
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioinformatics-guided discovery of biaryl-linked lasso peptides†
Hamada
Saad‡
*ab,
Thomas
Majer‡
a,
Keshab
Bhattarai‡
a,
Sarah
Lampe
a,
Dinh T.
Nguyen
b,
Markus
Kramer
c,
Jan
Straetener
d,
Heike
Brötz-Oesterhelt
 de,
Douglas A.
Mitchell
b and
Harald
Gross
*ae
de,
Douglas A.
Mitchell
b and
Harald
Gross
*ae
aDepartment of Pharmaceutical Biology, Institute of Pharmaceutical Sciences, University of Tübingen, Auf der Morgenstelle 8, 72076 Tübingen, Germany. E-mail: hamada.saad@pharm.uni-tuebingen.de; harald.gross@uni-tuebingen.de
bDepartment of Chemistry and the Carl R. Woese Institute for Genomic Biology, University of Illinois at Urbana–Champaign, Urbana, Illinois 61801, USA
cInstitute of Organic Chemistry, University of Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany
dDepartment of Microbial Bioactive Compounds, Interfaculty Institute of Microbiology and Infection Medicine, University of Tübingen, Auf der Morgenstelle 28, 72076 Tübingen, Germany
eCluster of Excellence: EXC 2124: Controlling Microbes to Fight Infection, University of Tübingen, Tübingen, Germany
First published on 30th October 2023
Abstract
Lasso peptides are a class of ribosomally synthesized and post-translationally modified peptides (RiPPs) that feature an isopeptide bond and a distinct lariat fold. A growing number of secondary modifications have been described that further decorate lasso peptide scaffolds. Using genome mining, we have discovered a pair of lasso peptide biosynthetic gene clusters (BGCs) that include cytochrome P450 genes. Using mass spectrometry, stable isotope incorporation, and extensive 2D-NMR spectrometry, we report the structural characterization of two unique examples of (C–N) biaryl-linked lasso peptides. Nocapeptin A, from Nocardia terpenica, is tailored with a Trp–Tyr crosslink, while longipepetin A, from Longimycelium tulufanense, features a Trp–Trp linkage. Besides the unusual bicyclic frame, a Met of longipepetin A undergoes S-methylation to yield a trivalent sulfonium, a heretofore unprecedented RiPP modification. A bioinformatic survey revealed additional lasso peptide BGCs containing P450 enzymes which await future characterization. Lastly, nocapeptin A bioactivity was assessed against a panel of human and bacterial cell lines with modest growth-suppression activity detected towards Micrococcus luteus.
Introduction
Ribosomally synthesized and post-translationally modified peptides (RiPPs) represent a structurally and functionally diverse group of natural products. Through the combined effects of improved bioinformatic algorithms, genome sequencing campaigns, and isolation/characterization projects, RiPPs feature an extraordinary array of post-translational modifications and architectural scaffolds that cover a broad range of biological functions.1Lasso peptides are one of nearly 50 described classes of RiPPs and display a structurally unique lariat conformation as the class-defining feature. The N-terminus of the core peptide and a side chain carboxylate of an Asp/Glu residing at position 7–9 form a macrolactam ring that is threaded by the C-terminal “tail” residues.2 Large, steric-locking residues adjacent to the plane of the ring and/or disulfide bridge(s) result in a kinetically trapped rotaxane conformation that endows most lasso peptides with extraordinary thermal and proteolytic stability.3
Lasso peptide biosynthesis starts with the ribosomal synthesis of a bipartite precursor peptide (A) that contains N-terminal leader and C-terminal core regions. The leader portion harbors the recognition sequence, which directs the enzymatic processing events via interaction with the RiPP precursor recognition element (RRE).4 Meanwhile, the core region receives all of the post-translational modifications (PTMs).5 Upon RRE binding, leader peptidolysis is initiated by the co-occurring (B) protein, releasing the core peptide as a substrate for the ATP-dependent lasso cyclase (C).3a,6 In some lasso peptide biosynthetic pathways, secondary modifications include disulfide bonds, C-terminal methylesterification, N-acetylation, citrullination, O-phosphorylation, glycosylation, epimerization, β-hydroxylation, and aspartimidation.2b,7 The combination of genome sequencing and enhanced genome-mining algorithms has revealed a large number of RiPP-associated PTMs, including lasso peptides.7c,8
Bacteria of the genus Nocardia were first studied mainly to understand their pathogenicity. However, metabolic and genomic studies later demonstrated the extraordinary potential of Nocardia to produce bioactive and structurally diverse secondary metabolites distinct from other Actinomycetota.9 In contrast to the well-established and prolific biosynthetic potential of Streptomyces, only a fraction of the predicted natural products for Nocardia have been discovered to date. Known RiPPs from Nocardia are currently restricted to the lipolanthines nocavionin,10a nocaviogua A and B,10b the thiopeptides nocathiacins10c and nocardithiocin,10d and the chimeric lanthipeptide nocathioamide.8d
Based on this promising potential,8d,9c,11 we mined the genomes of two highly similar Nocardia terpenica strains (i.e., IFM 0406 and 0706T) for RiPPs using RODEO.8a The Nocardia-specific bioinformatics analyses identified three putative lasso peptide biosynthetic gene clusters (BGCs) which were not linked to any reported natural product. One Nocardia BGC contained a member of protein family PF00067,12 a predicted cytochrome P450 protein. While P450-encoding genes are relatively rare in bacterial RiPP BGCs,13 they have yet to be reported in a lasso peptide BGC. Since RiPP-associated P450 proteins perform versatile and complex oxidative transformations,13,14 we envisioned such BGCs would yield a new type of lasso peptide. Thus, we characterized the products of this pathway, termed the nocapeptins, given the origin was Nocardia terpenica (taxonomic order: Corynebacteriales). Bioinformatic expansion outside of Nocardia led to the discovery and characterization of the longipeptins, from the phylogenetically distant actinomycete, Longimycelium tulufanense (taxonomic order: pseudonocardiales).
