
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium(V) terminal arylimido corroles: isolation, spectroscopic characterization and reactivity†
Ka-Pan
Shing‡
 a,
Lin
Qin‡
a,
Liang-Liang
Wu
a,
Jie-Sheng
Huang
*a and
Chi-Ming
Che
*ab
a,
Lin
Qin‡
a,
Liang-Liang
Wu
a,
Jie-Sheng
Huang
*a and
Chi-Ming
Che
*ab
aState Key Laboratory of Synthetic Chemistry, Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, China. E-mail: cmche@hku.hk
bHKU Shenzhen Institute of Research and Innovation, Shenzhen, China
First published on 2nd September 2023
Abstract
Terminal Ru(V)–imido species are thought to be as reactive to group transfer reactions as their Ru(V)–oxo homologues, but are less studied. With the electron-rich corrole ligand, relatively stable and isolable Ru(V)–arylimido complexes [Ru(tBu–Cor)(NAr)] (H3(tBu–Cor) = 5,15-diphenyl-10-(p-tert-butylphenyl)corrole, Ar = 2,4,6-Me3C6H2 (Mes), 2,6-(iPr)2C6H3 (Dipp), 2,4,6-(iPr)3C6H2 (Tipp), and 3,5-(CF3)2C6H3 (BTF)) can be prepared from [Ru(tBu–Cor)]2 under strongly reducing conditions. This type of Ru(V)–monoarylimido corrole complex with S = ½ was characterized by high-resolution ESI mass spectrometry, X-band EPR, resonance Raman spectroscopy, magnetic susceptibility, and elemental analysis, together with computational studies. Under heating/light irradiation (xenon lamp) conditions, the complexes [Ru(tBu–Cor)(NAr)] (Ar = Mes, BTF) could undergo aziridination of styrenes and amination of benzylic C(sp3)–H bonds with up to 90% product yields.
Introduction
Fe(V)- and Ru(V)–imido species are widely proposed but underexplored reactive intermediates in many Fe(III) and Ru(III)/Ru(IV)-catalysed C(sp3)–H amination reactions.1 Such reactive Fe(V)/Ru(V)–imido species can undergo atom-efficient functionalization of C–H bonds as their oxo-congeners (Fe(V)– and Ru(V)–oxo species),2 but are unstable and short-lived in solution.1b,3 Spectroscopic characterization and isolation of the Fe(V)/Ru(V)–imido complexes reactive towards hydrocarbons are essential for understanding their bonding, electronic structure, and reactivity. In the literature, although there are ample examples of well-characterized Ru(V)–oxo species,2a,4 terminal Ru(V)–imido species are sparse.5 Wilkinson and co-workers reported [nPr4N][Ru(NCy){OCEt2C(O)O}2],5d but the reactivity of this isolable Ru–imido complex towards hydrocarbons has not been documented. Literature examples of spectroscopically characterized M(V)–imido complexes that can aziridinate alkenes and/or aminate C(sp3)–H bonds, including Fe(V)–tosylimido species with the tetraamide ligand1b and Ru(V)–acylimido porphyrin1e complexes, are generated in solutions without isolation (Fig. 1a).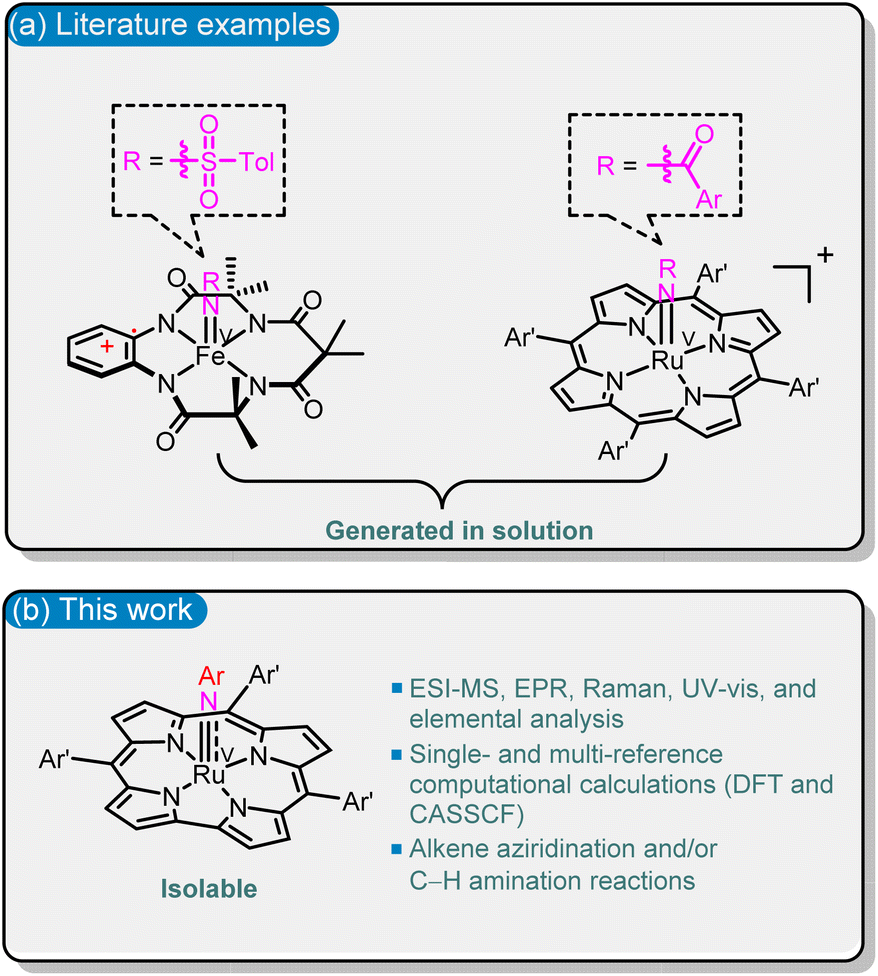 | ||
| Fig. 1 Examples of (a) in the literature and (b) in this work for spectroscopically characterized M(V)–imido species that can undergo alkene aziridination and/or C–H amination reactions. | ||
In order to obtain novel spectroscopically characterized M(V)–imido species reactive with hydrocarbons, and to improve the stability of such species, we attempted to focus on Ru(V) (considering the high reactivity of Ru(V)–acylimido porphyrin species1e) and the use of the trianionic corrole (Cor) ligand; the latter was previously shown to stabilize the [MV(Cor)(NAr)] (M = Cr, Mn, Re) species.6 One hurdle to overcome is that most of the reported Ru corroles are dimers with strong Ru![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Ru triple bonds,7 in contrast to the case of Ru porphyrins, where Ru2(II,II) porphyrin dimers are highly sensitive to oxygen and are useful precursors for Ru–carbene, –oxo or –imido species.8a,f Here we report reductive activation of Ru2(III,III) corrole dimers followed by reaction with aryl azides to give Ru(V)–arylimido (RuV(NAr)) complexes; the latter have been isolated and characterized by X-band EPR, resonance Raman (rR), HR-ESI-MS, UV-vis spectroscopy, elemental analysis, and/or DFT calculations. This type of RuV(NAr) complex can undergo alkene aziridination and C(sp3)–H amination reactions, both of which have not been reported for [MV(Cor)(NAr)] (M = Cr, Mn, Re).6
