
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSalt-stabilized alkylzinc pivalates: versatile reagents for cobalt-catalyzed selective 1,2-dialkylation†
Jie
Lin‡
a,
Kaixin
Chen‡
a,
Jixin
Wang
a,
Jiawei
Guo
a,
Siheng
Dai
a,
Ying
Hu
a and
Jie
Li
 *ab
*ab
aKey Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China. E-mail: jjackli@suda.edu.cn
bState Key Laboratory and Institute of Elemento-Organic Chemistry, Haihe Laboratory of Sustainable Chemical Transformations, College of Chemistry, Nankai University, Tianjin 300071, China
First published on 26th July 2023
Abstract
The construction of Csp3–Csp3 bonds through Negishi-type reactions using alkylzinc reagents as the pronucleophiles is of great importance for the synthesis of pharmaceuticals and agrochemicals. However, the use of air and moisture sensitive solutions of conventional alkylzinc halides, which show unsatisfying reactivity and limitation of generality in twofold Csp3–Csp3 cross-couplings, still represents drawbacks. We herein report the first preparation of solid and salt-stabilized alkylzinc pivalates by OPiv-coordination, which exhibit enhanced stability and a distinct advantage of reacting well in cobalt-catalyzed difluoroalkylation-alkylation of dienoates, thus achieving the modular and site-selective installation of CF2- and Csp3-groups across double bonds in a stereoretentive manifold. This reaction proceeds under simple and mild conditions and features broad substrate scope and functional group compatibility. Kinetic experiments highlight that OPiv-tuning on the alkylzinc pivalates is the key for improving their reactivity in twofold Csp3–Csp3 cross-couplings. Furthermore, facile modifications of bioactive molecules and fluorinated products demonstrate the synthetical utility of our salt-stabilized alkylzinc reagents and cobalt-catalyzed alkyldifluoroalkylation protocol.
Introduction
During the last decade, many significant advances have been witnessed in the efforts towards the development of transition metal-catalyzed selective dicarbofunctionalization of alkenes via cascade C–C bond formation, thus providing expedient access to the synthesis of carbogenic skeletons rapidly.1 Among them, most of the efforts focused on the selective diarylation2 and arylalkylation of alkenes.3 However, rather rare examples for achieving twofold Csp3–Csp3 cross-couplings across alkenes with two different alkyl moieties were reported. The early sporadic examples of catalytic alkene 1,2-dialkylations largely relied on the use of harsh alkylmagnesium halides as the nucleophiles.4 Otherwise the tethered alkenes bearing a Csp3–X (X = Br, I) center were utilized as the substrates for nickel-catalyzed intramolecular dialylation in the presence of alkylzinc reagents.5 These results demonstrated the viability of sequential dialkylation but there still remained drawbacks, such as limited substrate scope and poor functional group compatibility. Towards the possible reaction pathway of transition-metal-catalyzed alkene dialkylation, the challenging issues could be ascribed to (i) the hampered oxidative addition of transition metal-species into the alkyl–X bond;6 (ii) the facile propensity of other competitive side reactions via β-H elimination, atom-transfer radical addition and homocoupling pathways;7 (iii) the difficulty in controlling regio- and site selectivity (Scheme 1A).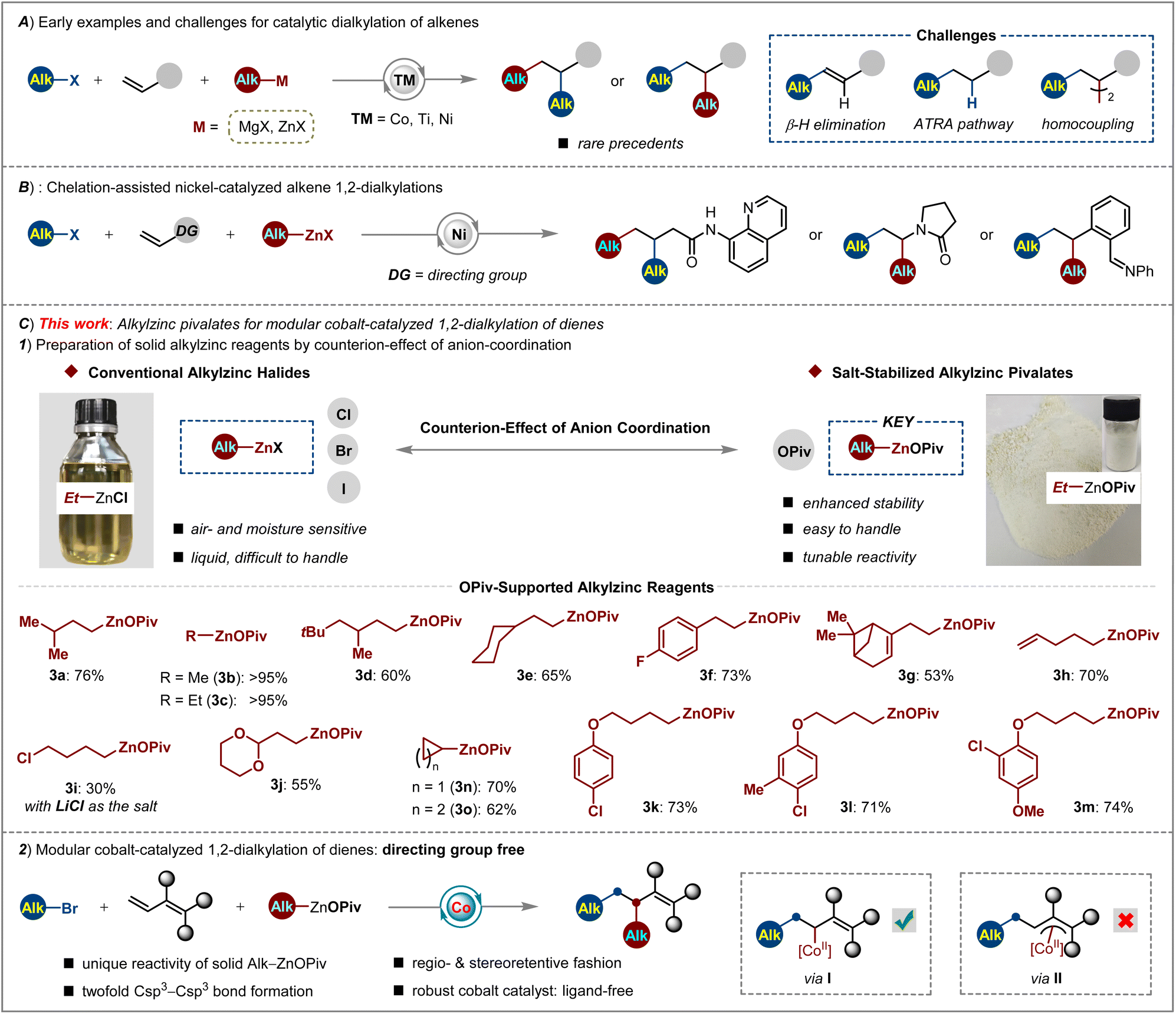 | ||
| Scheme 1 Current applications and limitations of alkylzinc reagents in sequence 1,2-dialkylation of alkenes. (A) Importance of the CF2-moiety in bioactive molecules. (B) Chelation-assisted strategy for alkene dialkylation. (C) Preparation of OPiv-supported alkylzinc reagents and their unique reactivity in modular cobalt-catalyzed 1,2-dialkylation of 1,3-dienes. | ||
To overcome these aforementioned challenges, several approaches have been achieved by nickel-catalyzed alkene dialkylation with the chelation-assistance of 8-aminoquinoline,8 enamide9 or imine.10 As such, these reactions displayed high chemo- and regioselectivity to install two Csp3-fragments across a double bond. Despite these major advances, the use of air and moisture sensitive solutions of alkylzinc halides (halides = Cl, Br, I), which are difficult to handle and should be stored under an inert atmosphere, as well as the unsatisfying reactivity and limitation of generality in cascade Csp3–Csp3/Csp3–Csp3 cross-couplings, still represent drawbacks (Scheme 1B).
Therefore, we hypothesized that the above issues can be solved by devising a new type of alkylzinc reagent with enhanced stability and tunable reactivity as the Csp3-nucleophile, as well as more a reactive catalytic system which operates under a radical relay process. Organozinc reagents are important intermediates with versatile reactivity in organic syntheses.11 Lei demonstrated that changing the anions of Ar–ZnX from Cl to Br or I will result in very different kinetics in palladium and nickel catalysis.12 Very recently, Knochel and us further disclosed the dramatic effects of carboxylate-coordination13 on improving the stability and reactivity of Ar–Zn14 and Si–Zn reagents.15 These observations gave us the perspective for tuning the physical and chemical properties of alkylzinc reagents by anion-regulation.
With this concept in mind, a new type of OPiv-supported alkylzinc reagents 3a–3m has been designed and prepared in moderate to excellent yields (30 to >95%) through transmetalation of alkylmagnesium bromides with Zn(OPiv)2. As compared to the poor stability and user-convenience of conventional alkylzinc reagents, these solid alkylzinc pivalates showed enhanced air and moisture stability by operationally simple protocols (Scheme 1C, top). Considering that fluorine-installation16 into bioactive molecules uniquely tunes their solubility, metabolic stability and bioavailability,17 we further illustrated the reactivity of this new type of alkylzinc pivalate by devising three-component alkyldifluoroalkylation of dienoates. Therefore, we envisioned capitalizing on the combination of (i) solid Alk–ZnOPiv as the nucleophile which exhibits providential reducibility and transmetalation rate, (ii) a practical, robust cobalt catalyst18 which favors rapid twofold Csp3–Csp3 cross-coupling and (iii) simple reaction conditions at 23 °C to achieve regio- and chemoselective difluoroalkylation-alkylation reaction of dienoates with broad difluoroalkyl halides and alkylzinc pivalates. Indeed, OPiv-coordination made Alk–ZnOPiv stand out among other salt-supported organozincs, which is the key to drive the highly active cobalt catalysis for realizing the modular, site-selective installation of CF2- and/or Csp3-groups across double bonds. Moreover, the formation of a flexible π-allylcobalt intermediate (II) was prohibited by steric interaction, thus ensuring the 1,2-difunctionalization of dienoates (I) in a stereoretentive fashion (Scheme 1C, bottom).
Results and discussion
We initiated our investigations by reacting dienoate 2a with bromodifluoroacetate 1a and OPiv-supported isopentylzinc reagent 3a-I (Table 1). Among the representative cobalt complexes, CoI2 has proven to be the choice of metal source for achieving 1,2-alkyldifluoroalkylation of dienoate via directing-group-free cobalt-catalyzed twofold Csp3–Csp3 cross-coupling, thus affording 4 in 78% yield of isolated product as the single regioisomer (entries 1–9). However, chelating ligands such as TMEDA, 1,10-phenanthrolines, as well as dppbz and dcype had negative effects, which decreased the yields of 4 to 31–68% (entries 10–13). As expected, no reaction was observed in the absence of a cobalt source (entry 14).|
|
|||||
|---|---|---|---|---|---|
| Entry | Variation | Yieldb (%) | Entry | Variation | Yieldb (%) |
| a Reaction conditions: 1a (2.0 equiv.), 2a (0.15 mmol, 1.0 equiv.), 3a-I (2.0 equiv.), [Co] (10 mol%), MeCN (1.5 mL), 25 °C, 16 h. b Isolated yields. c 1a (0.15 mmol, 1.0 equiv.), 2a (1.5 equiv.), 3a-I (2.5 equiv.). d TMEDA (30 mol%) as the ligand. e Ligand (11 mol%). | |||||
| 1 | CoCl2 | 57 | 8 | CoCl(PPh3)3 | Trace |
| 2 | CoBr2 | 58 | 9 | CoI2 | 78c |
| 3 | CoI2 | 62 | 10 | TMEDA | 68c,d |
| 4 | Co(acac)2 | 56 | 11 | 1,10-Phen | 54c,e |
| 5 | Co(acac)3 | 34 | 12 | Dppbz | 31c,e |
| 6 | CoBr2·DME | 49 | 13 | Dcype | 55c,e |
| 7 | Co(OAc)2 | 46 | 14 | Without [Co] | 0c |
These results encouraged us to further evaluate the unique anion-regulation of solid alkylzinc pivalate among that of other salt-supported alkylzinc reagents. To this end, a series of isopentyl zinc reagents 3a (I–VI) were prepared though transmetalation reactions of the corresponding isopentylmagnesium bromide with zinc salts (ZnX2; X = OPiv, Cl, Br, I). With these nucleophiles in hand, their different kinetics in the cobalt-catalyzed alkene dicarbofunctionalization were examined by performing parallel experiments under the described conditions (Scheme 2). Generally, all of these reactions were almost finished within 60 min. However, the conventional halide-supported isopentylzinc reagents 3a (II–IV) showed significantly poor reactivity and only furnished the desired product 4 in less than 20% conversions. In sharp contrast, on switching from halides to OPiv-anions, isopentylzinc pivalate 3a-I exhibited superior reactivity and steered the success of cobalt-catalyzed selective 1,2-dialkylation process. Similarly, the results of comparison experiments by using 3a-V and 3a-VI as the nucleophiles showed the superiority of the latter as well. These observations highlighted that the OPiv-coordination of organozincs is crucial to making these alkylzinc pivalates stand out among other salt-supported alkyl zinc reagents, thereby displaying unique kinetics in the 1,2-selective alkyldifluoroalkylation of dienoates via versatile cobalt-catalyzed twofold Csp3–Csp3 bond formation.