Results and discussion
Genome mining uncovers lasso peptide BGCs associated with cytochrome P450 enzymes
During an effort to discover new RiPPs from Nocardia, we uncovered an unusual lasso peptide BGC (nop, Fig. 1). The nop BGC is 5.4 kb in length and consists of six open-reading frames (ORFs), designated nopA-F. RODEO was used to examine the constituent gene products,8a,15 which predicted a class II lasso peptide encoded by nopA. Functional annotation of the local ORFs identified the requisite lasso cyclase (NopC, from protein family PF00733), leader peptidase (NopB, PF13471), and discretely encoded RRE domain (NopE, PF05402) (Fig. 1 and Table S1†). As observed for nearly all discrete RRE domains (as opposed to fused cases), the RRE binding to NopA is governed by a characteristic YxxP-containing recognition sequence.5c,16,24 Furthermore, the BGC includes a putative ABC transporter (NopD, PF00005). Notably, and as the principal criterion for target selection, a cytochrome P450 protein (NopF, PF00067) is locally encoded (Table S1†).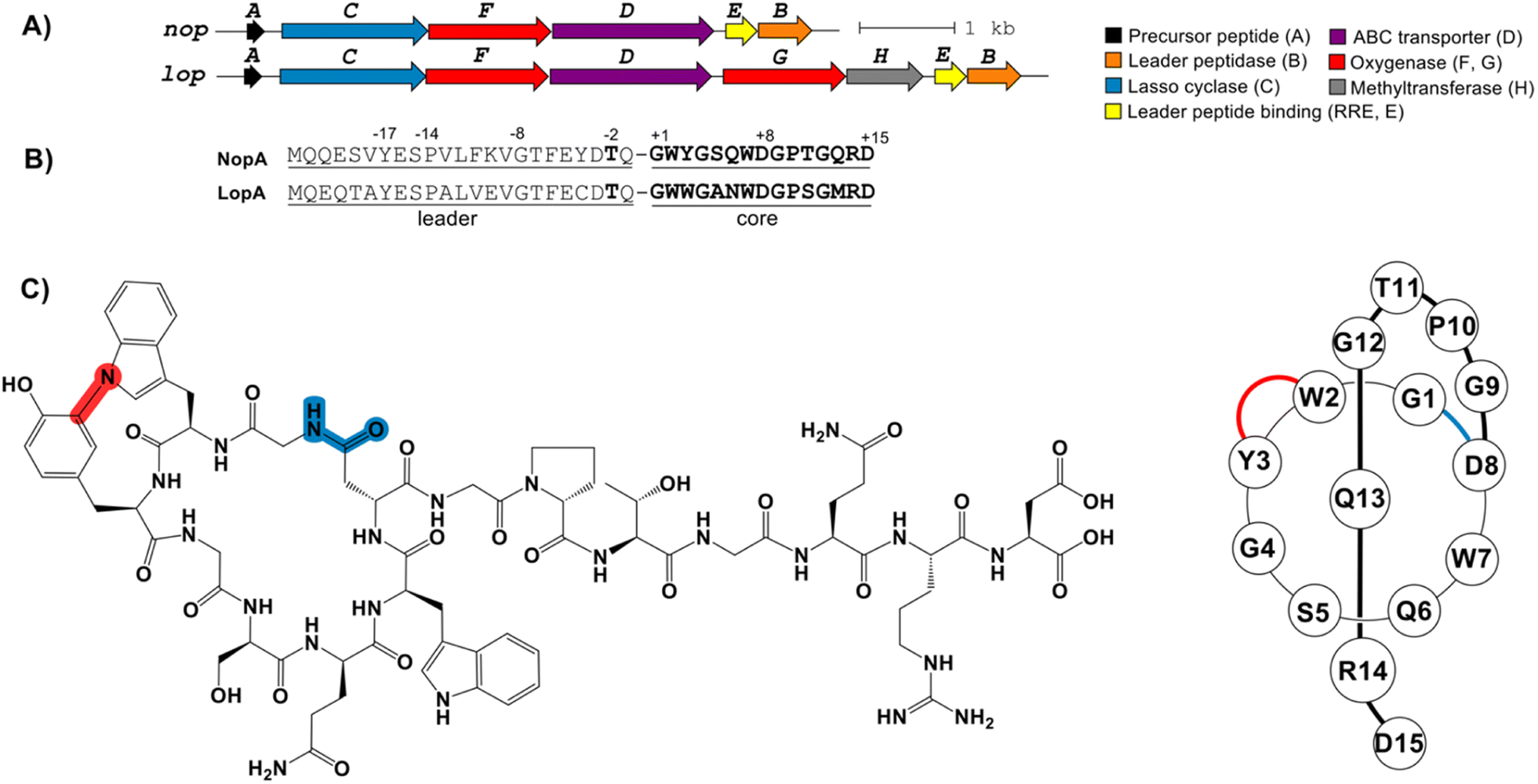 | ||
| Fig. 1 (A) The nop and lop BGCs produce nocapeptin A/B and longipeptin A–C, respectively. The A, C, D, E and B genes encode the precursor peptide, lasso cyclase, ABC transporter, RRE, and leader peptidase, respectively. The F and G genes encode two cytochrome P450 proteins while H encodes a hypothetical protein. (B) The leader and core regions of the precursor are indicated. Conserved leader peptides residues are numbered and bolded. (C) The structure formula of the bicyclic lasso peptide nocapeptin A (1, left); schematic drawing of nocapeptin A (1, right). | ||
A BLAST-P search of NopB/C followed by RODEO analysis unveiled a small number of similar BGCs (Fig. S2†). One such result was from Longimycelium tulufanense CGMCC 4.5737 and was termed lop BGC) BGC. The lop and nop BGCs exhibit considerable protein similarity and genetic synteny (Fig. 1 and Table S1†). However, L. tulufanense contains two additional genes, lopG and lopH, encoding a more divergent second cytochrome P450 protein and a protein of unknown function, respectively, suggesting that the resulting lasso peptide will receive additional modifications relative to the nop BGC product.
Based on the presence of a canonical Thr(-2), RODEO predicted that leader peptidolysis would occur at Gln/Gly for NopA and LopA, yielding 15-residue core peptides (Fig. 1). Macrocyclization was predicted between Gly1 and Asp8 with Arg14 being the most suitable candidate for a steric-locking residue. The P450 proteins encoded by the nop/lop BGCs suggested additional oxidations would be installed; however, bioinformatics cannot predict the specific modification(s) installed by these enzymes. Driven by the new core sequences and the unprecedented inclusion of P450 enzymes in the BGC, we initiated a media screening campaign to isolate and characterize the products of the nop/lop BGCs.
Metabologenomic identification of the lasso peptides
Culture extracts from N. terpenica IFM 0406 and L. tulufanense using 40 different growth media were screened by LC-MS, with particular attention given to metabolites matching the approximate size of the predicted core peptides (m/z 1500–2000).17 From the media screen, the MS profiles of IFM 0406 grown in a modified R4 medium at 32 °C exclusively afforded a pair of candidates with [M + 2H]2+ values of 845.3562 (major) and 861.3517 (minor), which were designated as nocapeptin A (1) and nocapeptin B (2), respectively (Fig. S3†). The neutral molecular formula prediction of 1 (C75H96N22O24) was consistent with macrolactam formation on the predicted core sequence and loss of 2H, suggesting an oxidative modification within the lasso peptide (Fig. S3†). Relative to 1, the molecular formula of 2 suggested two additional hydroxylation events (Fig. S4†).The collision-induced dissociation (CID) spectra of 1 and 2 showed a series of y and b ions confirming the NopA sequence (Fig. S4†). Despite the lower probability of observing internal fragments arising from two amide dissociation events, low-intensity y and b fragments were observed that corroborated the amino acid composition of the macrocycle and the residues forming the macrolactam (Tables S2 and S3†). A consistent −2.0157 Da deviation was found in most daughter ions, allowing localization of the secondary modification to the Gly1-Trp2-Tyr3 region.
Using a similar mass-targeted growth media screen,17L. tulufanense yielded candidate products with [M + 2H]2+ values of 851.3472 (major), 843.3501 (minor), and 844.3395 (trace), designated as longipeptins A–C (3–5), respectively (Fig. S7†). We hypothesized that the post-translational modification statues of the longipeptides would deviate from 1-2 owing to the presence of lopG and lopH in the BGC (Fig. 1). Using high-resolution tandem MS (HR-MS/MS), we confirmed the LopA sequence (Fig. S9–S11 and Tables S4–S6†). Besides the anticipated –2H loss, the neutral molecular formula of longipeptin A (3, C76H96N22O22S) was consistent with net addition of oxygen and methylation (+CH2) (Fig. S12 and S13†). While longipeptin B (4) retains methylation, it lacks hydroxylation, suggesting it may be a biosynthetic intermediate. On the other hand, longipeptin C (5) lacks methylation but retains net addition of oxygen (Fig. S12 and S13†).
Despite their structural similarity, the MS2 spectra of 3 and 4 differed significantly from 5 (Fig. S8†). To explore the discrepancy, and to determine if the net addition of oxygen was from hydroxylation, met oxidation, or another modification, we began with the analysis of the MS2 spectrum of 5. The resulting y/b ions agreed with the expected core sequence of LopA, a Gly1-Asp8 macrolactam, and loss of 2.0157 Da within the Gly1-Trp2-Trp3 region. Further MS2 inspection unveiled a new series of 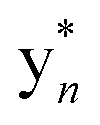 (yn-64 Da) ions consistent with ejection of Me2S and thus the presence of Met S-oxide (Fig. 2, S11 and Table S6†). Based on the trace amounts isolated, we conclude that 5 was likely formed by spontaneous air oxidation.
