
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomolecular interactions studied by low-field NMR using SABRE hyperpolarization†
Pierce
Pham
 and
Christian
Hilty
*
and
Christian
Hilty
*
Department of Chemistry, Texas A&M University, 3255 TAMU, College Station, TX 77843, USA. E-mail: chilty@tamu.edu
First published on 1st September 2023
Abstract
We demonstrate that low-field nuclear magnetic resonance provides a means for measuring biomacromolecular interactions without requiring a superconducting, or even a permanent magnet. A small molecule, 5-fluoropyridine-3-carboximidamide, is designed to be a specific ligand for the trypsin protein, while containing a fluorine atom as a nuclear spin hyperpolarizable label. With hyperpolarization by the parahydrogen based signal amplification by the reversible exchange method, fluorine NMR signals are detectable in the measurement field of 0.85 mT of an electromagnet, at a concentration of less than 100 μM. As a weak ligand for the protein, the hyperpolarized molecule can serve as a reporter for measuring the binding of other ligands of interest, illustrated by the determination of the dissociation constant KD of benzamidine from changes in the observed R2 relaxation rates. A signal enhancement of more than 106 compared to Boltzmann polarization at the measurement field indicates that this experiment is not feasible without prepolarization. The extended magnetic field range for the measurement of biomolecular interactions under near physiological conditions, with a protein concentration on the order of 10 μM or less, provides a new option for screening of ligand binding, measurement of protein–protein interactions, and measurement of molecular dynamics.
Introduction
Nuclear magnetic resonance is a de facto standard for the determination of the structure and interactions of biological macromolecules.1 Biomolecular NMR has risen to the challenge of characterizing macromolecules in large parts through the use of high magnetic fields. The resolving power of NMR spectroscopy increases with the magnetic field. Importantly, higher magnetic fields also improve the signal-to-noise ratio because the spin polarization is proportional to the magnetic field B0, combined with a dependence of the sensitivity of inductive NMR detectors on B01/2.2 The increased sensitivity makes it possible to measure NMR spectra of biological macromolecules at realistically achievable concentrations in the millimolar range or below.For the above reasons, biomolecular NMR is rarely performed at magnetic fields below approximately the Tesla range. This is despite the fact that a broader magnetic field range can give access to new information on molecular dynamics and interactions. The contributions of molecular motions and chemical exchange to the observed nuclear spin relaxation can be measured through field dependent relaxation dispersion. In field cycling experiments, a high magnetic field, such as on the order of 1 T or more, is used for generating spin polarization and detecting signals. Biomolecular applications of these techniques broadly include the measurement of protein dynamics, protein–lipid binding, protein folding, enzyme dynamics and others.3–5
Besides the information content of NMR parameters measured at different magnetic fields, a distinction of low-field NMR is the reduced complexity and cost of the spectrometer. An NMR spectrometer operating with a weak electromagnet can be realized at a cost that is orders of magnitude lower than that of high-field NMR. The main hindrance of applying low-field NMR measurements in a biological context is the insufficient spin polarization. The result of the sensitivity calculation can be drastically changed when a nuclear spin hyperpolarization method is applied. The hyperpolarization renders the level of the nuclear spin polarization independent of the magnetic field, in which signals are acquired. It enables the measurement of spectra of diluted samples at low field strengths, in the millitesla or even microtesla range.6,7
Here, we demonstrate the observation of biomacromolecular interactions by low-field NMR in an electromagnet at 0.85 mT. The measurement is enabled by a large sensitivity enhancement derived from parahydrogen. While high-resolution spectroscopy is precluded in the absence of chemical shifts at this low field, the binding of a ligand to the target protein can readily be detected by observing changes in the R2 relaxation rate.
The use of hyperpolarization for the detection of protein–ligand interactions and resulting applications in drug discovery have previously been proposed by our own group and others. Dissolution dynamic nuclear polarization (DNP) provided signal enhancements for 1H, 13C, or 19F, in the latter case enabling the detection of binding with micromolar ligand and nanomolar protein concentrations in a single scan.8–10 Alternative hyperpolarization methods proposed for the detection of ligand binding include triplet state DNP13 and chemically induced dynamic nuclear polarization (CIDNP).14 In hyperpolarized binding experiments, ligands with a wide range of affinities can be observed. Rapidly exchanging, weak ligands can provide a substantial boost in signal when present in excess. Weak ligands can further be used as reporters to detect competitively binding ligands of interest.11,12 Hyperpolarization provided by the signal amplification by reversible exchange (SABRE)15 method can simplify this experiment significantly because of the potential to enhance signals at zero or low magnetic fields.16,17 Parahydrogen can be produced inexpensively by cooling hydrogen gas and can be used for the detection of the binding of a hyperpolarized ligand, as well as other competing ligands.18,19 All such applications to date were performed with signal detection at high field. In this work, we make the case that parahydrogen, due to the ease with which it can produced, is ideally suited to provide spin polarization for expanding this application to low magnetic fields.