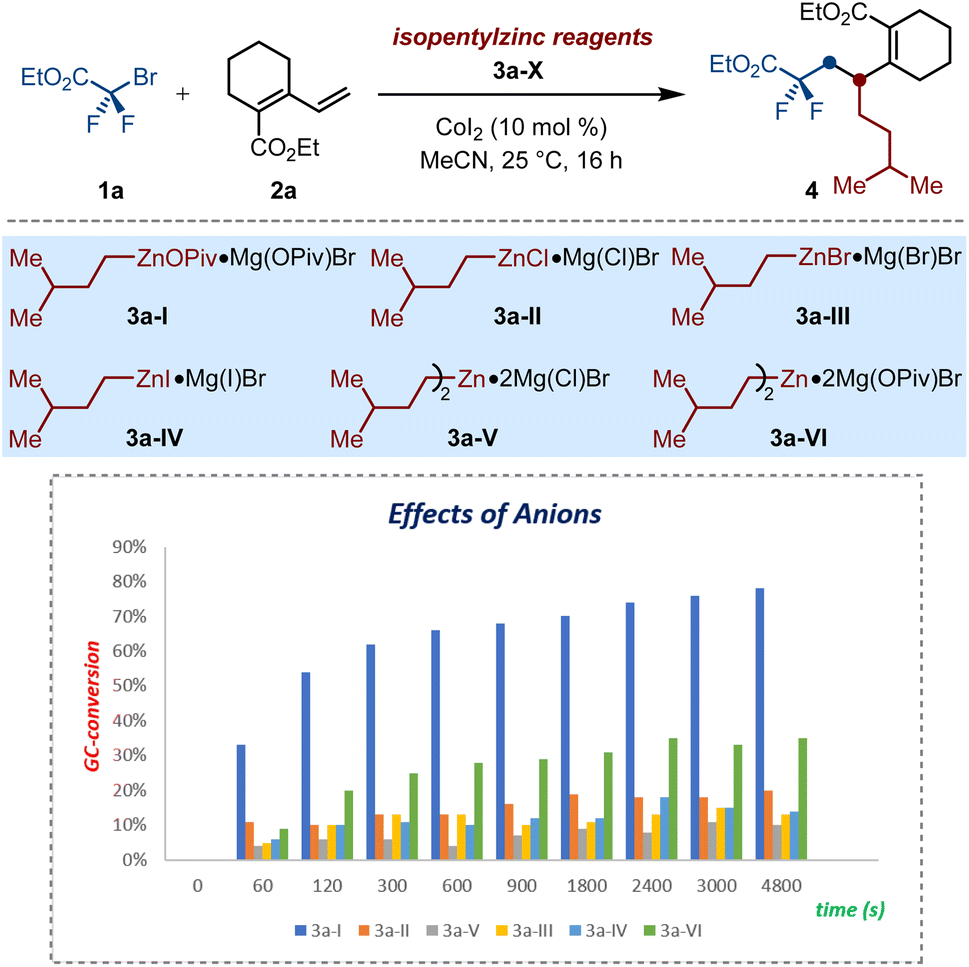 | ||
| Scheme 2 Kinetic experiments with alkylzinc reagents 3a-(I–VI). Reaction conditions: 1a (0.15 mmol, 1.0 equiv.), 2a (1.5 equiv.), 3a (2.5 equiv.), 0.5 to 80 min, biphenyl as the internal standard, detected by GC-analysis. | ||
By the use of OPiv-supported alkylzinc pivalates (3) as the typical Csp3-nucleophiles, we investigated the generality of our 1,2-selective dicarbofunctionalization of dienoates (2) (Scheme 3). Since the presence of stoichiometric LiCl in the solution of isopentylzinc pivalate resulted in a significantly decreased yield of 4, all alkylzinc pivalates were prepared from the corresponding alkyl bromides by I2-promoted Mg insertion in the absence of LiCl, followed by a transmetalation reaction with Zn(OPiv)2.19 We were pleased to find that various cyclic dienoates bearing ketal, gem-difluoro, dimethyl and aryl groups were employed as suitable substrates for the cascade cross-coupling to afford the products 4–7via 1,2-selective difunctionalization. The ring size of cyclic dienoates 2 had a slight influence on the envisioned transformations. Indeed, five- or seven-membered dienoates were well alkyldifluoroalkylated with 1a and isopentylzinc pivalate (3a-I), giving the corresponding compounds 8–9 in moderate to good yields. The more challenging acyclic dienoates smoothly underwent cobalt-catalyzed dicarbofunctionalization under identical reaction conditions. Notably, the simple cobalt catalysis showed excellent functional group compatibility and high regio- and stereoselectivity. Valuable electrophilic substituents, such as various aliphatic moieties bearing olefin, thienyl, ether, chloro and bromo groups were well tolerated and the stereoscopic configuration of the tetrasubstituted acrylates was completely retained in the catalytic system; the desired products 10–15 were obtained in 47–78% yields in a stereoretentive fashion. It is worth noting that various decorated 1,3-dienoate and 1,3-dienamide were also found to be viable substrates for the envisioned twofold Csp3–Csp3/Csp3–Csp3 bond formation, thereby delivering selective 1,2-dialkylated 16–17 as the sole products (Scheme 3A).
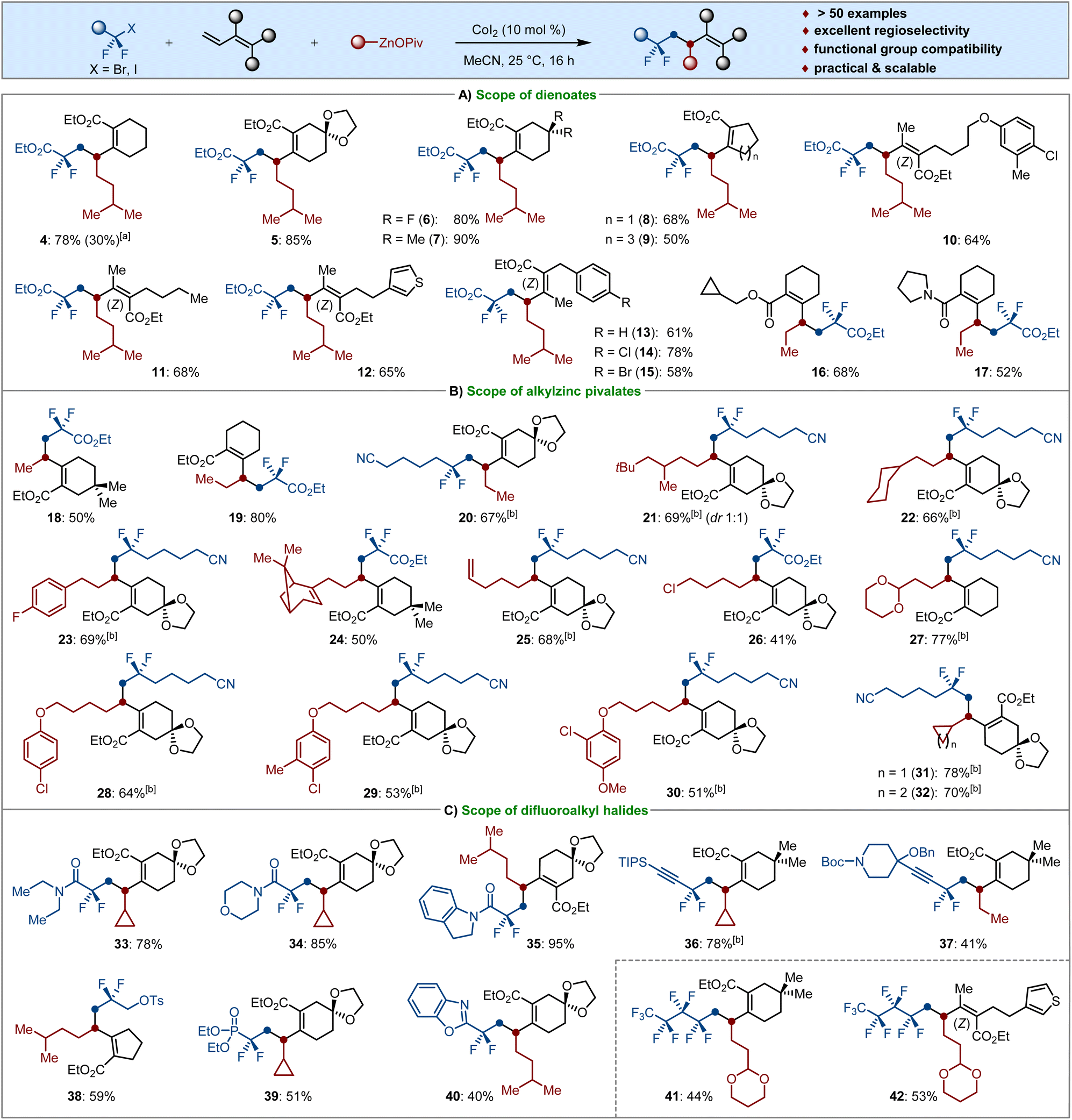 | ||
| Scheme 3 Substrate scope for the cobalt-catalyzed selective 1,2-dialkyldifluoroalkylation of dienoates. Reaction conditions: difluoroalkyl halides (1.5 equiv.), dienoates (0.15 mmol, 1.0 equiv.), Alk–ZnOPiv (2.5 equiv.), CoI2 (10 mol%), MeCN (1.5 mL), 25 °C, 16 h. [a] Alk–ZnOPiv·Mg(OPiv)Br·LiCl instead of 3a-I. [b] Difluoroalkyl halides (0.15 mmol, 1.0 equiv.), dienoates (1.5 equiv.), and TMEDA (30 mol%) were used. [c] 1.3 mmol scale. | ||
Thereafter, we next examined the sequential installation of CF2- and Csp3-groups across double bonds by using solid OPiv-supported alkylzinc reagents as the nucleophiles (Scheme 3B). Methy and ethyl groups are important pharmacophores in many bioactive molecules. Importantly, both Me–ZnOPiv and Et–ZnOPiv underwent cobalt-catalyzed difuloroalkylmethyl- and ethylation to provide the fluorinated products 18–20 in 50–80% isolated yields. Likewise, a variety of primary alkylzinc pivalates, as well as the functionalized organozincs are all competent Csp3-nucleophiles in the conjunctive cross-coupling reaction, giving the fluorinated carbogenic skeletons 21–30 in moderate to good yields under simple and practical reaction conditions. Moreover, increasing interest has been devoted to the introduction of strained carbocycles such as cyclopropanes and cyclobutanes during the past decade,20 which have found numerous applications in medicinal chemistry.21 As such, we prepared the corresponding solid alkylzinc pivalates and tested the cascade difluoroalkyl cyclopropanation and cyclobutanation of dienoates. It is worth noting that the expected products 31–32 were selectively obtained in 70–78% yields, which provides a perspective for installation of strained carbocycles and their further late-stage functionalization to afford highly complex molecules.