Ru triple bonds,7 in contrast to the case of Ru porphyrins, where Ru2(II,II) porphyrin dimers are highly sensitive to oxygen and are useful precursors for Ru–carbene, –oxo or –imido species.8a,f Here we report reductive activation of Ru2(III,III) corrole dimers followed by reaction with aryl azides to give Ru(V)–arylimido (RuV(NAr)) complexes; the latter have been isolated and characterized by X-band EPR, resonance Raman (rR), HR-ESI-MS, UV-vis spectroscopy, elemental analysis, and/or DFT calculations. This type of RuV(NAr) complex can undergo alkene aziridination and C(sp3)–H amination reactions, both of which have not been reported for [MV(Cor)(NAr)] (M = Cr, Mn, Re).6
Results and discussion
Synthesis of Ru(V)–arylimido complexes
The Ru2(III,III) corrole dimer [Ru(tBu–Cor)]2 (1, Scheme 1) was prepared in 65% yield and characterized by NMR, UV-vis, HR-ESI-MS, cyclic voltammetry, elemental analysis and X-ray crystallography (Fig. S1 in the ESI†). We attempted to generate Ru(V)–imido species by reaction of 1 with aryl azide, but the dimer was quantitatively recovered. We envisage weakening the RuRu bond of 1 by reducing this complex. Reduction of 1 with excess potassium graphite (KC8) in rigorously deoxygenated and dried THF, followed by overnight stirring at RT (Pyrex® Spinbar), afforded a species 2, tentatively the reduced dimer {[Ru(tBu–Cor)]2}2−. The effective magnetic moment of 2 (in situ generated) was determined to be 2.81μB using the Evans method, which indicates that 2 is paramagnetic with two unpaired electrons (S = 1). This is in contrast to the dimer 1 (S = 0) and mononuclear Ru(II) corrole species, which are expected to be diamagnetic, similar to the Ru(II) porphyrin complexes [Ru(Por)(L)2] (L = vacant or L = N2, CH3CN, THF, Et2O).8g Based on its magnetic moment (2.81μB), complex 2 is likely to be a Ru2(II,II) corrole dimer, whose electronic configuration (S = 1) is similar to that (σ2π4(δnb)4(π*)2) of the Ru2(II,II) porphyrin dimers reported by Collman and co-workers.8d Redox titration of 2 with [Cp2Fe]PF6 showed that ca. 2 equiv. of Cp2Fe and 1 equiv. of 1 were generated. In a control experiment, 1 was not able to reduce [Cp2Fe]PF6 under the same reaction conditions. The UV-vis spectrum of 2 in THF shows pairwise Soret bands at 411 and 445 nm, which are red-shifted from that of 1 at 329 and 399 nm (Fig. S4 in the ESI†). A Ru2(II,II) species, {[Ru(Cor)]2}2−, was previously proposed as the two-electron reduced product of [Ru(Cor)]2 by Guilard and co-workers.7b
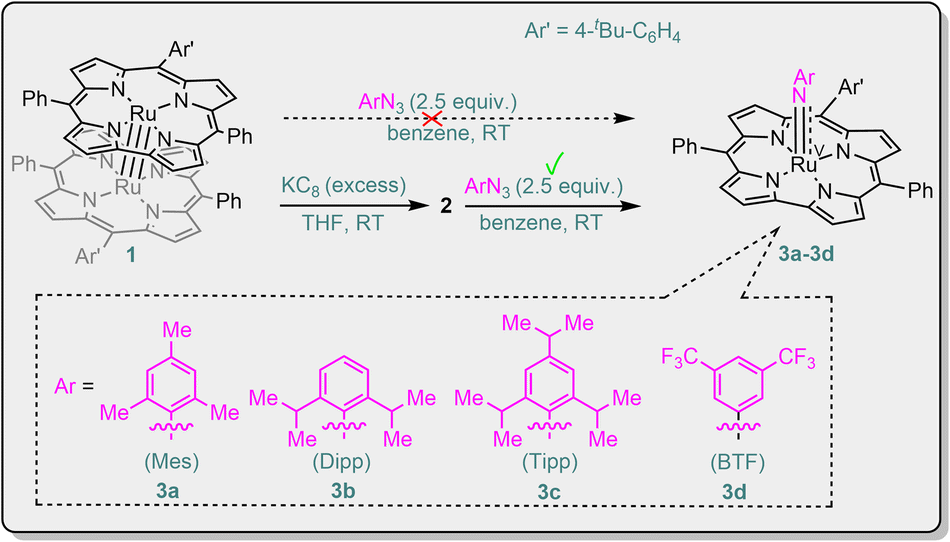 | ||
| Scheme 1 Synthesis of Ru(V)–arylimido corrole complexes 3a–3d. | ||
As shown in Scheme 1, treatment of 2 with aryl azides (ArN3, Ar = Mes, Dipp, Tipp, BTF) afforded a set of RuV(NAr) complexes [Ru(tBu–Cor)(NAr)] (3); [Ru(tBu–Cor)(NBTF)] (3d) bearing the CF3-substituted arylimido ligand is one of the rare examples of terminal imido complexes of late transition metals featuring strongly electron-withdrawing CF3 substituents on their imido ligands.9 The reactions using MesN3 (for 3a) and BTFN3 (for 3d) took only a few minutes, whereas the reactions with DippN3 (for 3b) and TippN3 (for 3c) bearing sterically more hindered aryl groups required stirring for 0.5 h at RT. Complexes 3 are likely formed via initial generation of [RuIV(tBu–Cor)(NAr)]− followed by disproportionation or one-electron oxidation. In the literature, Ru(IV)–oxo porphyrin was reported to undergo disproportionation;8f,10 Fe(IV)–oxo porphyrins and Mn(IV)–oxo corroles have been reported to convert into their one-electron oxidized counterparts in solution.11
MALDI-TOF MS was used to monitor the reaction. In initial trials using “1 + MesN3” in refluxing benzene solution, a cluster peak attributable to 3a was not observed. By using “2 + MesN3” (Fig. S6 in the ESI†), the ESI-MS spectrum of the reaction mixture showed a strong cluster peak at m/z 814.2479, which was attributed to the parent ion of [Ru(tBu–Cor)(NMes)] (3a, calcd for C50H42N5Ru: 814.2492; Fig. S12 in the ESI†), giving the simulated isotope pattern that matches that observed experimentally. As expected, the peak is isotopically sensitive, with the isotope pattern changing upon using a 50% 15N-enriched sample (14,15N–3a, prepared from 15N–MesN3; Fig. 2). Other Ru–arylimido complexes (3b–3d) have also been isolated and characterized by ESI-MS/MALDI-TOF MS. Complexes 3a–3d were found to gradually decompose within 24 h even under an inert atmosphere and at low temperature (argon filled glove box, refrigerator pre-equilibrated at −35 °C), as evidenced by UV-vis spectroscopy and ESI-MS analysis. Attempts to obtain diffraction-quality crystals of these complexes for X-ray crystal structure determination were not successful.
 | ||
Fig. 2 (a) HR-ESI-MS (positive-ion mode) spectrum of [Ru(tBu–Cor)(NMes)] (3a). (b) Experimental isotope pattern of “[Ru(tBu–Cor)(14NMes)]+ + [Ru(tBu–Cor)(15NMes)]+” in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (50% 15N enrichment). (c) Simulated pattern of (b). :1 ratio (50% 15N enrichment). (c) Simulated pattern of (b). | ||
EPR and electronic spectroscopy
After measuring their magnetic susceptibility (ca. 1.73μB using the Evans method), X-band EPR experiments were performed to elucidate the electronic structure of these Ru–arylimido corrole complexes 3. An anisotropic EPR signal for 3a (Fig. S16 in the ESI†) was found at frozen glass temperature (100 K, benzene). Upon treatment with 5 equiv. of PPh3, this EPR signal diminished, and [Ru(tBu–Cor)(PPh3)]+ (Fig. S8 in the ESI,†m/z 943.4) and MesN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PPh3 were found (Fig. S9 in the ESI;†m/z calcd: 396.1858, found: 396.1881; 31P NMR in CDCl3: −13.85 ppm).
PPh3 were found (Fig. S9 in the ESI;†m/z calcd: 396.1858, found: 396.1881; 31P NMR in CDCl3: −13.85 ppm).
The EPR spectrum of 3a was simulated using the parameters of gx = 2.010, gy = 2.005, gz = 1.880; Ax, Ay, Az = 53, 14, 14 MHz (14N). Simulation for the EPR spectrum of 3b (Fig. S17 in the ESI†) was also carried out, by employing the g and A values of gx = 2.000, gy = 1.980, gz = 1.870; Ax, Ay, Az = 50, 14, 14 MHz (14N). The EPR spectrum of 3c (Fig. 3a) is similar to that of its structurally similar complex 3b (g and A values for the simulated spectrum: same as those of 3b), and the signal pattern is assigned to the superhyperfine coupling to the 14N nucleus of the imido ligand, similar to that of Fe(V)–imido species.12 For the 50% 15N-enriched sample, 14,15N–3c (prepared from 15N–TippN3), the corresponding EPR spectrum (Fig. 3b) shows a signal which can be reasonably fitted by summing the simulated spectra of 3c and 15N–3c, with a 1:1 ratio (Fig. 3b; 14N: I = 1, 15N: I = ½). The EPR signals of 3a–3c are characterized by giso (gx + gy + gz)/3) values of 1.950–1.965, which can be assigned to Ru(V) complexes with S = ½.1e,2a These giso values are similar to those of Ru(V)–acylimido and Ru(V)–oxo porphyrin complexes (giso = 1.94,1e 1.970 (ref. 2a)), but smaller than those commonly observed for Ru(III) complexes (giso = 2.1–2.3 (ref. 2c and 13)).
 | ||
| Fig. 3 X-band EPR spectra of isolated samples of (a) 3c and (b) 14,15N–3c recorded at 100 K. | ||
Complexes 3a–3d each exhibit one Soret band (at 406–411 nm) in the UV-vis spectrum; like mononuclear [RuVI(Cor)(N)] complexes each complex also has one Soret band (at 417–419 nm),7d but unlike the Ru2(III,III) dimer 1, which exhibits two Soret bands (at 329 and 399 nm). In accord with the [RuV(Cor)(NAr)] formulation, the UV-vis spectra of 3a–3d (Fig. S5 in the ESI†) do not show typical absorption bands of corrole π–cation radical (Cor+˙) complexes (above 650 nm14). For example, a band at 676 nm was observed for [MnV(Cor+˙)(N)],15 the EPR spectrum of which (at 4 K) showed an isotropic radical signal, in contrast to the anisotropic EPR signals of 3a–3c.
Vibrational spectroscopy (IR and resonance Raman)
The vibrational spectra of 3a and its 50% 15N-enriched form 14,15N–3a were recorded. No bands over 3200 cm−1 were found in the FTIR spectra (isolated solid samples, KBr pellets), in agreement with the absence of the amino/amido group in the complex. The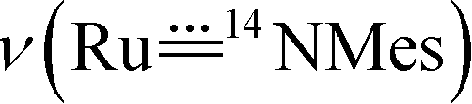 band could possibly be obscured by an intense band of the coordinated corrole ligand at 1016 cm−1; based on the FTIR spectroscopy, we tentatively assign the
band could possibly be obscured by an intense band of the coordinated corrole ligand at 1016 cm−1; based on the FTIR spectroscopy, we tentatively assign the 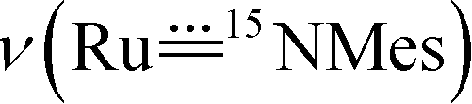 band of 14,15N–3a at 972 cm−1. To observe the
band of 14,15N–3a at 972 cm−1. To observe the  and
and  bands, we measured the resonance Raman (rR) spectra of 3a and 14,15N–3a (isolated samples dissolved in CH2Cl2). The rR spectrum of 14,15N–3a (Fig. 4b, λexcitation: 416 nm) shows pairwise bands (ca. 1:1 ratio due to 50% 15N-enrichment) at ∼949 and 921 cm−1 with Δν of ∼28 cm−1 agreeing with the value of 28 cm−1 estimated using Hooke's law. These two bands are assigned to
bands, we measured the resonance Raman (rR) spectra of 3a and 14,15N–3a (isolated samples dissolved in CH2Cl2). The rR spectrum of 14,15N–3a (Fig. 4b, λexcitation: 416 nm) shows pairwise bands (ca. 1:1 ratio due to 50% 15N-enrichment) at ∼949 and 921 cm−1 with Δν of ∼28 cm−1 agreeing with the value of 28 cm−1 estimated using Hooke's law. These two bands are assigned to  and
and  respectively, and the band at 921 cm−1 is absent in the rR spectrum of 3a (Fig. 4a). By considering literature examples of rR studies on metal–arylimido (M(NAr), e.g., Fe(NPh)16) complexes, other 15N-isotopically sensitive bands within 820–1000 cm−1 may be associated with the coordinated arylimido ligand, where 15N-labelling resulted in a downshift of the bands involving M–Nimido and aryl groups.
respectively, and the band at 921 cm−1 is absent in the rR spectrum of 3a (Fig. 4a). By considering literature examples of rR studies on metal–arylimido (M(NAr), e.g., Fe(NPh)16) complexes, other 15N-isotopically sensitive bands within 820–1000 cm−1 may be associated with the coordinated arylimido ligand, where 15N-labelling resulted in a downshift of the bands involving M–Nimido and aryl groups.
 | ||
| Fig. 4 rR spectra of the isolated samples (dissolved in dry CH2Cl2) of (a) 3a and (b) 14,15N–3a. (c) DFT-calculated Raman spectra of 3a, 15N–3a and 14,15N–3a. | ||
N-group transfer reactivities
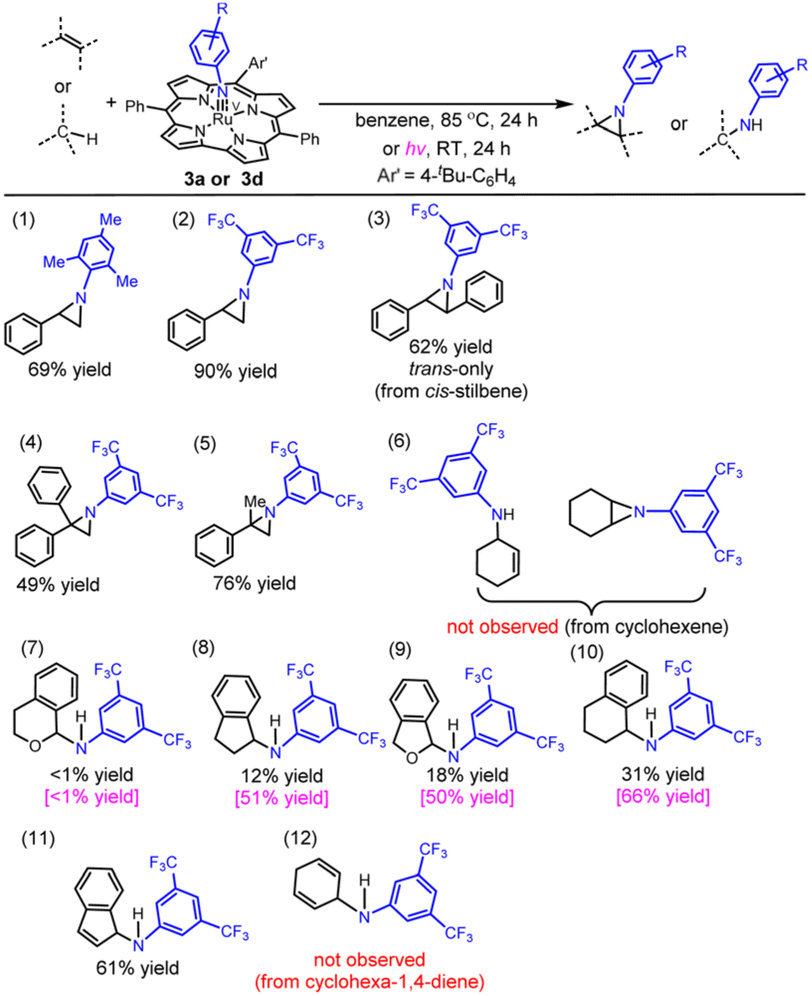
Complex 3a, which has a moderately hindered mesitylimido ligand with electron-donating Me groups, was reacted with styrene at 85 °C for 24 h (entry 1 in Table 1) to give 1-mesityl-2-phenylaziridine in 69% yield. Under the same conditions, the reaction of 3d bearing the 3,5-bis(trifluoromethyl)phenylimido ligand with styrene gave the aziridine product in much higher yield (90%; entry 2 in Table 1). This is attributed to the lower steric hindrance and higher electrophilicity of the 3,5-bis(trifluoromethyl)phenylimido ligand compared with the mesitylimido ligand. Reactive M(NAr) complexes with Ar = 3,5-bis(trifluoromethyl)phenyl (BTF) or other groups (e.g. C6F5, CF3–C6F4 and NO2–C6H4) with electron-withdrawing substituents are reactive intermediates involved in metal-catalysed N-group transfer reactions using the corresponding aryl azides.17 Reaction of 3d with cis-stilbene at 85 °C for 24 h gave trans-aziridine in 62% yield; cis-aziridine was not detected (entry 3 in Table 1). The reaction of 1,1-diphenylethylene with 3d gave diphenylaziridine in lower yield (49%, entry 4 in Table 1). This is attributed to the steric effect, as α-methylstyrene (which is sterically less hindered than 1,1-diphenylethylene) was found to react with 3d to give methylphenylaziridine in higher yield (76%, entry 5 in Table 1). 3d was reacted with excess cyclohexene at 85 °C for 24 h, and neither aziridination of the CC bond nor amination of the allylic C(sp3)–H bond was observed.
| a Ru(V)–arylimido complex (5.50 μmol) and the substrate (1.65 mmol) in benzene (1 mL); the product yields under UV-vis irradiation are presented in pink colour whereas the black ones refer to the yields obtained at 85 °C. |
|---|
|
We then examined the amination of benzylic C(sp3)–H bonds with 3d. Treatment of 3d with excess isochroman gave the amination product in <1% yield, whereas the reaction of indane and phthalane with 3d gave the amination product in 12% and 18% yields, respectively (entries 7–9 in Table 1; 85 °C for 24 h). The reaction of 3d with tetralin gave the amination product in 31% yield (entry 10 in Table 1; 29% yield for a 1 h reaction). Complex 3d exhibited a relatively high reactivity towards indene (61% yield of the amine product; entry 11 in Table 1). We found that light irradiation at RT (xenon lamp) promoted the 3d-mediated amination of indane, phthalane and tetralin as revealed by the higher product yields obtained (50–66% yield; entries 8–10 in Table 1). Indane and phthalane gave higher yield of the amination product than isochroman, which may be related to the statistical factor because isochroman has a smaller number of equivalent C–H bonds for the amination. In the amination of tetralin and indane by 3d, the product yield was found to decrease with increasing dissociation energy of the reactive C–H bond (cf. 82.9 and 85.3 kcal mol−1 for tetralin and indane, respectively18).
Catalytic N-group transfer reactions were examined using Ru corrole complexes 1 and 2 as catalysts. For the 1-catalysed reaction of styrene with aryl azide BTFN3 in benzene at 85 °C, the aziridination product was obtained in 5% yield. Upon changing the catalyst to 2, a much higher product yield of 46% was achieved (entries 1 and 2 in Table 2). Complex 2 also catalysed the aziridination of para-substituted styrenes to aziridines in 38–54% yields (entries 3–6 in Table 2). Using cis-stilbene as the substrate, the reaction afforded trans-aziridine as the major product (trans/cis ratio: 2.9:1.0, 48% yield, entry 8 in Table 2).

Regarding the mechanism of the 3d-mediated aziridination of alkenes, we conceive that the aziridination proceeds in a stepwise manner due to the formation of trans-aziridine from cis-stilbene (entry 3 in Table 1). For the aziridination of para-substituted styrenes by 3d, the plot of log(krel) against σmb and  using a dual parameter equation developed by Jiang and co-workers,19 shows a good linearity (R2 = 0.9979) featuring ρmb = −0.54 and
using a dual parameter equation developed by Jiang and co-workers,19 shows a good linearity (R2 = 0.9979) featuring ρmb = −0.54 and  (Fig. 5), with the linearity being better than that for the plot of log(krel) against σ+ (Fig. S15 in the ESI;†R2 = 0.9356). This is consistent with a stepwise mechanism in which a carboradical is involved. A positive value of
(Fig. 5), with the linearity being better than that for the plot of log(krel) against σ+ (Fig. S15 in the ESI;†R2 = 0.9356). This is consistent with a stepwise mechanism in which a carboradical is involved. A positive value of  indicates the spin density of the radical centre delocalized by the para-substituent, while a negative value of ρmb supports electrophilic attack of styrene by 3d.
indicates the spin density of the radical centre delocalized by the para-substituent, while a negative value of ρmb supports electrophilic attack of styrene by 3d.
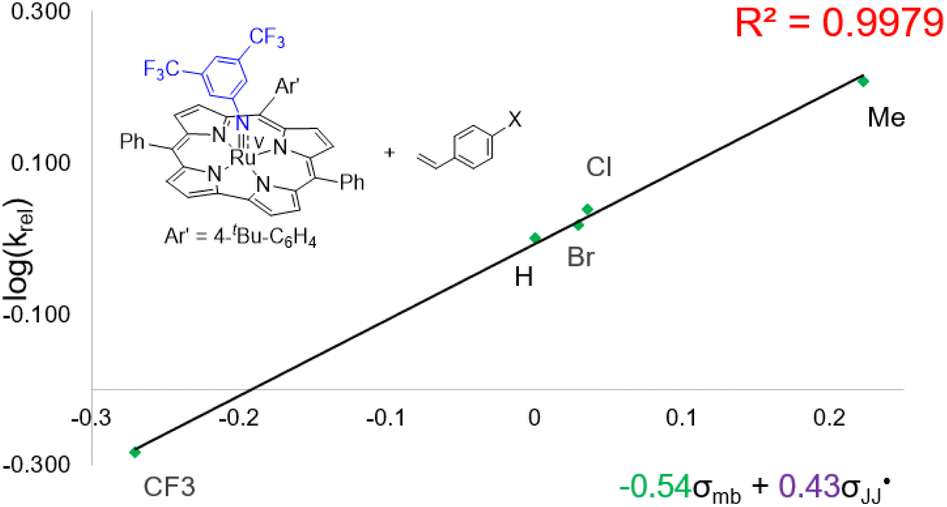 | ||
Fig. 5 Hammett correlation for the aziridination of para-substituted styrenes mediated by 3d using a dual-parameter method (σmb and  ). ). | ||
Furthermore, we examined the deuterium kinetic isotope effect (KIE) for 3d-mediated C(sp3)–H amination by using equimolar and excess of tetralin and d4-tetralin. The kH/kD value determined for such amination by 3d at 383 K is 4.0(2), comparable to that reported for the C(sp3)–H amination of ethylbenzene by [RuVI(TMP)(NNs)2] (kH/kD = 4.8) at 298 K.20 We propose that the C–H amination mediated by 3d involves a stepwise mechanism similar to that for the [RuVI(TMP)(NNs)2]-mediated amination,20 characterized by H atom abstraction followed by radical rebound.
Computational studies
We first calculated the Raman spectrum of 3a and 14,15N–3a to check the accuracy and reliability of the computational studies. The calculated energy bands of and
and  are located at 944 and 923 cm−1, respectively (Fig. 4c), which are consistent with the respective experimental values (∼949 and 921 cm−1). By converting the Raman shifts to bond distances, 3a has a calculated Ru–Nimido distance of 1.756 Å. The calculated Ru–Nimido distances for 3b and 3d are 1.779 and 1.763 Å, respectively. In terms of the Wiberg bond index (WBI), 3a, 3b and 3d have predicted Ru–Nimido bond orders of 2.05, 1.96 and 2.04, respectively, indicating a multiple bond character. This is in contrast to the cobalt complex with the imidyl ligand NDipp+˙, which is characterized by a calculated single Co–Nimidyl bond character (WBI ≈ 1.0).21
are located at 944 and 923 cm−1, respectively (Fig. 4c), which are consistent with the respective experimental values (∼949 and 921 cm−1). By converting the Raman shifts to bond distances, 3a has a calculated Ru–Nimido distance of 1.756 Å. The calculated Ru–Nimido distances for 3b and 3d are 1.779 and 1.763 Å, respectively. In terms of the Wiberg bond index (WBI), 3a, 3b and 3d have predicted Ru–Nimido bond orders of 2.05, 1.96 and 2.04, respectively, indicating a multiple bond character. This is in contrast to the cobalt complex with the imidyl ligand NDipp+˙, which is characterized by a calculated single Co–Nimidyl bond character (WBI ≈ 1.0).21
Similar to the previously reported Ru(V)–oxo porphyrin,2a both 3a and 3b adopt an S = 1/2 ground state with (dxy)2(dxz,dyz)1(dz2)0(dx2−y2)0 configuration. In both species, the singly occupied molecular orbital (SOMO) corresponds to dπ, while the π orbitals of the corrole ligand are ca. 0.4–0.5 eV lower than the SOMO, indicating that the corrole ligand is not redox-active (Fig. S18 in the ESI†). We calculated g values for 3a by DFT calculations; the calculated results (g = 2.010, 2.000, 1.890) are in good agreement with the values based on the experimental EPR spectrum (g = 2.010, 2.005, 1.880) (Table S3 in the ESI†). By DFT calculations, the % spin density on Ru/Nimido atom (% SDRu/N) of 3a, 3b, and 3d is 44/27, 35/36, and 53/19, respectively, and that on the coordinated arylimido ligand (% SDNAr) of 3a/3b/3d is 40/54/26 (Fig. 6).
 | ||
| Fig. 6 DFT-optimized spin density distributions (contour value: 0.005), bond angles, bond distances and out-of-plane deviations. | ||
The decrease of spin density on the arylimido ligand (% SDNAr) along the series 3b > 3a > 3d is in line with the decrease of the Ru–Nimido–CAr angle along the same series (160°, 146° and 138° for the DFT-optimized structures of 3b, 3a, and 3d, respectively; Fig. 6). The arylimido ligand of 3b bears bulky 2,6-substituents (iPr) and adopts a basically linear coordination mode, which minimizes steric conflicts between these substituents and the corrole ligand. Complex 3a has less bulky 2,6-substituents (Me); its arylimido ligand can accommodate a relatively bent coordination mode.22 A more bent coordination mode of the arylimido ligand corresponds to 3d which possesses no 2,6-substituents and bears electron-withdrawing 3,5-CF3 groups, like the case of [RuVI(TPP)(NBTF)2] (Ru–Nimido–CAr angles: 139.8(3)° and 143.7(4)°).23 The bending of the Ru–Nimido–CAr moiety would reduce spin delocalization along the z-axis (and into the aryl ring), thus decreasing the spin density on the arylimido group.
As the spin density on Nimido is the highest in 3b among 3a, 3b, and 3d, we performed multi-reference calculations for 3b using the complete active space self-consistent field (CASSCF) method to check if Ru(IV)-imidyl character is involved.17a,24 The calculation results revealed that 3b can be described as “85.9% of [RuV(tBu–Cor)(NDipp)] + 3.6% of [RuIV(tBu–Cor)(NDipp+˙)]” (Fig. 7). For the Ru–Nimido bond of 3b, a calculated bond order of 2.04 was obtained by considering the matrix of atomic orbital coefficients for all relevant molecular orbitals. Based on the occupation numbers of orbitals within the active space, the calculated Ru–Nimido bond order is 2.27, close to the formal bond order of 2.5 with σ2π4(π*)1 configuration. The spin population obtained from the CASSCF calculation is 0.55 on Ru and 0.35 on Nimido (Fig. S19 in the ESI†), indicating that 3b has a doublet ground state with the unpaired electron mainly residing on the Ru centre. This provides support for the formulation of 3b, and also 3a and 3d, as a formal Ru(V)–arylimido corrole species bearing considerable spin density on Nimido.
 | ||
| Fig. 7 Multi-configurational calculations (CASSCF method) using an active space of 11-electron, 9-orbital, i.e., CASSCF(11,9) to depict leading configurations of the doublet ground state of 3b. The remaining configurations each occupy <1% of the ground state. The natural orbitals associated with the assignment, occupation numbers (in parentheses) and atomic orbital contributions are shown in the upper panel (not according to relative energies). | ||
We compared the DFT calculation results for the Ru(NAr) complexes having an S = 1/2 ground state, including % spin density on Ru/NAr (% SDRu/NAr) and the Ru–NNAr distance (Table S4 in the ESI†). The % SDRu/NAr of [(SiPiPr3)Ru(N–p-CF3C6H4)]25a and [Ru(PNP)(NPh)] (PNP = [(tBu2PCH2SiMe2)2N]−)25b with different types of monoanionic supporting ligands is 40/54 and 30/68, respectively. In the case of complexes 3 with the same trianionic tBu–Cor supporting ligand, a direct comparison of % SDRu/NAr among various Ar groups can be made (3a: 44/40, 3b: 35/54, and 3d: 53/26); the % SDNAr decreases along the 3b > 3a > 3d series as discussed above. The Ru–NNAr distances in [(SiPiPr3)Ru(N–p-CF3C6H4)] and [Ru(PNP)(NPh)] (ca. 1.81–1.87 Å) are longer than in complexes 3 (ca. 1.76–1.78 Å).
The electronic structure of the Ru(V)–arylimido complex 3b calculated by CASSCF is different from that of the Fe(V)–tosylimido species [FeV(TAML)(NTs)]− calculated by DFT;1b,26 the properties of the M–Nimido π- and π*-bonding of these two complexes are different. For 3b with an essentially linear M–Nimido–CAr moiety (bond angle: 160°), two sets of π and π* orbitals between M-dxz/yz and N-px/y are involved, reminiscent of the corresponding CASSCF calculation results for other M(NAr) complexes with linear M–Nimido–CAr moieties.27 However, for [FeV(TAML)(NTs)]− having a bent Fe–Nimido–STs moiety (bond angle: 126° (ref. 1b)), only the π and π* orbitals between M-dyz and N-py are involved, and the M-dxz is non-bonding.26
DFT calculations were also carried out for the aziridination of cis-stilbene by 3d, which was experimentally found to favour the formation of the trans-aziridine product. The calculated reaction profile (Fig. S20 in the ESI†) involving the formation of a carboradical intermediate (Int-trans and Int-cis for trans- and cis-configuration, respectively) is consistent with the proposed stepwise mechanism. The ring-closure barrier for Int-trans and Int-cis is 14.1 and 13.3 kcal mol−1, respectively, both of which are larger than the barrier of 10.7 kcal mol−1 required for isomerization of Int-cis to the thermodynamically more favourable Int-trans by rotation about the CPh–CPh bond, allowing such rotation to take place prior to the ring closure, giving trans-aziridine as the major product.28
Conclusions
The electron-rich trianionic corrole ligand tBu–Cor is capable of stabilizing reactive M(V)–imido species, enabling the isolation and spectroscopic characterization of reactive Ru(V)–arylimido complexes capable of undergoing N-group transfer with hydrocarbons. The relatively stable Ru(V)–arylimido corroles [Ru(tBu–Cor)(NAr)] (3) isolated in this work have been characterized by X-band EPR, rR, ESI or MALDI-TOF MS, and elemental analysis. Their electronic structures were examined by single- (DFT) and multi-reference (CASSCF) computational studies. Complexes 3 could aziridinate alkenes and aminate benzylic C(sp3)–H bonds via a stepwise mechanism under heating/irradiation conditions, with product yields of up to 90% and 66%, respectively. The results of this work provide useful insights into the development of alkene and C(sp3)–H bond functionalization via reactive Ru(V)–arylimido intermediates.Author contributions
CMC conceived the idea and directed the project. KPS and LQ performed the experiments. LLW carried out the computational studies. KPS, LQ, JSH and CMC analysed the data. KPS, JSH and CMC wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Hong Kong Research Grants Council (HKU 17304019) and Basic Research Program-Shenzhen Fund (JCYJ20180508162429786). We thank Dr Xiao-Yong Chang for assistance in X-ray crystal structure determination of complex 1.Notes and references
- (a) K.-P. Shing, Y. Liu, B. Cao, X.-Y. Chang, T. You and C.-M. Che, Angew. Chem., Int. Ed., 2018, 57, 11947 CrossRef CAS PubMed; (b) S. Hong, K. D. Sutherlin, A. K. Vardhaman, J. J. Yan, S. Park, Y.-M. Lee, S. Jang, X. Lu, T. Ohta, T. Ogura, E. I. Solomon and W. Nam, J. Am. Chem. Soc., 2017, 139, 8800 CrossRef CAS PubMed; (c) Y.-D. Du, C.-Y. Zhou, W.-P. To, H.-X. Wang and C.-M. Che, Chem. Sci., 2020, 11, 4680 RSC; (d) W. Xiao, J. Wei, C.-Y. Zhou and C.-M. Che, Chem. Commun., 2013, 49, 4619 RSC; (e) D.-Y. Hong, Y. Liu, L. Wu, V. K.-Y. Lo, P. H. Toy, S.-M. Law, J.-S. Huang and C.-M. Che, Angew. Chem., Int. Ed., 2021, 133, 18619 CrossRef PubMed; (f) R. Vyas, G.-Y. Gao, J. D. Harden and X. P. Zhang, Org. Lett., 2004, 6, 1907 CrossRef CAS PubMed; (g) Y. Liu, G.-Q. Chen, C.-W. Tse, X. Guan, Z.-J. Xu, J.-S. Huang and C.-M. Che, Chem.–Asian J., 2015, 10, 100 CrossRef CAS PubMed.
- (a) K.-P. Shing, B. Cao, Y. Liu, H. K. Lee, M.-D. Li, D. L. Phillips, X.-Y. Chang and C.-M. Che, J. Am. Chem. Soc., 2018, 140, 7032 CrossRef CAS PubMed; (b) F. T. de Oliveira, A. Chanda, D. Banerjee, X. Shan, S. Mondal, L. Que Jr, E. L. Bominaar, E. Münck and T. J. Collins, Science, 2007, 315, 835 CrossRef PubMed; (c) C. Wang, K. V. Shalyaev, M. Bonchio, T. Carofiglio and J. T. Groves, Inorg. Chem., 2006, 45, 4769 CrossRef CAS PubMed; (d) E. Vanover, Y. Huang, L. Xu, M. Newcomb and R. Zhang, Org. Lett., 2010, 12, 2246 CrossRef CAS PubMed; (e) O. Y. Lyakin, A. M. Zima, N. V. Tkachenko, K. P. Bryliakov and E. P. Talsi, ACS Catal., 2018, 8, 5255 CrossRef CAS.
- (a) S. Hong, X. Lu, Y.-M. Lee, M. S. Seo, T. Ohta, T. Ogura, M. Clémancey, P. Maldivi, J.-M. Latour, R. Sarangi and W. Nam, J. Am. Chem. Soc., 2017, 139, 14372 CrossRef CAS PubMed; (b) K. M. Van Heuvelen, A. T. Fiedler, X. Shan, R. F. De Hont, K. K. Meier, E. L. Bominaar, E. Münck and L. Que Jr, Proc. Natl. Acad. Sci. U.S.A., 2012, 109, 11933 CrossRef CAS PubMed.
- (a) A. C. Dengel, W. P. Griffith, C. A. O'Mahoney and D. J. Williams, J. Chem. Soc., Chem. Commun., 1989, 1720 RSC; (b) C.-M. Che, V. W.-W. Yam and T. C. W. Mak, J. Am. Chem. Soc., 1990, 112, 2284 CrossRef CAS; (c) N. L. P. Fackler, S. Zhang and T. V. O'Halloran, J. Am. Chem. Soc., 1996, 118, 481 CrossRef CAS; (d) A. C. Dengel and W. P. Griffith, Inorg. Chem., 1991, 30, 869 CrossRef CAS; (e) J. J. Concepcion, J. W. Jurss, M. K. Brennaman, P. G. Hoertz, A. O. T. Patrocinio, N. Y. M. Iha, J. L. Templeton and T. J. Meyer, Acc. Chem. Res., 2009, 42, 1954 CrossRef CAS PubMed; (f) C.-M. Che, K.-Y. Wong and T. C. W. Mak, J. Chem. Soc., Chem. Commun., 1985, 988 RSC; (g) L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418 CrossRef CAS PubMed; (h) Y. Liu, S.-M. Ng, S.-M. Yiu, W. W. Y. Lam, X.-G. Wei, K.-C. Lau and T.-C. Lau, Angew. Chem., Int. Ed., 2014, 53, 14468 CrossRef CAS PubMed; (i) D. Lebedev, Y. Pineda-Galvan, Y. Tokimaru, A. Fedorov, N. Kaeffer, C. Copéret and Y. Pushkar, J. Am. Chem. Soc., 2018, 140, 451 CrossRef CAS PubMed; (j) D. Moonshiram, I. Alperovich, J. J. Concepcion, T. J. Meyer and Y. Pushkar, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 3765 CrossRef CAS PubMed; (k) C.-M. Che and K.-Y. Wong, J. Chem. Soc., Chem. Commun., 1986, 229 RSC.
- (a) W.-H. Leung, T. S. M. Hun, H.-w. Hou and K.-Y. Wong, J. Chem. Soc., Dalton Trans., 1997, 237 RSC; (b) T. Ishizuka, T. Kogawa, M. Makino, Y. Shiota, K. Ohara, H. Kotani, S. Nozawa, S.-i. Adachi, K. Yamaguchi, K. Yoshizawa and T. Kojima, Inorg. Chem., 2019, 58, 12815 CrossRef CAS PubMed; (c) R. P. Tooze, G. Wilkinson, M. Motevalli and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1986, 2711 RSC; (d) C. Redshaw, W. Clegg and G. Wilkinson, J. Chem. Soc., Dalton Trans., 1992, 2059 RSC.
- (a) A. B. Alemayehu, S. J. Teat, S. M. Borisov and A. Ghosh, Inorg. Chem., 2020, 59, 6382 CrossRef CAS PubMed; (b) R. A. Eikey, S. I. Khan and M. M. Abu-Omar, Angew. Chem., Int. Ed., 2002, 41, 3591 CrossRef; (c) N. Y. Edwards, R. A. Eikey, M. I. Loring and M. M. Abu-Omar, Inorg. Chem., 2005, 44, 3700 CrossRef CAS PubMed.
- (a) F. Jérôme, B. Billier, J.-M. Barbe, E. Espinosa, S. Dahaoui, C. Lecomte and R. Guilard, Angew. Chem., Int. Ed., 2000, 39, 4051 CrossRef; (b) K. M. Kadish, F. Burdet, F. Jérôme, J.-M. Barbe, Z. Ou, J. Shao and R. Guilard, J. Organomet. Chem., 2002, 652, 69 CrossRef CAS; (c) L. Simhovich, L. Luobeznova, I. Goldberg and Z. Gross, Chem.–Eur. J., 2003, 9, 201 CrossRef PubMed; (d) A. B. Alemayehu, H. Vazquez-Lima, K. J. Gagnon and A. Ghosh, Inorg. Chem., 2017, 56, 5285 CrossRef CAS PubMed.
- (a) P. J. Brothers and J. P. Collman, Acc. Chem. Res., 1986, 19, 209 CrossRef CAS; (b) J. P. Collman, P. J. Brothers, L. McElwee-White, E. Rose and L. J. Wright, J. Am. Chem. Soc., 1985, 107, 4570 CrossRef CAS; (c) J. A. Smieja, K. Shirzad, M. Roy, K. Kittilstved and B. Twamley, Inorg. Chim. Acta, 2002, 335, 141 CrossRef CAS; (d) J. P. Collman, C. E. Barnes, P. N. Swepston and J. A. Ibers, J. Am. Chem. Soc., 1984, 106, 3500 CrossRef CAS; (e) J. P. Collman, E. Rose and G. D. Venburg, J. Chem. Soc., Chem. Commun., 1993, 934 RSC; (f) J. T. Groves and K.-H. Ahn, Inorg. Chem., 1987, 26, 3831 CrossRef CAS; (g) M. J. Camenzind, B. R. James and D. Dolphin, J. Chem. Soc., Chem. Commun., 1986, 1137 RSC.
- (a) S. Fantauzzi, E. Gallo, A. Caselli, F. Ragaini, N. Casati, P. Macchi and S. Cenini, Chem. Commun., 2009, 3952 RSC; (b) Q. Liu, L. Long, P. Ma, Y. Ma, X. Leng, J. Xiao, H. Chen and L. Deng, Cell Rep. Phys. Sci., 2021, 2, 100454 CrossRef CAS; (c) J. Xiong, Q. Liu, B. Lavina, M. Y. Hu, J. Zhao, E. E. Alp, L. Deng, S. Ye and Y. Guo, Chem. Sci., 2023, 14, 2808 RSC.
- (a) W.-H. Leung and C.-M. Che, J. Am. Chem. Soc., 1989, 111, 8812 CrossRef CAS; (b) M. B. Ezhova and B. R. James, in Advances in Catalytic Activation of Dioxygen by Metal Complexes, ed. L. I. Simándi, Springer, Boston, 2003, vol. 26, ch. 1, p. 1 Search PubMed.
- (a) R. Zhang, J. H. Horner and M. Newcomb, J. Am. Chem. Soc., 2005, 127, 6573 CrossRef CAS PubMed; (b) J. T. Groves, Z. Gross and M. K. Stern, Inorg. Chem., 1994, 33, 5065 CrossRef CAS.
- M. Keilwerth, W. Mao, S. A. V. Jannuzzi, L. Grunwald, F. W. Heinemann, A. Scheurer, J. Sutter, S. DeBeer, D. Munz and K. Meyer, J. Am. Chem. Soc., 2023, 145, 873 CrossRef CAS PubMed.
- (a) P. Singh, A. K. Das, B. Sarkar, M. Niemeyer, F. Roncaroli, J. A. Olabe, J. Fiedler, S. Záliš and W. Kaim, Inorg. Chem., 2008, 47, 7106 CrossRef CAS PubMed; (b) J. T. Groves, M. Bonchio, T. Carofiglio and K. Shalyaev, J. Am. Chem. Soc., 1996, 118, 8961 CrossRef CAS; (c) B. R. James, D. Dolphin, T. W. Leung, F. W. B. Einstein and A. C. Willis, Can. J. Chem., 1984, 62, 1238 CrossRef CAS; (d) M. M. Taqui Khan, D. Srinivas, R. I. Kureshy and N. H. Khan, Inorg. Chem., 1990, 29, 2320 CrossRef.
- (a) R. Zhang and M. Newcomb, Acc. Chem. Res., 2008, 41, 468 CrossRef CAS PubMed; (b) L. Simkhovich, A. Mahammed, I. Goldberg and Z. Gross, Chem.–Eur. J., 2001, 7, 1041 CrossRef CAS PubMed; (c) D. N. Harischandra, R. Zhang and M. Newcomb, J. Am. Chem. Soc., 2005, 127, 13776 CrossRef CAS PubMed.
- H. Shi, R. Liang, D. L. Phillips, H. K. Lee, W.-L. Man, K.-C. Lau, S.-M. Yiu and T.-C. Lau, J. Am. Chem. Soc., 2022, 144, 7588 CrossRef CAS PubMed.
- M. P. Mehn, S. D. Brown, D. M. Jenkins, J. C. Peters and L. Que Jr, Inorg. Chem., 2006, 45, 7417 CrossRef CAS PubMed.
- (a) K. M. Carsch, I. M. DiMucci, D. A. Iovan, A. Li, S.-L. Zheng, C. J. Titus, S. J. Lee, K. D. Irwin, D. Nordlund, K. M. Lancaster and T. A. Betley, Science, 2019, 365, 1138 CrossRef CAS PubMed; (b) K.-H. Chan, X. Guan, V. K.-Y. Lo and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 2982 CrossRef CAS PubMed; (c) Y.-D. Du, Z.-J. Xu, C.-Y. Zhou and C.-M. Che, Org. Lett., 2019, 21, 895 CrossRef CAS PubMed; (d) J. Wei, W. Xiao, C.-Y. Zhou and C.-M. Che, Chem. Commun., 2014, 50, 3373 RSC; (e) L.-M. Jin, X. Xu, H. Lu, X. Cui, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2013, 52, 5309 CrossRef CAS PubMed; (f) F. Ragaini, A. Penoni, E. Gallo, S. Tollari, C. L. Gotti, M. Lapadula, E. Mangioni and S. Cenini, Chem.–Eur. J., 2003, 9, 249 CrossRef CAS PubMed.
- X.-S. Xue, P. Ji, B. Zhou and J.-P. Cheng, Chem. Rev., 2017, 117, 8622 CrossRef CAS PubMed.
- X.-K. Jiang, Acc. Chem. Res., 1997, 30, 283 CrossRef CAS.
- S. K.-Y. Leung, W.-M. Tsui, J.-S. Huang, C.-M. Che, J.-L. Liang and N. Zhu, J. Am. Chem. Soc., 2005, 127, 16629 CrossRef CAS PubMed.
- A. Reckziegel, M. Kour, B. Battistella, S. Mebs, K. Beuthert, R. Berger and C. G. Werncke, Angew. Chem., Int. Ed., 2021, 60, 15376 CrossRef CAS PubMed.
- (a) P. Barrie, T. A. Coffey, G. D. Forster and G. Hogarth, J. Chem. Soc., Dalton Trans., 1999, 4519 RSC; (b) M. J. Zdilla and M. M. Abu-Omar, J. Am. Chem. Soc., 2006, 128, 16971 CrossRef CAS PubMed; (c) V. C. Gibson, C. Redshaw, W. Clegg and M. R. J. Elsegood, Polyhedron, 2007, 26, 3161 CrossRef CAS.
- D. Intrieri, A. Caselli, F. Ragaini, P. Macchi, N. Casati and E. Gallo, Eur. J. Inorg. Chem., 2012, 569 CrossRef CAS.
- N. P. van Leest, M. A. Tepaske, J.-P. H. Oudsen, B. Venderbosch, N. R. Rietdijk, M. A. Siegler, M. Tromp, J. I. van der Vlugt and B. de Bruin, J. Am. Chem. Soc., 2020, 142, 552 CrossRef CAS PubMed.
- (a) A. Takaoka, L. C. H. Gerber and J. C. Peters, Angew. Chem., Int. Ed., 2010, 49, 4088 CrossRef CAS PubMed; (b) A. N. Walstrom, B. C. Fullmer, H. Fan, M. Pink, D. T. Buschhorn and K. G. Caulton, Inorg. Chem., 2008, 47, 9002 CrossRef CAS PubMed.
- M. Mahajan and B. Mondal, Inorg. Chem., 2023, 62, 5810 CrossRef CAS PubMed.
- For recent examples, see: (a) Y. Park, S. P. Semproni, H. Zhong and P. J. Chirik, Angew. Chem., Int. Ed., 2021, 60, 14376 CrossRef CAS PubMed; (b) W. Mao, Z. Zhang, D. Fehn, S. A. V. Jannuzzi, F. W. Heinemann, A. Scheurer, M. van Gastel, S. DeBeer, D. Munz and K. Meyer, J. Am. Chem. Soc., 2023, 145, 13650 CrossRef CAS PubMed.
- (a) S.-M. Au, J.-S. Huang, W.-Y. Yu, W.-H. Fung and C.-M. Che, J. Am. Chem. Soc., 1999, 121, 9120 CrossRef CAS; (b) Y. Tokita, K. Yamaguchi, Y. Watanabe and I. Morishima, Inorg. Chem., 1993, 32, 329 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Abbreviations, experimental procedures, characterization of compounds, computational details, Tables S1–S4, Fig. S1–S21, NMR spectra of compounds, and Cartesian coordinates from DFT calculations. CCDC 2214628. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02266h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |