
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew insights into the oxidation process from neutron and X-ray crystal structures of an O2-sensitive [NiFe]-hydrogenase†
Takeshi
Hiromoto
ab,
Koji
Nishikawa
b,
Seiya
Inoue
b,
Hideaki
Ogata
 b,
Yuta
Hori
c,
Katsuhiro
Kusaka
d,
Yu
Hirano
ae,
Kazuo
Kurihara
a,
Yasuteru
Shigeta
ac,
Taro
Tamada
*ae and
Yoshiki
Higuchi
*b
b,
Yuta
Hori
c,
Katsuhiro
Kusaka
d,
Yu
Hirano
ae,
Kazuo
Kurihara
a,
Yasuteru
Shigeta
ac,
Taro
Tamada
*ae and
Yoshiki
Higuchi
*b
aInstitute for Quantum Life Science, National Institutes for Quantum Science and Technology, 4-9-1 Anagawa, Inage, Chiba 263-8555, Japan. E-mail: tamada.taro@qst.go.jp
bGraduate School of Science, University of Hyogo, 3-2-1 Koto, Kamigori, Hyogo 678-1297, Japan. E-mail: hig@sci.u-hyogo.ac.jp
cCenter for Computational Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8577, Japan
dNeutron Industrial Application Promotion Center, Comprehensive Research Organization for Science and Society, 162-1 Shirakata, Tokai, Ibaraki 319-1106, Japan
eDepartment of Quantum Life Science, Graduate School of Science, Chiba University, 1-33 Yayoi, Inage, Chiba 263-8522, Japan
First published on 21st August 2023
Abstract
[NiFe]-hydrogenase from Desulfovibrio vulgaris Miyazaki F is an O2-sensitive enzyme that is inactivated in the presence of O2 but the oxidized enzyme can recover its catalytic activity by reacting with H2 under anaerobic conditions. Here, we report the first neutron structure of [NiFe]-hydrogenase in its oxidized state, determined at a resolution of 2.20 Å. This resolution allowed us to reinvestigate the structure of the oxidized active site and to observe the positions of protons in several short hydrogen bonds. X-ray anomalous scattering data revealed that a part of the Ni ion is dissociated from the active site Ni–Fe complex and forms a new square-planar Ni complex, accompanied by rearrangement of the coordinated thiolate ligands. One of the thiolate Sγ atoms is oxidized to a sulfenate anion but remains attached to the Ni ion, which was evaluated by quantum chemical calculations. These results suggest that the square-planar complex can be generated by the attack of reactive oxygen species derived from O2, as distinct from one-electron oxidation leading to a conventional oxidized form of the Ni–Fe complex. Another major finding of this neutron structure analysis is that the Cys17S thiolate Sγ atom coordinating to the proximal Fe–S cluster forms an unusual hydrogen bond with the main-chain amide N atom of Gly19S with a distance of 3.25 Å, where the amide proton appears to be delocalized between the donor and acceptor atoms. This observation provides insight into the contribution of the coordinated thiolate ligands to the redox reaction of the Fe–S cluster.
Introduction
[NiFe]-hydrogenases (H2ases) have a binuclear Ni–Fe complex in the active site to reversibly catalyze the oxidation of molecular hydrogen (H2), giving two electrons and two protons.1,2 The active site complex is buried deep in the catalytic unit composed of two subunits, called the ‘large’ and ‘small’, and is supported by cysteine residues from the large subunit (Fig. 1a), where a Ni ion coordinated by the two terminal cysteine residues and an Fe ion with one CO and two CN− ligands are bridged by the two cysteine thiolate ligands (Fig. 1b). Moreover, three Fe–S clusters (one [3Fe–4S]- and two [4Fe–4S]-type clusters), involved in electron transfer to external electron carriers, are arranged from the active site to the protein surface, passing through the small subunit.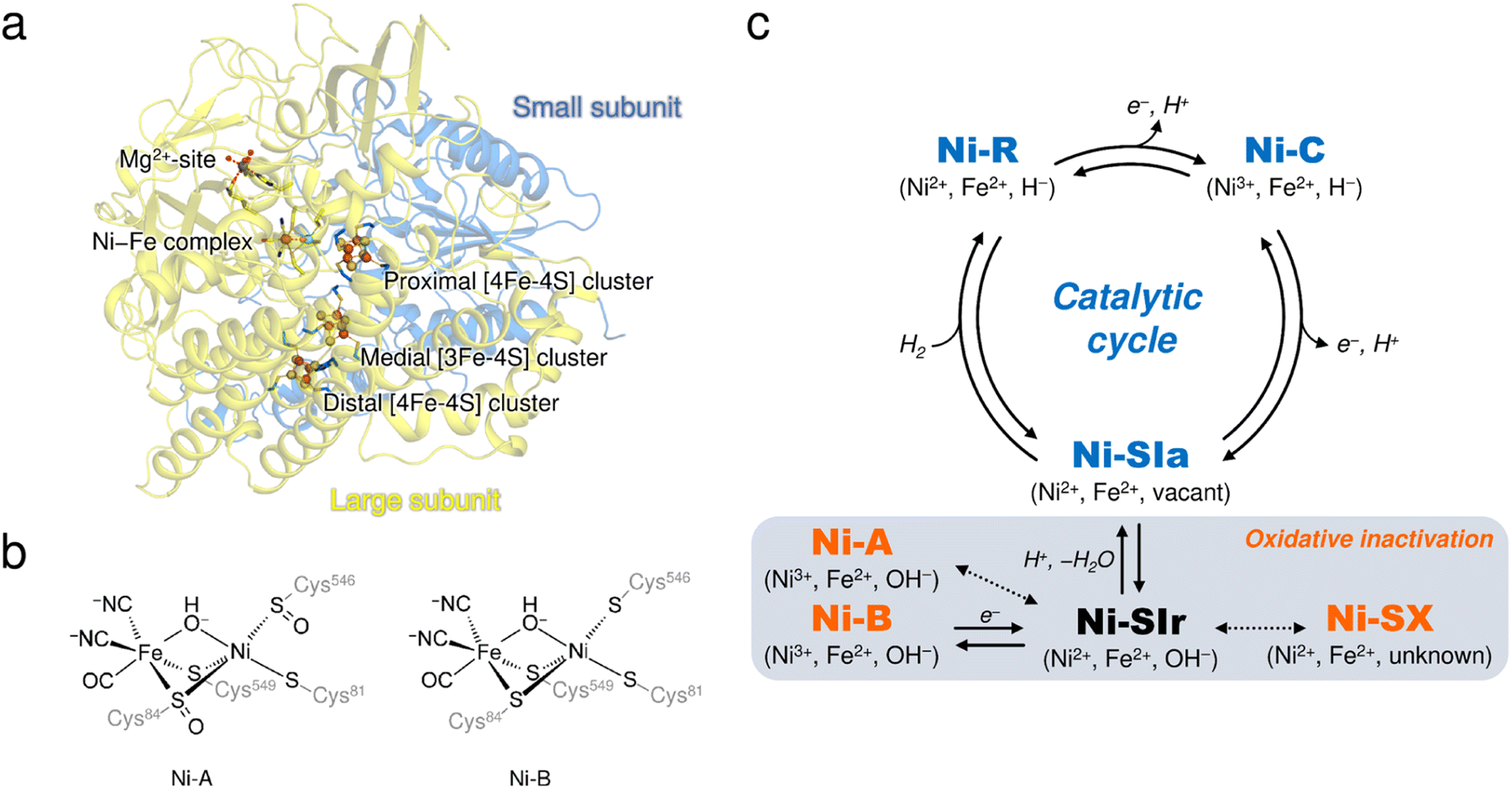 | ||
| Fig. 1 (a) Overall structure of DvMF-H2ase. The ribbon diagram shows that the heterodimeric enzyme is composed of a large (pale yellow) and a small (blue) subunit. The active site Ni–Fe complex and the three Fe–S clusters are labelled, and the constituent atoms are drawn as spheres: Fe, brown; Ni, bright green; O, red; S, yellow. The Mg2+ ion located in the large subunit is similarly depicted as a gray sphere. (b) Chemical structures of Ni-A and Ni-B. (c) The catalytically active and inactive states of the Ni–Fe complex. The active states are colored in blue and the inactive oxidized states are colored in orange. Oxidation states of the metal centers in each state and their bridging ligands are indicated in parentheses. Expected but not experimentally verified reactions are shown by dashed arrows.12 | ||
It is well known that most [NiFe]-H2ases are oxygen sensitive and are reversibly inactivated upon exposure to molecular oxygen (O2),3,4 which is one factor hindering their utilization for biological applications.5,6 The enzyme members can be classified into four groups, each containing multiple subgroups, based on the phylogenetic clustering of protein sequences and taking into account their biochemical characteristics.7 A membrane-bound [NiFe]-H2ase from Desulfovibrio vulgaris Miyazaki F (DvMF) belongs to group 1b, comprising 22 subgroups, and loses its catalytic activity at ambient O2 concentrations. This results in the formation of oxidized products of the active site complex.8,9 Atmospheric oxidation of the O2-sensitive enzymes generally leads to a mixture of two spectroscopically different states of the Ni–Fe complex, referred to as Ni-A and Ni-B (Fig. 1c).10,11 Ni-A requires considerable time for its reductive reactivation (‘unready’ state), whereas Ni-B is readily reactivated in a few seconds (‘ready’ state).3 Each binuclear complex in the oxidized enzyme exhibits paramagnetic behavior and is composed of a Ni ion in a formal oxidation state of +3 bridged by an oxygen-containing ligand to an Fe ion in the +2 oxidation state.8,12 Many spectroscopic and theoretical studies indicate that a hydroxide ion (μ2-OH−) bridges between the two metal centers in both oxidized forms.13,14 However, this has not been verified by protein X-ray crystallography because it remains challenging to visualize protons (H+).
Oxidative modification observed at the cysteine thiolate ligands (cys-sulfoxide (S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) or cys-sulfenic acid (S–OH)) could be a plausible explanation for the difference between Ni-A and Ni-B (Fig. 1b).8,14,15 A sulfenylation has been identified at the proposed catalytic base Cys546L,8 where the superscript L (or S) indicates that the residue is from the large (or small) subunit. Cys546L is assumed to accept a proton derived from the heterolytic cleavage of H2 at the active site and to pass the proton to the adjacent carboxylate group of Glu34L, connecting the proton to the protein surface via hydrogen-bonding networks.16 The oxidation of Cys546L to its sulfenate derivative inactivates the catalytic activity and thus this conversion must have occurred to form Ni-A, which requires a prolonged anaerobic incubation, as suggested by Volbeda et al.14 On the other hand, our recent FT-IR and EPR studies indicated that the aerobically isolated DvMF-H2ase contains mainly a paramagnetic Ni-B (Ni3+) with a minor amount of Ni-A (Ni3+) and an EPR-silent (Ni2+) state denoted as Ni-SX.17 Furthermore, X-ray crystallographic analysis suggested that the Ni ion occupancy in Ni-B was reduced by 20%, even though no reduction in occupancy was observed for the Fe ion or the CO and CN− ligands coordinated to Fe.15 Linking these two results (i.e., formation of the oxidized product with Ni2+ and Ni-elimination from the active site complex) requires an alternative experimental approach.
O) or cys-sulfenic acid (S–OH)) could be a plausible explanation for the difference between Ni-A and Ni-B (Fig. 1b).8,14,15 A sulfenylation has been identified at the proposed catalytic base Cys546L,8 where the superscript L (or S) indicates that the residue is from the large (or small) subunit. Cys546L is assumed to accept a proton derived from the heterolytic cleavage of H2 at the active site and to pass the proton to the adjacent carboxylate group of Glu34L, connecting the proton to the protein surface via hydrogen-bonding networks.16 The oxidation of Cys546L to its sulfenate derivative inactivates the catalytic activity and thus this conversion must have occurred to form Ni-A, which requires a prolonged anaerobic incubation, as suggested by Volbeda et al.14 On the other hand, our recent FT-IR and EPR studies indicated that the aerobically isolated DvMF-H2ase contains mainly a paramagnetic Ni-B (Ni3+) with a minor amount of Ni-A (Ni3+) and an EPR-silent (Ni2+) state denoted as Ni-SX.17 Furthermore, X-ray crystallographic analysis suggested that the Ni ion occupancy in Ni-B was reduced by 20%, even though no reduction in occupancy was observed for the Fe ion or the CO and CN− ligands coordinated to Fe.15 Linking these two results (i.e., formation of the oxidized product with Ni2+ and Ni-elimination from the active site complex) requires an alternative experimental approach.
[NiFe]-H2ases from Hydrogenovibrio marinus (Hm),18Ralstonia eutropha (Re),19 and Escherichia coli (Ec)20 are O2-tolerant enzymes classified into group 1d and can maintain their catalytic activities under air. The members harbour a proximal [4Fe–3S] cluster coordinated by six thiolate ligands, in contrast to the proximal [4Fe–4S] cluster coordinated by four thiolate ligands seen in the O2-sensitive enzymes. Under ambient O2 concentrations, the proximal [4Fe–3S] cluster of the O2-tolerant enzymes supplies surplus electrons to the Ni–Fe active site for the reduction of invading O2, accompanied by a large structural change with disruption of an internal Fe–S bond, which supports its super-oxidation.18,19 The O2-tolerant enzymes thus can protect the active site from the attack by reactive oxygen species which probably causes the formation of the unready states as in Ni-A.5,21,22 In contrast, the O2-sensitive enzymes do not have such an electron supply system from the proximal [4Fe–4S] cluster, but that would give us an advantage in investigating structural changes in different redox states of the proximal Fe–S cluster and in understanding the catalytic electron transfer.
Here, we report the first neutron structure of O2-sensitive DvMF-H2ase in its oxidized state at 2.20 Å resolution, allowing visualization of the protons characteristic of the oxidized enzyme. In determining the neutron structure by joint X-ray and neutron (XN) refinement, the high-resolution X-ray data collected at 1.04 Å revealed an oxidized product of the active site complex with a square-planar geometry, suggesting that it contains a Ni ion in the +2 oxidation state. We verified the presence of the square-planar Ni complex containing a cysteine sulfenate ligand via X-ray anomalous scattering data and quantum chemical calculations. Furthermore, the neutron structure enables us to observe a delocalized amide proton between the Cys17S thiolate Sγ atom coordinated to the proximal Fe–S cluster and the main-chain amide N atom of Gly19S, providing insight into the contribution of the coordinated thiolate ligands to the redox reaction of the Fe–S cluster.
Results and discussion
Reinterpretation of the active site structure in the oxidized enzyme
X-ray diffraction data collected here (up to 1.04 Å resolution) were used for further structure analysis to clarify the detailed oxidized structure of the active site Ni–Fe complex (Fig. 2a and S1a and Table S1†). Structure refinement with a model containing only Ni-B in the active site allowed visualization of the conformational changes of the coordinated cysteines as a mFo-DFc difference electron-density map (Fig. 2c and S1c†). In the model used for the map calculation (Fig. 2b), the occupancy for the Ni ion coordinating a hydroxide O atom was reduced to 68%, estimated by the program phenix.refine in PHENIX as described in ESI Text 1.† The occupancies for the coordinated Cys81L, Cys546L, and Cys549L were also reduced to 68% because significant negative electron densities for their Sγ atoms appeared, except for Cys84L.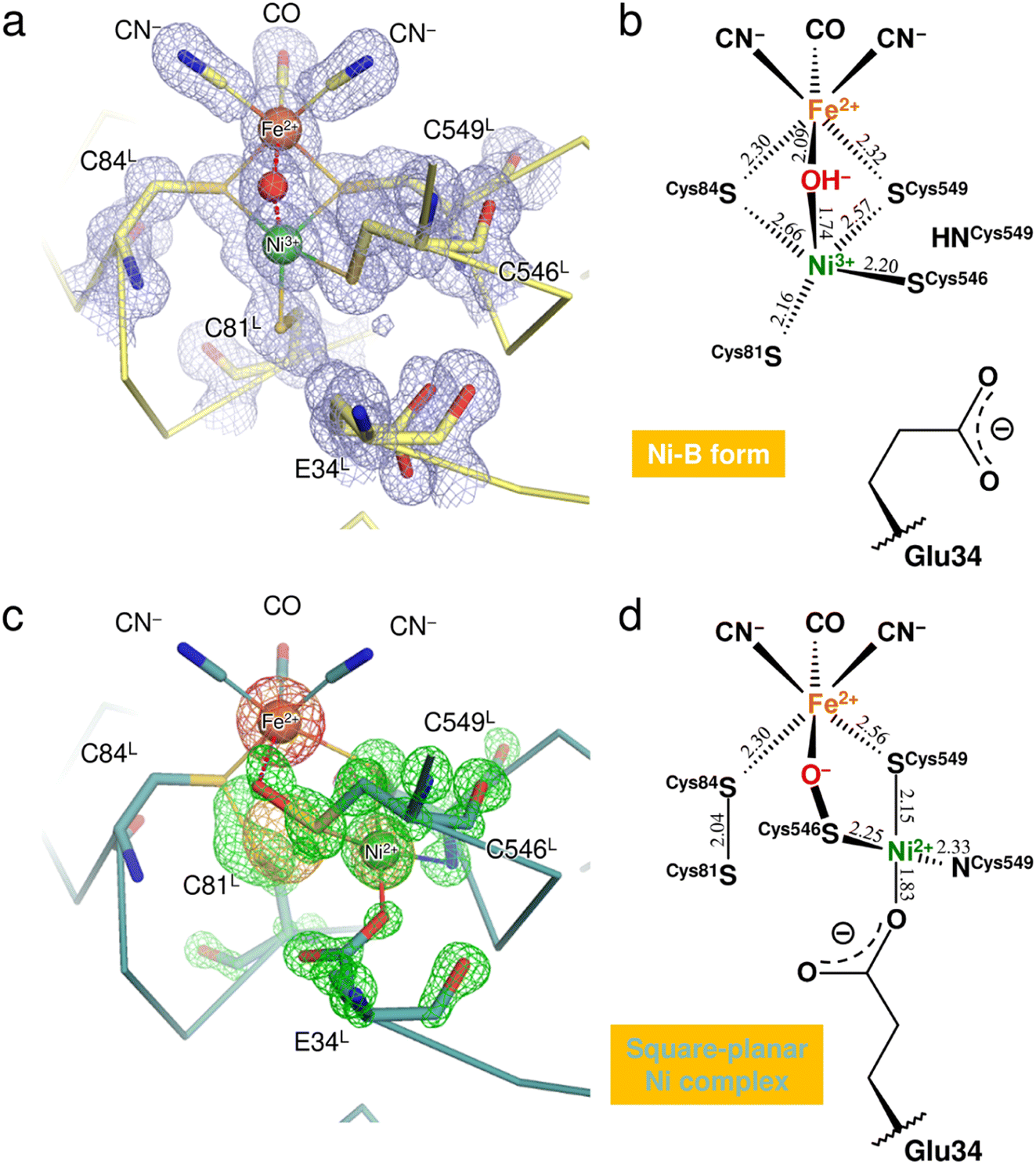 | ||
| Fig. 2 Structure determination of the two oxidized forms of the Ni–Fe complex. (a) The active site complex in Ni-B, superimposed with a 2mFo-DFc electron-density map (contoured at 1.0σ, pale blue mesh). The binuclear metal center and the bridging hydroxide ion are drawn as spheres: Fe, brown; Ni, green; O, red. Constituent residues of the active site are labeled with black letters. (b) Coordination structure of the active site complex in Ni-B. Selected bond lengths are given in Å and the coordinate uncertainties were estimated to be 0.014–0.018 Å from Cruickshank's DPI (diffraction-component precision index).56 (c) The square-planar Ni complex identified as the other oxidized form, superimposed with model-phased anomalous difference Fourier maps at wavelengths of 1.4800 Å (contoured at 4.0σ, bright orange mesh) and 1.4900 Å (contoured at 5.0σ, hot pink mesh). The green mesh shows an mFo-DFc electron-density map (contoured at 4.5σ), calculated for visualizing structural differences from the active site containing Ni-B with occupancy of 68%. (d) Coordination structure of the other oxidized form. The square-planar Ni complex is bridged to the Fe ion by the Cys546L sulfenate O atom. | ||
Anomalous scattering data were collected at 1.4800 Å and 1.4900 Å near the X-ray absorption K-edge of nickel (1.4879 Å). In the model-phased anomalous difference Fourier map for 1.4900 Å, only one strong peak was observed at the Fe site due to the lower energy side of the nickel edge (Fig. 2c and S1b†). The anomalous difference Fourier map for 1.4800 Å showed three strong peaks, two of which corresponded to the Fe and Ni sites of Ni-B. The third peak was close to the thiolate Sγ (Cys546L) and thus arose from the Ni ion dissociated from the active site complex by oxidation. The occupancy of the dissociated Ni ion was estimated to be 32% by comparing it with the Ni ion in Ni-B. This dissociated Ni site had been assigned as a sulfoxide or sulfenic acid O atom of Cys546L in the previous X-ray structure analyses.8,15
Simultaneous with the shift of the Ni ion, the Sγ atoms of Cys81L and Cys84L are released from their coordination of Ni-B and form a disulfide bond with each other, as assigned previously.15,23 Also, each alternative conformation of the Cys546L and Cys549L thiolate anions makes coordination bonds with the dissociated Ni ion at distances of 2.25 and 2.15 Å, respectively, to form a mononuclear Ni complex with a square-planar geometry (Fig. 2d). The deprotonated amide N (Cys549L) and the carboxylate Oε2 (Glu34L) are supplied as the other two ligands, with distances of 2.33 Å and 1.83 Å, respectively. In particular, the Glu34L side-chain is flipped to the dissociated Ni site by about 90° relative to its original position seen in Ni-B.
A residual mFo-DFc electron density appeared at the bridging ligand site of Ni-B in calculations with 68% occupancy of the hydroxide O atom (Fig. 2c and S1c†). Given that the occupancy for the bridging ligand site was estimated as 100% of a monooxygen atom (Table S2†), another monooxygen species remains present after the shift of the Ni ion. The Sγ (Cys546L) atom in the alternative conformation (32% occupancy) is sufficiently close with a distance of 1.65 Å to the bridging O site to form a single covalent bond, indicating the sulfenylation of Cys546L. In that case, the dissociated Ni ion is still bridged to the Fe ion by the sulfenic acid or sulfenate group of Cys546L (Fig. 2c). The Ni oxidation state of mononuclear complexes with a square-planar geometry is +2 in most inorganic complexes. This implies that the newly assigned oxidized form corresponds to an EPR-silent state,17 which we further examined by quantum chemical calculations.
Evaluation of the oxidized forms of the Ni–Fe complex by quantum chemical calculations
Quantum chemical calculations were conducted to verify the geometric and electronic structures of the two oxidized forms of the active site complex, i.e., the square-pyramidal Ni complex in Ni-B (Fig. 2b) and the square-planar Ni complex with a cysteine sulfenate ligand (Fig. 2d). The respective cluster models based on the crystal structure determined here (the Fe–Ni core with the CN− and CO ligands, the bridging X or Sγ–X species (X = O2− or OH−), Cys81L, Cys84L, Cys546L, Cys549L, and Glu34L) were used as calculation models. The computational details are described in ESI Text 1.† Possible combinations of the Ni charges (Ni2+ or Ni3+), the spin states (high or low spin), and the bridging monooxygen species (O2− or OH−) in each oxidized form were examined, and their optimized structures were explored by quantum chemical calculations. The root-mean-square deviations (RMSDs) of each optimized structure from the obtained crystal structure were then calculated to estimate the similarity of the calculated structures to the experimental data. The RMSD values of each optimized structure from the active site complex in Ni-B are summarized in Table 1. No optimized structure with Ni2+ was found because its model structure collapsed during the calculation. The calculation model with the smallest RMSD value confirmed that the active site in Ni-B has a Ni3+ ion in the low-spin state and a bridging μ2-OH− ligand (Fig. S2a†), in good agreement with the active site complex structure in Ni-B proposed experimentally.11,13 On the other hand, the calculated results for the other oxidized form show that the RMSD value between the calculation model possessing a Ni2+ ion in the low-spin state coordinated by a cysteine Sγ–O− ligand (Fig. S2b†) and the crystal structure is 0.27 Å (Table 1). This is the smallest of all the calculation models and validates our proposed geometric and electronic structures of the square-planar Ni complex in the active site.| Bridging species | Spin multiplicity | RMSD (Å) | |
|---|---|---|---|
| Ni3+ in Ni-B | O2− | Doublet | 0.91 |
| Quartet | 0.56 | ||
| OH− | Doublet | 0.41 | |
| Quartet | 0.46 | ||
| Square-planar Ni3+ complex | Sγ–O− | Doublet | 0.37 |
| Quartet | 0.46 | ||
| Sγ–OH | Doublet | 0.40 | |
| Quartet | 0.58 | ||
| Square-planar Ni2+ complex | Sγ–O− | Singlet | 0.27 |
| Sγ–OH | Singlet | 0.35 |
Insight into the oxidation process on the active site Ni–Fe complex
Cysteine can be oxidized to sulfenic acid by reacting with a two-electron oxidant such as hydrogen peroxide (H2O2) in biological systems.24,25 The cysteine sulfenate ligand was also identified at the active site in the O2-sensitive Desulfovibrio fructosovorans (Df) [NiFe]-H2ase,14 in which the sulfenylation was proposed to occur through a peroxide intermediate produced from invading O2.26,27 When looking at the square-planar Ni complex in the oxidized DvMF-H2ase (Fig. 2d), four electrons in total are consumed by the sulfenylation at the Cys546L Sγ-atom and the disulfide-bond formation between Cys81L and Cys84L. Considering that the reduction of O2 requires four electrons and additional protons, the square-planar Ni complex could be produced through the oxidation of the original Ni–Fe complex by O2 diffused from the protein exterior, which might be protonated leading to the formation of a reactive oxygen species prior to the sulfenylation of Cys546L.27Having said that, the major components of the active site structure in the oxidized enzyme have been thought to be the paramagnetic forms, Ni-A and Ni-B (Fig. 1b), which were not differentiated in this study. The square-planar Ni complex would be one candidate for an oxidized product in the ‘unready’ state because of its large structural differences from the Ni-B oxidized form in the ‘ready’ state. An EPR study on Dg-H2ase using 17O2 and H217O indicates that the bridging μ2-OH− ligand in Ni-A (and presumably in Ni-B) is derived from H2O, not from O2.28 A bridging μ2-OH− ligand at the active site complex was also proposed for the O2-tolerant Hm-H2ase that was anaerobically oxidized using the chemical oxidant ferricyanide,18 suggesting that one-electron oxidation at the Ni–Fe complex in Ni-SIa, leading to the formation of Ni-B, is achieved by electron release via the Fe–S clusters.4
Neutron structure analysis of DvMF-H2ase in its oxidized state
The neutron structure of the oxidized enzyme was determined at 2.20 Å resolution (Fig. S3†). The mFo-DFc neutron scattering length density map omitting the two Ni ions at the active site was calculated and presented in Fig. 3a. The neutron scattering length density for the dissociated Ni ion was relatively weak but observed at the corresponding site, indicating that the square-planar Ni complex was already produced prior to X-ray exposure, as the neutron diffraction data were first collected from the crystal without radiation damage problems. Since the main-chain amide proton of Cys549L in Ni-B was mainly occupied by H having a negative scattering length instead of D, the weakening of the neutron scattering length density for the dissociated Ni ion could be caused by overlapping of the Cys549L amide proton.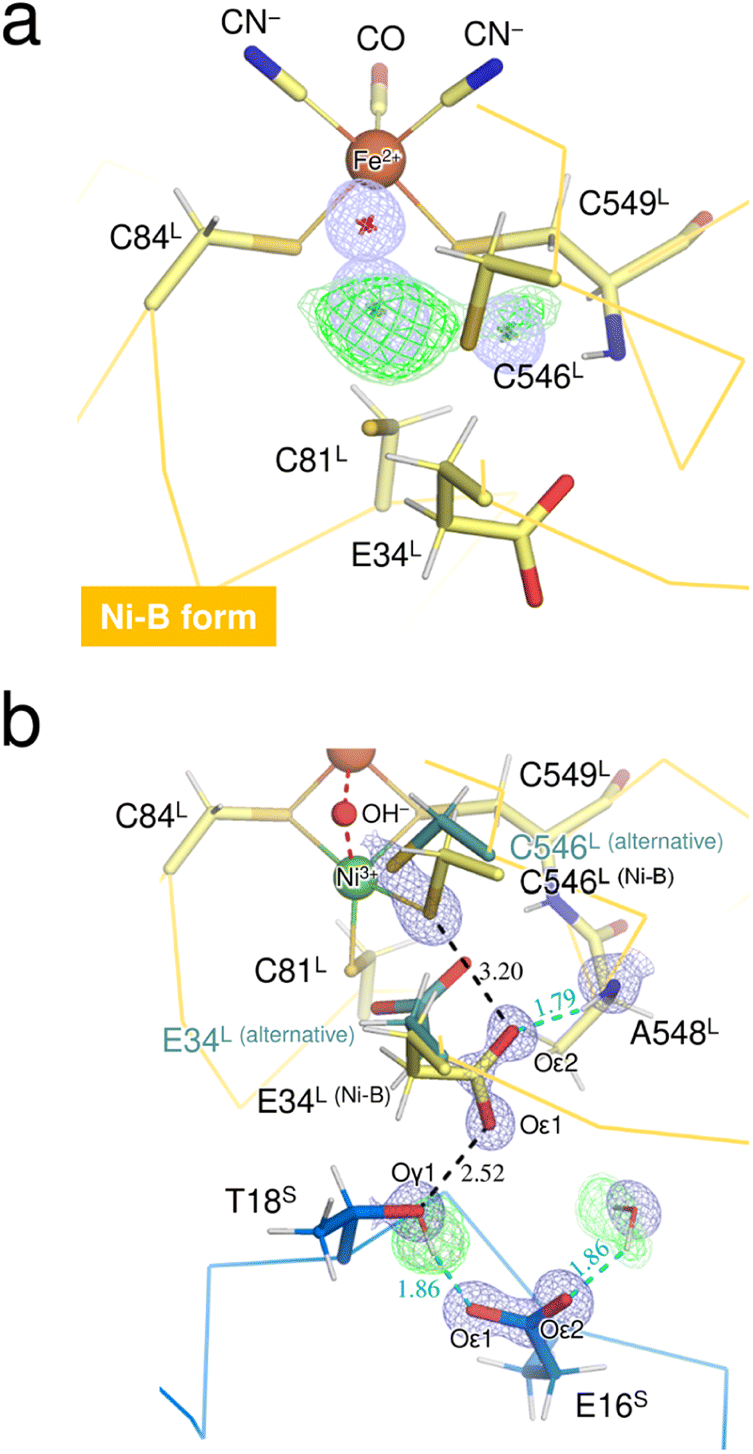 | ||
| Fig. 3 (a) The Ni–Fe complex in Ni-B, superimposed with a mFo-DFc omit electron-density map (contoured at 4.5σ, pale blue mesh) and with mFo-DFc omit neutron scattering length density maps (contoured at 3.2σ and 2.8σ, colored green and pale green, respectively). The bridging hydroxide ion and the two Ni sites are indicated by asterisks because their coordinates were omitted from the map calculations. The neutron scattering length density for the bridging ligand site occupied with 100% monooxygen species was not detected because of its cancellation by the negative scattering length of H. (b) The key residue Glu34L for proton transfer mediated by Cys546L, superimposed with a 2mFo-DFc electron-density map (contoured at 2.0σ, blue mesh) for selected atoms and with their mFo-DFc H/D-omit neutron scattering length density maps. Residues from the large subunit are colored pale yellow and those from the small subunit are colored blue. The positive peaks at 3.6σ, 3.2σ, and 2.8σ are colored dark green, green, and pale green, respectively. Hydrogen bonds are shown by cyan dashed lines, with the lengths shown in Å. The neutron scattering length density for the main-chain amide proton of Ala548L was not observed because of its partial H/D exchange or of having an alternative conformation. | ||
On the other hand, calculation of the mFo-DFc neutron scattering length density map omitting the bridging O species resulted in no significant density at the bridging ligand site (Fig. 3a). No neutron scattering length density was observed even for the oxygen moiety (with a total occupancy of 100%), suggesting its cancellation by the negative scattering length of H from the hydroxide ion. A similar cancellation effect is reported for a water molecule having multiple orientations in an organic macromolecule.29 Thus, the result implies that most of the molecules in the crystal contain an μ2-OH− (not OD−) at the bridging ligand site and that little H/D exchange occurs at the site, consistent with a previous report that the bridging ligand in Ni-A/B is not replaced by surrounding solvent molecules.28 Also, the hydrogen-bonding networks proposed for proton transfer are disconnected at the gate residue Glu34L in this oxidized enzyme as described below. This situation would lead to hinder further H/D exchange at the active site.
The thiolate Sγ atom of the putative catalytic base Cys546L is at a hydrogen-bond distance of 3.20 Å to the carboxylate Oε2 (Glu34L), assigned in Ni-B with 68% occupancy (Fig. 3b), but no neutron scattering length density was observed between Sγ (Cys546L) and Oε2 (Glu34L). The Glu34L carboxylate group is assumed to function as a gate for proton transfer.30 The formation of a short hydrogen bond (<2.6 Å) between Oε1 (Glu34L) and Oγ1 (Thr18S) could be advantageous for proton migration, as suggested previously based on a high-resolution X-ray structure of the reduced enzyme.16 Similarly, in our neutron structure of the oxidized enzyme, Oε1 (Glu34L) is adjacent to Oγ1 (Thr18S) at a distance of 2.52 Å. However, as shown in Fig. 3b, the hydroxyl proton of Thr18S is mainly directed toward Oε1 (Glu16S), indicating that there is no hydrogen bonding between Oγ1 (Thr18S) and Oε1 (Glu34L). The negative charge of the carboxylate group of Glu34L could be compensated by the positive charge of the active site Ni ion. Such an electrostatic interaction would be expected because side-chain flipping of Glu34L occurred in the other oxidized form to coordinate with the eliminated Ni ion.31 More details on potential proton transfer pathways are described in ESI Text 2.†
Three Fe–S clusters are aligned through the protein interior and provide the best route for electron transfer (Fig. 4a). Comparison of the X-ray structures of each cluster in this oxidized enzyme with those in the reduced enzyme shows that most bond lengths of internal Fe–S coordination are slightly shorter,32 as summarized in Tables S5–S7.† An unusual hydrogen bond was observed between the main-chain amide N (Gly19S) and Sγ (Cys17S) which is involved in the secondary coordination to the proximal cluster (Fig. 4b). The mFo-DFc H/D-omit neutron scattering length density map shows an elongated negative density between N (Gly19S) and Sγ (Cys17S), indicating that the amide proton is partially attracted to Sγ (Cys17S). Also, no H/D exchange appeared to occur at that site. The amide proton would be maintained quite tightly between the donor and acceptor atoms.
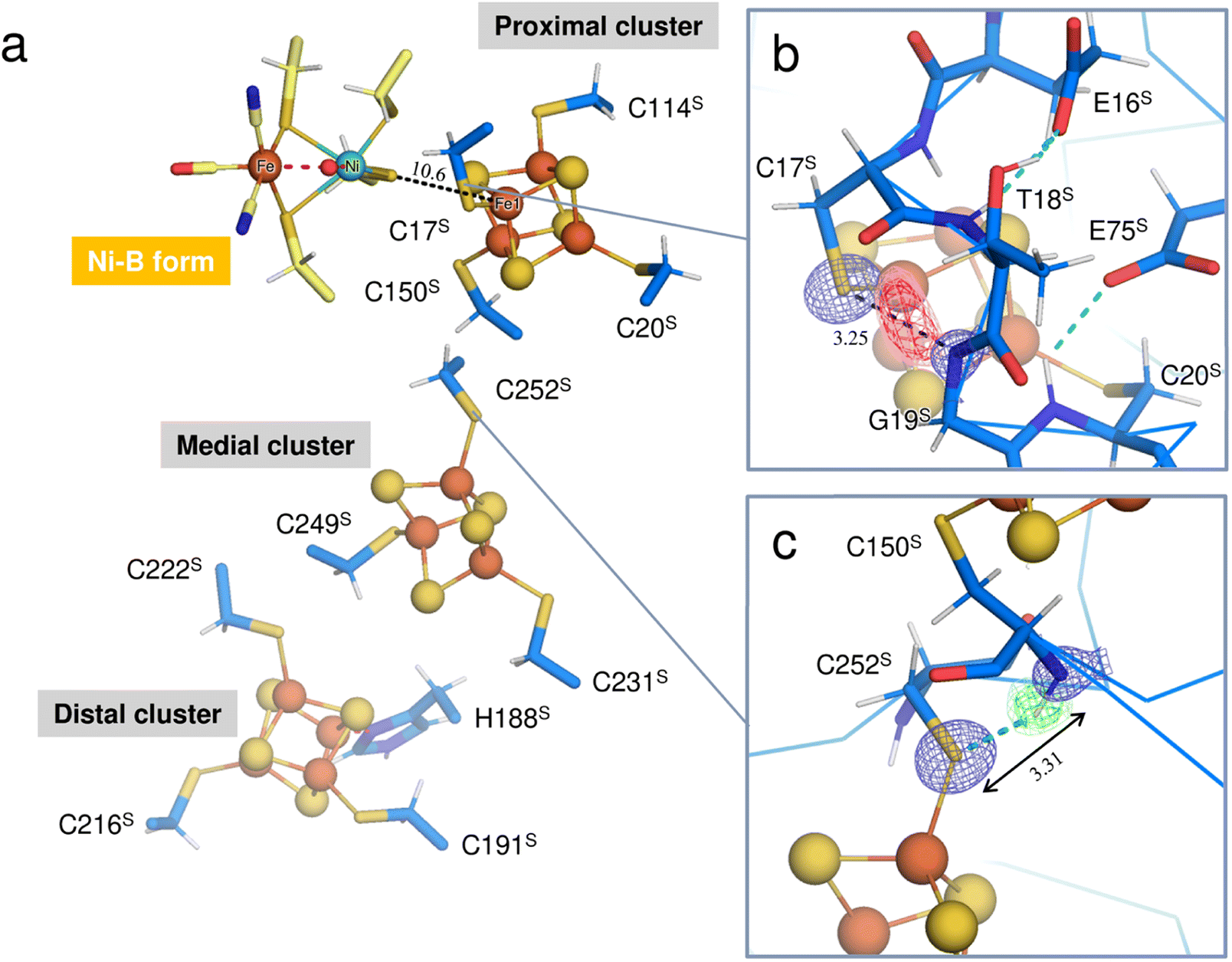 | ||
| Fig. 4 (a) Three Fe–S clusters in the oxidized enzyme. Constituent atoms of the Fe–S clusters are drawn as spheres (Fe, brown; S, yellow) and the coordinated cysteine residues are shown as sticks. The interatomic distance between Ni in the active site complex and Fe1 of the proximal cluster is indicated with a black dashed line. (b) Hydrogen bond between the main-chain amide N (Gly19S) and Sγ (Cys17S). A 2mFo-DFc electron-density map (contoured at 2.0σ) for the amide N and thiolate Sγ atoms is represented as a blue mesh. The mFo-DFc H/D-omit neutron scattering length density maps were calculated for visualizing the H and D positions and were superimposed on the atom pair, where the negative peaks at −3.2σ and −2.8σ are colored hot pink and pink, respectively. The distance between the amide N (Gly19S) and Sγ (Cys17S) is 3.25 Å. (c) Hydrogen bond between the main-chain amide N (Cys150S) and Sγ (Cys252S). A 2mFo-DFc electron-density map is depicted in the same way as in (b). In the mFo-DFc H/D-omit neutron scattering length density maps, the positive peaks at 3.2σ and 2.8σ are colored green and pale green, respectively. The distance between the amide N (Cys150S) and Sγ (Cys252S) is 3.31 Å. | ||
The amide N (Gly19S)–Sγ (Cys17S) distance of 3.25 Å is relatively short compared to NH⋯S hydrogen bonds in small compounds (3.25–3.55 Å).33 Two coordinated cysteine sulfur atoms of the medial [3Fe–4S] cluster and three of the distal [4Fe–4S] cluster form hydrogen bonds with nearby main-chain amide groups, with distances of over 3.3 Å (e.g., 3.31 Å for Cys252S (Fig. 4c), 3.37 Å for Cys191S (Fig. S5a†), and 3.44 Å for Cys222S (Fig. S5b†)). The H/D-omit neutron scattering length density maps confirmed that the protons in these hydrogen bonds are attached to the respective main-chain N atoms with covalent bond lengths, except for the N (Gly251S)–Sγ (Cys249S) and N (Tyr218S)–Sγ (Cys216S) hydrogen bonds, for which no neutron scattering length density was observed due to cancellation by partial H/D exchange.
Insight into the unusual NH⋯Sγ hydrogen bond in the secondary coordination sphere of the proximal [4Fe–4S] cluster
This neutron structure analysis shows that the Sγ (Cys17S) coordinated to the proximal [4Fe–4S] cluster is partially protonated by receiving a proton from the main-chain amide N (Gly19S) (Fig. 4b). The redox transition of the proximal cluster of DvMF-H2ase is (+2/+1) during the catalytic cycle.34,35 The oxidation of the cluster may somehow cause the polarization of the Fe1–Sγ (Cys17S) coordination bond, leading to the abstraction of the Gly19S amide proton (Fig. S6†). Alternatively, such proton transfer in a hydrogen bond is allowed when the hydrogen-bond donor and acceptor atoms have the same pKa values and thus the energy barrier between the two hydrogen positions is lowered.36,37 The pKa value of Sγ (Cys17S) could be modulated to be equal to that of N (Gly19S) by an increase in the oxidation state of the proximal cluster. Actually, the protonation at Sγ (Cys17S) makes it difficult to maintain its coordination with Fe1 of the cluster.38,39 Density functional simulations on O2-tolerant [NiFe]-H2ases suggest that protonation at the proximal cluster can weaken the internal Fe–S bond, leading to the large structural change of the cluster required for a transition to its super-oxidized form.40 Considering that Sγ (Cys17S) retained the coordination to the proximal cluster in this analysis, it is reasonable to infer that the proton in the N (Gly19S)–Sγ (Cys17S) hydrogen bond is delocalized between the respective protonated states, rather than being covalently bound to the thiolate Sγ atom (Fig. S6†).High-potential iron-sulfur proteins (HiPIPs) are among the most well structurally characterized [4Fe–4S]-type clusters.41,42 Typically, HiPIPs have one [4Fe–4S] cluster, with the (+3/+2) redox pair providing a redox potential of ≈+350 mV.43,44 This is fundamentally different from the redox potential of <−300 mV for the proximal cluster with the (+2/+1) redox pair in DvMF-H2ase. The cuboidal cluster is divided into two rhombic [2Fe–2S] subclusters, where each subcluster is bridged by four internal Fe–S bonds that are shorter than the other eight bonds. One subcluster responsible for storing an electric charge in HiPIP shows slightly shorter internal bond lengths upon oxidation of the cluster core, presumably due to the formation of a delocalized mixed-valence pair, giving two Fe2.5+ sites. Similarly, the proximal cluster of DvMF-H2ase is divided into the two subclusters Fe1–S2–Fe4–S3 and Fe2–S1–Fe3–S4 by four short bonds between 2.21 and 2.25 Å in length (Fig. 5 and Table S5†). Comparing the internal bond lengths of the oxidized cluster to those of the reduced cluster, the Fe1–S2–Fe4–S3 subcluster shows a greater difference than the Fe2–S1–Fe3–S4 subcluster, suggesting the larger effect of a one-electron redox reaction at the former subcluster. The Fe1–S2–Fe4–S3 subcluster is coordinated by two cysteine thiolate anions (Cys17S and Cys20S). There is no polar atom to form a hydrogen bond with Sγ (Cys20S) and so the effect of a one-electron redox reaction at the subcluster mainly extends to Sγ (Cys17S). Thus, the amide N (Gly19S)–Sγ (Cys17S) hydrogen bond would be involved in modulating the redox properties of the proximal cluster. Similarly, the reduction potential of rubredoxin is proposed to be correlated with the strength of NH⋯Sγ hydrogen bonds in the secondary coordination sphere.45
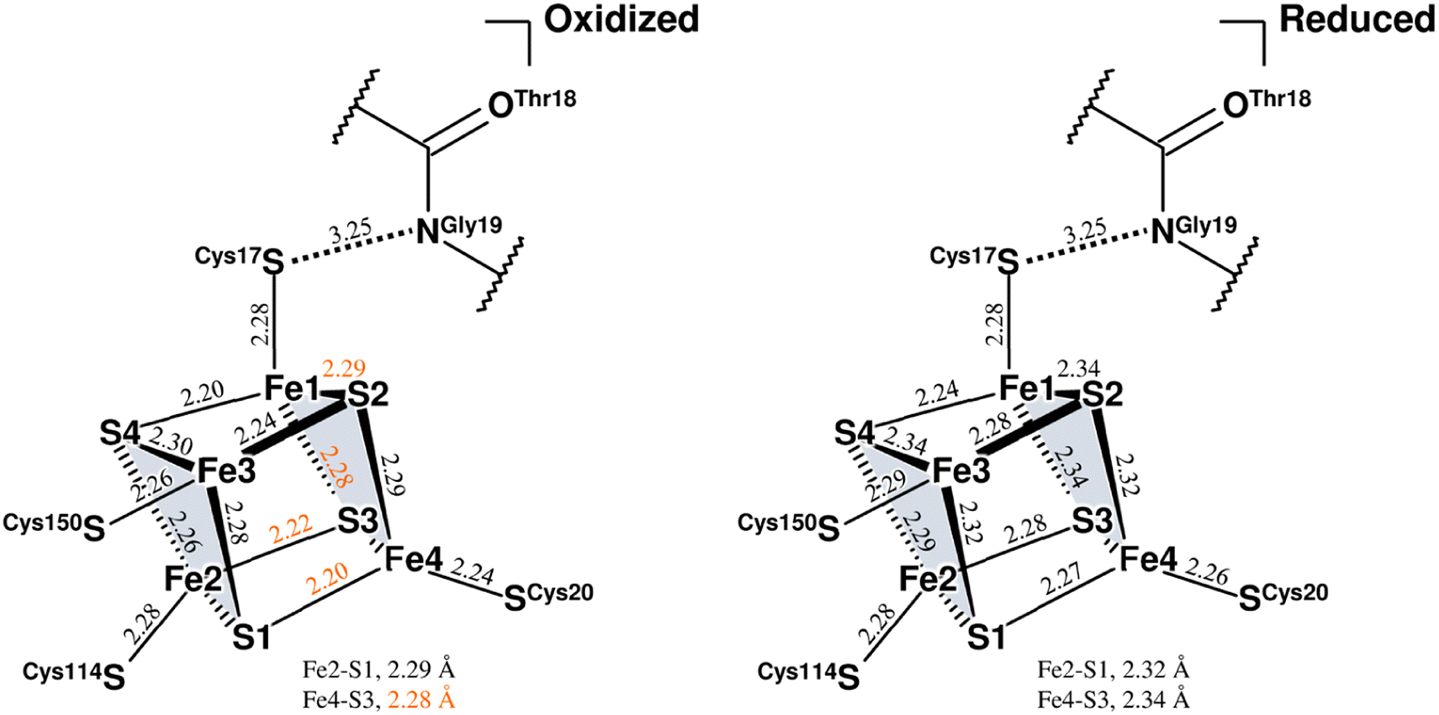 | ||
| Fig. 5 Coordination structures of the proximal cluster under different redox conditions. Structure of the oxidized proximal cluster observed in this study is shown on the left and that of the reduced cluster reported previously (PDB code 4U9H) is shown on the right. The constituent atoms are designated by numbering from Fe1 to Fe4 and from S1 to S4. The subclusters are colored in pale gray. Selected bond lengths are given in Å, where the coordinate uncertainties were estimated to be 0.014–0.016 Å for the oxidized cluster and 0.004 Å for the reduced cluster based on Cruickshank's DPI.56 From a comparison of both structures, bond lengths differing by 0.05 Å or more are indicated by orange numerical values. The interatomic distance between the main-chain amide N (Gly19S) and Sγ (Cys17S) is given in Å. | ||
The Sγ (Cys17S) atom is located on a straight-line connecting Ni of the active site complex and Fe1 of the proximal cluster. Given that a distance of 10.6 Å between the redox centers is sufficient to allow the direct tunneling of electrons,46,47 the amide N (Gly19S)–Sγ (Cys17S) hydrogen bond may also have something to do with the electron transfer.
Conclusions
In summary, we have revealed that the oxidized DvMF-H2ase possesses two types of oxidized products, the square-pyramidal Ni(III) complex in Ni-B and the square-planar Ni(II) complex with a cysteine sulfenate ligand. The Ni-B complex can be produced without O2 using the chemical oxidant ferricyanide.18 But the square-planar complex would be produced through an oxidation process different from that of Ni-B, because the cysteine sulfenylation requires a two-electron oxidant, presumably a reactive oxygen species derived from O2.26,27 As previously reported by Tai et al.,17 all of the inactive oxidized states of the active site can be reactivated under anaerobic conditions in the presence of H2. It would be interesting to see how H2 reacts with the oxidized forms and how the square-planer Ni(II) complex recovers its original active form.The neutron structure of DvMF-H2ase determined here leads to identifying an unusual NH⋯Sγ hydrogen bond in the secondary coordination sphere of the proximal Fe–S cluster.48 It is expected that the delocalized proton on the hydrogen bond could play roles in the modulation of the redox potentials of the cluster core, as suggested by Zheng et al.,45 and in the catalytic electron transfer reaction. Furthermore, it was confirmed that most protons involved in the potential proton transfer pathways containing water molecules were susceptible to H/D exchange. As a future perspective, the neutron structure analysis of DvMF-H2ase reduced in the presence of H2 will enable us to visually identify where protons migrate during catalysis, since the H and D atoms can be strictly distinguished by their negative and positive densities on neutron scattering length density maps.
Materials and methods
DvMF cells were cultured and harvested as described by Yahata et al.49 The cells were disrupted by sonication under aerobic conditions to obtain inactive oxidized enzymes. The membrane-bound enzymes were solubilized from the recovered pellet by digestion with trypsin (Sigma-Aldrich, St. Louis, MO, USA) and subsequently purified as described previously.50 Crystallization mother liquor was prepared in heavy water (99.9% D2O; Cambridge Isotope Laboratories, Cambridge, UK) containing 0.2 mM enzyme, 30% (v/v) 2-methyl-2,4-pentane-d12-diol, 10 mM D-glucose-d12 and 25 mM Tris-d11–DCl (pD 7.4). A tiny seed crystal was immersed into 90 μL of the mother liquor, which was then equilibrated at 283 K for six to eight weeks against a reservoir solution consisting of 40% (v/v) 2-methyl-2,4-pentanediol and 25 mM Tris-d11–DCl (pD 7.4) in D2O. Neutron diffraction from a resulting crystal was measured at 100 K using the IBARAKI Biological Crystal Diffractometer (iBIX) equipped with 30 2D detectors (Tokai, Japan).51 In preparation for joint XN refinement, X-ray diffraction data were collected on beamline BL26B1 at SPring-8 (Harima, Japan) from the same crystal. Anomalous scattering data were also collected over a total rotation range of 360° on beamline BL5A at Photon Factory (PF) (Tsukuba, Japan) using two wavelengths near the absorption K-edge of nickel (1.4879 Å). Processing of the diffraction data and structure refinement were performed as described in ESI Text 1.† The atomic coordinates of DvMF-H2ase in its oxidized state have been deposited in the RCSB Protein Data Bank (PDB) under the accession code 7YW6. All graphic images of molecular structures were generated using PyMOL (PyMOL molecular graphics system, version 1.8. Schrödinger, LLC). All quantum chemical calculations were performed using density functional theory at the B3LYP level with the 6-31g* basis set,52–54 implemented in the Gaussian 16 program package (Rev. C01).55 The computational details are described in ESI Text 1.†Data availability
All data that support the findings of this study are available within this paper and its ESI file,† or from the corresponding authors upon request.Author contributions
T. Hiromoto, K. Nishikawa, T. Tamada and Y. Higuchi designed this study. K. Nishikawa and S. Inoue prepared the protein sample and crystallized it. T. Hiromoto, K. Nishikawa, S. Inoue, H. Ogata and T. Tamada collected X-ray diffraction data and refined the X-ray structures. Y. Hori and Y. Shigeta performed quantum chemical calculations. K. Nishikawa, S. Inoue, K. Kusaka, Y. Hirano, K. Kurihara and T. Tamada collected, reduced and analyzed neutron diffraction data. T. Hiromoto and Y. Hirano performed joint XN refinement. T. Hiromoto, K. Nishikawa, H. Ogata, Y. Hori, Y. Shigeta, T. Tamada and Y. Higuchi wrote the original draft, and all authors edited and reviewed this paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by MEXT KAKENHI Grants-in-Aid for Scientific Research on Innovative Areas (Hydrogenomics and Biometal Sciences) 18H05516 (to Y. Higuchi), JP21H00014 (to Y. Hori), and JP20H05497 (to Y. Shigeta), and for Scientific Research (A) 19H00984 (to Y. Higuchi), (B) 19H03173 (to T. Tamada), (B) JP20H03215 (to H. Ogata), and (C) 16K07283 (to T. Tamada); a research grant from Hyogo Science and Technology Association (to T. Hiromoto); a Kaneko-Narita research grant from Protein Research Foundation (to T. Hiromoto); a JST CREST grant JPMJCR12M4 (to Y. Higuchi); Quantum Leap Flagship Program (Q-LEAP) JPMXS0120330644 (to T. Tamada and Y. Shigeta) from the Ministry of Education, Culture, Sports, Science and Technology. X-ray diffraction experiments were performed at SPring-8 (proposal no. 2014B1995 and 2018A2516) and at PF (proposal no. 2017G033). Neutron diffraction experiments using iBIX of J-PARC were performed under user programs (proposal no. 2014B0312 and 2015A0159). Some of the computations were performed using the computer facilities at the Research Institute for Information Technology, Kyushu University, and the Research Center for Computational Science, Okazaki, Japan. We thank Professor T. Yagi for his useful discussion and thank K. Hataguchi, K. Matsumoto, Y. Ikeda, and Y. Yamada for their technical assistance in this work.Notes and references
- P. M. Vignais and B. Billoud, Chem. Rev., 2007, 107, 4206–4272 CrossRef CAS PubMed.
- K. Nishikawa, H. Ogata and Y. Higuchi, Chem. Lett., 2020, 49, 164–173 CrossRef CAS.
- V. M. Fernandez, E. C. Hatchikian, D. S. Patil and R. Cammack, Biochim. Biophys. Acta, 1986, 883, 145–154 CrossRef CAS.
- A. L. De Lacey, E. C. Hatchikian, A. Volbeda, M. Frey, J. C. Fontecilla-Camps and V. M. Fernandez, J. Am. Chem. Soc., 1997, 119, 7181–7189 CrossRef CAS.
- P. P. Liebgott, F. Leroux, B. Burlat, S. Dementin, C. Baffert, T. Lautier, V. Fourmond, P. Ceccaldi, C. Cavazza, I. Meynial-Salles, P. Soucaille, J. C. Fontecilla-Camps, B. Guigliarelli, P. Bertrand, M. Rousset and C. Léger, Nat. Chem. Biol., 2010, 6, 63–70 CrossRef CAS PubMed.
- H. Tai and S. Hirota, ChemBioChem, 2020, 21, 1573–1581 CrossRef CAS PubMed.
- C. Greening, A. Biswas, C. R. Carere, C. J. Jackson, M. C. Taylor, M. B. Stott, G. M. Cook and S. E. Morales, ISME J., 2016, 10, 761–777 CrossRef CAS PubMed.
- H. Ogata, S. Hirota, A. Nakahara, H. Komori, N. Shibata, T. Kato, K. Kano and Y. Higuchi, Structure, 2005, 13, 1635–1642 CrossRef CAS PubMed.
- N. D. M. Noor, H. Matsuura, K. Nishikawa, H. Tai, S. Hirota, J. Kim, J. Kang, M. Tateno, K. S. Yoon, S. Ogo, S. Kubota, Y. Shomura and Y. Higuchi, Chem. Commun., 2018, 54, 12385–12388 RSC.
- A. L. De Lacey, V. M. Fernandez, M. Rousset and R. Cammack, Chem. Rev., 2007, 107, 4304–4330 CrossRef CAS PubMed.
- W. Lubitz, E. Reijerse and M. van Gastel, Chem. Rev., 2007, 107, 4331–4365 CrossRef CAS PubMed.
- H. Tai, Y. Higuchi and S. Hirota, Dalton Trans., 2018, 47, 4408–4423 RSC.
- J. L. Barilone, H. Ogata, W. Lubitz and M. van Gastel, Phys. Chem. Chem. Phys., 2015, 17, 16204–16212 RSC.
- A. Volbeda, L. Martin, E. Barbier, O. Gutiérrez-Sanz, A. L. De Lacey, P. P. Liebgott, S. Dementin, M. Rousset and J. C. Fontecilla-Camps, J. Biol. Inorg. Chem., 2015, 20, 11–22 CrossRef CAS PubMed.
- K. Nishikawa, S. Mochida, T. Hiromoto, N. Shibata and Y. Higuchi, J. Inorg. Biochem., 2017, 177, 435–437 CrossRef CAS PubMed.
- H. Ogata, K. Nishikawa and W. Lubitz, Nature, 2015, 520, 571–574 CrossRef CAS PubMed.
- H. Tai, L. Xu, S. Inoue, K. Nishikawa, Y. Higuchi and S. Hirota, Phys. Chem. Chem. Phys., 2016, 18, 22025–22030 RSC.
- Y. Shomura, K. S. Yoon, H. Nishihara and Y. Higuchi, Nature, 2011, 479, 253–256 CrossRef CAS PubMed.
- J. Fritsch, P. Scheerer, S. Frielingsdorf, S. Kroschinsky, B. Friedrich, O. Lenz and C. M. Spahn, Nature, 2011, 479, 249–252 CrossRef CAS PubMed.
- A. Volbeda, P. Amara, C. Darnault, J. M. Mouesca, A. Parkin, M. M. Roessler, F. A. Armstrong and J. C. Fontecilla-Camps, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 5305–5310 CrossRef CAS PubMed.
- J. A. Cracknell, A. F. Wait, O. Lenz, B. Friedrich and F. A. Armstrong, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20681–20686 CrossRef PubMed.
- L. Lauterbach and O. Lenz, J. Am. Chem. Soc., 2013, 135, 17897–17905 CrossRef CAS PubMed.
- A. Volbeda, P. Amara, M. Iannello, A. L. De Lacey, C. Cavazza and J. C. Fontecilla-Camps, Chem. Commun., 2013, 49, 7061–7063 RSC.
- M. L. Conte and K. S. Carroll, J. Biol. Chem., 2013, 288, 26480–26488 CrossRef PubMed.
- V. Gupta and K. S. Carroll, Biochim. Biophys. Acta, 2014, 1840, 847–875 CrossRef CAS PubMed.
- R. Breglia, C. Greco, P. Fantucci, L. De Gioia and M. Bruschi, Phys. Chem. Chem. Phys., 2018, 20, 1693–1706 RSC.
- S. Zacarias, A. Temporão, M. D. Barrio, V. Fourmond, C. Léger, P. M. Matias and I. A. Pereira, ACS Catal., 2019, 9, 8509–8519 CrossRef CAS.
- M. Carepo, D. L. Tierney, C. D. Brondino, T. C. Yang, A. Pamplona, J. Telser, I. Moura, J. J. Moura and B. M. Hoffman, J. Am. Chem. Soc., 2002, 124, 281–286 CrossRef CAS PubMed.
- M. Tadokoro, T. Ohhara, Y. Ohhata, T. Suda, Y. Miyasato, T. Yamada, T. Kikuchi, I. Tanaka, K. Kurihara, M. Oguni, K. Nakasuji, O. Yamamuro and R. Kuroki, J. Phys. Chem. B, 2010, 114, 2091–2099 CrossRef CAS PubMed.
- S. Dementin, B. Burlat, A. L. De Lacey, A. Pardo, G. Adryanczyk-Perrier, B. Guigliarelli, V. M. Fernandez and M. Rousset, J. Biol. Chem., 2004, 279, 10508–10513 CrossRef CAS PubMed.
- C. J. Kulka-Peschke, A.-C. Schulz, C. Lorent, Y. Rippers, S. Wahlefeld, J. Preissler, C. Schulz, C. Wiemann, C. C. M. Bernitzky, C. Karafoulidi-Retsou, S. L. D. Wrathall, B. Procacci, H. Matsuura, G. M. Greetham, C. Teutloff, L. Lauterbach, Y. Higuchi, M. Ishii, N. T. Hunt, O. Lenz, I. Zebger and M. Horch, J. Am. Chem. Soc., 2022, 144, 17022–17032 CrossRef CAS PubMed.
- H. Ohno, K. Takeda, S. Niwa, T. Tsujinaka, Y. Hanazono, Y. Hirano and K. Miki, PLoS One, 2017, 12, e0178183 CrossRef PubMed.
- J. Donohue, J. Mol. Biol., 1969, 45, 231–235 CrossRef CAS PubMed.
- M. Teixeira, I. Moura, A. V. Xavier, J. J. Moura, J. LeGall, D. V. DerVartanian, H. D. Peck, Jr. and B. H. Huynh, J. Biol. Chem., 1989, 264, 16435–16450 CrossRef CAS PubMed.
- M. Asso, B. Guigliarelli, T. Yagi and P. Bertrand, Biochim. Biophys. Acta, 1992, 1122, 50–56 CrossRef CAS PubMed.
- W. W. Cleland and M. M. Kreevoy, Science, 1994, 264, 1887–1890 CrossRef CAS PubMed.
- H. Ishikita and K. Saito, J. R. Soc., Interface, 2014, 11, 20130518 CrossRef PubMed.
- Y. Liu, P. Moënne-Loccoz, D. P. Hildebrand, A. Wilks, T. M. Loehr, A. G. Mauk and P. R. Ortiz de Montellano, Biochemistry, 1999, 38, 3733–3743 CrossRef CAS PubMed.
- H. Nakajima, E. Nakagawa, K. Kobayashi, S. I. Tagawa and S. Aono, J. Biol. Chem., 2001, 276, 37895–37899 CrossRef CAS PubMed.
- I. Dance, Chem. Sci., 2015, 6, 1433–1443 RSC.
- Y. Hirano, K. Takeda and K. Miki, Nature, 2016, 534, 281–284 CrossRef CAS PubMed.
- K. Takeda and K. Miki, FEBS J., 2017, 284, 2163–2166 CrossRef CAS PubMed.
- L. Noodleman, in Iron-Sulfur Clusters in Chemistry and Biology, ed. T. Rouault, De Gruyter, Berlin, 2017, vol. 1, pp. 21–75 Search PubMed.
- A. Sato, Y. Hori and Y. Shigeta, Inorg. Chem., 2023, 62, 2040–2048 CrossRef CAS PubMed.
- P. Zheng, S. J. Takayama, A. G. Mauk and H. Li, J. Am. Chem. Soc., 2012, 134, 4124–4131 CrossRef CAS PubMed.
- C. C. Page, C. C. Moser, X. Chen and P. L. Dutton, Nature, 1999, 402, 47–52 CrossRef CAS PubMed.
- M. E. Pandelia, W. Nitschke, P. Infossi, M. T. Giudici-Orticoni, E. Bill and W. Lubitz, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6097–6102 CrossRef CAS PubMed.
- S. T. Stripp, B. R. Duffus, V. Fourmond, C. Léger, S. Leimkühler, S. Hirota, Y. Hu, A. Jasniewski, H. Ogata and M. W. Ribbe, Chem. Rev., 2022, 122, 11900–11973 CrossRef CAS PubMed.
- N. Yahata, T. Saitoh, Y. Takayama, K. Ozawa, H. Ogata, Y. Higuchi and H. Akutsu, Biochemistry, 2006, 45, 1653–1662 CrossRef CAS PubMed.
- Y. Higuchi, N. Yasuoka, M. Kakudo, Y. Katsube, T. Yagi and H. Inokuchi, J. Biol. Chem., 1987, 262, 2823–2825 CrossRef CAS PubMed.
- T. Nakamura, M. Katagiri, T. Hosoya, K. Toh, M. Kitaguchi, M. Hino, T. Ebisawa, K. Sakasai and K. Soyama, IEEE Nucl. Sci. Symp. Med. Imaging Conf. Rec., NSS/MIC, 2012, pp. 222–225 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223–1229 CrossRef CAS.
- M. J. Frisch, et al., in Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, USA, 2016 Search PubMed.
- K. S. D. Kumar, M. Gurusaran, S. N. Satheesh, P. Radha, S. Pavithra, K. P. S. Thulaa Tharshan, J. R. Helliwell and K. Sekar, J. Appl. Crystallogr., 2015, 48, 939–942 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02156d |
| This journal is © The Royal Society of Chemistry 2023 |