Inspired by these findings, the scope with respect to various fluoroalkyl bromides was further tested. As shown in Scheme 3C, different bromodifluoroacetamides were successfully utilized for the cascade cross-coupling reactions with dienoates and alkylzinc pivalates to afford the corresponding products 33–35 in high yields. Importantly, gem-difluoropropargyl bromides bearing hindered triisopropylsilyl aliphatic substituents were proven to be viable substrates as well, leading to fluorinated alkynes 36–37 in 41–78% yields with no trace of carbometallated products. These results highlighted the excellent functional group tolerance and chemoselectivity of our cobalt catalyst. Therefore, the substrate scope could easily be extended to the use of α-bromodifluoromethyl-substituted tosylate (38), phosphonate (39) and benzoxazole as well (40). It was particularly noteworthy that perfluorobutyl iodide was found to be successful for the cobalt-catalyzed 1,2-dicarbofunctionalization of dienoates with [2-(1,3-dioxan-2-yl)ethyl]zinc pivalate, smoothly affording the polyfluorinated products 41–42 with high regio- and site-selectivity.
The substrate scope was not only limited to the difluoroalkyl halides; we further succeeded in examining alkyl halides. To our delight, various tertiary α-bromocarbonyl compounds were proven to be suitable electrophiles for the generation of α-Csp3 radicals in the presence of a cobalt source, thereby smoothly undergoing the selective 1,2-dialkylation process to afford the carbogenic skeletons 43–51 in 40–62% yields. Moreover, the secondary and primary α-bromocarbonyl compounds were proven to be suitable substrates as well, and the corresponding products 52–53 were obtained in 61% and 27% yields, respectively. Herein, a catalytic amount of TMEDA as the ligand is required for increasing the conversions to the desired products (Scheme 4).
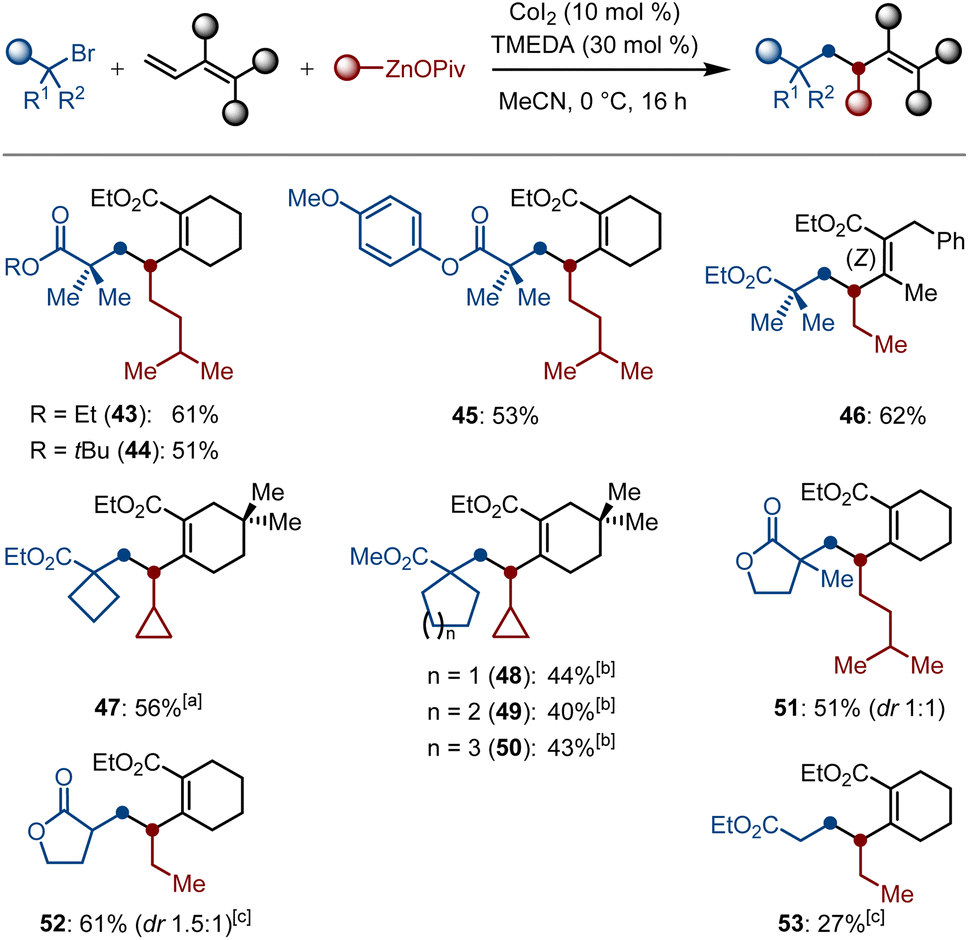 | ||
| Scheme 4 Substrate scope for the cobalt-catalyzed selective 1,2-dialkylation of dienoates. Reaction conditions: alkyl bromides (0.15 mmol, 1.0 equiv.), dienoates (1.5 equiv.), Alk–ZnOPiv (2.5 equiv.), CoI2 (10 mol%), TMEDA (30 mol%), MeCN (1.5 mL), 0 °C, 16 h. [a] 3 mmol scale. [b] Dienoates (0.15 mmol, 1.0 equiv.), alkyl bromides (2.0 equiv.), Alk–ZnOPiv (2.5 equiv.), CoI2 (10 mol%), MeCN (1.5 mL), 0 °C, 16 h. [c] w/o TMEDA, at 25 °C. | ||
Encouraged by the robustness of our cobalt-catalyzed sequential difluorolakylation-alkylation of functionalized dienoates, we became intrigued by the demonstration of its potential synthetical utility in the fluorine installation of druglike molecules and natural products (Scheme 5). Remarkably, dienoates derivatized from ibuprofen (54), citronellol (55), as well as difluoroalkyl bromides containing a complex molecule, such as estrone (56), L-menthol (57), vonoprazan (58), and duloxetine (59) derivatives, showed excellent compatibility to the direct twofold Csp3–Csp3 cross-coupling reaction by using OPiv-supported alkylzinc reagents as the nucleophilic partners. Thus, the high-yielding preparation of these fluorinated molecules should be proved instrumental for the discovery of novel drug precursors.
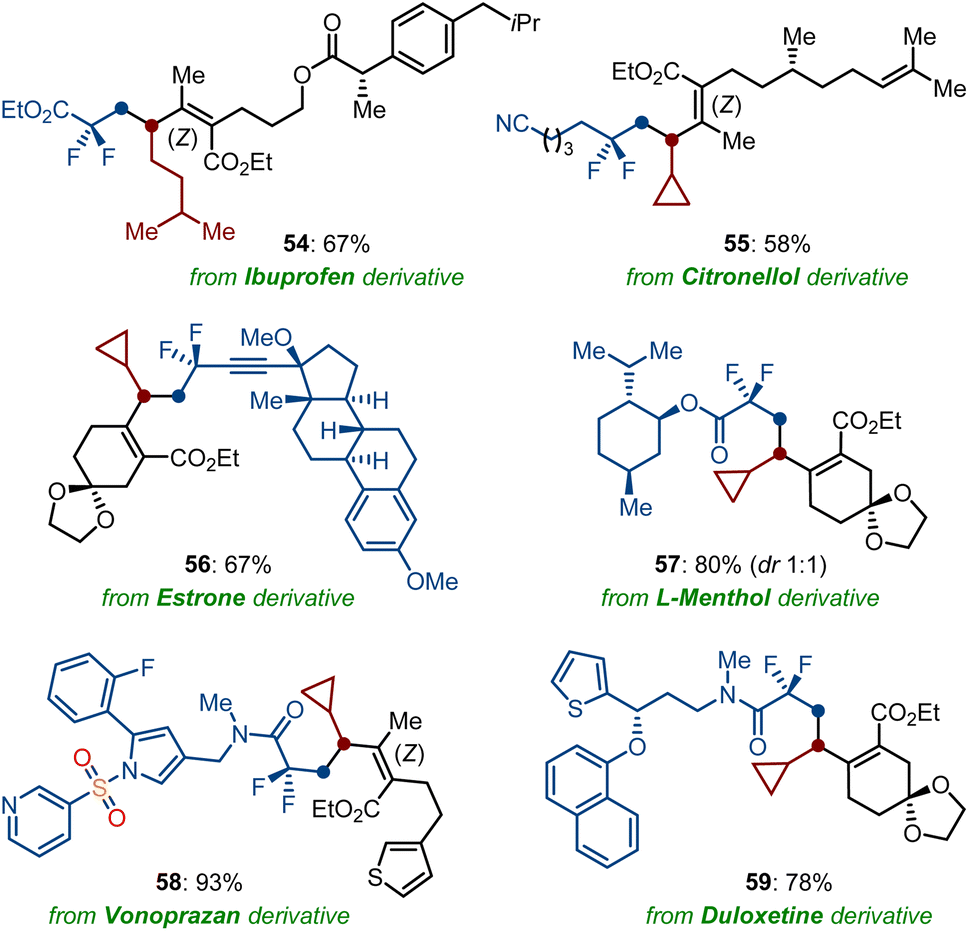 | ||
| Scheme 5 Late-stage fluorine installation of drugs and natural products. | ||
Intrigued by the outstanding catalytic activity of the cobalt catalyst, we sought to unravel the mechanism of this multicomponent cross-coupling. First, the cobalt-catalyzed difluoroalkylation-alkylation was completely inhibited by employing stoichiometric quantities of TEMPO as the radical scavengers. Moreover, a radical-clock experiment with the substrate α-cyclopropyl styrene 60 delivered the ring-opening products 61–62, which suggested a radical relay process in the reaction (Scheme 6A). We next investigated the stereoselective control of the reaction by setting parallel experiments with stereodefined (Z)- and (E)-dienoates. Notably, both (Z)-63 and (E)-63 smoothly underwent the envisioned cross-coupling process and generated the corresponding products 64 in complete control of stereoretentive fashion (Scheme 6B). These results indicated that the initial radical insertion formed a stable allylcobalt complex rather than a flexible π-allylcobalt intermediate, thereby ensuring the stereoselectivity of the targeted reaction. Beyond that, the ester group is crucial due to its conjugation effect on the active dienoates rather than a directing group. A set of cobalt-catalyzed dialkylations of exocyclic dienoate not only formed the desired product 9, but also delivered a hydrodifluoroalkylated compound 65 in 21% yield, which suggested that the reaction involves an initial 1,2-radical insertion (Scheme 6C). The well-defined cobalt(0) complex LnCo(dvtms) (66),22 was examined for the radical relay coupling reaction, delivering the desired product 4 with equal efficacy as compared to the CoI2/1,10-phen system. However, using CoCl(PPh3)3 as the catalyst only gave 4 in negligible conversion (Scheme 6D). These observations indicated that the low-valent Co(0)-species might be the catalytically active catalyst.
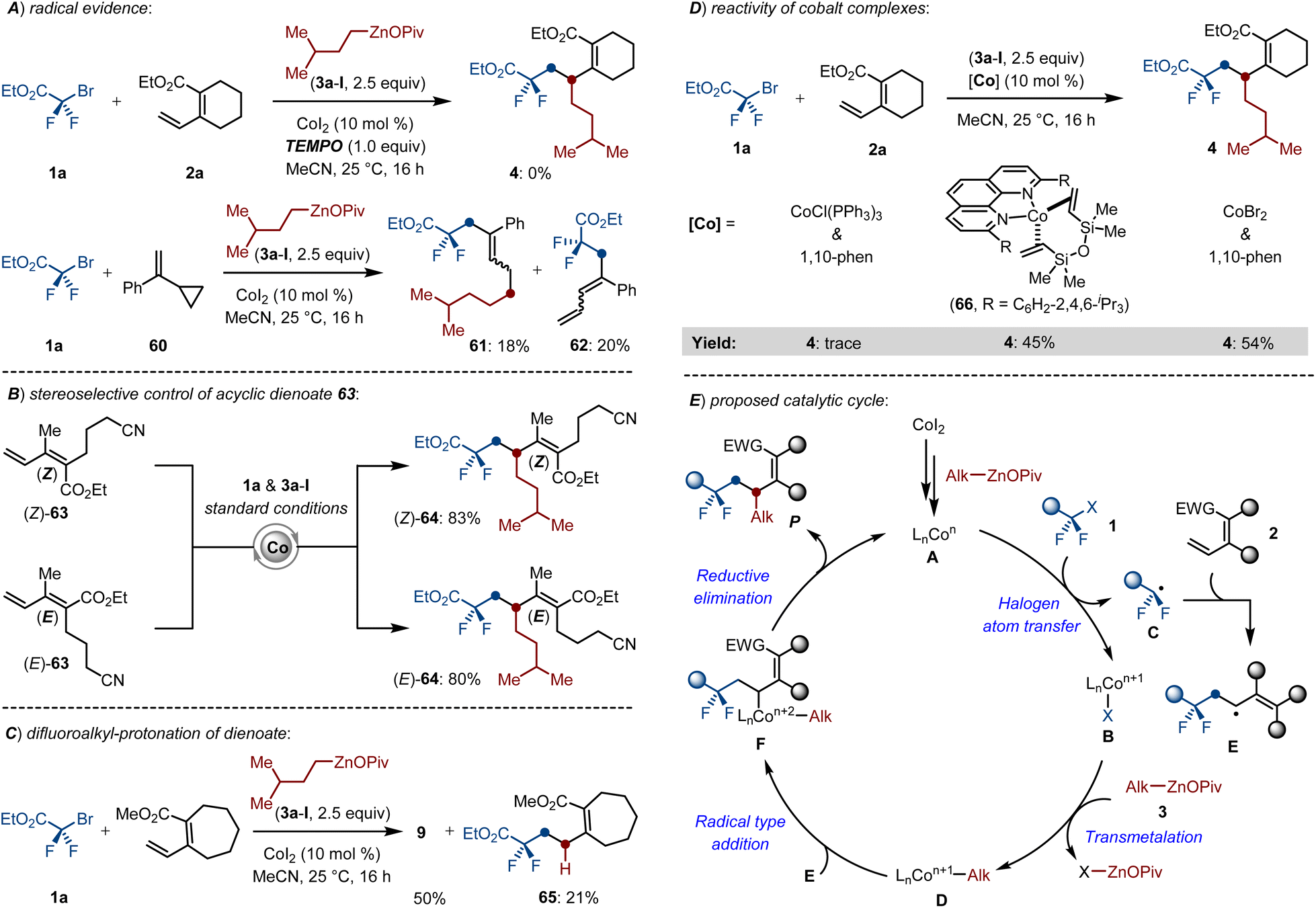 | ||
| Scheme 6 Mechanistic studies. | ||
Based on our findings and previous insights,3i,23 a plausible catalytic cycle has been proposed as shown in Scheme 6E. The initial reduction of CoI2 with alkylzinc pivalates formed the catalytically active LnCon-species (A), which promotes the intermolecular halogen atom transfer of difluoroalkyl bromides by SET to generate the difluoroalkyl radical B, then followed by a facile radical insertion of B into dienoates (2) to form the allyl radical E. Transmetalation between the newly formed LnCon+1-species and Alk–ZnOPiv (3) gave the alkyl-Con+1 (D). Subsequent radical type oxidation of D and E furnished the key intermediate F. Finally, reductive elimination delivered the 1,2-dialkylated products and regenerated the active LnCon-species.
Finally, the synthetic applications of functionalized difluoroalkylation-alkylation products were elaborated by various facile transformations (Scheme 7). First, the two ester groups could be selectively reduced by NaBH4 and LiAlH4, generating the corresponding alcohols 67 and 68, respectively. Facile base-mediated saponification of 4 gave a carboxylic acid, which can serve as a building block for the synthesis of a peptide with L-phenylalanine, leading to product 69 in 82% yield. Furthermore, we removed the protecting silyl group in the presence of TBAF, and performed a click-reaction of the newly formed terminal alkyne with benzyl azide and azido ester under copper catalysis, thus providing the polyfunctional triazoles 70–71 in good yields. Beyond this, the tertiary carboxylate moieties of the dialkylated compounds can be easily transformed into the N-hydroxyphthalimide esters 72–73, which displayed versatile transformations, such as the Giese reaction, reductive decarboxylation and borylation, leading to the corresponding products 74–76 in good yields.
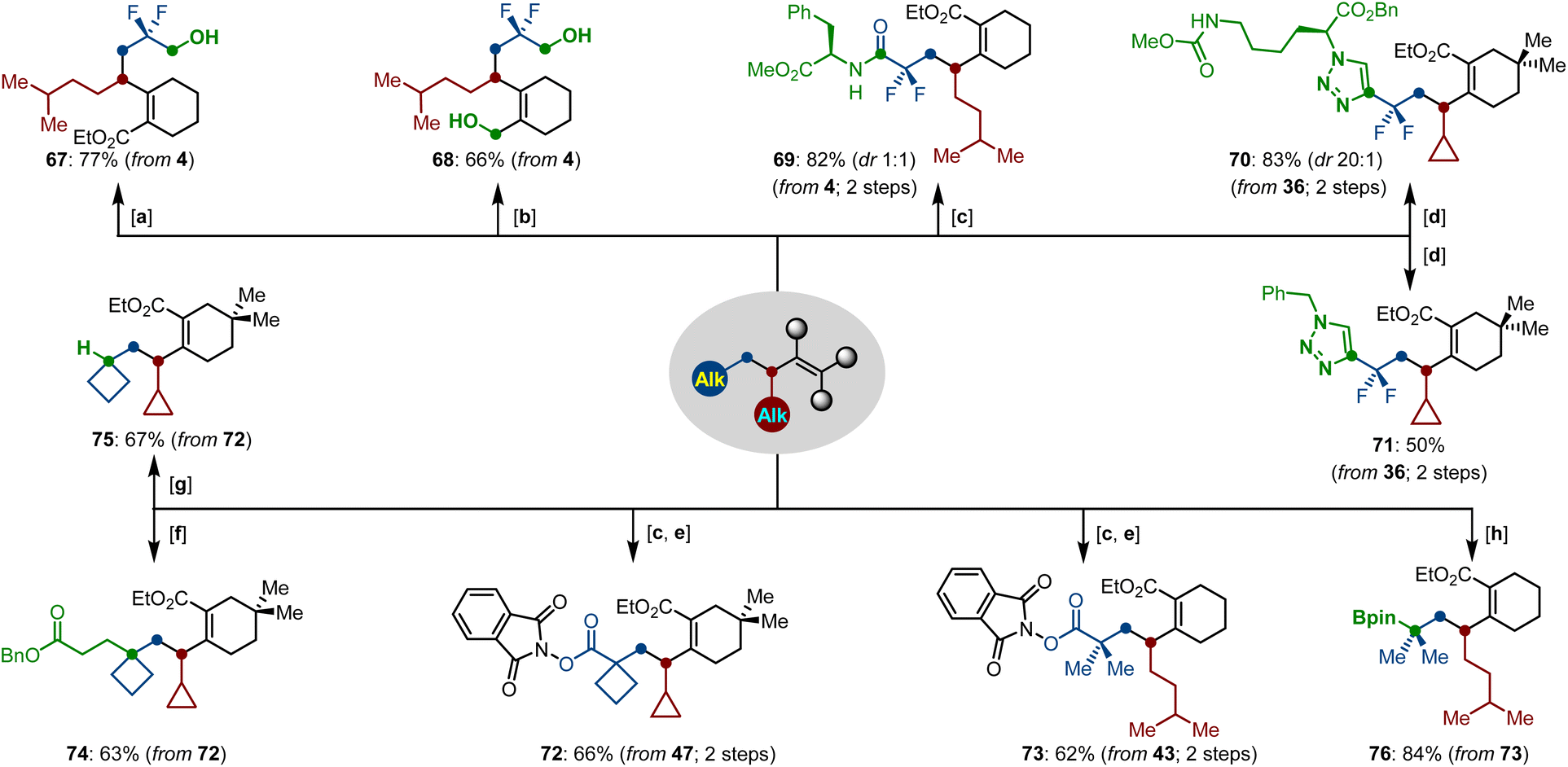 | ||
| Scheme 7 Facile late-stage functionalizations of dialkylated compounds. Reaction conditions: [a] NaBH4 (2.0 equiv.), EtOH, reflux, 4 h. [b] LiAlH4 (2.5 equiv.), THF, 23 °C, 4 h. [c] (i) LiOH (17 equiv.), THF/H2O, 23 °C, 2 h; (ii) L-phenylalanine (2.0 equiv.), PyBop (1.2 equiv.), Et3N (4.0 equiv.), THF, 23 °C, 12 h. [d] R–N3 (1.0 equiv.), CuI (10 mol%), DMF, 80 °C, 12 h. [e] N-Hydroxyphthalimide (1.2 equiv.), DIC (1.1 equiv.), DMAP (0.1 equiv.), CH2Cl2, 23 °C, 16 h. [f] Benzyl acrylate (1.0 equiv.), NiCl2·6H2O (20 mol%), Zn (2.0 equiv.), LiCl (3.0 equiv.), MeCN, 23 °C, 12 h. [g] PhSiH3 (1.5 equiv.), NiCl2·6H2O (10 mol%), Zn (0.5 equiv.), dtbbpy (20 mol%), THF/DMF/iPrOH, 40 °C, 1 h. [h] (MeB2pin2)Li (3.0 equiv.), NiCl2·6H2O (10 mol%), di-MeObipy (13 mol%), MgBr2·Et2O (1.5 equiv.), THF/DMF, 23 °C, 2 h. | ||
Conclusions
In summary, we have reported the preparation of a new type of solid and salt-stabilized alkylzinc pivalate, which shows enhanced stability and unique reactivity in the cobalt-catalyzed alkyldifluoroalkylation of dienoates under directing-group-free conditions, thus achieving the modular installation of CF2- and Csp3-groups across the double bond of dienoates via a cascade Csp3–Csp3/Csp3–Csp3 bond formation with complete control of the regioselectivity and stereoretentivity. Particularly noteworthy is the dramatic OPiv-tuning effect on Alk–ZnOPiv, which leads to superior stability and reactivity among other halide-supported alkylzincs. Moreover, the synthetic simplicity and utility of our 1,2-selective dicarbofuncationalization are well illustrated by the facile modifications of drug-like molecules and natural products, as well as the late-stage derivatizations of the fluorinated products. We believe that this work will provide a perspective for the design of novel organozinc reagents by the anion-regulation strategy and find wide applications in Negishi-type reactions.Data availability
The datasets supporting this article have been uploaded as part of the ESI material.†Author contributions
Jie Li conceptualized the project and wrote the paper; J. Lin and K. C. contributed equally to this work; J. Lin, K. C., and J. W. designed and performed the experiments; J. G., S. D., J. Lin, and Y. H. prepared the starting materials, all the authors were involved in the interpretation of the results presented in the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (22322108), Natural Science Foundation of Jiangsu Province (BK20221355), the start-up grant (NH10900422) of Soochow University, and a sponsored program by the Jiangsu Province for the Cultivation of Innovation (SR10900122).Notes and references
- Representative reviews: (a) Y.-C. Luo, C. Xu and X. Zhang, Chin. J. Chem., 2020, 38, 1371–1394 CrossRef CAS; (b) J. Diccianni, Q. Lin and T. Diao, Acc. Chem. Res., 2020, 53, 906–919 CrossRef CAS PubMed; (c) X. Qi and T. Diao, ACS Catal., 2020, 10, 8542–8556 CrossRef CAS PubMed; (d) J. Derosa, O. Apolinar, T. Kang, V. T. Tran and K. M. Engle, Chem. Sci., 2020, 11, 4287–4296 RSC; (e) R. Giri and S. Kc, J. Org. Chem., 2018, 83, 3013–3022 CrossRef CAS PubMed; (f) R. K. Dhungana, S. Kc, P. Basnet and R. Giri, Chem. Rec., 2018, 18, 1314–1340 CrossRef CAS PubMed and references cited therein..
- Selective examples for alkene diarylation: (a) B. Shrestha, P. Basnet, R. K. Dhungana, S. KC, S. Thapa, J. M. Sears and R. Giri, J. Am. Chem. Soc., 2017, 139, 10653–10656 CrossRef CAS; (b) J. Derosa, V. T. Tran, M. N. Boulous, J. S. Chen and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 10657–10660 CrossRef CAS; (c) W. Li, J. K. Boon and Y. Zhao, Chem. Sci., 2018, 9, 600–607 RSC; (d) S. Thapa, R. K. Dhungana, R. T. Magar, B. Shrestha, S. KC and R. Giri, Chem. Sci., 2018, 9, 904–909 RSC; (e) P. Gao, L.-A. Chen and M. K. Brown, J. Am. Chem. Soc., 2018, 140, 10653–10657 CrossRef CAS PubMed; (f) P. Basnet, R. K. Dhungana, S. Thapa, B. Shrestha, S. KC, J. M. Sears and R. Giri, J. Am. Chem. Soc., 2018, 140, 7782–7786 CrossRef CAS PubMed; (g) J. Derosa, R. Kleinmans, V. T. Tran, M. K. Karunananda, S. R. Wisniewski, M. D. Eastgate and K. M. Engle, J. Am. Chem. Soc., 2018, 140, 17878–17883 CrossRef CAS PubMed; (h) Y. Zhang, G. Chen and D. Zhao, Chem. Sci., 2019, 10, 7952–7957 RSC; (i) D. Anthony, Q. Lin, J. Baudet and T. Diao, Angew. Chem., Int. Ed., 2019, 58, 3198–3202 CrossRef CAS PubMed; (j) J. Derosa, T. Kang, V. T. Tran, S. R. Wisniewski, M. K. Karunananda, T. C. Jankins, K. L. Xu and K. M. Engle, Angew. Chem., Int. Ed., 2020, 58, 1202–1205 Search PubMed; (k) O. Apolinar, V. T. Tran, N. Kim, M. A. Schmidt, J. Derosa and K. M. Engle, ACS Catal., 2020, 10, 14234–14239 CrossRef CAS; (l) Z. Dong, Q. Tang, C. Xu, L. Chen, H. Ji, S. Zhou, L. Song and L.-A. Chen, Angew. Chem., Int. Ed., 2023, 62, e202218286 CAS and references cited therein..
- Selective examples for alkene arylalkylation: (a) T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801–805 CrossRef CAS PubMed; (b) A. García-Domínguez, Z. Li and C. Nevado, J. Am. Chem. Soc., 2017, 139, 6835–6838 CrossRef PubMed; (c) J.-W. Gu, Q.-Q. Min, L.-C. Yu and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 12270–12274 CrossRef CAS; (d) S. KC, R. K. Dhungana, B. Shrestha, S. Thapa, N. Khanal, P. Basnet, R. W. Lebrun and R. Giri, J. Am. Chem. Soc., 2018, 140, 9801–9805 CrossRef CAS PubMed; (e) L. Guo, H.-Y. Tu, S. Zhu and L. Chu, Org. Lett., 2019, 21, 4771–4776 CrossRef CAS PubMed; (f) A. García-Domínguez, R. Mondal and C. Nevado, Angew. Chem., Int. Ed., 2019, 58, 12286–12290 CrossRef; (g) S. Kc, R. K. Dhungana, N. Khanal and R. Giri, Angew. Chem., Int. Ed., 2020, 59, 8047–8051 CrossRef CAS PubMed; (h) C. Xu, R. Cheng, Y.-C. Luo, M.-K. Wang and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 18741–18747 CrossRef CAS; (i) X. Cheng, X. Liu, S. Wang, Y. Hu, B. Hu, A. Lei and J. Li, Nat. Commun., 2021, 12, 4366–4375 CrossRef CAS PubMed; (j) H.-Y. Tu, F. Wang, L. Huo, Y. Li, S. Zhu, X. Zhao, H. Li, F.-L. Qing and L. Chu, J. Am. Chem. Soc., 2020, 142, 9604–9611 CrossRef CAS PubMed; (k) V. Aryal, L. J. Chesley, D. Niroula, R. R. Sapkota, R. K. Dhungana and R. Giri, ACS Catal., 2022, 12, 7262–7268 CrossRef CAS and references cited therein..