(yn-64 Da) ions consistent with ejection of Me2S and thus the presence of Met S-oxide (Fig. 2, S11 and Table S6†). Based on the trace amounts isolated, we conclude that 5 was likely formed by spontaneous air oxidation.
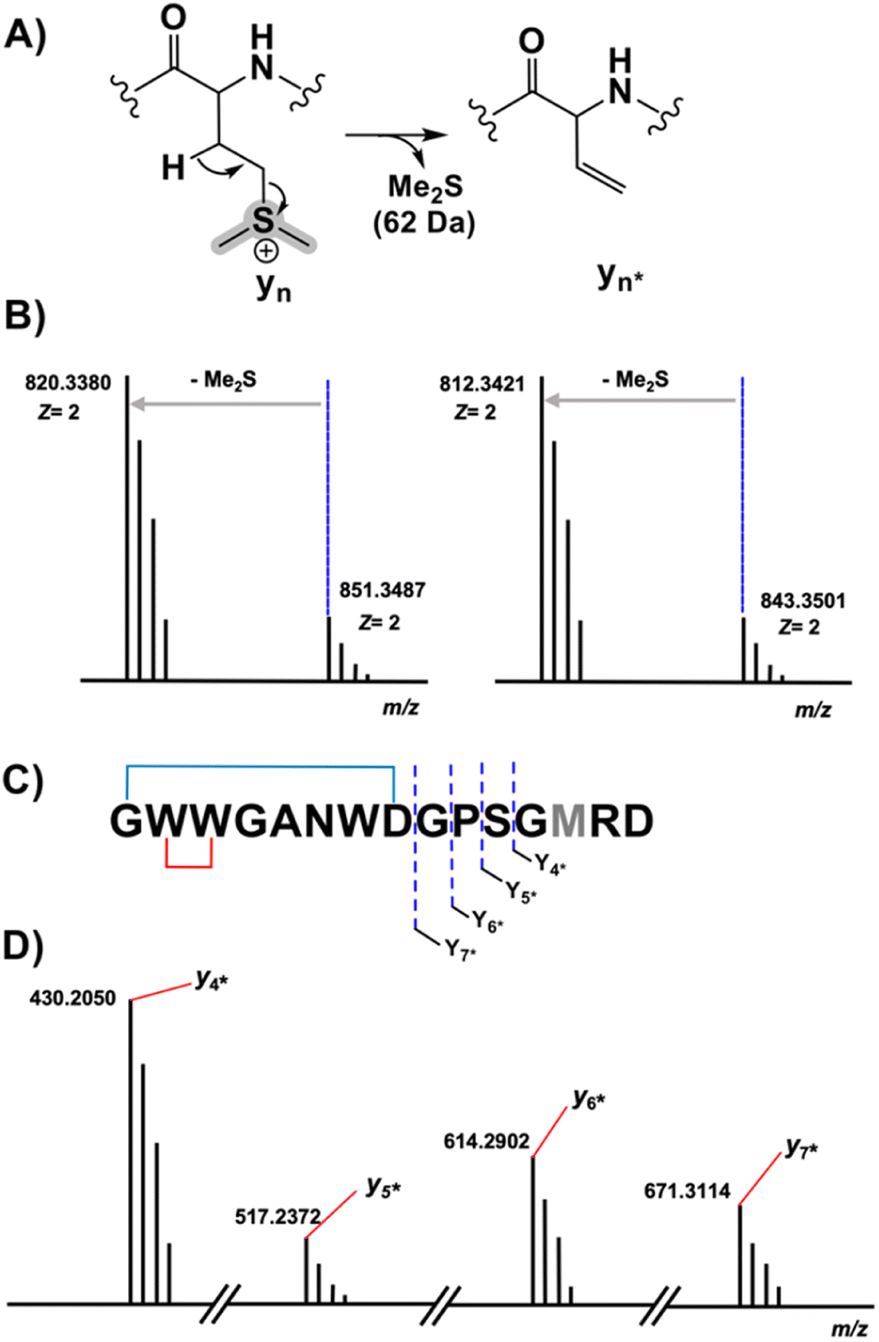 | ||
Fig. 2 MS characterization of the longipeptin sulfonium modification. (A) Proposed sulfonium fragmentation resulting in Me2S loss. (B) HR-MS/MS data of the [M + 2H]2+ of longipeptin A (left) and longipeptin B (right) exhibiting loss of Me2S. (C) Longipeptin core sequence detailing the 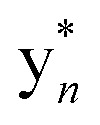 ions. (D) Annotated MS2 spectrum of longipeptin [M + 2H]2+ highlighting a new set of y ions ions. (D) Annotated MS2 spectrum of longipeptin [M + 2H]2+ highlighting a new set of y ions 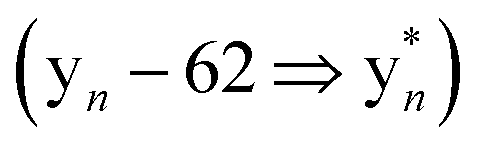 . . | ||
The b ion series of 3 and 4 supported the sequences and location of the previously noted modifications. In 3, the 2H loss and the exclusive hydroxylation were localized by the key b3 ion. Considering the spectral similarity of 3 and 4 (Fig. S8†) and the annotated b ions, methylation was localized to the Met13-Arg14-Asp15 C-terminal region. Considering several possibilities, S-methylation of Met13 was the most plausible and that such an assignment successfully dereplicated the full MS2 spectra of 3 and 4. The parent ions of 3 and 4 supported Met13 S-methylation by affording a characteristic M-62 ion, arising from a presumed β-elimination of dimethylsulfide (Fig. 2). In addition, the complete annotation of a new series of y fragments 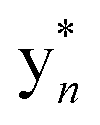 , (yn-62) including the most intense ions y4*–7*, corroborated the putative modification in 3 and 4 (Fig. S9–S10, Tables S4–S5†).
, (yn-62) including the most intense ions y4*–7*, corroborated the putative modification in 3 and 4 (Fig. S9–S10, Tables S4–S5†).
Stable isotope labeling of nocapeptin A
To evaluate the chemical nature of the −2 Da mass deviation localized to the first three residues of the nocapeptins (Gly-Trp-Tyr), feeding studies were conducted with [2H7] L-Tyr and [2H8] L-Trp.For nocapeptin A (1), the LC-MS profiles of IFM 0406 cultures supplemented with [2H7] L-Tyr and [2H8] L-Trp (all C–H protons replaced with deuterium) separately showed incorporation of +6 and +16 Da, respectively (Fig. 3, S5 and S6†). The [2H7] L-Tyr feeding data unambiguously demonstrated that the modification was not dehydrogenation, rather a single C–H of Tyr3 was involved in the mass deviation. The [2H8] L-Trp data showed the expected shift for Trp and allowed the conclusion that the C–H bonds of Trp2 and Trp7 were unaltered.
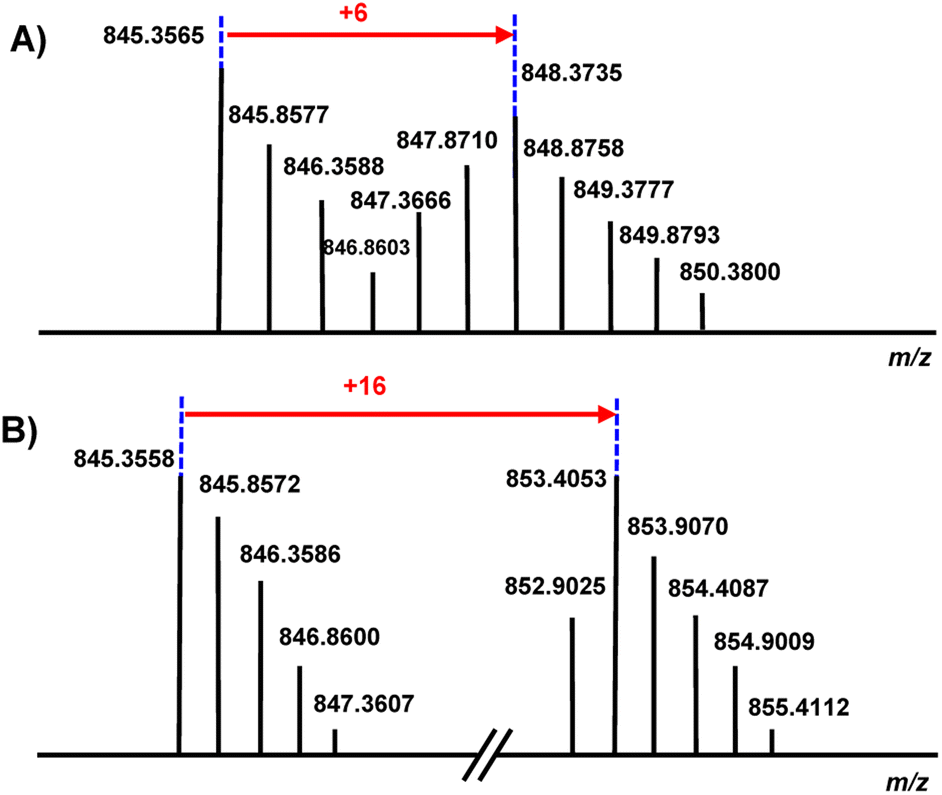 | ||
| Fig. 3 (A) The MS profile of nocapeptin A (1), [M + 2H]2+ upon culture supplementation with [2H7] L-Tyr. (B) The MS profile of nocapeptin A (1), [M + 2H]2+ upon culture supplementation with [2H8] L-Trp. | ||
Structure elucidation of nocapeptin A and longipeptin A
Larger scale cultures of IFM 0406 and L. tulufanense under the optimized expression conditions were performed to isolate the material required for structure determination. Subjecting n-butanol extracts of the supernatants to reversed phase C18 open column chromatography and pentafluorphenyl-phase-based HPLC, guided by LC-MS, enabled the isolation of nocapeptin A (1) and traces of longipeptin A (3). The elemental composition of 1 was determined as C75H96N22O24 with 39 ring/double bond equivalents. Extensive NMR analysis was then performed to elucidate the structure of nocapeptin A. 1H–1H COSY, 1H–1H TOCSY, 1H–13C HSQC, and 1H–13C HSQC-TOCSY of the exchangeable NH protons (δH 6.5–9.0) enabled assignment of nearly all spin systems (Fig. 4 and S16–S19†). These structural fragments were complemented with 1H–13C HMBC correlations between their side chain hydrogen and carbon atoms to deliver a complete set of amino acid moieties, 4xGly, 1xSer, 2xGln, 2xAsp, 1xPro, 1xThr and 1xArg (Fig. 4D and S20†). Furthermore, three aromatic residues, 2xTrp and 1xTyr, were mainly disclosed with the aid of 1H–13C HMBC and 1H–1H NOESY experiments (Fig. 4D, S28–S29 and S32†). In contrast to the typical AA'XX’ pattern for Tyr, an alternative ABX/AMX spin system with a characteristic upfield signal (δH 5.46, d ≈ 2 Hz)18 was observed, signifying a meta and para-disubstituted phenyl substructure. Making use of the 1H–13C HMBC, and 1H–1H NOESY couplings, such a signal was found to be the 2H-Tyr3 (Fig. 4C and S29†).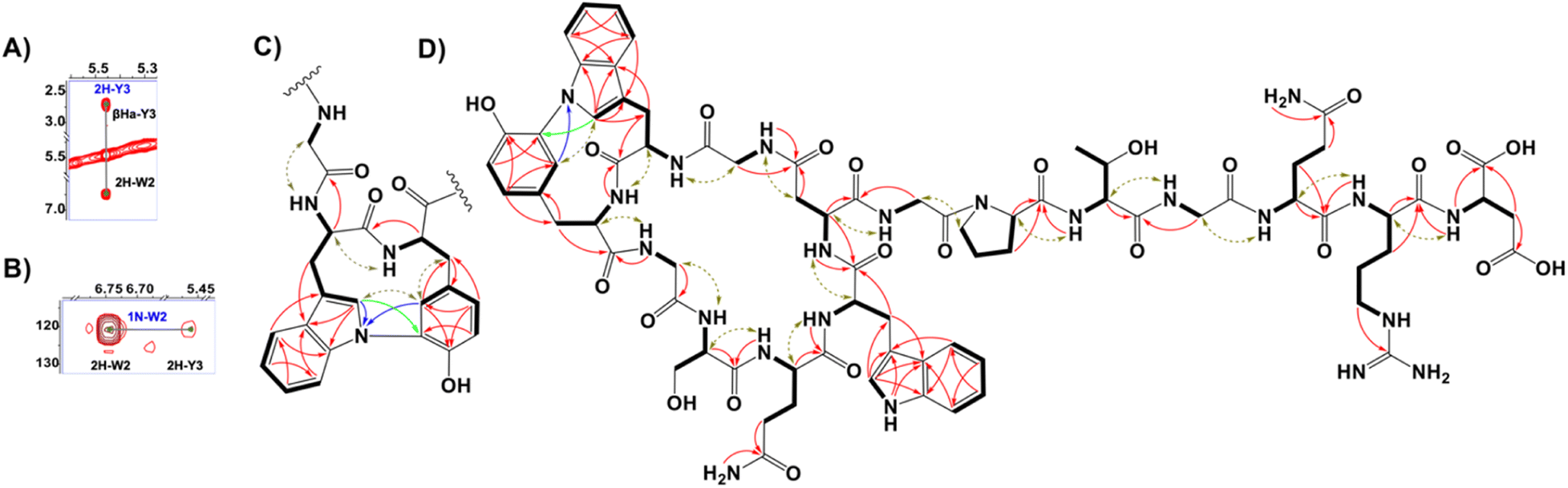 | ||
| Fig. 4 (A) 1H–1H NOESY spectrum highlighting the correlations between 2H-Trp2 and 2H-Tyr3 centers in nocapeptin A (1). (B) 1H–15N HMBC spectrum showing the correlations between the 1N-Trp2 and 2H-Tyr3 in nocapeptin A (1). (C) The key NMR correlations proving the elucidated biaryl fragment (1H–1H COSY and 1H–1H TOCSY: bold lines, 1H–1H NOESY: brown arrows, 1H–13C HMBC: red arrows, 1H–15N HMBC: blue arrows and 1H–13C LR-HSQMBC: green arrows). (D) The complete 2D-chemical structure of nocapeptin A (1) with the key NMR correlations. | ||
Notably, 1H-NMR analysis highlighted a discriminative resonance of a single aromatic NH of one Trp residue (δH 10.47, broad singlet), despite two Trp present in the peptide (Fig. S14†). 1H–13C HMBC and 1H–1H NOESY correlations assigned the unmodified Trp as Trp7 (Fig. S32†), and hence, Trp2 was expected to be modified at the indolic N1 position. The observable couplings obtained from a 1H–15N HMBC spectrum, between H2-Trp2 and H2-Tyr3 with N1-Trp2 (δN 120.80) in addition to 1H–13C LR-HSQMBC experiment,191H–1H NOESY relationships, and the isotope labeling experiments permitted the assignment of a biaryl connection between the N1 of Trp2 and C3 of Tyr3 (Fig. 4, S21, S23 and S29†). The polypeptide backbone of 1 was assigned via the sequential connectivity of the delineated fragments using 1H–13C HMBC cross-peaks from α-H resonances to the amide carbonyls of the neighboring amino acid. In addition, the detected Hα,β(i) → HN(i + 1) NOESY correlations supported the complete sequence. The macrolactam between Gly1 and Asp8 was validated similarly (Fig. 4D and S33†). Ultimately, a threaded conformation of 1 was evident with Gln13 and Arg14 assigned as upper and lower plugs, respectively, via NOESY correlations (Fig. S23†). The latter experimental findings were in good agreement with the calculated conformation model of the backbone structure, generated with the software LassoHTP.20
Using our stable isotope labeling results, we defined the absolute configuration of three amino acids. Thus, the interpretation of the MS-shift patterns showed that both Trp residues and the Tyr unit possess the L-configuration. In addition, L-Pro10 residue was deduced similarly and the stereochemistry of the remaining chiral residues of 1 were presumed to be L-configured. In analogy to other biarylic RiPPs,21 we expect that, due to the extra crosslink, atropisomery is given for nocapeptins. However, since in the 1H or 13C NMR spectra neither broad nor doubled resonances were observed, we assume that only one atropisomer is biosynthesized by nature.
Unlike nocapeptin A (1), the longipeptin A (3) titer was too low for a complete NMR-based structural elucidation. However, keeping in mind the anticipated modification of 3, the available NMR data permitted partial structural elucidation, including substructures containing the modifications. The annotated spin systems from 1H–1H COSY, 1H–1H TOCSY, and 1H–13C HSQC-TOCSY enabled the assignment of 1xAla, 1xPro, 1xSer, 1xArg, 1xAsn, 2xAsp, and 2xGly (Fig. S45–S47†).
Expectedly, the NMR data showed a significant downfield shift of Met γ-CH2 (δC 43.13 instead of the typical ∼30 ppm). Thus, in agreement with the MS data, we proposed this signal originated from S-methylation of Met13, forming a deshielded sulfonium (Fig. 5 and S47†). In addition, three candidate spin systems including the backbone amidic NH, α-H, and β-Ha+b were assembled in tandem with three aromatic systems to constitute the 3xTrp units (Fig. S47†).
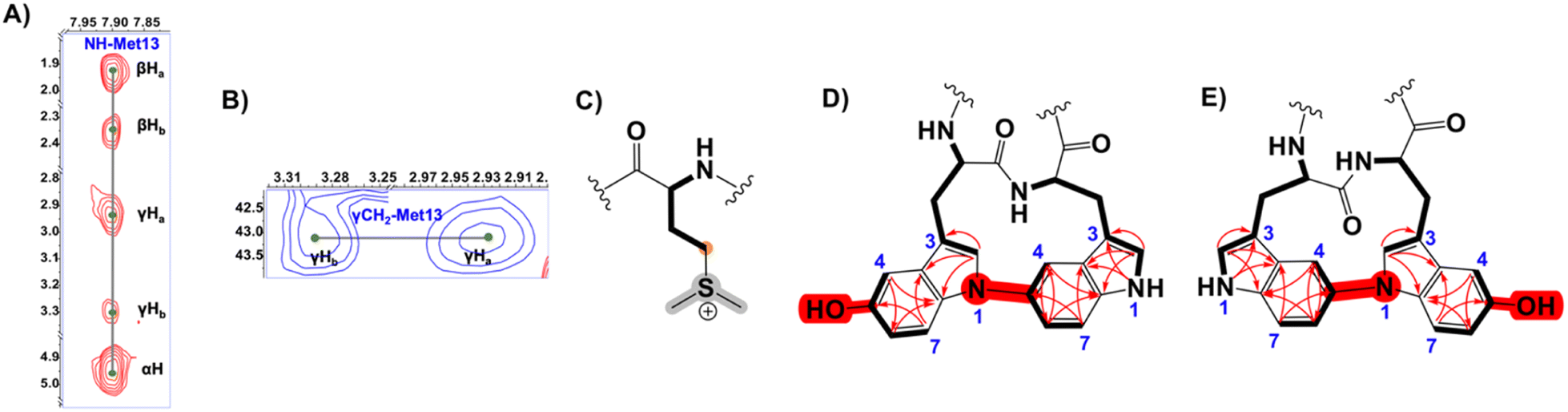 | ||
| Fig. 5 (A) 1H–1H TOCSY spectrum showing the Met13 spin system of longipeptin A (3) (B) 1H–13C HSQC spectrum highlighting the downfield cross-peaks of the γCH2 of S-methylated Met13 residue in longipeptin A (3) (C) Structure of the Met13 sulfonium elucidated by MS/NMR. (D and E) The proposed positional isomers of the (C–N) biaryl fragment of longipeptin A (3) with the key NMR couplings (1H–1H TOCSY, bold lines and 1H–13C HMBC, red arrows) that assembled the constituting structural units, Trp2 and Trp3. | ||
Unfortunately, we could not obtain an adequate 1H–1H NOESY spectrum, which prevented a confident connection of the Trp separate substructures even though weaker allylic 4J2H, βHa+b couplings, observed in the 1H–1H TOCSY spectrum suggested possible connectivities (Fig. S45 and S47†). The characteristic indolic NHs of 2xTrp as a pair of singlets (δH 10.18 and 10.55) (Fig. S41A†) and the 1H–13C HMBC correlations identified high-confidence locations of a crosslink and hydroxylation event in the biaryl-containing substructure (Fig. 5, S46 and S47†). Building on the MS data and comparable NMR shifts of 1, two possible isomers were proposed in which the (N1–C5) biaryl crosslink was adopted alternatively between Trp2 and Trp3 (Fig. 5 and S48†).
Evaluation of the biological activity and stability of nocapeptin A
Nocapeptin A (1) displayed no activity in the National Cancer Institute (NCI) cell-line cytotoxicity screen for antitumor agents. However, when assessed against a panel of bacteria, 1 exhibited growth suppression with an MIC of 16 μg mL−1 towards Micrococcus luteus (Tables S9 and 10†). We then tested the stability of 1, by heating the lasso peptide to 95 °C for 8 h (Fig. S49†) and by carboxypeptide Y or chymotrypsin treatment (Fig. S50†). Nocapeptin A was unaffected by these procedures, indicating high heat and proteolytic stability.Nocapeptins and longipeptins are uniquely tailored bicyclic lasso peptides
The lop BGC was expected to encode a lasso peptide with related chemical features. Unusually, longipeptin A (3) contained the biaryl linkage of interest in addition to indole hydroxylation and Met S-methylation. Despite the low titer, two positional isomers were presented for the (C–N) biaryl fragment (Fig. 5). In analogy to nocapeptins, we assume that N1 of Trp2 is coupled with C5 of Trp3.Considering their sequence similarity, we predict the P450 proteins NopF/LopF (76% identity, 87% similarity) form the biaryl linkages in 1 and 3, respectively. Given that Trp hydroxylation is unique to longipeptin, we anticipate LopG, a second P450 protein encoded by the lop BGC, performs this reaction. The greater sequence divergence of LopG relative to LopF/NopF (35% and 37% identity, respectively) supports this assignment.
Biarylic crosslinks are present in several distinct RiPP classes. Crocagin A22 is a tripeptide RiPP, in which two C-terminal Tyr–Trp residues undergo indole-backbone (C–N) cyclization by a dioxygenase. Atropeptides21b represent a P450-modified RiPP class that contains C–C and C–N linkages between Trp and Tyr residues. Biarylitides23 also contain P450-dependent C–C or C–N linkages between Tyr and His residues. While biarylitides display crosslinks between the first and third residues of the core region, 1 and 3 possess biaryl crosslinks at adjacent aromatic residues.
To shed light on the sequence-function relatedness of NopF/LopF versus known P450-modified RiPPs, a sequence similarity network (SSN) was constructed using the top 1000 non-redundant BLAST-P hits of NopF, predicted atropeptide- and biarylitide-associated cytochrome P450 proteins, and 883 P450 proteins encoded within 10 ORFs of an RRE domain.5c,21b,24 The SSN and similarity/identity analysis (Fig. S51 and S52†) suggested that NopF/LopF are rare examples of P450 proteins within lasso peptide BGCs. A comparative analysis of sequence space also demonstrates that NopF/LopF have significantly diverged from other RiPP-associated cytochrome P450 proteins.
Perhaps the most unusual PTM described in this study is the rare S-methylation of Met that affords a trivalent sulfonium. Given the other functional assignments, LopH is the most probable candidate Met S-methyltransferase. LopH shares modest sequence similarity (45%) with a hypothetical methyltransferase (WP_051757134.1, PF00145, Fig. S53†). To support or refute a tentative assignment of LopH as a methyltransferase, we obtained an AlphaFold-predicted structure and used DALI25 to identify structurally homologous matches from the Protein DataBank.26 The top hit was a methyltransferase domain-containing subunit of human 5,10-methylenetetrahydrofolate reductase (PDB code: 6fcx). This structure was crystallized with S-adenosylhomocysteine (SAH) bound (Fig. S54, S55 and S57†).27 A comparison of 6fcx and LopH found considerable similarity in the S-adenosylmethionine (SAM)-binding sites (specifically, 6fcx residues Glu463, Thr464, Thr481, Ser484, Thr560, and Thr573; which are equivalent to LopH Glu45, Thr46, Thr63, Ser66, Thr126, and Thr134) (Fig. S56 and S58†). As SAM is a common methyl donor for methyltransferases, the predicted structures and the sequence comparison support a tentative assignment of LopH in forming the sulfonium moiety in 3.28 Future experimental work will be necessary to confirm this prediction.
Sulfonium groups will readily react with nucleophiles if dealkylation or substitution is possible.29 The S-methylated Met residue of longipeptin A was stable to extraction and purification. The enhanced stability may be attributed to the position of Met within the lasso peptide structure as this position is equivalent to a steric plug residue established in nocapeptin A.
S-Methylated RiPPs are quite rare with only two cases of thiopeptides, Sch40832 (ref. 30) and the structurally related thioxamycin31 and thioactin.32S-Methylation has also been described for a proteusin that is proposed to contain a single S-methylated Cys.33 Thiol methylation was also illustrated only in a few NRPS cases. Echinomycin and thiocoraline represent NRPS-derived products where S-methylation is catalyzed by a SAM-dependent methyltransferase (Ecm18) or a bifunctional enzyme (TioN) with S-methyltransferase and amino acid adenylation domains.34 Maremycin A/B/G and FR900452 provide further examples of S-methylation from BGCs containing homologs of the methyltransferase MarQ.35
These examples highlight single S-methylation of Cys, thereby differing from the current report on S-dimethylmethionine. Unique examples for charged sulfonium units represent bleomycin A2 (ref. 36) and dimethylsulfoniopropionate (DMSP),37 respectively. Bleomycin A2 carries a 3-aminopropyl-dimethylsulfonium moiety as terminal amine at the Eastern side chain of the molecule. While the freestanding C domain BlmII is postulated to mediated the fusion of the charged side chain with the bleomycin aglycon, the actual biosynthetic origin of 3-aminopropyl-dimethylsulfonium moiety remains enigmatic. DMSPs are well known metabolites found in marine environments. Related compounds have been described, such as gonyol and gonydiol, which are DMSP-derived biosynthetic intermediates of malleicyprols.37 Generally, DMSP-related metabolites are part of the sulfur cycle, and some marine microorganisms, they protect against osmotic, oxidative, and thermal stresses. In this context, it is notable that the longipeptins producer, L. tulufanense, was isolated from a high-salinity lake38 which may connect the molecular structure of 3 with DMSP ecology.
Conclusions
In summary, we describe two lasso peptides, 1 and 3, tailored with four novel PTMs (C–N biaryl linkages between Trp–Trp and Trp–Tyr, S-methyl-Met, and 5-hydroxy-Trp), which are installed by a unique combination of RiPP biosynthetic enzymes. The current findings illustrate the value of targeted genome mining in prioritizing novel BGCs from underexplored/rare actinomycetes. As shown earlier, using tandem MS/isotopic incorporation facilitated the gene-to-molecule connection and also compensated for bioinformatic limitations in predicting the chemistry of the secondary PTM enzymes under investigation. Lasso peptides 1 and 3 represent highly tailored entities with 12/13 cyclic C–N biaryl systems fused with an 8-mer macrolactam cycle, respectively.The BGCs of 1 and 3 encode unique structural features, and the bioinformatic efforts in this study expand the range of PTMs associated with lasso peptide biosynthesis, suggesting that additional oxidative tailoring steps may yet be uncovered (Fig. S59†). To our surprise, the co-occurrence of cytochrome P450 proteins with further maturation enzymes in some retrieved candidate BGCs presents a roadmap to discover additional lasso peptides (Fig. S59†). Given the structural constraint installed by NopF/LopF, future work is warranted to reconstitute the enzymatic activity and substrate scope.39 Lastly, the prediction that LopH is a founding member of a new Met S-methyltransferase family suggests that biochemical characterization of LopH may result in diversifying the existing methylation panel with a sulfonium PTM that can be harnessed in different bioconjugation contexts.40
Data availability
The NMR and MS datasets generated and analysed during the current study are not publicly available but are available from the authors on request.Author contributions
H. S. conceptualized the project. H. S., T. M., K. B., S. L., D. T. N., M. K. and J. S. performed experiments. H. S., T. M., S. L., D. T. N., D. A. M. and H. G. analyzed data. H. S. wrote the first draft. H. S., H. B. O., D. A. M. and H. G. edited the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The NCI-60 anticancer screening service was generously provided by the National Cancer Institute (NCI) as part of the Development Therapeutics Program (DTP). We thank Dr D. Wistuba and her team (Mass Spectrometry Department, Institute for Organic Chemistry, University of Tübingen, Germany) for HR-MS measurements. We thank F. Mier (Pharmaceutical Institute, University of Tübingen) and Z. J. Yang and R. J. Juarez (Dept. of Chemistry, Vanderbilt University, TN, USA) for their help implementing the software LassoHTP. H. S. gratefully acknowledges the Ministry of Higher Education of Egypt (MOHE) for funding. K. B. gratefully acknowledges the funding for a PhD scholarship from the Deutscher Akademischer Austauschdienst (DAAD). D. A. M. acknowledges funding from the U.S. National Institute of General Medical Sciences (GM123998), while H. B.-O. acknowledges funding from the German Center for Infection Research (DZIF, TTU 09-818). Infrastructural support is from the Cluster of Excellence EXC 2124 (project ID 390838134) funded by the Deutsche Forschungsgemeinschaft (DFG).Notes and references
- (a) M. Montalbán-López, T. A. Scott, S. Ramesh, I. R. Rahman, A. J. van Heel, J. H. Viel, V. Bandarian, E. Dittmann, O. Genilloud, Y. Goto, M. J. Grande Burgos, C. Hill, S. Kim, J. Koehnke, J. A. Latham, A. J. Link, B. Martinez, S. K. Nair, Y. Nicolet, S. Rebuffat, H.-G. Sahl, D. Sareen, E. W. Schmidt, L. Schmitt, K. Severinov, R. D. Süssmuth, A. W. Truman, H. Wang, J.-K. Weng, G. P. van Wezel, Q. Zhang, J. Zhong, J. Piel, D. A. Mitchell, O. P. Kuipers and W. A. van der Donk, Nat. Prod. Rep., 2021, 38, 130–239 RSC; (b) Y. Li and S. Rebuffat, J. Biol. Chem., 2020, 295, 34–54 CrossRef CAS PubMed; (c) C. Ongpipattanakul, E. K. Desormeaux, A. DiCaprio, W. van der Donk, D. A. Mitchell and S. K. Nair, Chem. Rev., 2022, 122, 14722–14814 CrossRef CAS PubMed.
- (a) M. O. Maksimov, S. J. Pan and A. J. Link, Nat. Prod. Rep., 2012, 29, 996–1006 RSC; (b) J. D. Hegemann, K. J. Dit-Foque and X. Xie, in Comprehensive Natural Products III: Chemistry and Biology, ed. H.-W. Liu and T. P. Begley, Elsevier, 3rd edn, 2020, vol. 2, pp. 206–228 Search PubMed.
- (a) J. D. Hegemann, M. Zimmermann, X. Xie and M. A. Marahiel, Acc. Chem. Res., 2015, 48, 1909–1919 CrossRef CAS PubMed; (b) J. D. Hegemann, in Methods in Enzymology, ed. L. M. Hicks, Elsevier, 2021, vol. 663, pp. 177–204 Search PubMed; (c) J. D. Hegemann, ChemBioChem, 2020, 21, 7–18 CrossRef CAS PubMed.
- (a) S. Zhu, C. D. Fage, J. D. Hegemann, A. Mielcarek, D. Yan, U. Linne and M. A. Marahiel, Sci. Rep., 2016, 6, 35604 CrossRef CAS PubMed; (b) T. Sumida, S. Dubiley, B. Wilcox, K. Severinov and S. Tagami, ACS Chem. Biol., 2019, 14, 1619–1627 CrossRef CAS PubMed; (c) A. Alfi, A. Popov, A. Kumar, K. Y. J. Zhang, S. Dubiley, K. Severinov and S. Tagami, ACS Synth. Biol., 2022, 11(6), 2022–2028 CrossRef CAS PubMed; (d) A. M. Kretsch, M. G. Gadgil, A. J. DiCaprio, S. E. Barrett, B. L. Kille, Y. Si, L. Zhu and D. A. Mitchell, Biochem, 2023, 62, 956–967 CrossRef CAS PubMed.
- (a) B. J Burkhart, G. A. Hudson, K. L Dunbar and D. A. Mitchell, Nat. Chem. Biol., 2015, 11, 564–570 CrossRef PubMed; (b) J. Koehnke, G. Mann, A. F. Bent, H. Ludewig, S. Shirran, C. Botting, T. Lebl, W. E. Houssen, M. Jaspars and J. H. Naismith, Nat. Chem. Biol., 2015, 11, 558–563 CrossRef CAS PubMed; (c) A. M. Kloosterman, K. E. Shelton, G. P. van Wezel, M. H. Medema and D. A. Mitchell, mSystems, 2020, 5, e00267 CrossRef CAS PubMed.
- (a) K. P. Yan, Y. Li, S. Zirah, C. Goulard, T. A. Knappe, M. A. Marahiel and S. Rebuffat, ChemBioChem, 2012, 13, 1046–1052 CrossRef CAS PubMed; (b) A. J. DiCaprio, A. Firouzbakht, G. A. Hudson and D. A. Mitchell, J. Am. Chem. Soc., 2019, 141, 290–297 CrossRef CAS PubMed; (c) J. D. Koos and A. J. Link, J. Am. Chem. Soc., 2019, 141, 928–935 CrossRef CAS PubMed.
- (a) C. Zhang and M. R. Seyedsayamdost, ACS Chem. Biol., 2020, 15, 890–894 CrossRef CAS PubMed; (b) L. Cao, M. Beiser, J. D. Koos, M. Orlova, H. E. Elashal, H. V. Schröder and A. J. Link, J. Am. Chem. Soc., 2021, 143, 11690–11702 CrossRef CAS PubMed; (c) L. A. Harris, P. M. B. Saint-Vincent, X. Guo, G. A. Hudson, A. J. DiCaprio, L. Zhu and D. A. Mitchell, ACS Chem. Biol., 2020, 15, 3167–3175 CrossRef CAS PubMed.
- (a) J. I. Tietz, C. J. Schwalen, P. S. Patel, T. Maxson, P. M. Blair, H.-C. Tai, U. I. Zakai and D. A. Mitchell, Nat. Chem. Biol., 2017, 13, 470–478 CrossRef CAS PubMed; (b) W. L. Cheung-Lee and A. J. Link, J. Ind. Microbiol. Biotechnol., 2019, 46, 1371–1379 CrossRef CAS PubMed; (c) D. Y. Travin, D. Bikmetov and K. Severinov, Front. Genet., 2020, 11, 26 CrossRef PubMed; (d) H. Saad, S. Aziz, M. Gehringer, M. Kramer, J. Straetener, A. Berscheid, H. Brötz-Oesterhelt and H. Gross, Angew. Chem., Int. Ed., 2021, 60, 16472–16479 CrossRef CAS PubMed.
- (a) D. Männle, S. M. K. McKinnie, S. S. Mantri, K. Steinke, Z. Lu, B. S. Moore, N. Ziemert and L. Kaysser, mSystems, 2020, 5, e00125 CrossRef PubMed; (b) D. Dhakal, V. Rayamajhi, R. Mishra and J. K. Sohng, J. Ind. Microbiol. Biotechnol., 2019, 46, 385–407 CrossRef CAS PubMed; (c) A. Engelbrecht, H. Saad, H. Gross and L. Kaysser, Microb. Physiol., 2021, 31, 217–232 CrossRef PubMed; (d) Q. Luo, S. Hiessl and A. Steinbüchel, Environ. Microbiol., 2014, 16, 29–48 CrossRef CAS PubMed.
- (a) V. Wiebach, A. Mainz, M. A. J. Siegert, N. A. Jungmann, G. Lesquame, S. Tirat, A. Dreux-Zigha, J. Aszodi, D. Le Beller and R. D. Süssmuth, Nat. Chem. Biol., 2018, 14, 652–654 CrossRef CAS PubMed; (b) S. Chang, Y. Luo, N. He, X. Huang, M. Chen, L. Yuan and Y. Xie, Front. Chem., 2023, 11, 1233938 CrossRef CAS PubMed; (c) J. E. Leet, W. Li, H. A. Ax, J. A. Matson, S. Huang, R. Huang, J. L. Cantone, D. Drexler, R. A. Dalterio and K. S. Lam, J. Antibiot., 2003, 56, 232–242 CrossRef CAS PubMed; (d) A. Mukai, T. Fukai, Y. Hoshino, K. Yazawa, K. I. Harada and Y. Mikami, J. Antibiot., 2009, 62, 613–619 CrossRef CAS PubMed.
- (a) J. Chen, A. Frediansyah, D. Männle, J. Straetener, H. Brötz-Oesterhelt, N. Ziemert, L. Kaysser and H. Gross, ChemBioChem, 2020, 21, 2205–2213 CrossRef CAS PubMed; (b) A. Botas, M. Eitel, P. N. Schwarz, A. Buchmann, P. Costales, L. E. Núñez, J. Cortés, F. Morís, M. Krawiec, M. Wolanski, B. Gust, M. Rodriguez, W. N. Fischer, B. Jandeleit, J. Zakrzewska-Czerwińska, W. Wohlleben, E. Stegmann, P. Koch, C. Méndez and H. Gross, Angew. Chem., Int. Ed., 2021, 60, 13536–13541 CrossRef CAS PubMed.
- J. Mistry, S. Chuguransky, L. Williams, M. Qureshi, G. A Salazar, E. L. L. Sonnhammer, S. C. E. Tosatto, L. Paladin, S. Raj, L. J. Richardson, R. D. Finn and A. Bateman, Nucleic Acids Res., 2021, 49, D412–D419 CrossRef CAS PubMed.
- (a) G. Zhong, ACS Bio Med Chem Au, 2023, 3, 371–388 CrossRef CAS PubMed; (b) S. Kunakom, H. Otani, D. W. Udwary, D. T. Doering and N. J. Mouncey, J. Ind. Microbiol. Biotechnol., 2023, 50, kuad005 CrossRef CAS PubMed.
- (a) J. D. Rudolf, C. Y. Chang, M. Ma and B. Shen, Nat. Prod. Rep., 2017, 34, 1141–1172 RSC; (b) M. C. Tang, Y. Zou, K. Watanabe, C. T. Walsh and Y. Tang, Chem. Rev., 2017, 117(8), 5226–5333 CrossRef CAS PubMed.
- C. J. Schwalen, G. A. Hudson, B. Kille and D. A. Mitchell, J. Am. Chem. Soc., 2018, 140, 9494–9501 CrossRef CAS PubMed.
- (a) X. Yang and W. A. van der Donk, Chem.–Eur. J., 2013, 19, 7662–7677 CrossRef CAS PubMed; (b) B. J. Burkhart, G. A. Hudson, K. L. Dunbar and D. A. Mitchell, Nat. Chem. Biol., 2015, 11, 564–570 CrossRef CAS PubMed.
- H. Gross, Appl. Microbiol. Biotechnol., 2007, 75, 267–277 CrossRef CAS PubMed.
- T. P. Wyche, A. C. Ruzzini, L. Schwab, C. R. Currie and J. Clardy, J. Am. Chem. Soc., 2017, 139, 12899–12902 CrossRef CAS PubMed.
- R. T. Williamson, A. V. Buevich, G. E. Martin and T. Parella, J. Org. Chem., 2014, 79, 3887–3894 CrossRef CAS PubMed.
- R. J. Juarez, Y. Jiang, M. Tremblay, Q. Shao, A. J. Link and Z. J. Yang, J. Chem. Inf. Model., 2023, 63, 522–530 CrossRef CAS PubMed.
- (a) J. J. Hug, J. Dastbaz, S. Adam, O. Revermann, J. Koehnke, D. Krug and R. Müller, ACS Chem. Biol., 2020, 15, 2221–2231 CrossRef CAS PubMed; (b) P. Nanudorn, S. Thiengmag, F. Biermann, P. Erkoc, S. D. Dirnberger, T. N. Phan, R. Fürst, R. Ueoka and E. J. N. Helfrich, Angew. Chem., Int. Ed., 2022, 61, e2022083 CrossRef PubMed.
- K. Viehrig, F. Surup, C. Volz, J. Herrmann, A. Abou Fayad, S. Adam, J. Köhnke, D. Trauner and R. Müller, Angew. Chem., Int. Ed., 2017, 56, 7407 CrossRef CAS PubMed.
- (a) M. M. Zdouc, M. M. Alanjary, G. S. Zarazúa, S. I. Maffioli, M. Crüsemann, M. H. Medema, S. Donadio and M. Sosio, Cell Chem. Biol., 2021, 28, 733–739 CrossRef CAS PubMed; (b) J. J. Hug, N. A. Frank, C. Walt, P. Senica, F. Panter and R. Müller, Molecules, 2021, 26, 7483 CrossRef CAS PubMed.
- K. E. Shelton and D. A. Mitchell, Methods Enzymol., 2023, 679, 191–233 CAS.
- L. Holm, Nucleic Acids Res., 2022, 50, W210–W215 CrossRef CAS PubMed.
- S. K. Burley, C. Bhikadiya, C. Bi, S. Bittrich, L. Chen, G. V. Crichlow, C. H. Christie, K. Dalenberg, L. D. Costanzo, J. M. Duarte, S. Dutta, Z. Feng, S. Ganesan, D. S. Goodsell, S. Ghosh, R. K. Green, V. Guranović, D. Guzenko, B. P. Hudson, C. L. Lawson, Y. Liang, R. Lowe, H. Namkoong, E. Peisach, I. Persikova, C. Randle, A. Rose, Y. Rose, A. Sali, J. Segura, M. Sekharan, C. Shao, Y. P. Tao, M. Voigt, J. D. Westbrook, J. Y. Young, C. Zardecki and M. Zhuravleva, Nucleic Acids Res., 2021, 49, D437–D451 CrossRef CAS PubMed.
- D. S. Froese, J. Kopec, E. Rembeza, G. A. Bezerra, A. E. Oberholzer, T. Suormala, S. Lutz, R. Chalk, O. Borkowska, M. R. Baumgartner and W. W. Yue, Nat. Commun., 2018, 9, 2261 CrossRef PubMed.
- A. W. Struck, M. L. Thompson, L. S. Wong and J. Micklefield, ChemBioChem, 2012, 13, 2642–2655 CrossRef CAS PubMed.
- E. G. Gharakhanian and T. J. Deming, Chem. Commun., 2016, 52, 5336–5339 RSC.
- M. S. Puar, T. M. Chan, V. Hegde, M. Patel, P. Bartner, K. J. Ng, B. N. Pramanik and R. D. MacFarlane, J. Antibiot., 1998, 51, 221–224 CrossRef CAS PubMed.
- M. Matsumoto, Y. Kawamura, Y. Yasuda, T. Tanimoto, K. Matsumoto, T. Yoshida and J. Shoji, J. Antibiot., 1989, 42, 1465–1469 CrossRef CAS PubMed.
- B. S. Yun, T. Hidaka, K. Furihata and H. Seto, J. Antibiot., 1994, 47, 1541–1545 CrossRef CAS PubMed.
- M. J. Helf, A. Jud and J. Piel, ChemBioChem, 2017, 18, 444–450 CrossRef CAS PubMed.
- (a) K. Watanabe, K. Hotta, A. P. Praseuth, K. Koketsu, A. Migita, C. N. Boddy, C. C. C. Wang, H. Oguri and H. Oikawa, Nat. Chem. Biol., 2006, 2, 423–428 CrossRef CAS PubMed; (b) A. H. Al-Mestarihi, G. Villamizar, J. Fernandez, O. E. Zolova, F. Lombo and S. Garneau-Tsodikova, J. Am. Chem. Soc., 2014, 136, 17350–17354 CrossRef CAS PubMed.
- T. Huang, Y. Duan, Y. Zou, Z. Deng and S. Lin, ACS Chem. Biol., 2018, 13, 2387–2391 CrossRef CAS PubMed.
- B. Shen, L. Du, C. Sanchez, D. J. Edwards, M. Chen and J. M. Murrell, J. Nat. Prod., 2002, 65, 422–431 CrossRef CAS PubMed.
- (a) C. Liao and F. P. Seebeck, Angew. Chem., Int. Ed., 2019, 58, 3553–3556 CrossRef CAS PubMed; (b) F. Trottmann, J. Franke, I. Richter, K. Ishida, M. Cyrulies, H.-M. Dahse, L. Regestein and C. Hertweck, Angew. Chem., Int. Ed., 2019, 58, 14129–14133 CrossRef CAS PubMed; (c) F. Trottmann, K. Ishida, M. I. Ito, H. Kries, M. Groll and C. Hertweck, Nat. Chem., 2022, 14, 884–890 CrossRef CAS PubMed.
- Z. F. Xia, T. W. Guan, J. S. Ruan, Y. Huang and L. L. Zhang, Int. J. Syst. Evol. Microbiol., 2013, 63, 2813–2818 CrossRef CAS PubMed.
- Y. Zhao, E. Marschall, M. Treisman, A. McKay, L. Padva, M. Crüsemann, D. R. Nelson, D. L. Steer, R. B. Schittenhelm, J. Tailhades and M. J. Cryle, Angew. Chem., Int. Ed., 2022, 61, e202204957 CrossRef CAS PubMed.
- (a) D. Guan, F. Chen, Y. Qiu, B. Jiang, L. Gong, L. Lan and W. Huang, Angew. Chem., Int. Ed., 2019, 131, 6750–6754 CrossRef; (b) J. Zang, Y. Chen, W. Zhu and S. Lin, Biochem, 2019, 59, 132–138 CrossRef PubMed; (c) N. L. Kjærsgaard, T. B. Nielsen and K. V. Gothelf, ChemBioChem, 2022, 23, e202200245 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02380j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |