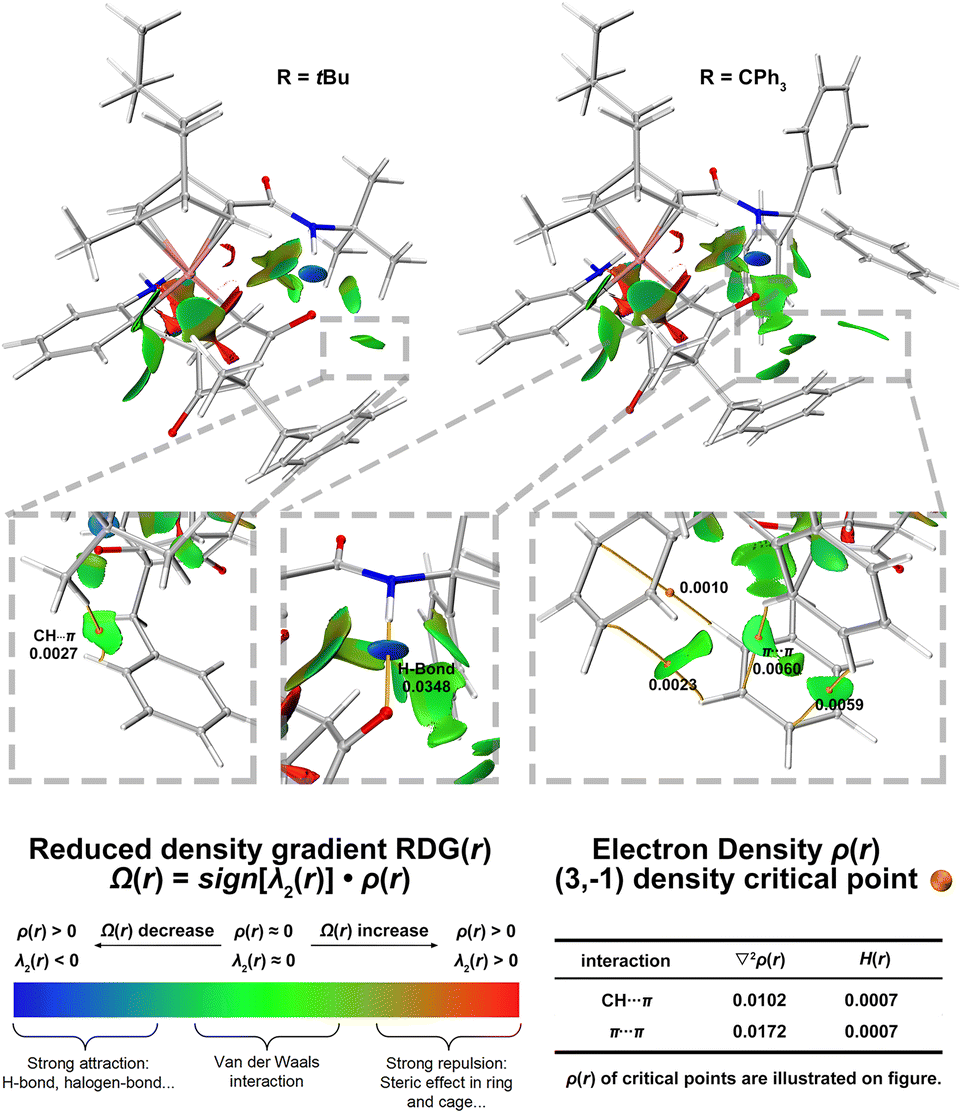 | ||
| Fig. 2 The non-covalent interaction analysis within the sign(λ2)ρ colored RDG plot and the topology analysis of the electron density within the framework of QTAIM for ligand L8 (R = tBu) and L15 (R = CPh3). | ||
Eyring analysis was also performed for different ligands (Table 2).28 For ligand L4 (R = Me), the differential activation entropy ΔΔS‡ is the factor dominating enantioselectivity. As we explained above, the rotation of the benzyl group in the unfavorable transition state is hindered, which is entropically unfavorable due to the steric repulsion of the ligand. For ligand L8 (R = tBu) and ligand L14 (R = CHPh2), the differential activation enthalpy ΔΔH‡ becomes an increasingly important factor for enantiocontrol, which is consistent with our observation that there are additional non-covalent interactions in the favorable transition states. For L14, though the existence of enthalpy–entropy compensation reduces the favorable differential activation entropy ΔΔS‡ to some extent, the significant favorable differential activation enthalpy ΔΔH‡ greatly enhances the enantioselectivity.29
|
|
|||
|---|---|---|---|
| Entry | Ligand | ΔΔH‡ (kcal mol−1) | ΔΔS‡ (cal mol−1 K−1) |
| a Reaction conditions: 1a (0.10 mmol), 2a (0.20 mmol), and Rh catalyst (3 mol% Rh), in the EtOH (1 mL) at T °C for 12 h. | |||
| 1 | L4 (R = Me) | −1.224 | 0.445 |
| 2 | L8 (R = tBu) | −1.671 | 0.128 |
| 3 | L14 (R = CHPh2) | −2.412 | −1.573 |
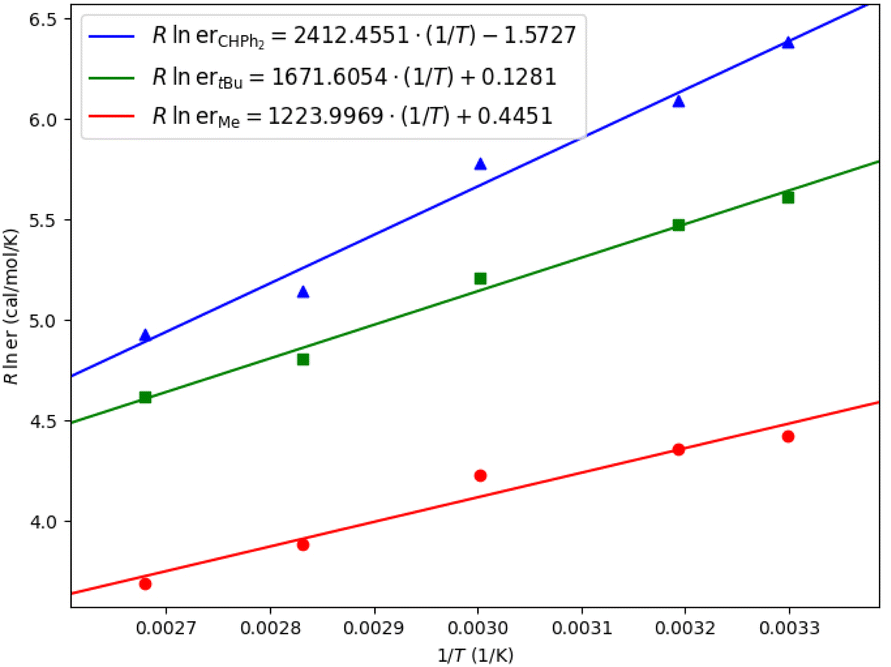
The role of amide–diene ligand in controlling enantioselectivity can be summarized as follows: (1) the rigid skeleton of the ligand provided steric repulsion to control the enantioselectivity, which is important in ligands with small substituents; (2) the amide moiety of the ligand provided hydrogen bonding interaction to stabilize the transition state; (3) the substituent moiety of the ligand provided dispersion interactions to stabilize the favourable transition state, which is important in ligands with large substituents.
Conclusions
In conclusion, we have developed a new type of asymmetric indolization reaction based on a desymmetrizing [3 + 2] annulation strategy, and a range of valuable cyclopenta[b]indoles bearing an all-carbon quaternary stereocenter were obtained in high yields with good enantioselectivities via a rhodium-catalyzed enantioposition-selective addition/5-exo-trig cyclization/dehydration cascade.30 In this study, the optimal chiral ligand that can effectively control the formation of the remote stereocenter was identified based on a QSSR model, and further studies such as DFT calculation, non-covalent interaction analysis and Eyring analysis were performed to understand the multiple functions of the ligand.Data availability
All experimental procedures, characterization, and computational details for this study can be found in the ESI.†Author contributions
X. D. conceived the project. C. W. conducted the study under X. D.'s supervision. Z. C. conducted the initial investigation. C. P., C. B., and J. X. assisted with the experiments and the analytical characterization. C. W. performed the theoretical calculations and modelling. X. D. and C. W. wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support from the Natural Science Foundation of Jiangsu Province (BK20200080) and the State Key Laboratory of Coordination Chemistry, Nanjing University. The authors thank the Hefei Advanced Computing Center for the computational resources.Notes and references
- (a) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489–4497 CrossRef CAS PubMed; (b) N. K. Kaushik, N. Kaushik, P. Attri, N. Kumar, C. H. Kim, A. K. Verma and E. H. Choi, Molecules, 2013, 18, 6620–6662 CrossRef CAS PubMed; (c) T. V. Sravanthi and S. L. Manju, Eur. J. Pharm. Sci., 2016, 91, 1–10 CrossRef CAS PubMed; (d) M. G. Ciulla and K. Kumar, Tetrahedron Lett., 2018, 59, 3223–3233 CrossRef CAS; (e) Y. Wan, Y. Li, C. Yan, M. Yan and Z. Tang, Eur. J. Med. Chem., 2019, 183, 111691 CrossRef CAS PubMed.
- (a) U. Berens, J. M. Brown, J. Long and R. Selke, Tetrahedron: Asymmetry, 1996, 7, 285–292 CrossRef CAS; (b) T. Benincori, E. Brenna, F. Sannicol, L. Trimarco, P. Antognazz, E. Cesarotti, F. Demartin, T. Pilati and G. Zotti, J. Organomet. Chem., 1997, 529, 445–453 CrossRef CAS; (c) T. Benincori, O. Piccolo, S. Rizzo and F. Sannicolò, J. Org. Chem., 2000, 65, 8340–8347 CrossRef CAS PubMed; (d) T. Baumann and R. Brückner, Angew. Chem., Int. Ed., 2019, 58, 4714–4719 CrossRef CAS PubMed; (e) T. He, L. Peng, S. Li, F. Hu, C. Xie, S. Huang, S. Jia, W. Qin and H. Yan, Org. Lett., 2020, 22, 6966–6971 CrossRef CAS PubMed.
- (a) G. Bartoli, G. Bencivenni and R. Dalpozzo, Chem. Soc. Rev., 2010, 39, 4449–4465 RSC; (b) R. Dalpozzo, Chem. Soc. Rev., 2015, 44, 742–778 RSC; (c) J.-B. Chen and Y.-X. Jia, Org. Biomol. Chem., 2017, 15, 3550–3567 RSC; (d) Y.-C. Zhang, F. Jiang and F. Shi, Acc. Chem. Res., 2020, 53, 425–446 CrossRef CAS PubMed; (e) M.-S. Tu, K.-W. Chen, P. Wu, Y.-C. Zhang, X.-Q. Liu and F. Shi, Org. Chem. Front., 2021, 8, 2643–2672 RSC.
- B.-M. Yang, X. Q. Ng and Y. Zhao, Chem Catal., 2022, 2, 3048–3076 CrossRef CAS.
- (a) N. Ototake, Y. Morimoto, A. Mokuya, H. Fukaya, Y. Shida and O. Kitagawa, Chem.–Eur. J., 2010, 16, 6752–6755 CrossRef CAS PubMed; (b) L. Sun, H. Chen, B. Liu, J. Chang, L. Kong, F. Wang, Y. Lan and X. Li, Angew. Chem., Int. Ed., 2021, 60, 8391–8395 CrossRef CAS PubMed; (c) Y. Wang, X. Zhou, W. Shan, R. Liao, Y. Deng, F. Peng and Z. Shao, ACS Catal., 2022, 12, 8094–8103 CrossRef CAS; (d) Z.-S. Wang, L.-J. Zhu, C.-T. Li, B.-Y. Liu, X. Hong and L.-W. Ye, Angew. Chem., Int. Ed., 2022, 61, e202201436 CAS.
- (a) L. Peng, K. Li, C. Xie, S. Li, D. Xu, W. Qin and H. Yan, Angew. Chem., Int. Ed., 2019, 58, 17199–17204 CrossRef CAS PubMed; (b) Y.-P. He, H. Wu, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2020, 59, 2105–2109 CrossRef CAS PubMed; (c) W.-C. Yang, X.-B. Chen, K.-L. Song, B. Wu, W.-E. Gan, Z.-J. Zheng, J. Cao and L.-W. Xu, Org. Lett., 2021, 23, 1309–1314 CrossRef CAS PubMed; (d) D. Xu, S. Huang, F. Hu, L. Peng, S. Jia, H. Mao, X. Gong, F. Li, W. Qin and H. Yan, CCS Chem., 2022, 4, 2686–2697 CrossRef CAS.
- (a) M. Tian, D. Bai, G. Zheng, J. Chang and X. Li, J. Am. Chem. Soc., 2019, 141, 9527–9532 CrossRef CAS PubMed; (b) S. Lu, J.-Y. Ong, H. Yang, S. B. Poh, X. Liew, C. S. D. Seow, M. W. Wong and Y. Zhao, J. Am. Chem. Soc., 2019, 141, 17062–17067 CrossRef CAS PubMed; (c) C.-S. Wang, L. Wei, C. Fu, X.-H. Wang and C.-J. Wang, Org. Lett., 2021, 23, 7401–7406 CrossRef CAS PubMed; (d) X. Li, L. Zhao, Z. Qi and X. Li, Org. Lett., 2021, 23, 5901–5905 CrossRef CAS PubMed; (e) S. Jia, Y. Tian, X. Li, P. Wang, Y. Lan and H. Yan, Angew. Chem., Int. Ed., 2022, 61, e202206501 CAS; (f) H. Yang, H.-R. Sun, R.-Q. He, L. Yu, W. Hu, J. Chen, S. Yang, G.-G. Zhang and L. Zhou, Nat. Commun., 2022, 13, 1–9 Search PubMed.
- (a) K.-W. Chen, Z.-H. Chen, S. Yang, S.-F. Wu, Y.-C. Zhang and F. Shi, Angew. Chem., Int. Ed., 2022, 61, e202116829 CAS; (b) P. Zhang, Q. Xu, X.-M. Wang, J. Feng, C.-J. Lu, Y. Li and R.-R. Liu, Angew. Chem., Int. Ed., 2022, 61, e202212101 CAS; (c) Z.-H. Chen, T.-Z. Li, N.-Y. Wang, X.-F. Ma, S.-F. Ni, Y.-C. Zhang and F. Shi, Angew. Chem., Int. Ed., 2023, 62, e202300419 CrossRef CAS PubMed; (d) L.-Y. Wang, J. Miao, Y. Zhao and B.-M. Yang, Org. Lett., 2023, 25, 1553–1557 CrossRef CAS PubMed.
- (a) S. Müller, M. J. Webber and B. List, J. Am. Chem. Soc., 2011, 133, 18534–18537 CrossRef PubMed; (b) A. Martínez, M. J. Webber, S. Müller and B. List, Angew. Chem., Int. Ed., 2013, 52, 9486–9490 CrossRef PubMed; (c) L. Kötzner, M. J. Webber, A. Martínez, C. De Fusco and B. List, Angew. Chem., Int. Ed., 2014, 53, 5202–5205 CrossRef; (d) B. Ghosh, R. Balhara, G. Jindal and S. Mukherjee, Angew. Chem., Int. Ed., 2021, 60, 2–9 CrossRef.
- (a) J. Chen, X. Han and X. Lu, Angew. Chem., Int. Ed., 2017, 56, 14698–14701 CrossRef CAS PubMed; (b) Y.-P. He, J. Cao, H. Wu, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2021, 60, 7093–7097 CrossRef CAS PubMed; (c) A. Whyte, J. Bajohr, R. Arora, A. Torelli and M. Lautens, Angew. Chem., Int. Ed., 2021, 60, 20231–20236 CrossRef CAS PubMed; (d) G. Yang, J. Pan, Y.-M. Ke, Y. Liu and Y. Zhao, Angew. Chem., Int. Ed., 2021, 60, 20689–20694 CrossRef CAS PubMed; (e) G. Wang, J.-C. Li, Y.-G. Zhou and Z.-S. Ye, Org. Lett., 2021, 23, 802–807 CrossRef CAS PubMed; (f) X.-D. Hu, Z.-H. Chen, J. Zhao, R.-Z. Sun, H. Zhang, X. Qi and W.-B. Liu, J. Am. Chem. Soc., 2021, 143, 3734–3740 CrossRef CAS PubMed.
- During the preparation of this manuscript, a copper-catalyzed asymmetric radical cascade reaction reaction was disclosed by Leifert and Studer group to realize the asymmetric Fukuyama indole synthesis, see: T. Drennhaus, D. Leifert, J. Lammert, J. P. Drennhaus, K. Bergander, C. G. Daniliuc and A. Studer, J. Am. Chem. Soc., 2023, 145, 8665–8676 CAS.
- J. Li and J. M. Cook in Name Reactions in Heterocyclic Chemistry, ed. J. J. Li, Wiley, Hoboken NJ, 2005, pp. 154–158 Search PubMed.
- (a) X. Dou, Y. Huang and T. Hayashi, Angew. Chem., Int. Ed., 2016, 55, 1133–1137 CrossRef CAS PubMed; (b) L. Yin, J. Xing, Y. Wang, Y. Shen, T. Lu, T. Hayashi and X. Dou, Angew. Chem., Int. Ed., 2019, 58, 2474–2478 CrossRef CAS PubMed; (c) B. Ye, J. Yao, C. Wu, H. Zhu, W. Yao, L. Jin and X. Dou, ACS Catal., 2022, 12, 2434–2440 CrossRef CAS.
- (a) T. Yasukawa, H. Miyamura and S. Kobayashi, Acc. Chem. Res., 2020, 53, 2950–2963 CrossRef CAS PubMed; (b) M. M. Heravi, M. Dehghani and V. Zadsirjan, Tetrahedron: Asymmetry, 2016, 27, 513–588 CrossRef CAS; (c) P. Tian, H.-Q. Dong and G.-Q. Lin, ACS Catal., 2012, 2, 95–119 CrossRef CAS; (d) T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829–2844 CrossRef CAS PubMed; (e) K. Fagnou and M. Lautens, Chem. Rev., 2003, 103, 169–196 CrossRef CAS PubMed.
- (a) J. Horn, S. P. Marsden, A. Nelson, D. House and G. G. Weingarten, Org. Lett., 2008, 10, 4117–4120 CrossRef CAS PubMed; (b) H. Y. Li, J. Horn, A. Campbell, D. House, A. Nelson and S. P. Marsden, Chem. Commun., 2014, 50, 10222–10224 RSC; (c) H. Lam, J. Tsoung and M. Lautens, J. Org. Chem., 2017, 82, 6089–6099 CrossRef CAS PubMed.
- T. Das, ChemistrySelect, 2020, 5, 14484–14509 CrossRef CAS.
- X.-P. Zeng, Z.-Y. Cao, Y.-H. Wang, F. Zhou and J. Zhou, Chem. Rev., 2016, 116, 7330–7396 CrossRef CAS PubMed.
- Y. Huang and T. Hayashi, Chem. Rev., 2022, 18, 14346–14404 CrossRef PubMed.
- (a) G. Pattison, G. Piraux and H. W. Lam, J. Am. Chem. Soc., 2010, 132, 14373–14375 CrossRef CAS PubMed; (b) I. D. Roy, A. B. Burns, G. Pattison, B. Michel, A. J. Parker and H. W. Lam, Chem. Commun., 2014, 50, 2865–2868 RSC; (c) M. Hatano and T. Nishimura, Angew. Chem., Int. Ed., 2015, 127, 11099–11102 CrossRef; (d) F. Xue and T. Hayashi, Angew. Chem., Int. Ed., 2018, 57, 10368–10372 CrossRef CAS PubMed.
- For the pioneering work and reviews on quantitative structure–selectivity relationship, see: (a) K. C. Harper and M. S. Sigman, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2179–2183 CrossRef CAS PubMed; (b) A. F. Zahrt, S. V. Athavale and S. E. Denmark, Chem. Rev., 2020, 120, 1620–1689 CrossRef CAS PubMed; (c) J. M. Crawford, C. Kingston, F. D. Toste and M. S. Sigman, Acc. Chem. Res., 2021, 54, 3136–3148 CrossRef CAS PubMed; (d) W. L. Williams, L. Zeng, T. Gensch, M. S. Sigman, A. G. Doyle and E. V. Anslyn, ACS Cent. Sci., 2021, 7, 1622–1637 CrossRef CAS PubMed.
- (a) M. R. Smith III, R. Bisht, C. Haldar, G. Pandey, J. E. Dannatt, B. Ghaffari, R. E. Maleczka Jr and B. Chattopadhyay, ACS Catal., 2018, 8, 6216–6223 CrossRef PubMed; (b) S. M. Preshlock, D. L. Plattner, P. E. Maligres, S. W. Krska, R. E. Maleczka Jr and M. R. Smith III, Angew. Chem., Int. Ed., 2013, 52, 12915–12919 CrossRef CAS PubMed.
- J. M. Lopchuk, I. L. Green, J. C. Badenock and G. W. Gribble, Org. Lett., 2013, 15, 4485–4487 CrossRef CAS PubMed.
- (a) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed; (b) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed; (c) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- For more information on Hayashi's stereochemical model, see: (a) G. Berthon and T. Hayashi in Catalytic Asymmetric Conjugate Reactions, ed. A. Córdova, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010, pp. 1–70 Search PubMed; (b) E. A. B. Kantchev, Chem. Sci., 2013, 4, 1864–1875 RSC.
- For the pioneering work on surface distance projection maps, see: (a) A. Poater, F. Ragone, R. Mariz, R. Dorta and L. Cavallo, Chem.–Eur. J., 2010, 16, 14348–14353 CrossRef CAS PubMed; (b) L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286–2293 CrossRef CAS; (c) L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- For the pioneering work and reviews on non-covalent interaction analysis and topology analysis, see: (a) E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 18, 6498–6506 CrossRef PubMed; (b) R. Laplaza, F. Peccati, R. A. Boto, C. Quan, A. Carbone, J.-P. Piquemal, Y. Maday and J. Contreras-García, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 11, e1497 Search PubMed; (c) R. F. W. Bader, Atoms in molecules: a quantum theory, Clarendon Press, Oxford, 1994, pp. 1–432 Search PubMed; (d) P. L. A. Popelier in The Chemical Bond: Fundamental aspects of chemical bonding, ed. G. Frenking and S. Shaik, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010, pp. 271–308 Search PubMed.
- For reviews on dispersion effects, see: (a) J. P. Wagner and P. R. Schreiner, Angew. Chem., Int. Ed., 2015, 54, 12274–12296 CrossRef CAS PubMed; (b) M. Stöhr, T. V. Voorhisb and A. Tkatchenko, Chem. Soc. Rev., 2019, 48, 4118–4154 RSC; (c) K. L. Mears and P. P. Power, Acc. Chem. Res., 2022, 55, 1337–1348 CrossRef CAS PubMed.
- For the pioneering work and reviews on Eyring analysis, see: (a) M. Palucki, N. S. Finney, P. J. Pospisil, M. L. Güler, T. Ishida and E. N. Jacobsen, J. Am. Chem. Soc., 1998, 120, 948–954 CrossRef CAS; (b) R. R. Knowles, S. Lin and E. N. Jacobsen, J. Am. Chem. Soc., 2010, 132, 5030–5032 CrossRef CAS PubMed; (c) E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, United States of America, 2006, pp. 370–372 Search PubMed.
- For reviews on enthalpy–entropy compensation, see: (a) W. Linert, Chem. Soc. Rev., 1994, 23, 429–438 RSC; (b) L. Liu and Q.-X. Guo, Chem. Rev., 2001, 101, 673–696 CrossRef CAS PubMed; (c) A. Pan, T. Biswas, A. K. Rakshit and S. P. Moulik, J. Phys. Chem. B, 2015, 119, 15876–15884 CrossRef CAS PubMed.
- Two other pathways, i.e., rhodium-catalyzed enantioposition-selective imine formation/Rh-catalyzed intramolecular addition/isomerization pathway or reversable imine formation/Rh-catalyzed dynamic kinetic intramolecular addition/isomerization pathway, are unlikely but not impossible. These pathways could not be ruled out at this stage.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization, and computational details. CCDC 2260854 (for 3aa). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02474a |
| This journal is © The Royal Society of Chemistry 2023 |