Results and discussion
We designed a ligand for the trypsin protein, 5-fluoropyridine-3-carboximidamide (FPCA; Fig. 1a), for hyperpolarization by SABRE. This molecule is based on benzamidine, a known ligand for the protein. It includes a binding site for the iridium containing polarization transfer catalyst, as well as a 19F label to receive spin polarization and produce strongly enhanced signals that are distinct from 1H. As a weak ligand for the protein, it is suitable to serve as a reporter11,12 to measure the binding of other competitive ligands of interest. We envisage that a single purpose designed ligand of this type, weakly binding to a target protein, can be used for screening of ligand binding or for biophysical investigations involving any number of small-molecule ligands or macromolecular binding partners.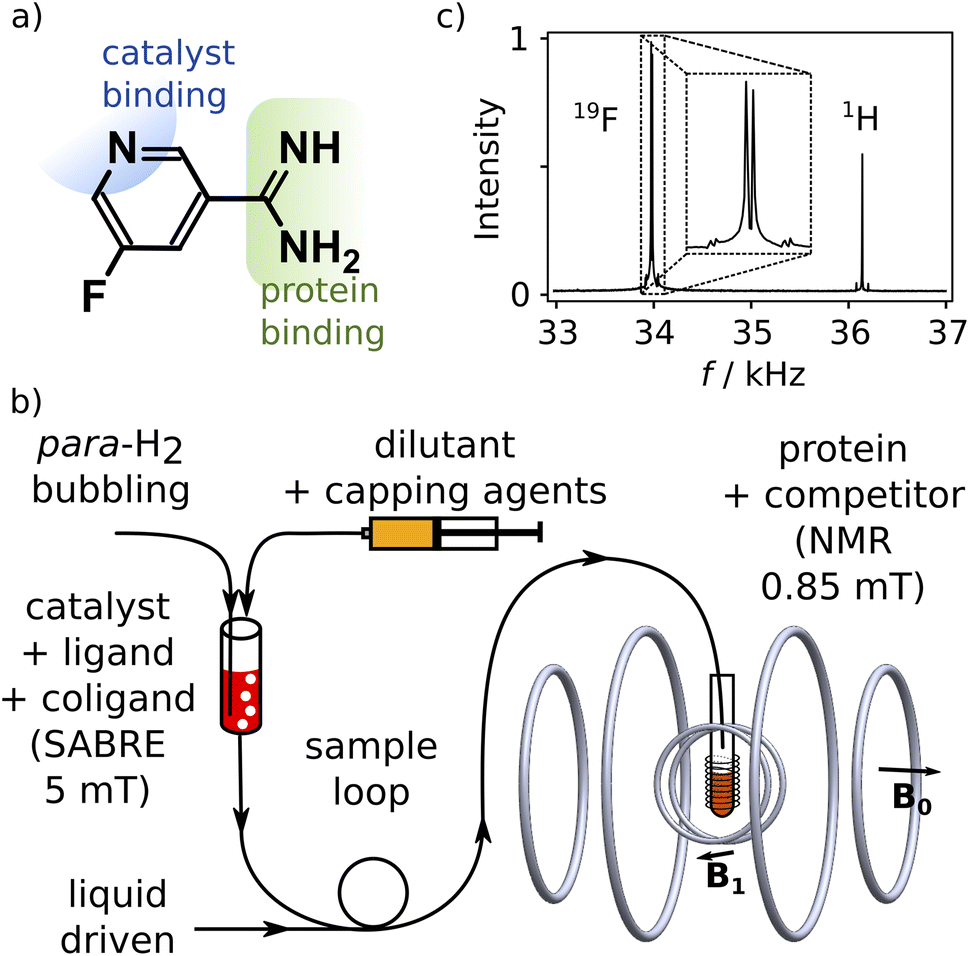 | ||
| Fig. 1 (a) Chemical structure of 5-fluoropyridine-3-carboximidamide (FPCA). The binding sites to SABRE catalysts and trypsin are indicated. (b) Low-field NMR spectrum of SABRE hyperpolarized FPCA, showing the 19F and 1H frequencies at 34 and 36.2 kHz, respectively. (c) Illustration of the SABRE hyperpolarization procedure and the low-field NMR instrument. The SABRE polarized reporter ligand is injected into the NMR measurement field and mixed with a solution of protein and competing ligand in a 10 mm NMR tube. | ||
Hyperpolarization of FPCA occurred in a small solenoid producing a 5 mT magnetic field, using methanol as solvent (Fig. 1b and ESI†). During this process, both 19F and 1H spin polarization became enhanced. The hyperpolarized sample is mixed with the capping agent 2,2′-bipyridine.20 This agent strongly binds to the SABRE polarization transfer catalyst, deactivates the catalyst, and hinders the binding of the previously hyperpolarized substrate. An NMR spectrum measured after the transfer of the molecule into a homogeneous magnetic field of 0.85 mT and dilution at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15.3 with phosphate buffer (pH 7.6) is shown in Fig. 1c. This field is less than 20 times that of the earth, and 104 times smaller than typical high fields used for NMR, resulting in a substantial simplification of the experiment that is further described below. The signals of 1H and 19F spins are observed simultaneously, respectively at 36.2 kHz and 34.0 kHz (Fig. 1c). The relative signal intensities of the two nuclei are a function of the original hyperpolarization mediated through the network of J-couplings in the molecule, relaxation losses occurring before the signal acquisition, and the bandwidth of the detector, which in this experiment was centered at 34.0 kHz.
:15.3 with phosphate buffer (pH 7.6) is shown in Fig. 1c. This field is less than 20 times that of the earth, and 104 times smaller than typical high fields used for NMR, resulting in a substantial simplification of the experiment that is further described below. The signals of 1H and 19F spins are observed simultaneously, respectively at 36.2 kHz and 34.0 kHz (Fig. 1c). The relative signal intensities of the two nuclei are a function of the original hyperpolarization mediated through the network of J-couplings in the molecule, relaxation losses occurring before the signal acquisition, and the bandwidth of the detector, which in this experiment was centered at 34.0 kHz.
With the goal of measuring biological interactions, the R2 relaxation of the ligand 19F spin was determined from single-scan Carr–Purcell–Meiboom–Gill (CPMG) experiments (Fig. 2a). The low-field NMR signals were acquired simultaneously with the application of the pulses (Fig. 2b–d). The decay of the echo intensities due to the R2 relaxation is prominently visible when applying a digital band-pass filter (33.9–34.1 kHz) to the time domain data. After the Fourier transform of each echo signal to yield spectra similar to Fig. 1c, the relaxation decay can be analyzed from a series of 19F frequency peaks (Fig. 2e). The corresponding R2 relaxation rate is obtained by fitting these integrated signals to a single exponential function (Fig. 2f).
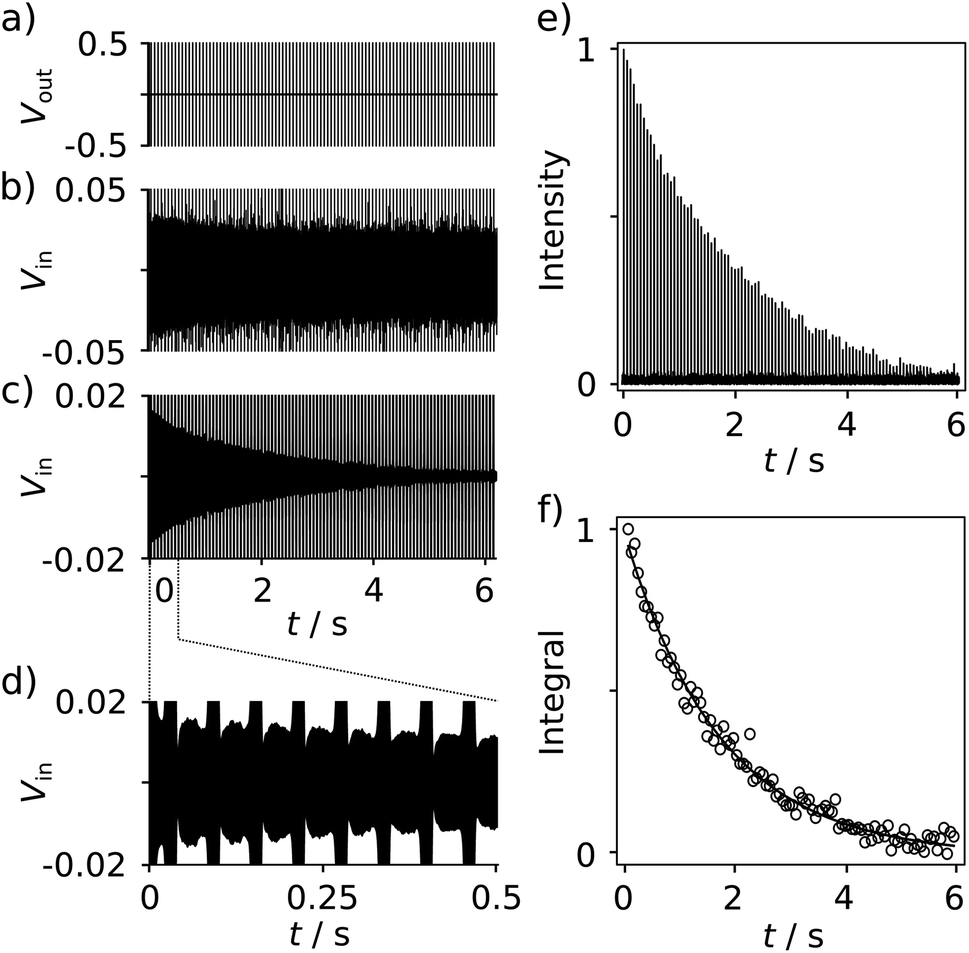 | ||
| Fig. 2 (a) NMR Pulse sequence for measuring the R2 relaxation rate of reporter ligand spins. A π/2 (0.665 ms) pulse is followed by 100 π pulses (1.33 ms) spaced by 2 × τCPMG = 60 ms. (b) Time-domain trace measured from a sample of 655 μM of SABRE hyperpolarized FPCA, showing the NMR signals in-between the pulses. (c) Time-domain signals from the data in (b) processed with a digital band-pass filter (Butterworth; 33.9–34.1 kHz) to illustrate the R2 decay of the 19F signal. (d) The expanded region shows the first 8 echoes. (e) Series of Fourier-transformed 19F signals from each echo in (b). (f) Integrated 19F signals, fitted with a single exponential to obtain R2 relaxation rates. | ||
Comparing R2 relaxation traces measured in the absence and presence of the target protein trypsin reveals a shorter relaxation of the ligand signal when the ligand binds to the protein under fast exchange (Fig. 3a, left and center). In competition with benzamidine (BA or competitor), the relaxation rate again becomes slower (Fig. 3a, right). In the comparison of R2 relaxation rates, no significant relaxation contribution due to the binding of FPCA to the polarization transfer catalyst is expected, as the catalyst is deactivated by the strongly binding bivalent capping ligand during injection. Indeed, previous high-field NMR studies with a related ligand indicated that after the addition of a capping ligand, the expected R2 relaxation rate changes due to protein binding were observed.18,19 Thus, the following calculations consider a binding equilibrium only between the hyperpolarized reporter ligand, the protein, and, where present, the competitor. Here, the competitor BA serves as an example of a ligand of interest in a screening experiment, which partially displaces the reporter ligand from the protein binding site. The experiments are performed at a concentration of 131 ± 6 μM FPCA, 5.7 ± 0.3 μM trypsin, and 42.5 ± 3.8 μM BA. These concentrations are at or below the concentrations used for typical ligand screening by high-field NMR.21
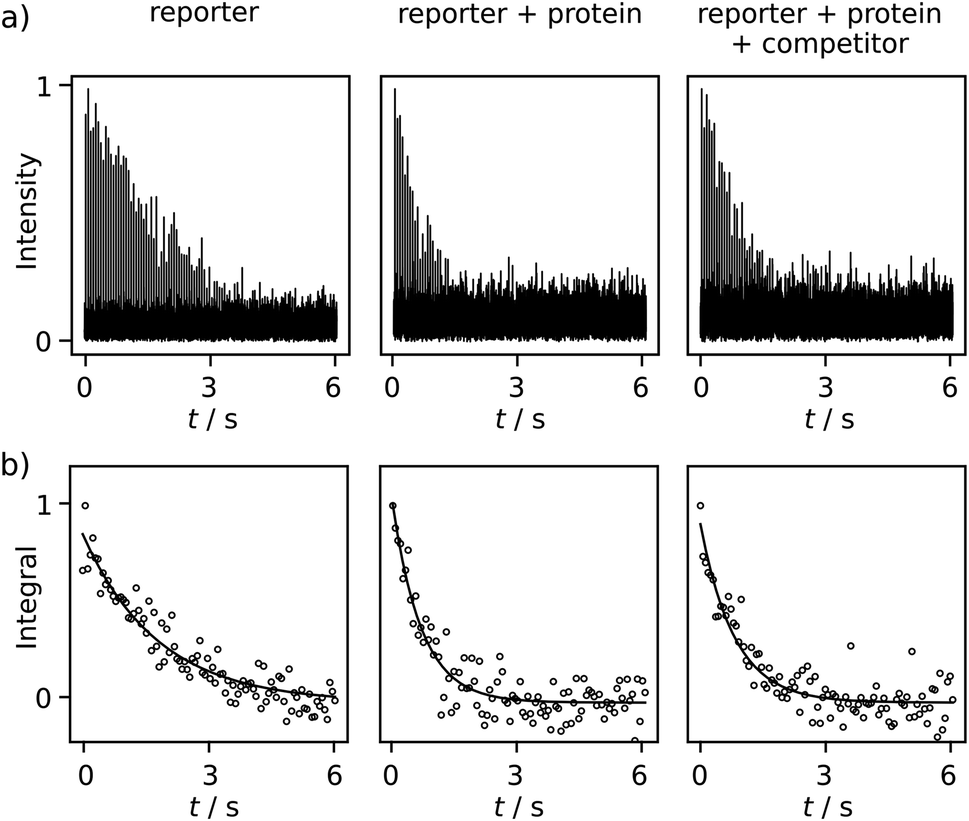 | ||
| Fig. 3 (a) One set of serial Fourier-transformed spectra for the CPMG experiments of free ligand (131 ± 6 μM reporter only), non-competition (reporter and 5.7 ± 0.3 μM protein), and competition (reporter, protein, and 42.5 ± 3.8 μM competitor). (b) The fitting results of R2 relaxation rates for these experiments. The fitted rates for these data sets are 0.556 s−1, 1.43 s−1 and 1.23 s−1 from left to right. | ||
Additional data sets are shown in Fig. S12 and S13.† The R2 rates are 0.557 ± 0.002, 1.54 ± 0.11, 1.12 ± 0.11 s−1 for the respective experiments of free reporter ligand, reporter binding without competition, and reporter binding with competition. The distinctive difference among these R2 values, after comparing with the uncertainty, indicates BA is a trypsin inhibitor. Because BA does not have a binding site to iridium, it cannot be hyperpolarized and directly observed in the low field. The determination of the KD of BA becomes possible via the quantification of the R2 rate change, using equations for a fast exchanging reporter.11,12,19 The equations require knowledge of the protein–reporter dissociation constant, KD,r = 179 ± 12 μM, which was determined from an NMR titration experiment (ESI†). These parameters yield a competitor KD of 35 ± 17 μM, which agrees with the range of 16–39 μM found in the literature for BA. These values include pure aqueous buffers or buffers with <10% alcohol.12,19,22 In the present experiments, the final fraction of methanol after dilution with phosphate buffer was approximately 13%, which was determined from pre-established dilution factors. This alcohol content is not expected to significantly alter trypsin integrity, as buffers with up to 30% methanol were previously shown to preserve enzymatic efficacy in the time frame of the experiment.18 This observation is further corroborated by the close agreement of measured KD values, as described above.
The specific limits on accuracy and signal-to-noise ratio of the experiment are illustrated by additional data sets measured at higher and lower concentrations. Data obtained with 655 μM FPCA are shown in Fig. S10 and S11,† and one measurement is plotted and fitted in Fig. 2e and f. The SNR for these spectra reaches as high as 100, which increases the accuracy in KD determination. The average R2 rate of FPCA was 0.562 ± 0.005 s−1, indicating a high reproducibility with a deviation as low as 1%. The average R2 rates for the non-competition and competition experimenters are 1.22 ± 0.05 and 0.929 ± 0.015 s−1, respectively. The respective deviations of 4% and 2% are most likely caused by concentration differences from injections. These deviations are minor in further determining the protein affinity of BA, which results in KD = 41 ± 8 μM. When the concentrations of FCPA, trypsin, and BA are reduced to 65.5 ± 3.1, 5.7 ± 0.3, and 28.6 ± 2.5 μM respectively, an SNR of 8 is obtained, which is clearly sufficient to differentiate the ligand signal from background noise. However, the noise contribution becomes more significant, as shown in Fig. S14 and S15,† and leads to a KD determined as 37 ± 35 μM.
Based on the achieved accuracy, the detection limit for the FPCA reporter ligand is below 100 μM when measured in an NMR tube of 8 mm inner diameter at the low field. The signals achieved in this experiment can be compared with DNP experiments, where a detection limit near 1 μM was obtained when using NMR tubes with an inner diameter of 4 mm in a high-field, 9.4 T magnet.9,12
Absolute polarization levels and signal enhancements can be calculated by comparison with a reference sample. The comparison with trifluoroacetic acid pre-polarized at 1 T indicates that SABRE of FCPA achieved an estimated 19F spin polarization of 1.09%. This polarization corresponds to signal enhancements of 3.97 × 106, 3380, and 359 fold in comparison to the Boltzmann polarization at 0.85 mT, 1 T, and 9.4 T, respectively (calculation in ESI†). These signal enhancements compare not unfavorably to signal enhancements of several thousand-fold at 9.4 T obtained in the DNP experiments, given the substantially reduced complexity of the SABRE experiment.
The large enhancement factor at the low field is a direct consequence of the low Boltzmann polarization that would otherwise be present at that field strength. It provides a striking illustration of the infeasibility of measuring Boltzmann polarized signals of dilute samples under these conditions.
The limit of detection may in the future be further reduced by instrumental improvements such as optimizing the B0 field and Q-factor of the receiver coil,2 and by increasing the level of hyperpolarization. The 19F enhancement using SABRE-SHEATH may be up to 10-fold better than SABRE in millitesla fields.23 These improvements would readily reach a detection limit in the range of several micromolar. On the other hand, it would be informative to consider the limiting factors in acquiring signals with smaller detection coils, of a size that would be commonly used in high-field NMR. The SNR per unit sample volume is inversely proportional to the coil diameter, when comparing solenoidal coils of identical length-to-diameter ratios.24 The sample of lower concentration tested in the present experiments, 65.5 μM FCPA, in a corresponding coil with 2× smaller diameter (8× lower volume of 440 μL) would result in an estimated SNR of 2 with the current polarization levels. A sample in a coil with 3× smaller diameter (130 μL volume) would have an SNR of 0.9. These two sample sizes approximately correspond to nominal NMR sample sizes of 5 mm and 3 mm, respectively. Therefore, the experiment would be possible using somewhat higher sample concentrations or with other improvements in SNR. Previously described NMR experiments routinely inject hyperpolarized samples into 5 mm tubes,18,25 whereby smaller samples could be measured in flow cells.26
SABRE polarization of substrates requires individual optimization to obtain a catalyst–ligand exchange rate resulting in the highest signal enhancement. For example, when the hyperpolarization of FPCA was attempted using the typical SABRE catalyst chloro(1,5-cyclooctadiene)[4,5-dimethyl-1,3-bis(2,4,6-trimethylphenyl)-imidazol-2-ylidene]iridium(I), no signal was observed. With chloro(1,5-cyclooctadiene)[1-methyl-3-(2,4,6-trimethylphenyl)-imidazol-2-ylidene]iridium(I), an SNR of only 22 was obtained. In comparison, an SNR as high as 100 was observed when the first catalyst was used with dimethyl sulfoxide (DMSO) as a coligand. The low polarization in the former experiments is likely due to the presence of the electron-withdrawing fluorine and amidine moieties causing weaker binding to iridium catalysts. The inclusion of DMSO stabilizes the active SABRE complex,27 bringing the exchange rate into a favorable range. Other parameters including the temperature, magnetic field, duration of hydrogen introduction, etc., were further optimized (ESI Fig. S5–S7†).
The requirement for the design of a ligand with a binding site for the polarization transfer catalyst and subsequent SABRE optimizations lends appeal to the idea of using such a ligand as a reporter. Only one reporter ligand needs to be designed and optimized for each protein. This ligand can be used for various biochemical and biophysical studies, such as screening for the binding of other ligands, the determination of protein–protein interactions, enzyme–substrate interactions, and others.
A simulation19 using 131 μM FPCA and 5.7 μM protein indicated that with appropriate adjustment of competitor concentrations, the present experiment can be used to determine their corresponding KD in a range of 0.1 μM to 1 mM (Fig. S20†). Other methods for detecting protein–ligand interactions may further be applied. In experiments similar to intermolecular ligand–ligand NOE (ILOE)28 and interligand NOE for pharmacophore mapping (INPHARMA),29 the 1H hyperpolarization from the first ligand may for instance be transferred to a 19F label of the second ligand. The simultaneous detection of both nuclei in the same spectrum may be used to derive the binding kinetics of these ligands. Because magnetic susceptibility variations have less severe effects, the low field is ideal for working with immobilized proteins, as in target immobilized NMR screening (TINS).30 Mixtures of water-soluble or heterogeneous catalysts,31,32 ligands, and proteins may further be developed for the direct hyperpolarization, making multi-scan experiments possible simply by re-introducing fresh parahydrogen. This approach would facilitate 2D NMR to correlate different nuclei or different coupling patterns.33
Different ligand designs may be selected for high-field NMR using DNP and SABRE. FPCA is chosen for this work due to the three mentioned key factors of SABRE efficiency, protein binding affinity, and 19F label. Different trypsin ligand designs, from previous investigations9,12,18 and FPCA, still have similar KD in a range of 140–180 μM, which emphasizes the feasibility of altering weak ligands to include required hyperpolarization properties, without significantly impacting their affinity. The 19F NMR measurements exclude the interference of other proton signals originating from either SABRE-hyperpolarization or thermal contributions.
Low-field NMR detection of biological interactions, using an apparatus as shown in Fig. 1b, demonstrates multiple advantages in economy, effectiveness, and simplicity. First, the cost of a low-field NMR spectrometer is a fraction of that of even a traditional benchtop NMR spectrometer, not to mention a commercial high-field instrument. At low field, minor bubble formation does not affect the measurement, which further simplifies the injection procedure. Low-field NMR detection also does not suffer from interference of signals from non-hyperpolarized components. The SABRE signal enhancement of >106 compared to the Boltzmann polarization in the millitesla field is a much larger factor than for high-field NMR. With well-established 1H SABRE methods, low-field NMR may further be used for 1H detection by eliminating signals even from water solvent, which is present at a concentration that is >50000 times larger than that of hyperpolarized ligands. The use of 1H polarization will require the distinction of signals from other hyperpolarized 1H species, which in the absence of chemical shift resolution may be achieved by detecting characteristic heteronuclear coupling constants or by an experimental procedure that avoids other hyperpolarized species.
Conclusions
In summary, we demonstrated the use of 19F SABRE hyperpolarization to observe protein–ligand interactions in a low-cost, low-field NMR spectrometer. This capability was illustrated with the measurement of the binding affinity of benzamidine for the trypsin protein through the observation of R2 relaxation changes of a reporter ligand. Key aspects of the ligand design that enable this application include a binding motif for the SABRE polarization transfer catalyst, the 19F spin probe, and a binding affinity resulting in fast exchange with the protein bound form. Here, the SABRE-hyperpolarized ligand was detected at a concentration below 70 μM. This concentration can be further lowered to the level of several micromolar, with the discussed improvements in instrumentation and hyperpolarization, concomitant with a reduction in the residual concentration of organic solvent from the hyperpolarization. Once a suitable ligand for SABRE hyperpolarization is identified that interacts with a target protein, it can be used to screen for the binding of other ligands or to measure interactions involving macromolecular binding interfaces or cellular components. The method thus broadens the application range of SABRE hyperpolarization, facilitating the measurement of interactions in drug discovery and other biochemical and biophysical problems.Experimental
5-Fluoropyridine-3-carboximidamide hydrochloride (FPCA) was synthesized from 5-fluoropyridine-3-carbonitrile (Ambeed, Arlington Heights, IL), sodium methoxide (Alfa Aesar, Ward Hill, MA), and ammonium chloride (Alfa Aesar), using a general synthesis procedure from ref. 34 with modifications described in the ESI† (Caution: sodium methoxide is flammable and corrosive). The reaction product was characterized using 13C, 1H and 19F NMR. Samples for parahydrogen polarization were prepared consisting of 0.2 mM or 1 mM chloro(1,5-cyclooctadiene)[4,5-dimethyl-1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene]iridium(I) (Strem, Newburyport, MA), referred to as precatalyst, 2 mM or 10 mM FPCA, and 2 mM or 10 mM dimethyl sulfoxide (DMSO) (Alfa Aesar, Ward Hill, MA) in methanol-d4 (Cambridge Isotope Laboratories, Andover, MA). Parahydrogen was enriched to ∼95% using a cryocooler (Advanced Research Systems, Macungie, PA) operated at 29 K. 0.5 mL sample aliquots were pressurized with 120 psi of parahydrogen gas (Caution: hydrogen gas is flammable and forms explosive mixtures with air). The catalyst was activated under the parahydrogen atmosphere in 5 min at 25 °C. Hyperpolarization was produced by bubbling parahydrogen in a solenoidal electromagnet at a field of 5 mT as described.18 After hyperpolarization, the catalyst was deactivated using 100 mM pyridine (Sigma-Aldrich, St. Louis, MO) and 100 mM 2,2′-bipyridine (Sigma-Aldrich) in methanol (Fisher Scientific, Waltham, MA) added to the sample at 1:1 v/v ratio using a syringe pump (Nexus 6000, Chemyx, Stafford, TX) (Caution: Methanol is toxic and flammable, pyridine is flammable, bipyridine is toxic). The sample was then pushed into a 10 mm NMR tube with water at a flow rate of 170 mL min−1 (1000D syringe pump, Teledyne ISCO, Lincoln, NE). The NMR tube was pre-installed in the low-field NMR spectrometer. The NMR tube contained 1 mL of the non-hyperpolarized sample component, consisting of 50 mM phosphate buffer, pH = 7.6, optionally containing 20 μM or 41 μM trypsin, and optionally containing 0.15 or 0.5 mM benzamidine hydrochloride as competing ligand. A low-field NMR spectrometer with an electromagnet producing a field of 0.85 mT was used for signal acquisition.7 The spectrometer was fitted with a coil insert accommodating a 10 mm NMR tube in vertical orientation. The π/2 pulse length was 0.665 ms. A Carr–Purcell–Meiboom–Gill (CPMG) pulse sequence was used to measure R2 relaxation rates. The pulse train contained 100 π pulses and a delay between pulses of 2 × τCPMG = 60 ms. For obtaining accurate R2 rates, the τCPMG delay should be chosen sufficiently short to refocus magnetization under residual field inhomogeneity and diffusion and any convective motions in the sample. Signals were sampled with a rate of 800 kHz using a multifunction data acquisition board (PCIe-6259, NI, Austin, TX). Individual spin echoes were Fourier transformed from time domain data extending over 48 ms centered between the pulses. Signal acquisition and data processing were performed using Python (Python Software Foundation, https://www.python.org).
Data availability
Data for this paper, including R2 relaxation data, are available at Texas A&M University OAKTrust at https://hdl.handle.net/1969.1/198459.Author contributions
CH designed the project. PP prepared samples and performed experiments. All authors interpreted results and wrote manuscript.Conflicts of interest
Texas A&M University has filed a patent application covering parts of this work.Acknowledgements
Financial support from the National Science Foundation (Grant CHE-1900406) and the Welch Foundation (Grant A-1658) is gratefully acknowledged. The authors thank Olga Korzh and Ratnamala Mandal for useful discussions.References
-
K. Wüthrich, NMR of Proteins and Nucleic Acids, Wiley, New York, 1986 Search PubMed
.
- D. I. Hoult and R. E. Richards, J. Magn. Reson., 1976, 24, 71–85 Search PubMed
- G. Parigi, E. Ravera, M. Fragai and C. Luchinat, Prog. Nucl. Magn. Reson. Spectrosc., 2021, 124–125, 85–98 CrossRef CAS PubMed
- M. Pu, A. Orr, A. G. Redfield and M. F. Roberts, J. Biol. Chem., 2010, 285, 26916–26922 CrossRef CAS PubMed
- M. M. Rosenberg, T. Yao, G. C. Patton, A. G. Redfield, M. F. Roberts and L. Hedstrom, Biochemistry, 2020, 59, 2359–2370 CrossRef CAS PubMed
- P. Štěpánek, C. Sanchez-Perez, V.-V. Telkki, V. V. Zhivonitko and A. M. Kantola, J. Magn. Reson., 2019, 300, 8–17 CrossRef PubMed
- Y. Zhu, C.-H. Chen, Z. Wilson, I. Savukov and C. Hilty, J. Magn. Reson., 2016, 270, 71–76 CrossRef CAS PubMed
- M. H. Lerche, S. Meier, P. R. Jensen, H. Baumann, B. O. Petersen, M. Karlsson, J. Ø. Duus and J. H. Ardenkjær-Larsen, J. Magn. Reson., 2010, 203, 52–56 CrossRef CAS PubMed
- Y. Lee, H. Zeng, S. Ruedisser, A. D. Gossert and C. Hilty, J. Am. Chem. Soc., 2012, 134, 17448–17451 CrossRef CAS PubMed
- Y. Wang, J. Kim and C. Hilty, Chem. Sci., 2020, 11, 5935–5943 RSC
- C. Dalvit, M. Flocco, S. Knapp, M. Mostardini, R. Perego, B. J. Stockman, M. Veronesi and M. Varasi, J. Am. Chem. Soc., 2002, 124, 7702–7709 CrossRef CAS PubMed
- Y. Kim and C. Hilty, Angew. Chem., Int. Ed., 2015, 54, 4941–4944 CrossRef CAS PubMed
- K. Miyanishi, T. Sugiki, T. Matsui, R. Ozawa, Y. Hatanaka, H. Enozawa, Y. Nakamura, T. Murata, A. Kagawa, Y. Morita, T. Fujiwara, M. Kitagawa and M. Negoro, J. Phys. Chem. Lett., 2023, 14, 6241–6247 CrossRef CAS PubMed
- F. Torres, M. Bütikofer, G. R. Stadler, A. Renn, H. Kadavath, R. Bobrovs, K. Jaudzems and R. Riek, J. Am. Chem. Soc., 2023, 145, 12066–12080 CrossRef CAS PubMed
- R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. P. Elliott, S. B. Duckett, G. G. R. Green, I. G. Khazal, J. Lopez-Serrano and D. C. Williamson, Science, 2009, 323, 1708–1711 CrossRef CAS PubMed
- S. Lehmkuhl, M. Suefke, A. Kentner, Y.-F. Yen, B. Blümich, M. S. Rosen, S. Appelt and T. Theis, J. Chem. Phys., 2020, 152, 184202 CrossRef CAS PubMed
- T. Theis, P. Ganssle, G. Kervern, S. Knappe, J. Kitching, M. P. Ledbetter, D. Budker and A. Pines, Nat. Phys., 2011, 7, 571–575 Search PubMed
- R. Mandal, P. Pham and C. Hilty, Chem. Sci., 2021, 12, 12950–12958 RSC
- R. Mandal, P. Pham and C. Hilty, Anal. Chem., 2022, 94, 11375–11381 CrossRef CAS PubMed
- R. E. Mewis, M. Fekete, G. G. R. Green, A. C. Whitwood and S. B. Duckett, Chem. Commun., 2015, 51, 9857–9859 RSC
- A. D. Gossert and W. Jahnke, Prog. Nucl. Magn. Reson. Spectrosc., 2016, 97, 82–125 CrossRef CAS PubMed
- D. Rauh, S. Reyda, G. Klebe and M. T. Stubbs, Biol. Chem., 2005, 383, 1309–1314 Search PubMed
- N. V. Chukanov, O. G. Salnikov, R. V. Shchepin, A. Svyatova, K. V. Kovtunov, I. V. Koptyug and E. Y. Chekmenev, J. Phys. Chem. C, 2018, 122, 23002–23010 CrossRef CAS PubMed
- T. L. Peck, R. L. Magin and P. C. Lauterbur, J. Magn. Reson., Ser. B, 1995, 108, 114–124 CrossRef CAS PubMed
- S. Bowen and C. Hilty, Angew. Chem., Int. Ed., 2008, 47, 5235–5237 CrossRef CAS PubMed
- H.-Y. Chen and C. Hilty, ChemPhysChem, 2015, 16, 2646–2652 CrossRef CAS PubMed
- P. J. Rayner, J. P. Gillions, V. D. Hannibal, R. O. John and S. B. Duckett, Chem. Sci., 2021, 12, 5910–5917 RSC
- M. F. Rega, B. Wu, J. Wei, Z. Zhang, J. F. Cellitti and M. Pellecchia, J. Med. Chem., 2011, 54, 6000–6013 CrossRef CAS PubMed
- V. M. Sánchez-Pedregal, M. Reese, J. Meiler, M. J. J. Blommers, C. Griesinger and T. Carlomagno, Angew. Chem., Int. Ed., 2005, 44, 4172–4175 CrossRef PubMed
- S. Vanwetswinkel, R. J. Heetebrij, J. van Duynhoven, J. G. Hollander, D. V. Filippov, P. J. Hajduk and G. Siegal, Chem. Biol., 2005, 12, 207–216 CrossRef CAS PubMed
- L. B. Bales, K. V. Kovtunov, D. A. Barskiy, R. V. Shchepin, A. M. Coffey, L. M. Kovtunova, A. V. Bukhtiyarov, M. A. Feldman, V. I. Bukhtiyarov, E. Y. Chekmenev, I. V. Koptyug and B. M. Goodson, J. Phys. Chem. C, 2017, 121, 15304–15309 CrossRef CAS PubMed
- F. Shi, P. He, Q. A. Best, K. Groome, M. L. Truong, A. M. Coffey, G. Zimay, R. V. Shchepin, K. W. Waddell, E. Y. Chekmenev and B. M. Goodson, J. Phys. Chem. C, 2016, 120, 12149–12156 CrossRef CAS PubMed
- L. S. Lloyd, R. W. Adams, M. Bernstein, S. Coombes, S. B. Duckett, G. G. R. Green, R. J. Lewis, R. E. Mewis and C. J. Sleigh, J. Am. Chem. Soc., 2012, 134, 12904–12907 CrossRef CAS PubMed
- J. B. Medwid, R. Paul, J. S. Baker, J. A. Brockman, M. T. Du, W. A. Hallett, J. W. Hanifin, R. A. Hardy and M. E. Tarrant, J. Med. Chem., 1990, 33, 1230–1241 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02365f |
| This journal is © The Royal Society of Chemistry 2023 |