- (a) K. Mizutani, H. Shinokubo and K. Oshima, Org. Lett., 2003, 5, 3959–3961 CrossRef CAS PubMed; (b) J. Terao, Y. Kato and N. Kambe, Chem.–Asian J., 2008, 3, 1472–1478 CrossRef CAS.
- (a) V. B. Phapale, E. Buñuel, M. García-Iglesias and D. J. Cárdenas, Angew. Chem., Int. Ed., 2007, 46, 8790–8795 CrossRef CAS ; also see a Ni-catalyzed dicarbofunctionalization of vinyl–Bpin: ; (b) M. Chierchia, P. Xu, G. J. Lovinger and J. P. Morken, Angew. Chem. Int. Ed., 2019, 58, 14245–14249 CrossRef CAS PubMed.
- (a) D. J. Cárdenas, Angew. Chem., Int. Ed., 2003, 42, 384–387 CrossRef PubMed; (b) A. C. Frisch and M. Beller, Angew. Chem., Int. Ed., 2005, 44, 674–688 CrossRef CAS PubMed; (c) A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656–2670 CrossRef CAS.
- (a) L. Capdevila and X. Ribas, PATAI'S Chemistry of Functional Groups: Cobalt-catalyzed Cross-Coupling Reactions, John Wiely & Sons, 2022 Search PubMed; (b) Y. Ikeda, T. Nakamura, H. Yorimitsu and K. Oshima, J. Am. Chem. Soc., 2002, 124, 6514–6515 CrossRef CAS PubMed.
- (a) J. Derosa, V. A. van der Puyl, V. T. Tran, M. Liua and K. M. Engle, Chem. Sci., 2018, 9, 5278–5283 RSC ; also see reductive dialkylation: ; (b) T. Yang, Y. Jiang, Y. Luo, J. J. H. Lim, Y. Lan and M. J. Koh, J. Am. Chem. Soc., 2020, 142, 21410–21419 CrossRef CAS PubMed; (c) J.-X. Zhang and W. Shu, Org. Lett., 2022, 24, 3844–3849 CrossRef CAS PubMed.
- C. Xu, Z.-F. Yang, L. An and X. Zhang, ACS Catal., 2019, 9, 8224–8229 CrossRef CAS.
- R. K. Dhungana, R. R. Sapkota, L. M. Wickham, D. Niroula and R. Giri, J. Am. Chem. Soc., 2020, 142, 20930–20936 CrossRef CAS PubMed.
- (a) Handbook of Functionalized Organometallics: Applications in Synthesis, ed. P. Knochel, Wiley, 2005 Search PubMed; (b) D. Haas, J. M. Hammann, R. Greiner and P. Knochel, ACS Catal., 2016, 6, 1540–1552 CrossRef CAS; (c) P. Eckert, S. Sharif and M. G. Organ, Angew. Chem., Int. Ed., 2021, 60, 12224–12241 CrossRef CAS.
- (a) L. Jin, C. Liu, J. Liu, F. Hu, Y. Lan, A. S. Batsanov, J. A. K. Howard, T. B. Marder and A. Lei, J. Am. Chem. Soc., 2009, 131, 16656–16657 CrossRef CAS PubMed; (b) L. Jin, J. Xin, Z. Huang, J. He and A. Lei, J. Am. Chem. Soc., 2010, 132, 9607–9609 CrossRef CAS PubMed; (c) J. Li, L. Jin, C. Liu and A. Lei, Chem. Commun., 2013, 49, 9615–9617 RSC.
- A. Hernán-Gómez, E. Herd, E. Hevia, A. R. Kennedy, P. Knochel, K. Koszinowski, S. M. Manolikakes, R. E. Mulvey and C. Schnegelsberg, Angew. Chem., Int. Ed., 2014, 53, 2706–2710 CrossRef PubMed.
- (a) S. Bernhardt, G. Manolikakes, T. Kunz and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 9205–9209 CrossRef CAS; (b) C. I. Stathakis, S. Bernhardt, V. Quint and P. Knochel, Angew. Chem., Int. Ed., 2012, 51, 9428–9432 CrossRef CAS; (c) T. J. Greshock, K. P. Moore, R. T. McClain, A. Bellomo, C. K. Chung, S. D. Dreher, P. S. Kutchukian, Z. Peng, I. W. Davies, P. Vachal, M. Ellwart, S. M. Manolikakes, P. Knochel and P. G. Nantermet, Angew. Chem., Int. Ed., 2016, 55, 13714–13718 CrossRef CAS.
- (a) J. Wang, Z. Duan, X. Liu, S. Dong, K. Chen and J. Li, Angew. Chem., Int. Ed., 2022, 61, e202202379 CAS; (b) Y. Hu, J. Peng, B. Hu, J. Wang, J. Jing, J. Lin, X. Liu, X. Qi and J. Li, Nat. Commun., 2023, 14, 1454–1465 CrossRef CAS PubMed.
- (a) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed; (b) M.-C. Belhomme, T. Besset, T. Poisson and X. Pannecoucke, Chem. – Eur. J., 2015, 21, 12836–12865 CrossRef CAS PubMed; (c) C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2015, 54, 3216–3221 CrossRef CAS; (d) Z. Feng, Y.-L. Xiao and X. Zhang, Acc. Chem. Res., 2018, 51, 2264–2278 CrossRef CAS PubMed; (e) D.-Q. Dong, H. Yang, J.-L. Shi, W.-J. Si, Z.-L. Wang and X.-M. Xu, Org. Chem. Front., 2020, 7, 2538–2575 RSC.
- (a) X.-S. Hu, J.-S. Yu and J. Zhou, Chem. Commun., 2019, 55, 13638–13648 RSC; (b) J. B. I. Sap, C. F. Meyer, N. J. W. Straathof, N. Iwemene, C. W. am Ende, A. A. Trabanco and V. Gouverneur, Chem. Soc. Rev., 2021, 50, 8214–8247 RSC.
- (a) C. Gosmini, J.-M. Begouin and A. Moncomble, Chem. Commun., 2008, 3221–3233 RSC; (b) G. Cahiez and A. Moyeux, Chem. Rev., 2010, 110, 1435–1462 CrossRef CAS PubMed; (c) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef CAS; (d) A. Guérinot and J. Cossy, Acc. Chem. Res., 2020, 53, 1351–1363 CrossRef PubMed; (e) J. Li, Top. Organomet. Chem., 2023 DOI:10.1007/3418_2023_83.
- Alk–ZnOPv·Mg(OPiv)Br was abbreviated as Alk–ZnOPiv for the sake of clarity.
- (a) P. Ren, L.-A. Stern and X. Hu, Angew. Chem., Int. Ed., 2012, 51, 9110–9113 CrossRef CAS PubMed; (b) C. Andersen, V. Ferey, M. Daumas, P. Bernardelli, A. Guérinot and J. Cossy, Org. Lett., 2019, 21, 2285–2289 CrossRef CAS PubMed.
- (a) H.-U. Reissig, in The Chemistry of the Cyclopropyl Group, ed. Z. Rappoport, Wiley, 1987 Search PubMed; (b) J. Pietruszka, Chem. Rev., 2003, 103, 1051–1070 CrossRef CAS PubMed.
- Y. Ueda, H. Tsurugi and K. Mashima, Angew. Chem., Int. Ed., 2020, 59, 1552–1556 CrossRef CAS PubMed.
- X. Liu, H. Chen, D. Yang, B. Hu, Y. Hu, S. Wang, Y. Lan, A. Lei and J. Li, ACS Catal., 2023, 13, 9254–9263 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02345a |
| ‡ J. Lin and K. Chen contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |