
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOuter coordination sphere influences on cofactor maturation and substrate oxidation by cytochrome P460†
Melissa M.
Bollmeyer
 ,
Sean H.
Majer
,
Rachael E.
Coleman
and
Kyle M.
Lancaster
*
,
Sean H.
Majer
,
Rachael E.
Coleman
and
Kyle M.
Lancaster
*
Baker Laboratory, Department of Chemistry and Chemical Biology, Cornell University, 162 Sciences Drive, Ithaca, NY 14853, USA. E-mail: kml236@cornell.edu
First published on 19th July 2023
Abstract
Product selectivity of ammonia oxidation by ammonia-oxidizing bacteria (AOB) is tightly controlled by metalloenzymes. Hydroxylamine oxidoreductase (HAO) is responsible for the oxidation of hydroxylamine (NH2OH) to nitric oxide (NO). The non-metabolic enzyme cytochrome (cyt) P460 also oxidizes NH2OH, but instead produces nitrous oxide (N2O). While both enzymes use a heme P460 cofactor, they selectively oxidize NH2OH to different products. Previously reported structures of Nitrosomonas sp. AL212 cyt P460 show that a capping phenylalanine residue rotates upon ligand binding, suggesting that this Phe may influence substrate and/or product binding. Here, we show via substitutions of the capping Phe in Nitrosomonas europaea cyt P460 that the bulky phenyl side-chain promotes the heme-lysine cross-link forming reaction operative in maturing the cofactor. Additionally, the Phe side-chain plays an important role in modulating product selectivity between N2O and NO during NH2OH oxidation under aerobic conditions. A picture emerges where the sterics and electrostatics of the side-chain in this capping position control the kinetics of N2O formation and NO binding affinity. This demonstrates how the outer coordination sphere of cyt P460 is tuned not only for selective NH2OH oxidation, but also for the autocatalytic cross-link forming reaction that imbues activity to an otherwise inactive protein.
Introduction
Ammonia oxidizing bacteria (AOB) derive their energy for life from the oxidation of ammonia (NH3) to nitrite (NO2−).1 In each step of this primary metabolism, enzymes must control product formation and avoid off-pathway reactions that lead to energy loss. The integral-membrane Cu protein ammonia monooxygenase (AMO) is responsible for the first step in which NH3 is hydroxylated using oxygen to form hydroxylamine (NH2OH). This step requires the input of two electrons, which are hypothesized to be supplied from the oxidation of NH2OH to nitric oxide (NO) by the multi-heme enzyme hydroxylamine oxidoreductase (HAO).2 The third electron from NH2OH oxidation can then be used for cellular respiration. It is both noteworthy and imperative that HAO catalyzes such a selective oxidation owing to the highly reactive nature of NH2OH, especially with Fe.3–7 Indeed, NH2OH will readily disproportionate to a variety of products including NO, N2, NH3, and nitrous oxide (N2O).8 For AOB to survive, HAO must minimize these non-productive reactions. The selective nature of HAO may in part be attributed to its use of the cytochrome (cyt) P460 metallocofactor, the only heme known to oxidize its Fe-bound substrate. Besides the HAOs, the cyt P460 family comprises the only other enzymes known to contain the heme P460 cofactor. These enzymes can oxidize NH2OH but their product distribution features a large fraction of N2O under aerobic conditions.9 Under anaerobic conditions, cyt P460 exclusively oxidizes NH2OH to N2O.9 Production of N2O from NH2OH is an energetically futile process because only two electrons are generated per equivalent of NH2OH. These electrons would only be sufficient to drive AMO catalysis, leaving no electrons to enter the respiratory electron transport chain. For this reason, we have proposed that cyt P460 is not a part of the primary metabolic ammonia oxidation pathway.9 Plausible roles include NH2OH and/or NO detoxification,10 although this remains speculative.Although cyt P460 and HAO both oxidize NH2OH using heme P460 cofactors, they share little overall structural similarity. HAO is a homotrimeric protein with seven c-type electron-transfer hemes and one heme P460 per each ca. 65 kDa subunit.11 In addition to the two cysteine cross-links formed by the canonical cyt c CXXCH motif, heme P460 of HAO contains additional cross-links from the C3 atom and phenol oxygen of a tyrosine to the α-meso carbon and adjacent pyrrole carbon of the porphyrin, respectively.12 In contrast, cyt P460 is a much smaller homodimeric protein with only one heme P460 per ca. 16 kDa subunit.13,14 Like HAO, the heme P460 cofactor of cyt P460 contains an additional cross-link, but instead features a covalent attachment from the nitrogen of a lysine residue to the γ-meso carbon of the porphyrin.14 Spectroscopic studies of HAO's heme P460 have been limited because contributions from this metallocofactor are often occluded by the seven other c-type hemes. Moreover, HAO containing the tyrosine cross-link has not been recombinantly expressed, making it challenging to study the roles of residues involved in catalysis. Our group has gleaned insight into the mechanism of NH2OH oxidation by studying cyt P460, which has afforded us an unobscured spectroscopic handle and access to mutagenesis.
The catalytic cycles of HAO and cyt P460 likely feature the same initial steps: NH2OH binds and then the resulting FeIII–NH2OH adduct is oxidized to a six-coordinate (6c) {FeNO}6 (Enemark–Feltam notation)15via a 6c {FeNO}7 intermediate (Fig. 1).16 In the presence of O2 HAO produces NO2−, although this was attributed to the non-enzymatic reaction of NO with O2.2 Further oxidation to NO2− does not occur in the absence of O2, nor is there subsequent reactivity with additional equivalents of NH2OH, supporting the contention that NO is the enzymatic product.2 In contrast, the {FeNO}6 intermediate of cyt P460 can undergo attack by a second equivalent of NH2OH to form N2O. Under aerobic conditions, cyt P460 produces a mixture of N2O and NO2−, where NO2− production is the result of NO dissociation followed by a non-enzymatic reaction with O2. The reaction between NO and O2 drives the equilibrium towards NO release, whereas under anaerobic conditions cyt P460 only produces N2O.9 Product selectivity between NO and N2O is therefore a matter of competition between the rates of NO release and N2O formation.
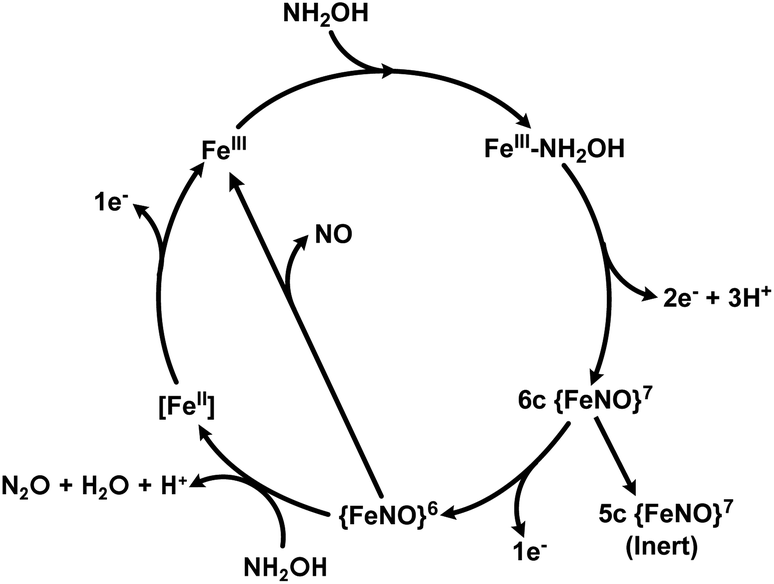 | ||
| Fig. 1 The working catalytic cycle of cytochrome P460. Adapted from ref. 16. | ||
To better understand the mechanistic details of biological NH2OH oxidation, our focus has been drawn to the outer coordination sphere of cyt P460. In one of our prior studies, we showed that a distal glutamate is required to serve as a proton relay during oxidation of NH2OH, demonstrating that a properly configured outer coordination sphere is essential to biological NH2OH oxidation.17Nitrosomonas europaea cyt P460 contains this distal Glu and is competent for NH2OH oxidation, whereas Nitrosomonas sp. AL212 cyt P460 has an alanine residue in the position of Glu and is unable to oxidize NH2OH despite its ability to form, via shunts, all previously observed intermediates of the cyt P460 catalytic cycle. Mutation of Ala to Glu restored NH2OH oxidation activity. Crystal structures showed that in addition to this Glu residue rotating in and out to interact with NH2OH, a capping phenylalanine rotates to accommodate substrate binding (Fig. 2). This implicated a role for the capping Phe residue in controlling substrate and/or product binding.
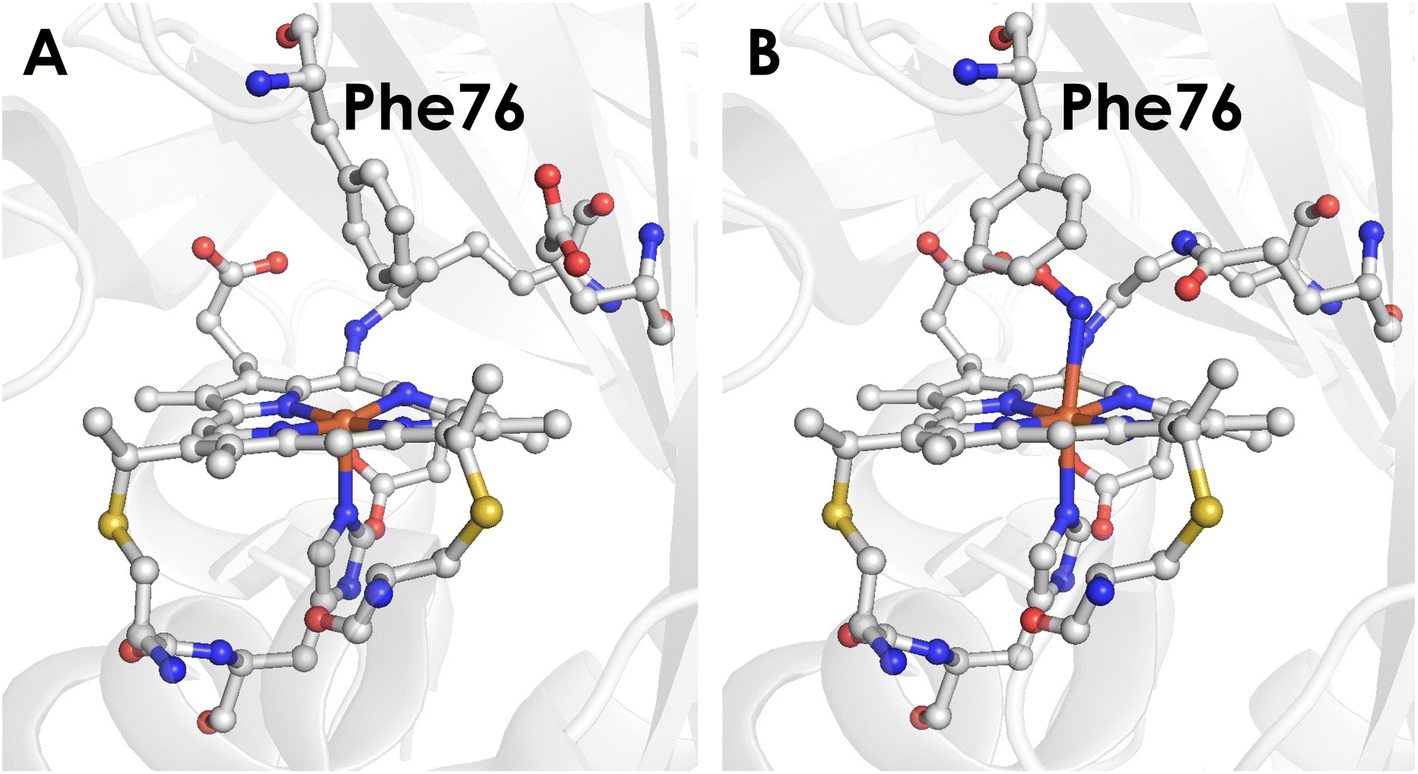 | ||
| Fig. 2 (A) Crystal structure of the Nitrosomonas sp. AL212 cyt P460 Ala131Glu mutant (PDB 6E0X) showing the capping Phe side-chain directly above the Fe. (B) Crystal structure of the Nitrosomonas sp. AL212 cyt P460 Ala131Gln mutant (PDB 6E0Y) showing the re-orientation of Phe to accommodate NH2OH binding. | ||
If the capping Phe does provide steric influence on the active site and reactive intermediates, it could play an important role in the mechanism of cyt P460. We now show via mutagenesis of this capping Phe that it plays a critical role in both heme-lysine cross-link formation as well as NH2OH oxidation. Notably, removing the steric hindrance of the Phe side-chain results in a change in the rate-determining step of NH2OH oxidation that impacts product selectivity. This study highlights some of the key features of the cyt P460 active site that control N2O versus NO formation.
Results
Expression of Phe41Ala cyt P460 results in cross-link deficient protein
Growth and purification of the N. europaea cyt P460 Phe41Ala mutant following the same procedure as WT cyt P460 yielded a red protein, in contrast to the British racing green of WT cyt P460. The UV/vis absorption spectrum of the as-isolated mutant exhibited a sharp Soret band with a maximum at 403 nm and Q-bands at 627 nm and 501 nm (Table S1†) consistent with a cross-link deficient (CLD) cyt P460 (Fig. 3A).19 There was also a shoulder at 436 nm that was indicative of ca. 40% cross-linked (CL) protein in the sample. Unlike CLD variants generated by mutation of the cross-linking lysine residue, the Phe41Ala mutant still contains this lysine. It is instead similar to the recently reported CLD mutant Arg44Ala whose crystal structure showed the uncross-linked Lys forming a salt-bridge with Glu97.20 While the Arg44Ala mutant did not contain any CL component the Phe41Ala mutant was purified as a mixture, suggesting that cross-link formation may be more facile in the Phe41Ala mutant compared to the Arg44Ala mutant.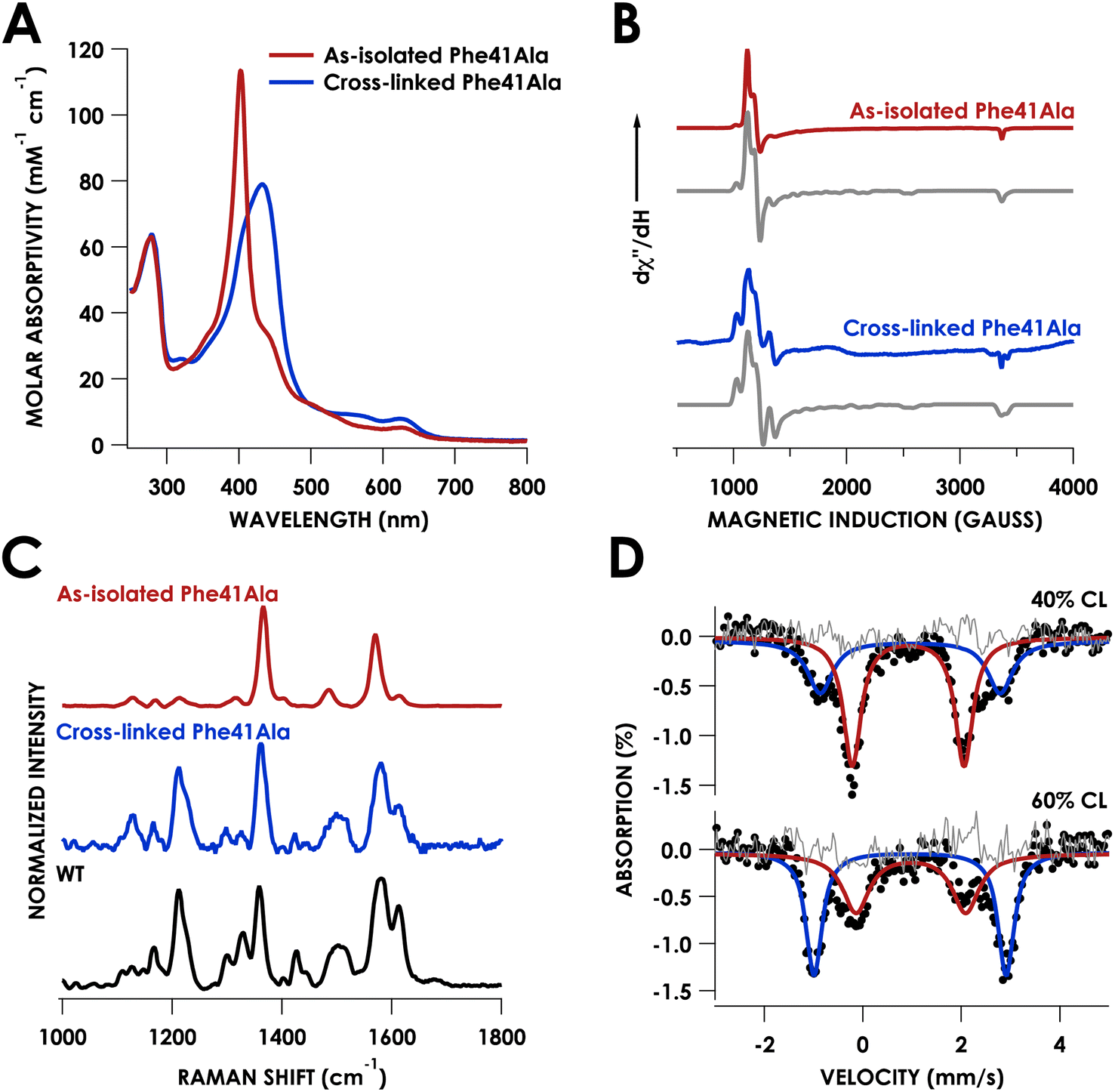 | ||
| Fig. 3 Characterization of Phe41Ala cyt P460. (A) UV/vis absorption spectra of the as-isolated (red) and cross-linked enzyme (blue). (B) CW X-band EPR spectra collected at 10 K of the as-isolated (red) and cross-linked enzyme (blue). Grey traces are the corresponding simulations. (C) Resonance Raman spectra obtained via excitation at 405 nm for as-isolated Phe41Ala cyt P460 (red), cross-linked Phe41Ala cyt P460 (blue), and WT cyt P460 (black). (D) Mössbauer spectra for Phe41Ala cyt P460 containing either 40% cross-linked (CL) character (top) or 60% cross-linked character (bottom). Raw data is shown as black dots, the cross-link deficient (CLD) component is shown in red, the CL component is shown in blue, and the residual is shown in grey. | ||
Characterization of the as-isolated Phe41Ala mutant by X-band electron-paramagnetic resonance (EPR) spectroscopy at 10 K revealed two high-spin FeIII components (Fig. 3B). The major component accounted for 81% of the signal and was fit to g-values of 5.99, 5.55, 1.99 with E/D = 0.01 (Fig. S1†). This component resembled that of the CLD WT proenzyme (Fig. S2†), which had g-values of 6.02, 5.54, and 1.99 (Table S2†).20 The second component was a more rhombic signal with an E/D = 0.03 and g-values of 6.54, 5.05, and 1.97, consistent with those of CL WT cyt P460.9 The resonance Raman spectrum obtained with laser excitation at 405 nm was also consistent with previously characterized CLD mutants with an oxidation state marker band (ν4) at 1367 cm−1 and spin state marker band (ν3) at 1485 cm−1 (Table S3†). The positions of these bands fall within the range of six-coordinate S = 5/2 ferric hemes. Comparison of the ν3 position to other CLD mutants shows a slight shift to lower frequency compared to WT proenzyme and a greater shift compared to the Lys70Tyr mutant, which could be attributed to the more open active site of the Phe41Ala mutant allowing coordination of water.
Mössbauer spectroscopy provided further characterization of the mixed CL and CLD forms of the Phe41Ala mutant. Two samples of Phe41Ala cyt P460 enriched with 57Fe were purified containing different ratios of CL to CLD forms (Table S4†). The percentage of CL protein in the sample was quantified by UV/vis absorption spectroscopy, where the extinction coefficient at 436 nm was used to obtain the concentration of the CL protein and the extinction coefficient at 280 was used to obtain total protein concentration. The first sample contained 40% CL protein by UV/vis quantification while the second contained 60% CL protein. Mössbauer spectra of the reduced samples of Phe41Ala cyt P460 obtained at 85 K and zero field contained two doublets that varied in intensity (Fig. 3D). For the sample with 40% CL protein, the minor component had an isomer shift of 0.96 ± 0.02 mm s−1 and quadrupole splitting of 3.65 ± 0.03 mm s−1 and resembled that of WT, which in our study had an isomer shift of 0.96 ± 0.01 mm s−1 and quadrupole splitting of 3.71 ± 0.01 mm s−1 collected at 4 K (Fig. S3†). The doublet corresponding to the CL component accounted for 36% of the sample, close to the composition obtained from UV/vis quantification. The major component had an isomer shift of 0.92 ± 0.01 mm s−1 and quadrupole splitting of 2.27 ± 0.02 mm s−1. In the sample that contained 60% CL protein, this second component was still present but in a smaller percentage, allowing us to assign it to the CLD protein. It is also consistent with other S = 2 c-type heme proteins such as deoxyhemoglobin and deoxymyoglobin.21 Meanwhile, the Mössbauer spectrum of the reduced Lys70Tyr obtained at 4 K and 0 field had an isomer shift of 0.46 ± 0.01 mm s−1 and quadrupole splitting of 1.04 ± 0.02 mm s−1, consistent with an S = 0 system (Fig. S3B†). The decrease in temperature could explain the change in spin compared to CLD Phe41Ala. Alternatively, the Tyr could promote binding of water, resulting in a low-spin, 6c FeII heme.
Influence of Phe41 on cross-link formation driven by peroxide
We previously showed that cross-link formation in either the WT proenzyme or an Arg44Ala mutant is driven by peroxide, which motivated us to determine whether the Phe41Ala mutant could undergo the same maturation process.20 Indeed, reaction of 10 μM Phe41Ala cyt P460 with 3 equivalents of Li2O2 (30 μM) resulted in the immediate decay of the 403 nm Soret and isosbestic conversion to a broad Soret centered at 436 nm consistent with a cross-linked ferryl-type compound II product observed previously for WT cyt P460 (Fig. 4). There were no observable intermediates, in contrast to the 413 nm ferryl intermediate observed during maturation of the WT proenzyme. After quenching the reaction with sodium dithionite (Na2S2O4) the product exhibited a sharp Soret maximum at 460 nm characteristic of FeII CL cyt P460. Re-oxidation of the product gave a UV/vis spectrum with a Soret maximum at 436 nm and Q-bands at 628 nm and 570 nm (Fig. 3A).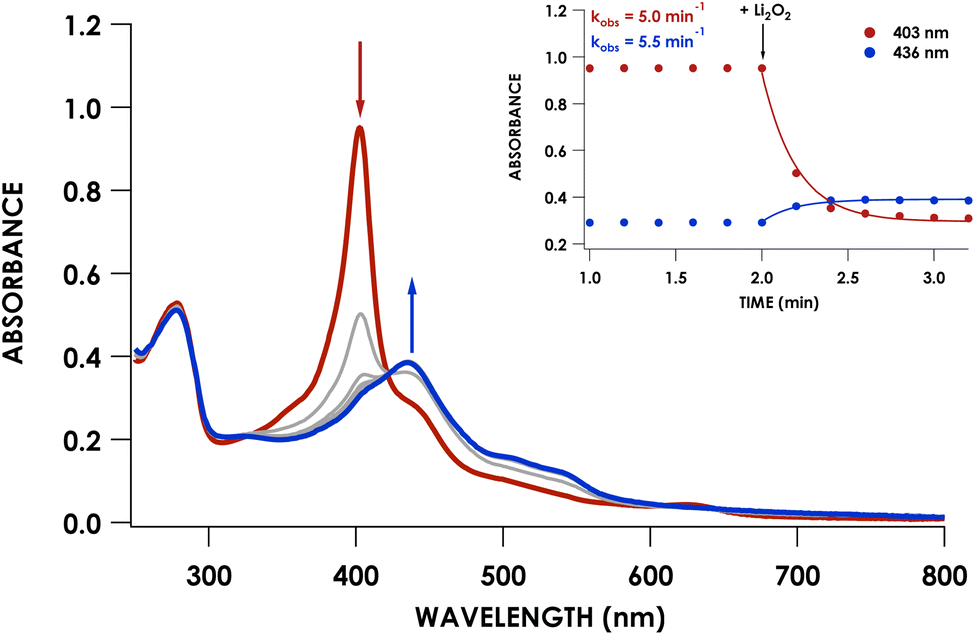 | ||
| Fig. 4 Reaction of 10 μM as-isolated Phe41Ala cyt P460 (red) with 30 μM Li2O2 in 20 mM Tris/200 mM NaCl buffer at pH 8.0 and 25 °C. The blue trace represents the final scan and grey traces were recorded every 0.2 minutes. Time courses of the absorbances corresponding to the proenzyme (403 nm) and product (436 nm) are shown in the inset. | ||
The lack of accumulation of the ferryl intermediate indicated a change in either the mechanism or kinetics of cross-link formation. To determine whether Phe41Ala cyt P460 is matured by the same mechanism as WT proenzyme, the cross-link reaction was repeated using 8 μM Phe41Ala cyt P460 with 8 equivalents (64 μM) Li2O2 in the presence of 2 mM guaiacol. Guaiacol is commonly used in peroxidase assays and is oxidized by ferryl species, forming a 470 nm absorbance attributed to oxidized guaiacol.22 We previously used this assay to show that cross-link formation of the WT proenzyme proceeds through a ferryl intermediate.20 When guaiacol was present during the cross-link reaction of Phe41Ala cyt P460, there was rapid formation of a 470 nm absorbance consistent with guaiacol oxidation (Fig. S4†). Importantly, cross-link formation was inhibited, indicating that the mechanism of cross-link formation likely proceeds through a ferryl intermediate similar to that of WT proenzyme.
To capture the ferryl intermediate, stopped-flow UV/vis reactions were carried out on the ms–s timescale using several concentrations of Li2O2. The reaction of 10 μM Phe41Ala cyt P460 with 30 μM Li2O2 resulted in decay of the 403 nm Soret without formation of the intermediate. When the concentration of Li2O2 was increased to 40 μM a 413 nm intermediate was observed (Fig. 5A). The decay of the CLD Soret at 403 nm could not be fit to a single exponential and was better described by a double exponential (Table S5†). Full-wavelength scans of the reaction show an initial rapid decay of the 403 nm Soret to form the 413 nm intermediate with some overlap of the intermediate at 403 nm. Subsequent decay of the intermediate to the final product accounts for the second, slower exponential.
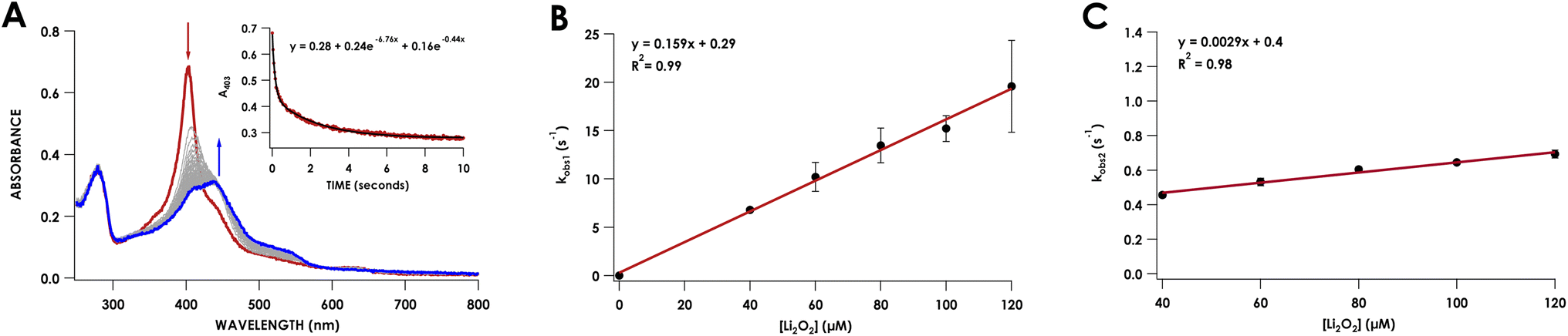 | ||
| Fig. 5 (A) Stopped-flow UV/vis reaction of 10 μM as-isolated Phe41Ala cyt P460 and 40 μM Li2O2 in 20 mM Tris/200 mM NaCl (pH 8.0) recorded over 10 seconds. The red trace is the first scan, and the blue trace is the final scan. Scans were recorded every 4 ms and grey traces are average scans every 20 ms. The inset is the absorbance at 403 nm recorded every 4 ms fit to a double exponential. (B) Plot of kobs1 obtained from fitting the absorbance at 403 nm to a double exponential versus the concentration of Li2O2. (C) Plot of kobs2versus the concentration of Li2O2. | ||
Plotting the observed rate corresponding to the initial decay of the 403 nm Soret to form the 413 nm intermediate, kobs1, against the concentration of Li2O2 showed a linear dependence with peroxide (Fig. 5B). Comparing this rate to the rate of WT proenzyme did not show any significant changes, with the rate for Phe41Ala cyt P460 being 0.16 ± 0.02 s−1 (μM Li2O2)−1 and that of WT proenzyme being 0.13 ± 0.02 s−1 (μM Li2O2)−1.20 Plotting kobs2versus Li2O2 concentration also showed a linear dependence with peroxide, albeit a small dependence at only 0.17 min−1 (μM Li2O2)−1. At 40 μM Li2O2, the observed rate of consumption of the 413 nm intermediate was 2.8 min−1 (μM p460)−1, whereas the WT proenzyme had a constant rate of 0.42 min−1 (μM p460)−1. It therefore appears that Phe hinders the conversion of the ferryl intermediate to the CL species. This change in rate is insufficient to explain the lack of accumulation of the ferryl species at lower Li2O2 concentrations given that the rate of its formation is significantly faster than its consumption. At lower concentrations of Li2O2, the highly reactive intermediates may be prone to off-pathway reactivity with solvent, preventing accumulation of the ferryl species. While accumulation of the ferryl intermediate was not necessary for full conversion to CL protein, its greater susceptibility to non-productive reactivity with solvent could explain the isolation of a CLD protein from recombinant expression. In vivo, off-pathway reactivity may out-compete cross-link formation without sufficiently high peroxide concentrations to drive the reaction to completion. In AOB, cyt P460 must be able to efficiently form the cross-link to function in NH2OH oxidation. Thus, the capping Phe appears to promote cross-link formation.
Because CLD variants of cyt P460 are catalytically inactive, the fully CL form of the Phe41Ala mutant was necessary to study the role of Phe in NH2OH oxidation. To avoid heme degradation during maturation with Li2O2, the Phe41Ala mutant was cross-linked by reaction with O2-saturated buffer (generated by bubbling O2 for 20 min) in the presence of 10 mM NH2OH. Under these conditions, the 403 nm Soret isosbestically converts to the 442 nm Soret of NH2OH-bound CL protein (Fig. S5†). These conditions allow for the formation of a NH2OH-bound CL protein as the final product rather than a CL compound II species that is prone to degradation. Without the addition of NH2OH the cross-link still forms, but its Soret band quickly loses intensity, indicative of heme degradation. NH2OH has previously been shown to prevent heme degradation in the presence of peroxide for both cyt P460 and HAO, likely by binding to the Fe and blocking access of peroxide.23,24
After washing the matured Phe41Ala cyt P460 to remove NH2OH, the UV/vis absorption spectrum was consistent with that obtained following maturation with Li2O2. The X-band EPR spectrum measured at 10 K had two high-spin components with a more axial component (E/D = 0.01) and a more rhombic component (E/D = 0.03) (Fig. 1B). While the EPR spectrum of WT N. europaea cyt P460 contains only one rhombic high-spin component, the presence of several components is reminiscent of Nitrosomonas sp. AL212 cyt P460 variants in which the distal alanine (Ala131) was replaced by either glutamate or glutamine.17 WT, Ala131Glu, and Ala131Gln Nitrosomonas sp. AL212 cyt P460 all contained two high-spin rhombic components with different ratios to each other (Table S2†). The existence of two EPR signals can be explained by an equilibrium between two heme conformations. Previously we attributed the two components in Nitrosomonas sp. AL212 cyt P460 to the different orientations of the capping Phe. In structures of N. europaea cyt P460, the loop containing the capping Phe is missing from the density, which can be attributed to flexibility of the loop. Greater flexibility of the loop in N. europaea cyt P460 compared to that of Nitrosomonas sp. AL212 cyt P460 may allow the heme to exist in the conformation with greater rhombicity. This greater rhombicity could arise due to asymmetry imposed by distortion of the heme away from planarity. Replacing the bulky Phe residue with Ala could limit this flexibility and drive the heme towards a more symmetric component associated with lower rhombicity. The resonance Raman spectrum obtained using laser excitation at 405 nm had intensity ratios that resembled WT cyt P460, namely a less intense ν4 band (Fig. 1C). The ν10 band, which is sensitive to out-of-plane distortions of the porphyrin, occurred at 1614 cm−1 which was the same as WT cyt P460, suggesting the Phe41Ala mutant remains ruffled.
Having established a consistent reaction to form the fully CL species without heme degradation, we could use this protein to study the influence of Phe41 on the oxidation of NH2OH. In the following experiments, the fully CL form of Phe41Ala cyt P460 obtained by reaction with O2 and NH2OH is used.
Influence of Phe41 on the product selectivity of NH2OH oxidation
Under anaerobic conditions, cyt P460 stoichiometrically converts two equivalents of NH2OH to N2O.9 In the presence of O2, a mixture of N2O and NO2− are observed. We previously showed that NO2− formation is the result of the enzymatic product NO reacting non-enzymatically with O2.9 Reaction of NO with O2 thus drives the equilibrium of the {FeNO}6 towards NO release. At higher starting concentrations of NH2OH, the bimolecular reaction of the {FeNO}6 with NH2OH is accelerated and results in a greater ratio of N2O to NO production. To determine whether the Phe41Ala mutation affected product selectivity, N2O and NO2− concentrations were measured following reactions run under turnover conditions with 5 μM WT or Phe41Ala cyt P460, 2 mM [Ru(NH3)6]Cl3, 6 μM phenazine methosulfate (PMS), and varying amounts of NH2OH under both anaerobic and aerobic conditions (Tables S6–S8†). Consistent with WT cyt P460 under anaerobic conditions, all of the NH2OH oxidized by either cyt P460 variant was accounted for by N2O production measured by GC-MS (Fig. S6†). Aerobic conditions led to a mixture of N2O and NO2− as measured by headspace GC-MS and Griess diazotization assays, respectively. Accounting for the higher background detection of NO2− in the reactions with Phe41Ala cyt P460 compared to WT cyt P460, there was no significant difference in NO2− production at lower initial NH2OH concentrations. As the initial NH2OH concentration was increased to 500 μM and 1000 μM, NO2− production by Phe41Ala cyt P460 dropped below that of WT cyt P460 (Fig. 6). The remaining NH2OH was accounted for by N2O production. The shift in product formation towards more N2O indicates either the rate of NO release has decreased, or the rate of N2O formation has increased. Hydroxylamine concentration dependence points to the latter explanation.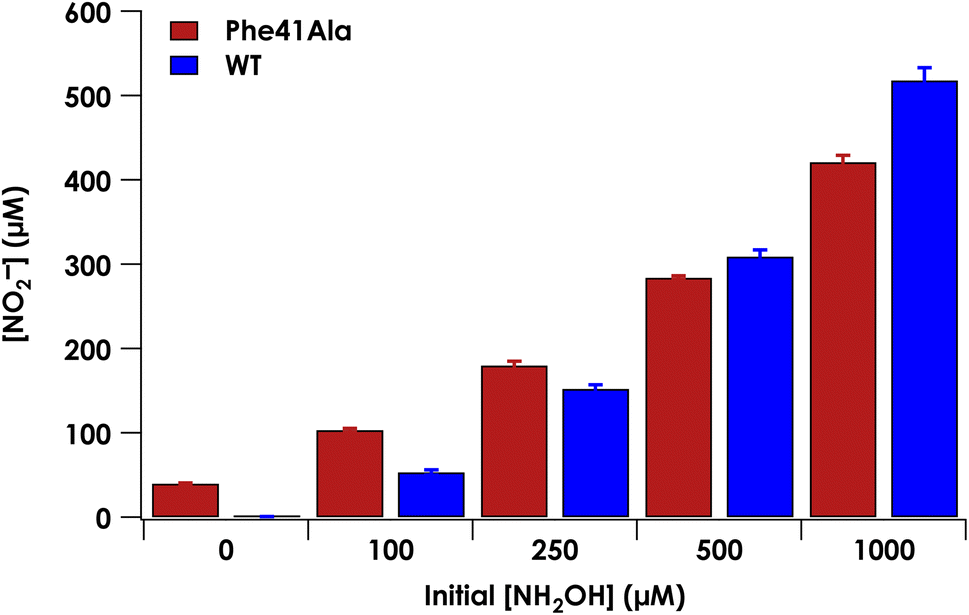 | ||
| Fig. 6 Nitrite production measured by a Griess diazotization assay of the aerobic reaction of 5 μM CL Phe41Ala (red) or WT (blue) cyt P460, 6 μM PMS, 2 mM [Ru(NH3)6]Cl3, and varying concentrations of NH2OH. Nitrite concentrations reflect NO production, where NO produced from cyt P460 reacted with O2 to form NO2−. Error bars represent standard deviations of three trials. | ||
Influence of Phe41 on substrate binding
Given the influence of Phe41 on product partitioning, we sought to determine its role in substrate binding. The 298 K Kd for NH2OH of CL Phe41Ala cyt P460 in 50 mM HEPES (pH 8.0) was 22 ± 8 mM (Fig. S7†), which was within error of the Kd for WT determined in this study (30.0 ± 0.7 mM). The 298 K Kd for NO was 16 ± 3 μM (Fig. S8†), a modest four-fold increase from that of WT determined in this study (3.6 ± 0.4 μM). Additionally, full conversion to the {FeNO}6 could not be obtained in phosphate buffer, suggesting a role of Phe41 in protecting the intermediate from solvent exposure. A study of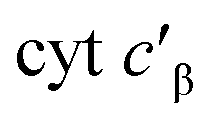 from Methylococcus capsulatus, which contains an analogous capping Phe residue (Phe32), suggested that a quadrupolar interaction between Phe32 and NO results in a modest destabilization of the Fe–NO bond.18 While we do not observe that effect in cyt P460, there may be other factors influencing Fe–NO bonding. The increase in Kd could be the result of either an increase in the off-rate or a decrease in the on-rate (eqn (1)).
from Methylococcus capsulatus, which contains an analogous capping Phe residue (Phe32), suggested that a quadrupolar interaction between Phe32 and NO results in a modest destabilization of the Fe–NO bond.18 While we do not observe that effect in cyt P460, there may be other factors influencing Fe–NO bonding. The increase in Kd could be the result of either an increase in the off-rate or a decrease in the on-rate (eqn (1)).| Kd = koff/kon | (1) |
To determine whether the NO on-rate or off-rate was influenced by the Phe41Ala mutation, we obtained the off-rates using FeIIEDTA to trap NO and drive the {FeNO}6 to release. Because FeIIEDTA reacts rapidly with NO (kon = (1.23 ± 0.06) × 108 M−1 s−1 at 298 K, pH 7.0), the rate of decay of the {FeNO}6 species is assumed to equal the NO off-rate.25 Fitting the decay of the {FeNO}6 Soret band at 455 nm to an exponential (Fig. S9†), the NO off-rate of WT cyt P460 was 0.35 ± 0.05 s−1. Using the same conditions, the off rate of Phe41Ala cyt P460 was 0.30 ± 0.03 s−1 (Fig. S10†), within error of that observed for WT cyt P460. The on-rates were then determined by the relationship between the Kd and the association/dissociation rates (Table 1). The decrease in NO binding affinity upon removal of the bulky Phe side-chain can be explained by a decrease in the on-rate. Although it might be expected that removal of the steric hindrance imposed by the bulky phenyl group would increase the on-rate, a more plausible scenario is that NO must compete with water binding to Fe in the active site. Gatekeeping Phe residues commonly appear in proteins and play an important role in binding affinities.26–28 In Shewanella frigidimarina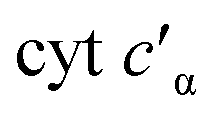 , a distal Phe residue lies nearly parallel to the heme and about 3.3 Å away from the Fe atom.29 It was shown to exist in two conformations and could displace bound NO allowing for controlled NO release. The positioning of the Phe side-chain is critical in determining its function as a gatekeeping residue. In contrast to
, a distal Phe residue lies nearly parallel to the heme and about 3.3 Å away from the Fe atom.29 It was shown to exist in two conformations and could displace bound NO allowing for controlled NO release. The positioning of the Phe side-chain is critical in determining its function as a gatekeeping residue. In contrast to  , the distal Phe in cyt P460 is perpendicular to the heme and likely prevents solvent molecules from entering the active site. When NO is bound, Phe is nearly parallel to the N–O bond in the ferric heme–nitrosyl complex. Rather than putting steric strain on the bound NO, Phe41 could protect NO from interactions with solvent analogous to protection of the intermediates during cross-link formation. If Phe were to displace NO, cyt P460 would be expected to produce greater amounts of NO compared to N2O, making it more like HAO.
, the distal Phe in cyt P460 is perpendicular to the heme and likely prevents solvent molecules from entering the active site. When NO is bound, Phe is nearly parallel to the N–O bond in the ferric heme–nitrosyl complex. Rather than putting steric strain on the bound NO, Phe41 could protect NO from interactions with solvent analogous to protection of the intermediates during cross-link formation. If Phe were to displace NO, cyt P460 would be expected to produce greater amounts of NO compared to N2O, making it more like HAO.
| Cyt P60 variant | K d (M) | k off (s−1) | k on (M−1 s−1) |
|---|---|---|---|
| WT | (3.6 ± 0.4) x 10−6 | 0.35 ± 0.05 | 9.8 × 104 |
| Phe41Ala | (16 ± 3) x 10−6 | 0.30 ± 0.03 | 1.9 × 104 |
A more open active site changes the rate-determining step
We have previously shown that CLD cyt P460 is not competent for NH2OH oxidation because the cross-link positions the distal Glu for proton-relay.30 As-isolated Phe41Ala cyt P460 is a mixture of the CLD and CL species, so the cross-link forming reaction is expected to restore activity upon conversion of inactive CLD protein to active CL protein. To assess the steady-state kinetics of the Phe41Ala mutation before and after cross-link formation, a DCPIP/PMS-coupled assay with varying concentrations of NH2OH was used to obtain a steady-state activity plot (Fig. S11†). As-isolated protein was indeed slower than the CL protein with specific activities of 3.7 ± 0.4 μM DCPIP μM−1 cyt P460 mM−1 NH2OH min−1 and 6.4 ± 0.4 μM DCPIP μM−1 cyt P460 mM−1 NH2OH min−1, respectively. The percentage of the CL species in the as-isolated protein was quantified using the extinction coefficient at 436 nm. With the assumption that there is minimal contribution to the absorbance at 436 nm from the CLD species, which is true for the CLD mutant Lys70Tyr, CL Phe41Ala accounted for 56% of the as-isolated protein. Quantification using this method was also consistent with the Mössbauer quantification, indicating this is a good approximation. Correcting the specific activity of the as-isolated protein to account for only the CL species, the specific activity becomes 6.6 μM DCPIP μM−1 cyt P460 mM−1 NH2OH min−1. This is within error of that obtained for the fully CL Phe41Ala protein, indicating that the reaction completely restores activity.With the knowledge that cross-link formation does restore activity to Phe41Ala cyt P460, we then sought to compare the kinetics of CL Phe41Ala to WT cyt P460. Using the same conditions, WT cyt P460 had a specific activity of 10.3 ± 0.4 μM DCPIP μM−1 cyt P460 mM−1 NH2OH min−1. The higher value in WT than previously reported is attributed to the use of stirring during the reaction rather than inversion.9 If Phe does provide steric hindrance, removing it might be expected to increase the rate of NH2OH oxidation. However, the slower rate of the Phe41Ala mutant indicates that there must be another effect of Phe on the kinetics. Turning to the full-wavelength spectral time-course of anaerobic turnover conditions, there is a striking difference between Phe41Ala and WT cyt p460 (Fig. 7). WT cyt P460 was shown to accumulate the {FeNO}6 intermediate, establishing the rate-determining step as the bimolecular reaction of the {FeNO}6 intermediate with NH2OH.9 The Phe41Ala mutant did not accumulate this 455 nm intermediate and instead accumulated a 442 nm intermediate consistent with the NH2OH-adduct. Removing the bulky Phe side-chain must therefore have altered the kinetics such that formation of the {FeNO}6 is slower or consumption of the {FeNO}6 is faster.
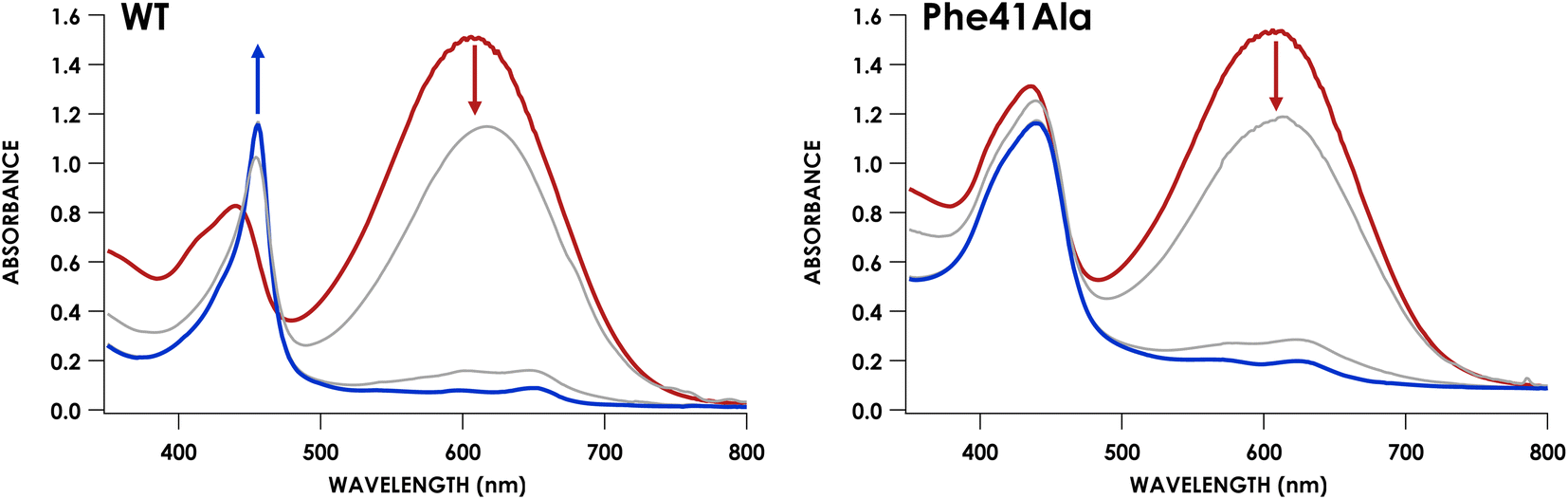 | ||
| Fig. 7 Full-wavelength UV/vis absorption traces showing the reactions of 12 μM WT (left) or Phe41Ala (right) cyt P460 with 70 μM DCPIP and 10 mM NH2OH. The red trace is before NH2OH addition and the blue trace is the end of the reaction. Grey traces represent scans after NH2OH addition collected every 6 seconds. | ||
The influence of the Phe41Ala mutation on the kinetics of consumption of the {FeNO}6 was investigated by reacting a shunted {FeNO}6 species with NH2OH. Pseudo first-order kinetics for WT cyt P460 were obtained using 5 μM cyt P460, 5 μM NO and NH2OH concentrations of 800 μM, 2 mM, 6 mM, or 12 mM. Plotting the initial rates versus NH2OH concentration showed Michaelis–Menten behaviour (Fig. S12†). To fit to the Michaelis–Menten equation, the {FeNO}6 was treated as the enzyme species and the initial concentration was calculated using the Kd. The apparent kcat and KM were 0.22 min−1 and 1.8 mM, respectively. For Phe41Ala cyt P460, the NO concentration was increased to 50 μM to account for the lower binding affinity. The apparent kcat was 0.42 min−1 and the KM was 0.8 mM, indicating that the rate of NH2OH attack on the {FeNO}6 is faster for the Phe41Ala mutant compared to WT. Removing the steric hindrance of the phenyl group likely allows more facile access of NH2OH to the {FeNO}6 intermediate, increasing the rate of N–N bond formation. While the rate of {FeNO}6 consumption increased, the overall turnover frequency is slower than that of WT cyt P460. This can be explained by a change in the rate-determining step from consumption of the {FeNO}6 to the initial proton-coupled electron transfer reaction from the FeIII–NH2OH adduct (Fig. 8). Such a change in rate-determining step is consistent with the observation of the FeIII–NH2OH intermediate accumulating during turnover instead of the {FeNO}6. Moreover, this change in rate-determining step can explain the shift in product partitioning towards N2O observed during aerobic turnover conditions. Because the NO off-rate did not change, formation of N2O can better compete with NO release. This becomes more evident at higher NH2OH concentrations where the bimolecular rate of NH2OH attack on the {FeNO}6 reaches its maximum.
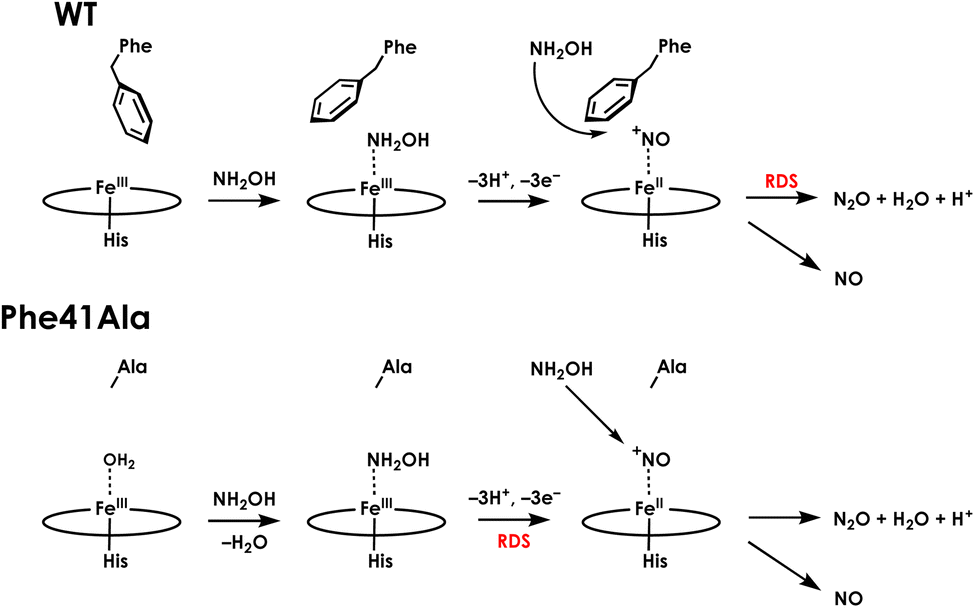 | ||
| Fig. 8 Summary schematic of the effects of the Phe41Ala mutant on NH2OH oxidation by cyt P460. | ||
The Phe41Arg mutation influences FeIII–NO binding
With the knowledge that product partitioning between N2O and NO is affected by steric access to the {FeNO}6 intermediate, we sought to push product selectivity towards greater NO release in order to better understand how the primary metabolic enzyme HAO is biased toward NO production. One of the ways in which this could be achieved is by blocking access to the {FeNO}6 to a greater extent than the native Phe residue. Mutation of Phe41 to Trp would be expected to slow the rate of N2O formation and could lead to greater NO release. However, expression and purification of the Phe41Trp mutant yielded a CLD protein that could not be fully converted to CL protein (see ESI†).An alternative approach to shifting product partitioning to NO is by destabilizing the Fe–NO bond to promote more facile NO release. A DFT study of {FeNO}6 porphyrins suggested that the ground state is the FeII–NO+ form characterized by strong Fe–NO and N–O bonds.31 The excited state, which is close in energy to the ground state, was characterized as a low-spin FeIII–NO˙ state with a significantly weaker Fe–NO bond. A distal positive charge could destabilize the FeII–NO+ form by repulsive electrostatic interactions and favor the FeIII–NO˙ state, leading to a weaker Fe–NO bond. This was shown to occur in Klebsiella pneumoniae and Dechloromonas aromatica chlorite dismutase, which both have unusually weak Fe–NO bonds attributed to a distal arginine residue.32 In cyt P460 the FeII–NO+ state is expected to be the predominant form to allow for more facile attack of NH2OH on a more electrophilic NO species. Fourier-transform infrared (FTIR) spectroscopy of WT cyt P460 treated with excess NO revealed a new band at 1912 cm−1 compared to the spectrum of the FeIII resting state (Fig. S16†). Treatment with 15NO resulted in a red-shift of this band to 1871 cm−1. This band can be assigned to an N–O stretching mode that is consistent with values reported for other ferric-heme nitrosyl species containing an axial His residue.33–35 Moreover, it is consistent with an FeII–NO+ configuration for WT cyt P460. Introduction of a positively charged side-chain in the position of Phe41 that is in close proximity to the bound NO could destabilize this configuration and result in greater FeIII–NO˙ character that would have two effects: weakening of the Fe–NO bond and disfavoring NH2OH attack on the {FeNO}6. To test whether a positive charge could shift product partitioning, we generated the Phe41Arg mutant.
Expression and purification of the Phe41Arg mutant also produced a mixture of CLD and CL protein. This mutant behaved similarly to Phe41Ala cyt P460 and could be converted to fully CL protein by reaction with O2 and NH2OH (Fig. S17†). The CL form exhibits a Soret maximum at 442 nm. Titration of the fully CL Phe41Arg mutant with NO (Fig. S18†) yielded a Kd of 86 ± 14 μM, 22-fold greater than that of WT cyt P460. Gratifyingly, the introduction of a distal positive charge in the position of Phe41 does weaken the NO binding affinity. However, under aerobic turnover conditions the proportion of NH2OH converted to NO relative to N2O was unchanged from WT cyt P460. Despite the weakened NO binding affinity, other factors such as steric influences are likely playing a role. For HAO, the active site contains more polar residues including an Asp/His pair and Tyr that are involved in a H-bond network with bound NH2OH (Fig. S19†).11 Differences in the electrostatic environment of the outer coordination sphere could play a role in promoting NO release and preventing N2O formation. Moreover, heme P460 in HAO exhibits a greater degree of ruffling compared to that of N. europaea cyt P460 (2.4 Å vs. 0.7 Å), which has been predicted to favor the FeIII–NO˙ configuration.36 This configuration would not only lead to more facile NO release but would also shut down attack by a second equivalent of NH2OH to form N2O. Thus, it may be the case that HAO's product distribution is biased toward NO (and thus, establishing net electron flow for cellular respiration) by inhibiting the {FeNO}6 reaction with NH2OH to form N2O, affording time for NO release and/or transnitrosylation.
Conclusions
The active site of cyt P460 is unique in that it must first be able to catalyze its own post-translational modification: a heme-lysine cross-link formation that is required for proper enzyme function. It must also carry out controlled NH2OH oxidation. To do so, the outer coordination sphere is fine-tuned to control the highly reactive intermediates of these reactions. This was highlighted by mutation of a capping Phe residue, which influenced both cross-link formation and NH2OH oxidation. Removing the steric hindrance of Phe leaves the reactive intermediates prone to off-pathway reactivity with solvent, while introduction of the bulkier Trp residue blocks access of peroxide to the active site. If reactive oxygen species are limited in the cell, cyt P460 must be able to efficiently react with peroxide to form the cross-link, and the active site is optimized to do so using the capping Phe residue.Factors that influence the different product selectivity between HAO and cyt P460 have been difficult to study owing to the spectroscopic obstruction of the heme P460 signal in HAO and lack of a recombinant expression system. This study provides insight into the selectivity for cyt P460 controlled by the outer coordination sphere. The position of the capping Phe plays an important role in this selectivity by providing steric hindrance to the {FeNO}6 intermediate. Removing this steric hindrance increases the rate of NH2OH attack on the {FeNO}6 species and ultimately changes the product selectivity towards greater N2O production. Introducing a positive charge in this position by mutation to Arg weakened the binding affinity for NO but was not enough to influence the product partitioning. Control over the product partitioning between N2O and NO thus requires an interplay between access to the {FeNO}6 and influence over the Fe–NO bond strength.
Data availability
Data supporting this article have been uploaded as ESI.†Author contributions
KML and MMB designed the research project. MMB, SHM, and REC carried out experiments under supervision of KML. KML and MMB wrote the manuscript. All authors contributed to editing and revision of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
KML gratefully acknowledges the National Institutes of Health for support in the form of a Maximizing Investigators' Research Award (MIRA) (R35-GM124908). MMB and SHM gratefully acknowledge the National Science Foundation Graduate Research Program (DGE-1650441). EPR data were collected at ACERT, which is supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number P41GM103521. We thank Siddarth Chandrasekharan for assistance with EPR data collection.References
- J. K. Zorz, J. A. Kozlowski, L. Y. Stein, M. Strous and M. Kleiner, Front. Microbiol., 2018, 9, 938 CrossRef.
- J. D. Caranto and K. M. Lancaster, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 8217–8222 CrossRef CAS PubMed.
- I.-K. Choi, Y. Liu, Z. Wei and M. D. Ryan, Inorg. Chem., 1997, 36, 3113–3118 CrossRef CAS PubMed.
- S. E. Bari, V. T. Amorebieta, M. M. Gutiérrez, J. A. Olabe and F. Doctorovich, J. Inorg. Biochem., 2010, 104, 30–36 CrossRef CAS PubMed.
- A. B. McQuarters, L. E. Goodrich, C. M. Goodrich and N. Lehnert, Z. Anorg. Allg. Chem., 2013, 639, 1520–1526 CrossRef CAS.
- J. R. Stroka, B. Kandemir, E. M. Matson and K. L. Bren, ACS Catal., 2020, 10, 13968–13972 CrossRef CAS.
- D. A. Bazylinski, R. A. Arkowitz and T. C. Hollocher, Arch. Biochem. Biophys., 1987, 259, 520–526 CrossRef CAS PubMed.
- G. E. Alluisetti, A. E. Almaraz, V. T. Amorebieta, F. Doctorovich and J. A. Olabe, J. Am. Chem. Soc., 2004, 126, 13432–13442 CrossRef CAS PubMed.
- J. D. Caranto, A. C. Vilbert and K. M. Lancaster, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14704–14709 CrossRef CAS PubMed.
- L. Y. Stein, in Methods in Enzymology, Elsevier, 2011, vol. 486, pp. 131–152 Search PubMed.
- W. J. Maalcke, A. Dietl, S. J. Marritt, J. N. Butt, M. S. M. Jetten, J. T. Keltjens, T. R. M. Barends and B. Kartal, J. Biol. Chem., 2014, 289, 1228–1242 CrossRef CAS PubMed.
- P. Cedervall, A. B. Hooper and C. M. Wilmot, Biochemistry, 2013, 52, 6211–6218 CrossRef CAS PubMed.
- M. Numata, T. Saito, T. Yamazaki, Y. Fukumori and T. Yamanaka, J. Biochem., 1990, 108, 1016–1021 CrossRef CAS.
- A. R. Pearson, B. O. Elmore, C. Yang, J. D. Ferrara, A. B. Hooper and C. M. Wilmot, Biochemistry, 2007, 46, 8340–8349 CrossRef CAS PubMed.
- J. H. Enemark and R. D. Feltham, Coord. Chem. Rev., 1974, 13, 339–406 CrossRef CAS.
- A. C. Vilbert, J. D. Caranto and K. M. Lancaster, Chem. Sci., 2018, 9, 368–379 RSC.
- M. A. Smith, S. H. Majer, A. C. Vilbert and K. M. Lancaster, Chem. Sci., 2019, 10, 3756–3764 RSC.
- H. R. Adams, D. A. Svistunenko, M. T. Wilson, S. Fujii, R. W. Strange, Z. A. Hardy, P. A. Vazquez, T. Dabritz, G. J. Streblow, C. R. Andrew and M. A. Hough, J. Biol. Chem., 2023, 104742 CrossRef CAS PubMed.
- A. C. Vilbert, J. D. Caranto and K. M. Lancaster, Chem. Sci., 2018, 9, 368–379 RSC.
- M. M. Bollmeyer, R. E. Coleman, S. H. Majer, S. D. Ferrao and K. M. Lancaster, J. Am. Chem. Soc., 2023 DOI:10.1021/jacs.3c03608.
- T. A. Kent, K. Spartalian, G. Lang, T. Yonetani, C. A. Reed and J. P. Collman, Biochim. Biophys. Acta, 1979, 580, 245–258 CrossRef CAS PubMed.
- B. Chance and A. C. Maehly, Methods in Enzymology, Elsevier, 1955, vol. 2, pp. 764–775 Search PubMed.
- A. B. Hooper and K. R. Terry, Biochemistry, 1977, 16, 455–459 CrossRef CAS PubMed.
- F. N. Liew, M. A. Brandys, S. Biswas, J. N. Nguyen, M. Rahmawati, M. Nevala, B. O. Elmore, M. P. Hendrich and H. J. Kim, Biochemistry, 2020, 59, 704–716 CrossRef CAS PubMed.
- J. F. Demmink, I. C. F. van Gils and A. A. C. M. Beenackers, Ind. Eng. Chem. Res., 1997, 36, 4914–4927 CrossRef CAS.
- R. E. Joseph and A. H. Andreotti, Biochemistry, 2011, 50, 221–229 CrossRef CAS PubMed.
- Y.-M. Zhang, J. Hurlbert, S. W. White and C. O. Rock, J. Biol. Chem., 2006, 281, 17390–17399 CrossRef CAS PubMed.
- J. Carro, P. Amengual-Rigo, F. Sancho, M. Medina, V. Guallar, P. Ferreira and A. T. Martínez, Sci. Rep., 2018, 8, 8121 CrossRef PubMed.
- A. Manole, D. Kekilli, D. A. Svistunenko, M. T. Wilson, P. S. Dobbin and M. A. Hough, J. Biol. Inorg. Chem., 2015, 20, 675–686 CrossRef CAS PubMed.
- R. E. Coleman, A. C. Vilbert and K. M. Lancaster, Biochemistry, 2020, 59, 2289–2298 CrossRef CAS PubMed.
- V. K. K. Praneeth, F. Paulat, T. C. Berto, S. D. George, C. Näther, C. D. Sulok and N. Lehnert, J. Am. Chem. Soc., 2008, 130, 15288–15303 CrossRef CAS.
- Z. Geeraerts, A. K. Heskin, J. DuBois, K. R. Rodgers and G. S. Lukat-Rodgers, J. Inorg. Biochem., 2020, 211, 111203 CrossRef CAS.
- L. M. Miller, A. J. Pedraza and M. R. Chance, Biochemistry, 1997, 36, 12199–12207 CrossRef CAS PubMed.
- Y. Wang and B. A. Averill, J. Am. Chem. Soc., 1996, 118, 3972–3973 CrossRef CAS.
- X. D. Ding, A. Weichsel, J. F. Andersen, T. Kh. Shokhireva, C. Balfour, A. J. Pierik, B. A. Averill, W. R. Montfort and F. A. Walker, J. Am. Chem. Soc., 1999, 121, 128–138 CrossRef CAS.
- F. Walker, J. Inorg. Biochem., 2005, 99, 216–236 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental materials and methods, and supplementary figures and tables. See DOI: https://doi.org/10.1039/d3sc02288a |
| This journal is © The Royal Society of Chemistry 2023 |