
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTCMBank: bridges between the largest herbal medicines, chemical ingredients, target proteins, and associated diseases with intelligence text mining
Qiujie
Lv†
 a,
Guanxing
Chen†
a,
Haohuai
He†
a,
Ziduo
Yang
a,
Lu
Zhao
bc,
Hsin-Yi
Chen
a and
Calvin Yu-Chian
Chen
*adef
a,
Guanxing
Chen†
a,
Haohuai
He†
a,
Ziduo
Yang
a,
Lu
Zhao
bc,
Hsin-Yi
Chen
a and
Calvin Yu-Chian
Chen
*adef
aArtificial Intelligence Medical Research Center, School of Intelligent Systems Engineering, Shenzhen Campus of Sun Yat-sen University, Shenzhen, Guangdong 518107, P. R. China. E-mail: chenyuchian@mail.sysu.edu.cn
bDepartment of Clinical Laboratory, The Sixth Affiliated Hospital, Sun Yat-sen University, Guangzhou, Guangdong 510655, P. R. China
cBiomedical Innovation Center, The Sixth Affiliated Hospital, Sun Yat-sen University, Guangzhou, Guangdong 510655, P. R. China
dDepartment of Medical Research, China Medical University Hospital, Taichung 40447, Taiwan
eDepartment of Bioinformatics and Medical Engineering, Asia University, Taichung 41354, Taiwan
fGuangdong L-Med Medicine Biotechnology Co., Ltd, Meizhou, Guangdong 514699, P. R. China
First published on 8th August 2023
Abstract
Traditional Chinese Medicine (TCM) has long been viewed as a precious source of modern drug discovery. AI-assisted drug discovery (AIDD) has been investigated extensively. However, there are still two challenges in applying AIDD to guide TCM drug discovery: the lack of a large amount of standardized TCM-related information and AIDD is prone to pathological failures in out-of-domain data. We have released TCM Database@Taiwan in 2011, and it has been widely disseminated and used. Now, we developed TCMBank, the largest systematic free TCM database, which is an extension of TCM Database@Taiwan. TCMBank contains 9192 herbs, 61![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 966 ingredients (unduplicated), 15179 targets, 32529 diseases, and their pairwise relationships. By integrating multiple data sources, TCMBank provides 3D structure information of ingredients and provides a standard list and detailed information on herbs, ingredients, targets and diseases. TCMBank has an intelligent document identification module that continuously adds TCM-related information retrieved from the literature in PubChem. In addition, driven by TCMBank big data, we developed an ensemble learning-based drug discovery protocol for identifying potential leads and drug repurposing. We take colorectal cancer and Alzheimer's disease as examples to demonstrate how to accelerate drug discovery by artificial intelligence. Using TCMBank, researchers can view literature-driven relationship mapping between herbs/ingredients and genes/diseases, allowing the understanding of molecular action mechanisms for ingredients and identification of new potentially effective treatments. TCMBank is available at https://TCMBank.CN/.
966 ingredients (unduplicated), 15179 targets, 32529 diseases, and their pairwise relationships. By integrating multiple data sources, TCMBank provides 3D structure information of ingredients and provides a standard list and detailed information on herbs, ingredients, targets and diseases. TCMBank has an intelligent document identification module that continuously adds TCM-related information retrieved from the literature in PubChem. In addition, driven by TCMBank big data, we developed an ensemble learning-based drug discovery protocol for identifying potential leads and drug repurposing. We take colorectal cancer and Alzheimer's disease as examples to demonstrate how to accelerate drug discovery by artificial intelligence. Using TCMBank, researchers can view literature-driven relationship mapping between herbs/ingredients and genes/diseases, allowing the understanding of molecular action mechanisms for ingredients and identification of new potentially effective treatments. TCMBank is available at https://TCMBank.CN/.
1 Introduction
Traditional Chinese medicine (TCM) has enjoyed widespread use throughout Asia for millennia and has shown curative effects in improving people's health and preventing various diseases in long-term practice.1–4 The ingredients of TCM have long provided abundant resources for the discovery and development of modern medicines and it is estimated that about one-third of all medicines are derived from natural herbs.5,6 For example, Tu Youyou from China was awarded the Nobel Prize in Physiology and Medicine in 2015 for the discovery of artemisinin (Qing Hao Su) from Artemisia annua as a treatment for malaria.7 The ephedrine, extracted from Ephedrae herba (Ma Huang), is an anti-asthmatic medication.8 Developing an effective ingredient from TCM into a medication involves two important aspects: determining the active ingredients in herbs and further mastering the therapeutic mechanism of the interaction between the active compounds and protein targets at the molecular level.9 However, the curative effect of most TCM is based on thousands of years of folk practice and is not documented in a uniform standard. Thus, many of their ingredients and targets still remain elusive, which severely hinders the modernization of TCM.TCM modernization has been investigated extensively, especially with the rise of Artificial intelligence (AI) technology.10 AI has played an important role in transforming industries and scientific research.11–13 The popular machine learning algorithms in AI have strong fitting abilities.14 They learn patterns and rules from data and use them to predict new data. With a significant quantity of labeled data, machine learning algorithms can learn hidden patterns and complex relationships in the data and even capture patterns that are difficult for humans to perceive. Machine learning algorithms are applied to quantitative structure–activity relationship (QSAR) modeling, molecular optimization and drug repurposing (DR), and accelerating the process of drug discovery.15–17 Note that AI-assisted drug discovery (AIDD) is heavily influenced by data. The performance of AIDD is heavily reliant on the size of the training dataset, as larger sample sizes generally produce more accurate models.18 In addition, the data utilized for drug discovery are derived from scientific literature, drug patents, or multiple laboratories across the globe, which may contain discrepancies and errors in their data standards. The utilization of such incongruent data inherently presents systemic risks.19
Overall, researchers using machine learning technology to guide rational modern drug discovery still face two challenges. The first challenge is the lack of a large amount of standardized TCM-related information, such as information on active ingredients in herbs, the association between ingredients and target proteins, and so on. In the past few decades, substantial efforts have been dedicated to the isolation of active compounds from herbs and research on their potential targets, resulting in a wealth of novel data on both active compounds and their targets.20 However, the sources of new data in TCM are diversely scattered in books and journals, making it difficult for researchers to collect comprehensive information about ingredients and their targets.21,22 Having a profile of the ingredients and identifying the target and its mechanism of action are basic elements of Chinese medicine research, and incomplete data information may lead to deviations in research results. While it is challenging to organize these TCM data with a unified standard, the creation of a non-commercial, and high-quality TCM database is imminent.
There are several databases that provide valuable resources for research in Chinese medicine and drug discovery, such as HIT,23 TCM Database@Taiwan,24 SymMap V2.0,25 TCMID V2.0,26,27 TCMSP V2.3,28 ETCM (2018 Oct. 26),29 TCM-ID (2021 Oct. 18),30 and HERB (2020 Dec. 02).31 Currently, there are problems with these TCM-related databases: some are difficult to access or have very limited entries, while others lack information on the association between herbs/ingredients and targets/diseases. These problems make it impossible for TCM researchers to carry out comprehensive systematic analysis.
The second challenge is that AIDD is prone to pathological failures in out-of-domain data, and most methods lack wet experimental validation. Single models tend to have certain fragility or its dependency on certain data points.32 AIDD adapts to the training data by minimizing a loss function, but this may cause the model to fail to generalize to new data. This may be related to the activity cliff or the chemical space region of the new molecule is different from the training set. Activity cliffs refer to the sharp changes in compound activity caused by seemingly minor structural modifications.33 The features of some structures may be more representative than others, leading the model to learn the higher weight on these structures. The single model may be too sensitive to certain structures. If the structure of a new molecule is similar to a molecule in the training set, the single model may follow the pattern of the molecule in the training set, leading to a decrease in prediction accuracy. Ensemble learning (EL) constructs multiple predictive learning models through a combination of certain strategies, which helps to obtain better predictive performance.34,35 Furthermore, wet lab experiments generally refer to experiments performed in actual physics laboratories, using real data to evaluate the performance and accuracy of models. Wet lab experiments can verify the correctness of the AI method, which is an essential step.
For the first challenge, Our TCM Database@Taiwan,24 established in 2011, provides a massive amount of information and 3D structure on commonly used herbs/ingredients. In that year, Nature Medicine reported that TCM Database@Taiwan was slightly larger than the Chem-TCM launched by researchers at King's College London in collaboration with the Shanghai Institute of Materia Medica.36 TCM Database@Taiwan has been widely disseminated and heavily cited and has been incorporated into the ZINC database.37
Now, expanding off our TCM Database@Taiwan, we have developed TCMBank (https://TCMBank.CN/), a free and comprehensive Chinese medicine database, which contains standardized information on herbs, ingredients, targets, diseases, and many other resources. TCMBank is a repository containing 9191 herbs, 61966 unduplicated ingredients, 15179 gene targets, 32529 diseases, and useful information on their relationships. TCMBank increased the number of compounds in herbs from 32364 to 61966 (unduplicated) and added two new data fields, targets and diseases. The number of connected herbs and connected ingredients is 9010 and 54676 respectively, and their average number of connections is 16.05 and 5.26. TCMBank also provides 3D structure information of compounds in mol2 format for convenient adoption in virtual screening38 or molecular simulation. Additionally, TCMBank's intelligent document identification module (IDIM) employs selenium39 to regularly download the recent articles from PubChem40 and further uses pdfplumber,41 optical character recognition (OCR),42 automatic summarization and keyword extraction in natural language processing (NLP),43,44 and optical structure recognition (OSRA)45 to extract TCM-related information. This allows constant updates of the database, and information will be verified twice before being integrated into the TCMBank database.
For the second challenge, benefiting from a large amount of high-quality association information of ingredients/components and targets in TCMBank, we attempted to develop components with the activity of inhibiting or activating key components/proteins in pathogenic pathways by using machine learning methods. Here, we proposed an EL-based drug discovery framework for identifying potentially effective lead and drug repurposing, which significantly improves the efficiency of virtual screening by seeking consensus among prediction methods. The EL-based drug discovery framework is composed of 4 primary steps: (1) the molecular docking46 is employed to identify possible interaction patterns between the active component and protein target. (2) The ligand-based EL model learns the mapping between the molecular structure and physicochemical properties and predicts the biological activity of compounds based on this relationship. (3) The hybrid neural network-based (HNN-based) EL model is used to predict the drug-target affinity (DTA), which quantifies the binding strength between the drug and target. (4) The kinetic properties and interactions of the protein–ligand complexes are assessed through molecular dynamics (MD) simulations, in vitro scratch and transwell experiments. In vitro assays allow for the study of living cells under controlled conditions, providing valuable insights into in vivo behavior and lending credibility to AI-based protocols. The advancement of AI technology has elevated the modernization of TCM to a higher level, with its ultimate goal of promoting human health.
Currently, TCMBank is the largest free TCM database with the most systematic and comprehensive data. Fig. 1 highlights that TCMBank has the largest data size compared to other TCM databases. The development of TCMBank is of great significance and will provide new insight into the modernization of TCM. TCMBank offers several advantages:
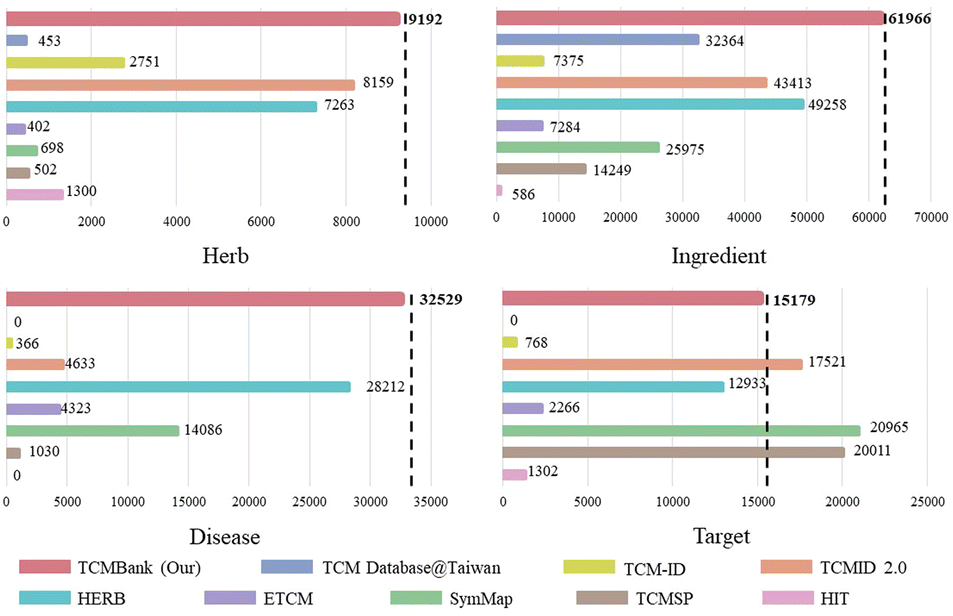 | ||
| Fig. 1 TCMBank stands out as the most comprehensive free downloadable TCM database, surpassing other TCM databases in terms of data size. | ||
(1) TCMBank is currently the largest free database that provides standard information on targets and diseases through intelligent recognition of published references and books.
(2) TCMBank offers the most systematic list, detailed and relational information about herbs/ingredients/targets/diseases and provides jump links to public data sources.
(3) TCMBank uses IDIM to intelligently identify newly published references and continuously provides the latest TCM-related information, which alleviates the lag of data updates in the TCM database.
(4) We proposed an EL-based drug discovery protocol for identifying potential lead and drug repurposing, which has the potential to accelerate drug discovery.
2 Materials and methods
2.1 Comprehensive data collection and processing using AI
TCMBank records TCM-related information, collected from applying text mining methods on books, published papers, and TCM-related databases (TCMID, TCMSP, SymMap, TCM-ID, HERB, and ETCM), to establish a heterogeneous network of six pairs of relationships between TCM herbs and their corresponding diseases, compounds, and relevant targets. Section 4.2 introduces automatic summarization and keyword extraction in the intelligent document recognition module. Together with text mining and manual curation, volunteers obtained lists or basic information related to TCM from books and fill in detailed information and cross-references from public databases. To guarantee the reliability of TCMBank, all information must undergo manual verification at least twice before it is updated in the database. Fig. 2 provides a visual representation of the TCMBank establishment process, including biased LexRank, an intelligent document identification module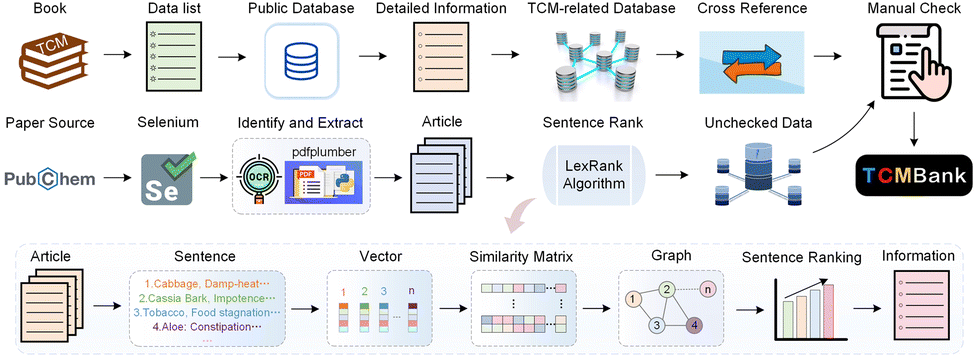 | ||
| Fig. 2 A schematic diagram of the data processing framework and objectives in TCMBank. Part of the concept appears in published ref. 63. | ||
TCMBank characterizes 9191 herbs, 61966 unduplicated ingredients, and their relationships. We use NLP and knowledge graphs to intelligently identify a large amount of herbs/ingredient-related information from published references, public databases, and verifiable Chinese medical books and dictionaries,47–54 such as the Encyclopedia of Traditional Chinese Medicines,47 Shennong Ben Cao Jing Shu,50etc. TCMBank contains general information about the herbs, including name, properties, meridian tropism, function, indication, therapeutic class, etc. It also contains the physical and chemical properties of the ingredients, including the name, SMILES, ADMET, distribution coefficient (logD), AlogP, solubility, volume of compounds, etc. The 3D structure of TCM ingredients was energy minimized in the MM2 force field and constructed using ChemBioOffice 2008 (CambridgeSoft, Cambridge, MA).
TCMBank also records the detailed information of 15179 targets and 32529 diseases, with these entries mainly originating from public databases (e.g. Online Mendelian Inheritance in Man (OMIM, April 2018 Release, https://www.omim.org),55 HUGO Gene Nomenclature Committee (HGNC, https://www.genenames.org/),56 Medical Subject Headings (MeSH, https://www.nlm.nih.gov/mesh/meshhome.html),57 ENsembl (https://asia.ensembl.org/),58 Disease Ontology (DO, https://disease-ontology.org/),59 Human Phenotype Ontology (HPO, March 2018 Release, https://hpo.jax.org/app/),60 Gene–Disease Association Database (DisGeNET v5.0, https://www.disgenet.org/home/),61etc.).
Moreover, TCMBank has also consolidated the TCM-related databases (TCMID, TCMSP, SymMap, TCM-ID, HERB, and ETCM) and combined the herbs/ingredients with the same English/Chinese name or alias to avoid redundancy. We also provide jump links for herbs/ingredients to provide access to pertinent information in external public data sources, such as other TCM databases, disease public databases, DrugBank (https://www.drugbank.ca/),62 CAS (https://sso.cas.org/), and PubChem (https://pubchem.ncbi.nlm.nih.gov/)40 and so on.
The linking between targets/diseases and herbs/ingredients is determined by the overlap between disease-associated targets and the potential protein targets of the herbs/ingredients. With the aid of AI text mining, we linked 15179 targets and 32529 diseases to 9191 herbs and 61966 ingredients in TCMBank. The mapping of the herb/ingredient to the target/disease is done through a final manual examination to establish a highly reliable relationship. In addition, TCMBank also integrates the TCM-related databases (TCMID, TCMSP, SymMap, TCM-ID, HERB, and ETCM) to determine the ingredient-target associations and cross-references to the external database page that contains this information. A thorough inspection was conducted to address any discrepancies in gene or disease ID across different resources.
2.2 Building an intelligent document identification module
The rapid accumulation of new references from laboratory and clinical studies on herbs, ingredients and targets has been observed. Existing TCM databases are no longer updated or updated once in a long time after release. The lag of data updates hinders the development of TCM modernization.Here, we build an intelligent document identification module (IDIM) to continuously add TCM-related information in TCMBank. The purpose of establishing the AI-based IDIM module is to assist volunteers to extract TCM-related information from published literature and improve the efficiency of human inspection. Our IDIM uses AI techniques including selenium,39 pdfplumber,41 optical character recognition (OCR),42 optical structure recognition (OSRA),45 biased LexRank based on feature fusion for automatic summarization, and biased LexRank based on prior graph for keyword extraction in NLP for literature mining and is divided into 5 stages: regular download, PDF parsing, intelligent retrieval, manual checking, and storage (Fig. 2).
First, selenium, a web browser automation tool, is used to regularly download the latest PDF documentation from PubChem. The browser imitates the operation of a real user, and automatically clicks, enters, opens, and validates according to the script code. Then, we use pdfplumber and OCR to parse PDF documents and obtain detailed information about tree structures, text characters, graphs, and tables. OSRA, an open-source tool developed by the National Institutes of Health, is used to identify chemical structures of molecular graphs in the literature and convert them into SMILES or Structural Data (SD) representations.
The format and text of published biomedical literature usually have a relatively fixed structure, which is a semi-structured text. We hope to use AI-based technology to summarize and condense the text, so as to assist humans to recognize TCM-related information quickly and accurately. Next, we preprocess the text content, including removing stop words, removing punctuation marks, unifying it into lowercase letters, words stemming, tagging part of speech of words, calculating term frequency-inverse sentence frequency (TF-ISF) of words, and identifying named entity, etc., and dividing the text content into two basic units: sentences and words. The biased LexRank-based on feature fusion is used to extract summaries, which is introduced in Section 4.2.1. The biased LexRank based on the prior graph is used for keyword extraction, which is introduced in Section 4.2.2. After obtaining abstracts and keywords, volunteers identified herb/ingredient/target/disease-related information and their relationship by combining other information such as images and SMILES.
Finally, the TCM-related information extracted by IDIM requires at least two manual verifications to guarantee the credibility of the TCM resources. The establishment of a public dataset requires highly reliable data information. It is inevitable that the data information in TCMBank ultimately relies on human judgment.
2.3 Biased LexRank based on feature fusion for automatic summarization extraction
For the automatic summary extraction task, the proposed biased LexRank based on the feature fusion model is divided into two stages: (1) we construct a multi-layer perceptron (MLP) classifier to determine whether a sentence is selected as a summary and use this classifier to compute prior probability score of all sentences that are chosen as summaries. (2) To construct the graph, each sentence in the text is represented as a node, and the prior score is taken as the initial score of the node. We score each node using biased LexRank and select high-scoring sentences to generate summaries. The details of the biased LexRank based on the feature fusion framework are shown in Fig. 3.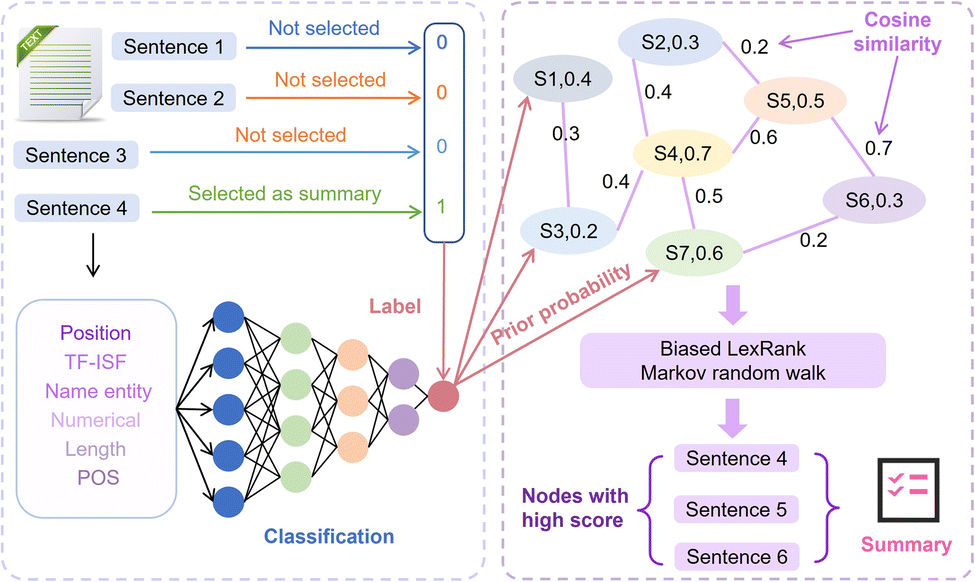 | ||
| Fig. 3 The overall framework of biased LexRank based on feature fusion for automatic summarization extraction. The red dashed box on the left is the first stage of the model. The classifier uses the six feature vectors to compute the prior probability score of all sentences being chosen as summaries. The blue dashed box on the right is the second stage. After the complete document is transformed into a graph, biased LexRank is used to calculate the score of each node through Markov random walks. Finally, a summary is generated using a few high-scoring sentences. | ||
Specifically, in the first phase, we approach the summarization generation task as a binary classification task. Sentences selected as summaries in the text in the public dataset are considered positive samples while the remaining sentences are regarded as negative samples. Then we extracted 6 features in the text and used MLP as a classifier. The 6 features are TF-ISF, named entity, numeric attributes, parts of speech (POS), position and length of sentences. Finally, the multilayer perceptron calculates the prior score of sentences being chosen as summaries in new text, and this score is used as the initial weight of the node in the next stage of the graph.
In the second stage, first we consider sentences as nodes, the correlation between sentences as edges, and the whole article is represented as a graph. The initial scores of the nodes are the prior scores outputted by the classifier in the previous stage. Cosine similarity is commonly employed to measure the relationship between nodes, as shown in eqn (1).
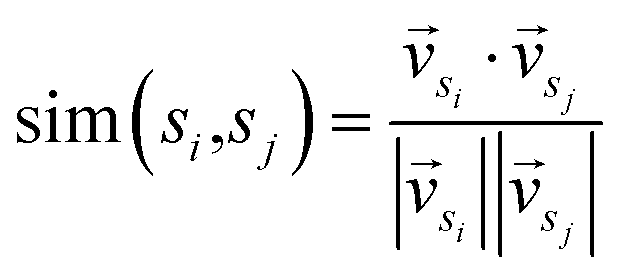 | (1) |
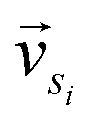 and
and 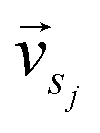 represent the vectors of two sentences, respectively, and | | represents the module of the vector. Then, the weight of each node is calculated by Markov random walk, as shown in eqn (2).
represent the vectors of two sentences, respectively, and | | represents the module of the vector. Then, the weight of each node is calculated by Markov random walk, as shown in eqn (2). | (2) |
| Pt = [dM + (1 − d)B]TPt−1 | (3) |
Compared with classical LexRank,64 the difference of our proposed biased LexRank based on feature fusion is the default weight of nodes. During the selection process of random walk to sentences, it is easier for the model to choose sentences with high scores. The initial score of each node is set as the prior probability obtained from the classifier, which makes the model combines the high-dimensional features of the sentence and more comprehensively evaluates the importance of the sentence selected as the summary.
2.4 Biased LexRank based on prior graphs for keyword extraction
Similar to automatic summary extraction, the proposed biased LexRank based on prior graphs for keyword extraction also introduces prior knowledge. The overall framework of biased LexRank based on the prior graph for keywords extraction is shown in Fig. 4. The original LexRank network initializes all nodes with the same weight, without considering the original difference in the importance of keyword nodes. The proposed biased LexRank based on the prior graph consists of two steps. First, public dictionary data are used to construct a word graph network with prior knowledge. In the second stage, we integrate the prior information into the biased LexRank network and introduce the node rank value equation to iteratively calculate the weight scores of keyword nodes. The top k nodes with higher weight values are selected as keywords.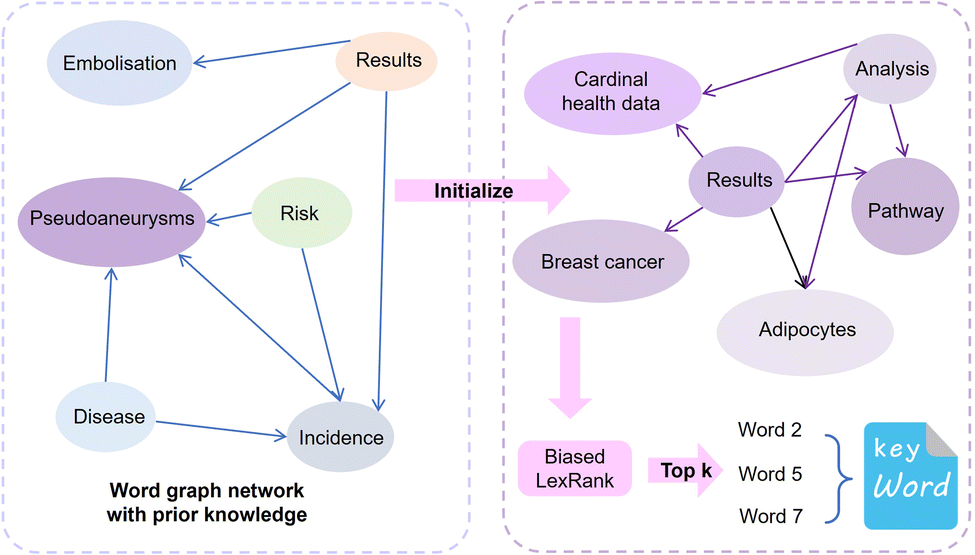 | ||
| Fig. 4 The overall framework of biased LexRank based on prior graphs for keyword extraction. The red dashed box on the left (first stage) indicates that public dictionary data are used to construct a word graph network with prior knowledge. The blue dashed box on the right (second stage) indicates that the prior information is used as the initialization value of the selected keyword importance, and the node ranking value is updated iteratively. The top k node words with higher node values are selected as keywords. | ||
In the first stage, the Schutz 2008 (ref. 43) and PubMed65 public dataset was used as the source of public dictionary data. The entries in Schutz 2008 and PubMed are respectively selected from 1231 and 500 papers in PubMed Central, which are distributed in 254 journals and are authoritative. We perform preprocessing such as word segmentation and stop word removal on public dictionaries. The word is used as the node of the graph network, and the relationships between words are represented as the edges to construct the network. The weight of the edge is the degree of co-occurrence between two words in the sliding window. The final importance weight of the node is calculated iteratively through eqn (4).
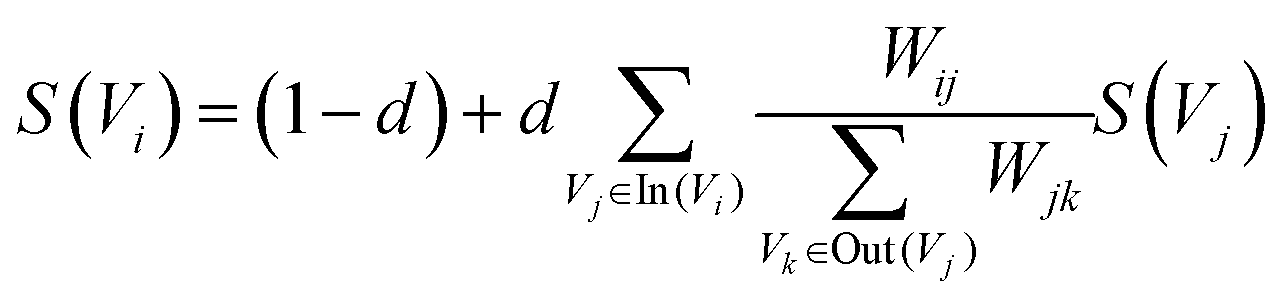 | (4) |
In the second stage, the importance of candidate words is initialized with the prior information of the word graph network constructed from public datasets. Usually, the definition of the word i includes the word j, and the word j may be a relatively basic word. The base word is less likely to be a keyword. Based on this phenomenon, for each node word, we introduce a possible degree of whether it is a base word using eqn (5).
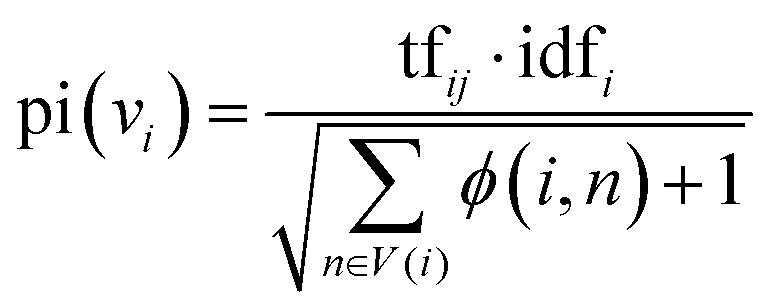 | (5) |
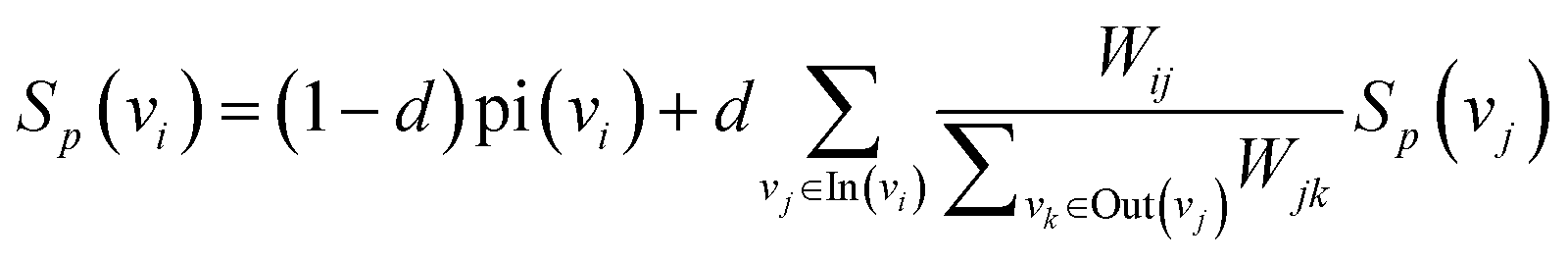 | (6) |
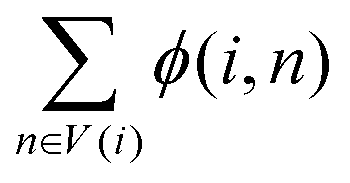 represents the weight sum of n nodes. The equation shows that the more times word i explains other words, the less important the candidate word i is.
represents the weight sum of n nodes. The equation shows that the more times word i explains other words, the less important the candidate word i is.
We iteratively update the node rank value through eqn (6) and select the top k node words with higher ranking as keywords. There are many professional terms in biomedical texts and graph-based biased LexRank can effectively enhance the weight of professional terms by utilizing prior knowledge.
2.5 EL-based drug discovery protocol
The ligand-based EL model is an integrated regression estimator whose architecture is shown in Fig. 5, including feature dimensionality reduction, basic regression model and voting averaging algorithm. The SMILES strings of inhibitor molecules are used as input for the EL model. The negative logarithm of their half-maximal inhibitory concentration (IC50), pIC50, was used as the regression label. IC50 is the amount of a substance required to inhibit a particular biological function or compound by 50% in vitro. First, the SMILES of the drug molecule is transformed into a 3D structure and energy minimized by the CHARMm. We calculated 204 genetic function approximation (GFA) features of inhibitors using DS software and further applied feature dimensionality reduction for feature selection. Features with missing values above 60%, correlation below 98%, and cumulative importance below 99% were discarded.
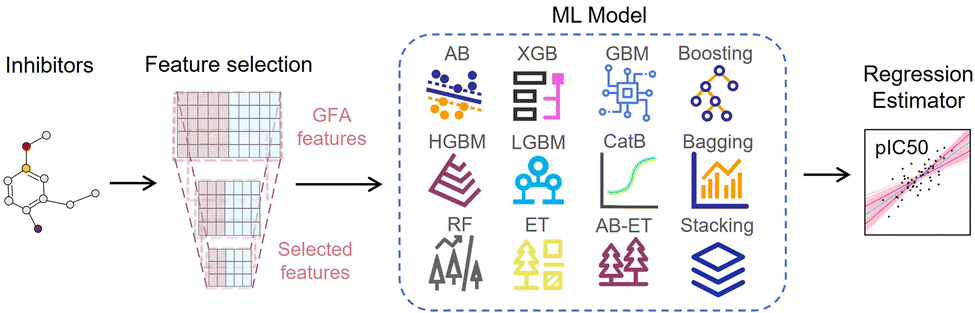 | ||
| Fig. 5 The overall framework of the ligand-based EL model for molecular activity prediction, including extraction and selection of GFA features, EL model composed of 12 regression estimators, and the voting average algorithm. | ||
Then, these features are fed into an integrated regression estimator, including boosting, bagging, and stacking algorithms. Boosting is an EL model mainly used to reduce bias and variance, which can convert a series of weak learners into strong learners. It has 6 classic variant regression algorithms: adaptive boosting (AB), extreme gradient boosting (XGB), gradient boosting machine (GBM), categorical boosting (CatB), histogram-based gradient boosting (HGBM), and light gradient boosting machine (LGBM). Bagging is also an ensemble meta-algorithm applied to decision tree methods, which reduces variance and helps to avoid overfitting, and is used to improve the stability and reliability of the model. The random forests (RF), extra trees (ET), and AdaBoosted extra trees (AB-ET) are extensions of bagging and are used as regression learners to construct ligand-based EL models. Stacking is also an EL algorithm, which takes the prediction results of multiple basic algorithms as input for comprehensive prediction. All 12 regression algorithms use the same training and test sets. The ligand-based EL model is an ensemble regression estimator that performs vote-average integration of the results produced by multiple basic models and obtains the predicted pIC50, the calculation formula is as follows:
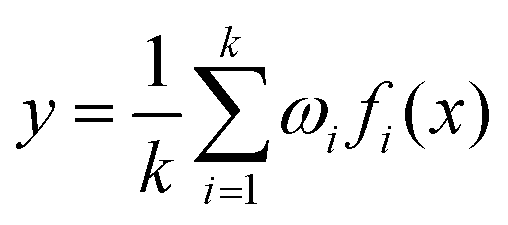 | (7) |
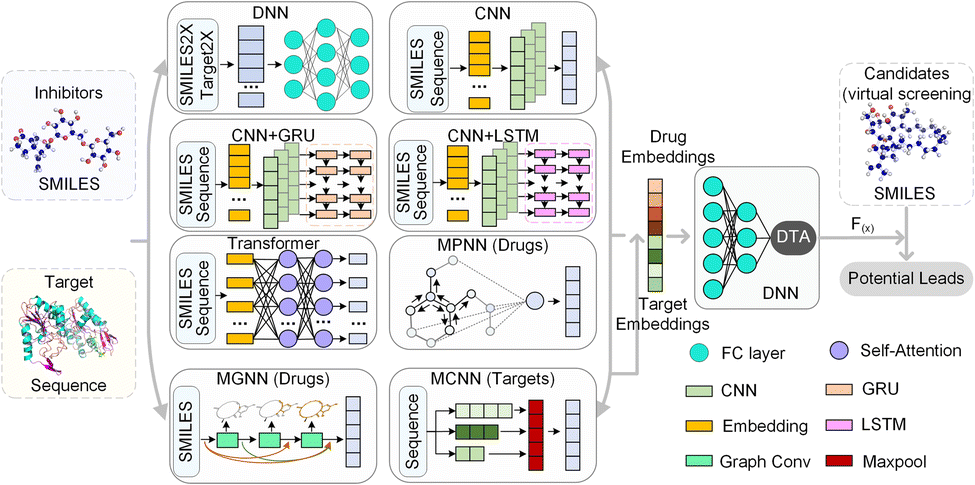 | ||
| Fig. 6 The framework of the HNN-based EL model for DTA prediction. SMILES2X and Target2X represent 4 different descriptors of drugs and targets respectively. The HNN-based EL model integrates multiple basic models to effectively extract embedding features of drugs and targets and predict their DTA. The trained HNN is used to further screen and identify drug candidates for potential leads to activate or inhibit targets. | ||
HNN-based EL model mainly consists of multiple basic models and a comprehensive prediction module. For drug molecules, there are 10 basic models to extract embedding vectors, where the DNN module contains 4 types of molecular descriptors, and 6 basic models including CNN, CNN-GRU, CNN-LSTM, transformer, MPNN, and our MGNN. For the target protein, the HNN-based EL model has 9 basic models, where the DNN module also contains 4 different lengths vector of amino acid composition, and 5 basic models including CNN, CNN-GRU, CNN-LSTM, transformer, and our MCNN.
The DNN module preprocesses the SMILE string of the drug and the amino acid sequence of the target, converting them into feature maps through chemical descriptors. The 4 drug molecular descriptors include 1024-bit extended connection fingerprints derived from the Morgan algorithm, 881-bit substructure-based PubChem fingerprints, 2048-bit daylight fingerprints, and 200-bit RDKit-2D descriptors. For target proteins, the DNN module contains 4 descriptor mapping modules, including a 100-bit quasi-sequence-order descriptor, a 343-bit conjoint triad descriptor, a 30-bit pseudo amino acid composition, and an 8420-bit amino acid composition up to 3-mers. These descriptor features are then entered into DNN to automatically extract the embedding vector.
The CNN module is a deep fully convolutional network, mainly composed of embedding layers, convolutional layers and max pooling layers. The CNN directly accepts the SMILES and the amino acid sequence as input and maps the embedded features through the embedding layer. These embedded features are continuously encoded by deep convolutional layers, and the final convolution layer outputs the final feature vector. CNN-GRU and CNN-LSTM are basic models connected by CNN with GRU and LSTM respectively. Similar to the CNN module, the SMILES and the amino acid sequence are first embedded and encoded by CNN. Then, the generated feature vectors are used as the input of GRU and LSTM respectively. The output of GRU or LSTM is used as the embedding vector of CNN-GRU and CNN-LSTM modules, respectively, and is waiting to be sent to the decoder.
In the transformer module, the module used to encode the drug has an 8-layer network with 8 attention heads, and the feature extraction module of the target is a 2-layer network with 4 attention heads. The SMILES representation and amino acid sequence are directly taken as input and fed into the embedding layer. Following the embedding layer, an encoding layer with a self-attention mechanism is employed to iteratively process the input layer by layer. The encoding layer weights the correlations between embedding vectors to generate output encodings. Each encoding layer passes its encoding as input to the next encoding layer, and the final encoding layer outputs the final encoded feature vector.
MPNN module is a general computing framework for graph neural networks, including two stages, message passing and readout. In the message passing stage, MPNN generates information according to the chemical information of atomic nodes and edges and transmits the information according to the topology structure of the network. Atomic node features include symbol, degree, hybridization, chirality type, etc., and bond features include type, conjugation, ring, etc. Then, the node-level representations are aggregated by a readout function to obtain the embedding vector of the drug molecule. Note that MPNN is used to encode only drug molecules and does not process the target protein.
Further, we have introduced a novel multi-scale graph neural network (MGNN) with 27 graph convolutional layers, arranged in a dense connection fashion is used to learn the overall structure of the compound, while preserving the local structure to learn better representations of compounds,68 as shown at the top of Fig. 7. MGNN contains 3 multi-scale blocks, each of which has a transition layer behind it. In the multi-scale block, each layer is connected to every other layer by a dense connection, allowing all layers to update parameters directly according to the gradient calculated by the loss function. Two adjacent multi-scale blocks are connected by a transition layer to reduce the computational cost by reducing the channel numbers to half of the input. Finally, a readout layer is utilized to encapsulate the whole molecule into a single map vector to represent the drug. Similarly, a multi-scale convolutional neural network (MCNN) is proposed to learn the multi-scale characteristics of a protein, as shown at the bottom of Fig. 7. Specifically, there are three branches with different receptive fields in MCNN to recognize protein residues in local environments of different sizes. They expand the receptive field by stacking multiple 3 × 3 convolutional layer, and each additional convolutional layer expands the receptive field by 2. Note that achieving complete coverage of the entire protein sequence with an expanded receptive field is not essential, as only specific regions of a protein (near to binding pocket) have contributions to the binding.
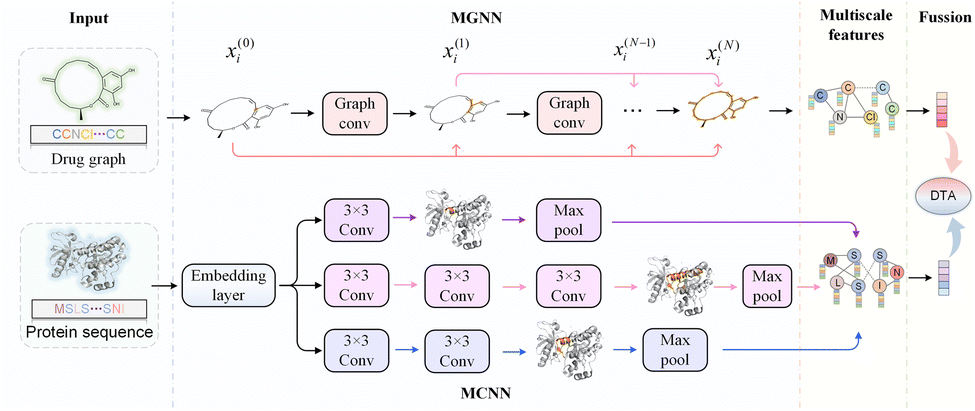 | ||
| Fig. 7 The framework of MGNN and MCNN for drug–target affinity prediction. Deep MGNN can capture both the local and global structure of the compounds. MCNN is proposed to extract the multi-scale features of a protein. | ||
Finally, we concatenated the drug and target embedding vectors output by the above basic models separately and feed them into a decoder composed of fully connected layers to achieve DTA prediction.
3 Results and discussion
3.1 Web interface
TCMBank offers a user-friendly website that allows users to easily access detailed information and relationships between herbs, ingredients, gene targets, and associated diseases. TCMBank's homepage is designed for easy navigation with an introduction, a news section, and a navigation bar with different search categories. Using the search box, the user can select the search category: herbs, ingredients, targets, diseases, or all. Keyword search can be performed in Chinese, Pinyin, English, or Alias name, and a prompt will appear with the search subject that can direct users to the details page when clicked on.Users can go to the Herbs page by searching, browsing, or clicking a hyperlink to an item in the navigation bar. The detailed information on the Herbs page includes the statistical pie chart and detailed table of herbs (Fig. 8A). The statistical pie chart includes classification statistics for herb types, herb properties, and herb meridian tropism. Users can move the mouse cursor to the leaves of the pie chart to know the proportion of the part. Each sector of the pie chart represents a different classification, and the user can filter the full list to get a sublist corresponding to that sector's classification. After clicking, the table below is updated with the information for that category. For example, when the mouse cursor is clicked on the cold fan blade in the herb property, herbs with cold properties such as “Prepared Tortoise plastron” appear in the table below. Additionally, searches for a herb or category of herbs can be performed in Chinese, Pinyin, English, or the Alias name. By clicking on a herb name (blue text) on the list page, users can jump to the view of the herb's information in the corresponding details page. Fig. 8B shows an example of the detailed information page of the herb pomegranate fruit (SHI LIU). The detail page displays detailed information, external links, relationship networks, and lists of associations with the other three categories.
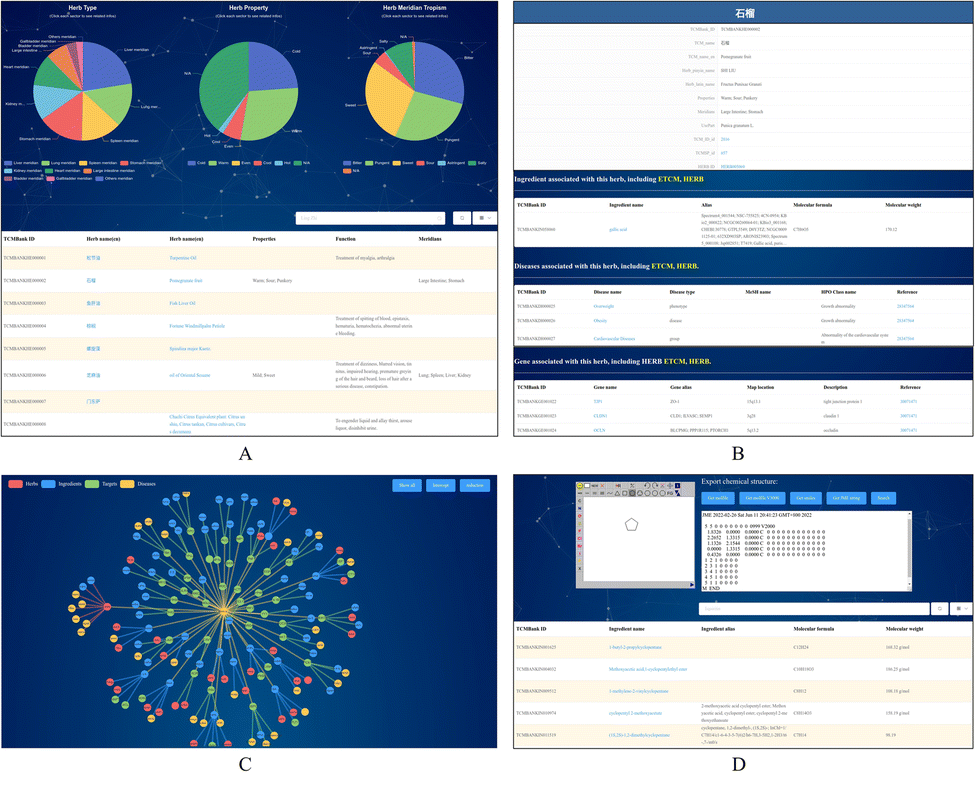 | ||
| Fig. 8 TCMBank provides a user-friendly web page. (A) Pie chart by category and a full list of herbs in TCMBank. Click any sector in the pie chart to filter the list of eligible herbal medicines. (B) The detailed and TCM-related lists information on herb pomegranate fruit (SHI LIU). (C) Examples of herb-ingredientdisease networks constructed by the systematic analysis function of TCMBank. (D) Example of structural matching and structural similarity search on ingredient page. | ||
We developed a network-based tool (Fig. 8C) that presents the intricate relationships between herbs, ingredients, their potential targets, and diseases. This tool offers a user-friendly way to explore these relationships and identify potential targets, thus facilitating the inference of their therapeutic mechanisms. Hovering over a node, users can access details about the node and highlights other nodes connected to it. Users can also expand details by clicking on a node to observe the relationship network diagram centered on this node and restore the initial minimized map by clicking the reduction button. The presence of interactions between ingredients in herbs and disease-related targets suggests potential mechanisms for treating the disease with the ingredient. Moreover, if a herb ingredient shares a target gene with a known drug, it may suggest a potential mechanism of the ingredient for treating the disease corresponding to the target.
To facilitate the study of the action mechanism of ingredients and targets, users can explore and screen ingredients based on substructures or structural similarity. TCMBank also provides structural matching and structural similarity searches on the ingredients page (Fig. 8D). JSME is a free molecule editor written in JavaScript.71 JSME editor provides keyboard shortcut menus for commonly used functions and uses a friendly view to create or edit molecular structures. The JSME editor allows users to export molecular structures in multiple formats such as simplified molecular input line entry system (SMILES), molfile files, and text representations. The editor can be utilized as an input tool for querying TCMBank databases. The applet can also search the structure by entering SMILES in the text box on the right to obtain the list of ingredients containing this structure. Users can refer to the help page if they have trouble navigating through the database, and once they find the information needed, they can customize and download the required data through the download page.
3.2 Performance comparison of biased LexRank based on feature fusion
In this section, we present a performance comparison of biased LexRank based on feature fusion with other baselines for automatic summarization extraction. The datasets used are DUC2001 (ref. 72) and DUC2002 (ref. 73) corpora. DUC2001 contains 30 clusters, 309 documents, and 11026 sentences. DUC2002 contains 59 clusters, 576 documents, and 14370 sentences. The resulting summaries are evaluated by the ROUGE metric based on n-gram co-occurrence. ROUGE is a recall-based metric for fixed-length summaries, commonly used are ROUGE-1, ROUGE-2, and ROUGE-SU4. We calculate ROUGE-N using eqn (8).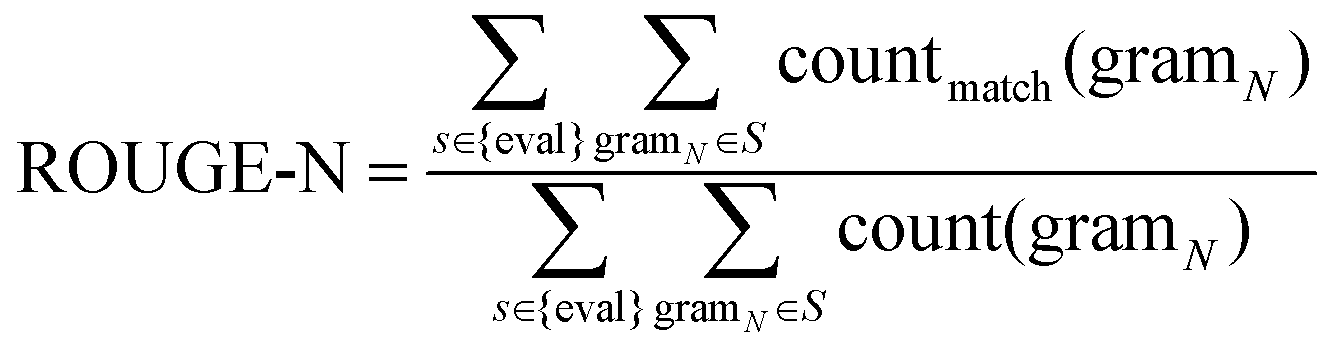 | (8) |
The comparative performance of the proposed method and the baselines on DUC2001 and DUC2002 is depicted in Fig. 9. Lead baseline directly extracting the initial N words of the document as a summary, where N equals 100. LSA is a model based on latent semantic analysis.74 The biased LexRank model generates summaries of better quality than LexRank or any of the other baselines. The biased LexRank obtains prior knowledge from sentence features and adds it to the graph initialization weights. This modeling method can obtain better summaries than before. It can be inferred that enhancing the initialization weights of sentence nodes in the document graph can improve the quality of the summarization results generated by the model.
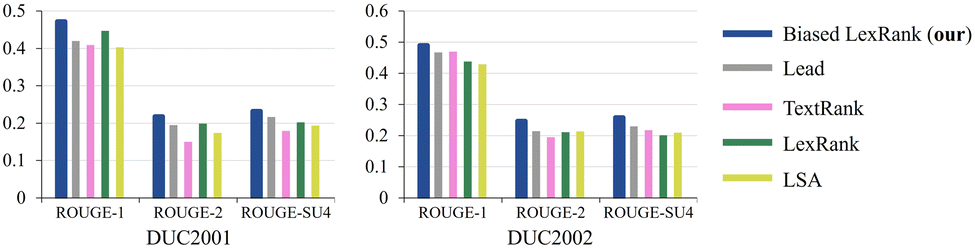 | ||
| Fig. 9 The performance of the proposed method is compared with the baseline in terms of multiple metrics on DUC2001 and DUC2002. | ||
After obtaining an excellent automatic summary generation model, biased LexRank, we applied it to the IDIM of TCMBank. Biased LexRank based on feature fusion can summarize and condense published literature, assist volunteers to quickly extract TCM-related information, and improve work efficiency.
3.3 Performance comparison of biased LexRank based on prior graphs
For the keyword extraction task, we perform performance evaluation on two datasets, SemEval2010 (ref. 75) and SemEval2017.76 SemEval2010 is a widely used dataset for evaluating keyword extraction, which includes 244 complete scientific papers obtained from the ACM Digital Library. SemEval2017 consists of 500 research articles extracted from ScienceDirect journals, with even distribution across the fields of computer science, materials science, and physics. In the evaluation setting, two metrics are used: Mean Precision at Top 5 (P @ 5) and Mean Reciprocal Rank (MRR).Fig. 10 illustrates the result comparison of the proposed biased LexRank based on the prior graph and baselines for keyword extraction on SemEval2010 and SemEval2017. The proposed biased LexRank achieves the best performance among all the models evaluated on both datasets. The proposed biased LexRank (P @ 5 = 0.228) is slightly lower than PositionRank (P @ 5 = 0.232) on the P @ 5 metrics of SemEval2010. Since there are many professional terms in the published literature in the test set, biased LexRank uses the prior knowledge of the word graph constructed from the scientific literature in the public data set, which can effectively increase the weight of professional terms. Compared with the LexRank method, the performance of biased LexRank improves by 0.06, 0.139 on SemEval2010, and 0.014, 0.018 on SemEval2017. This reflects the effectiveness of introducing prior knowledge from word graphs constructed from public datasets.
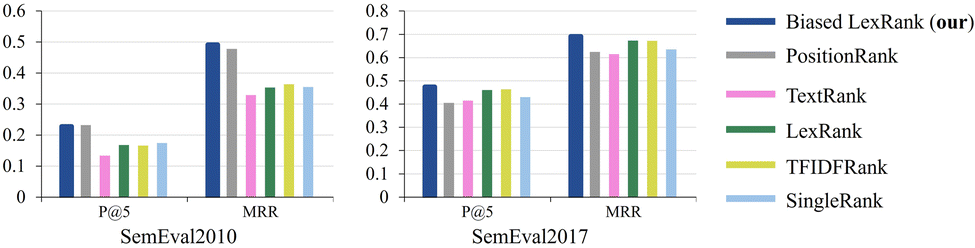 | ||
| Fig. 10 The result of the proposed biased LexRank is compared with the baseline using various metrics on SemEval2010 and SemEval2017. | ||
3.4 TCMBank's big data-driven AI accelerates drug discovery
Biophysiologists identify the gene targets involved in targeting specific diseases by unraveling the pathogenesis. The workflow of modern drug development is based on gene targets. In order to find potential leads of a certain target to develop drugs for treating corresponding diseases, we designed the EL-based drug discovery framework by integrating molecular docking, machine learning, ensemble learning, molecular dynamics simulations and wet experiments. In this section, we present the workflow of the EL-based drug discovery framework (Fig. 11) and demonstrate its effectiveness using colorectal cancer and Alzheimer's disease as cases.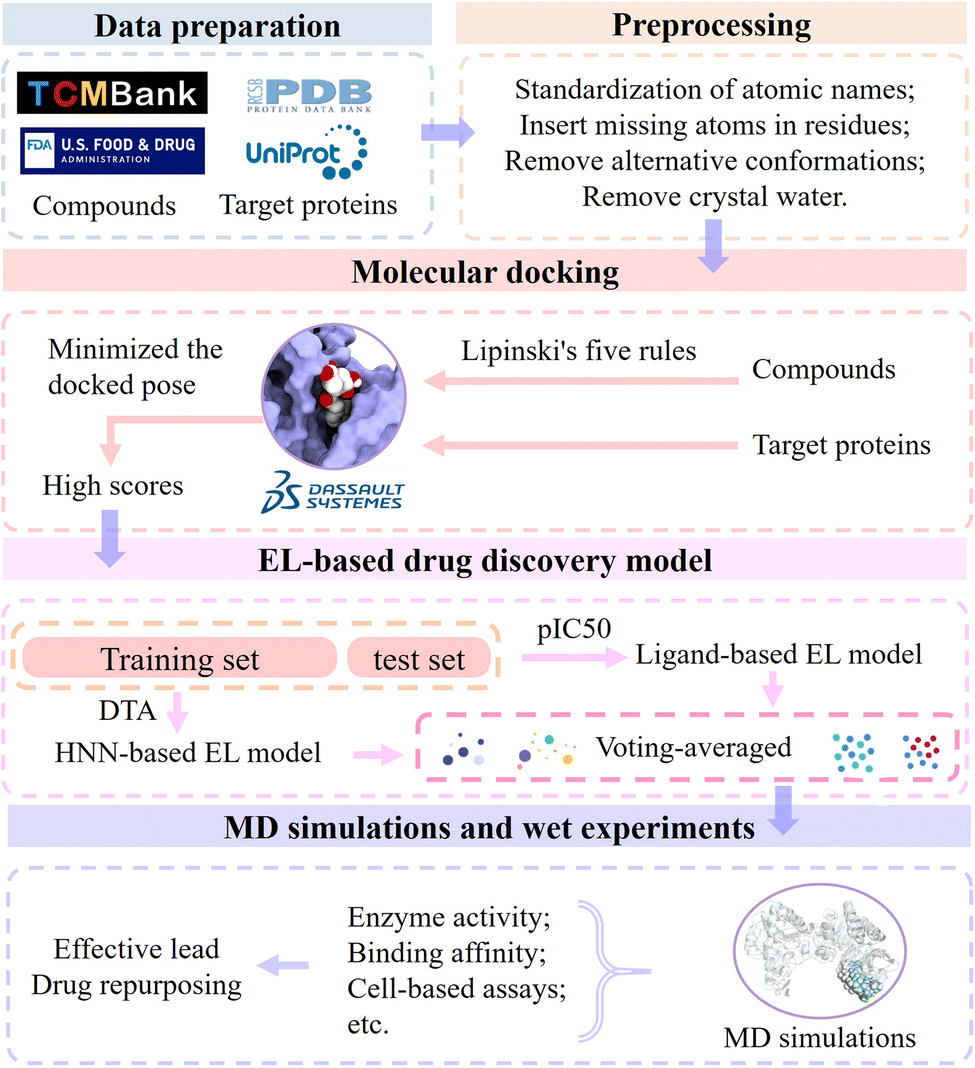 | ||
| Fig. 11 Flowchart of the EL-based drug discovery framework, including molecular docking, ligand-based EL models for pIC50 prediction, HNN-based EL models for DTA prediction, molecular dynamics simulations, and wet experiments. | ||
First, the sequence and crystal structure of the target protein were obtained from UniProt Knowledgebase and Protein Data Bank, respectively. We used molecular collections of 61966 ingredient compounds in TCMBank, FDA-approved compounds, and non-FDA compounds in the ZINC database for drug development and drug repurposing. Discovery Studio 2017 R2 client was used for initial screening, and its molecular docking program evaluates the interaction between the compound and the target to identify potential drug candidates. All molecules require preprocessing prior to docking, including standardization of atomic names, insertion of missing atoms in residues, and removal of alternative conformations and crystal water. Next, the molecular collection was filtered by Lipinski's five rules, and selected compounds were prepared for docking simulations. We docked the calculated ligand conformations using LibDock and minimized the docked pose using CHARMm. A batch of candidate molecules with the highest scores were selected for further study.
Then, the molecular set is randomly partitioned into a training set and a test set in a ratio of 8:2. We use two trained models, the ligand-based EL model and the HNN-based EL model, to predict pIC50 and DTA, respectively. The evaluation of candidate molecules was conducted by employing voting scoring rules that combined molecular docking scores, predicted pIC50, and predicted DTA. The maximum score is 10, and the rest are scored proportionally. Note that we generally select a ligand near the key site of the target as the control group during the virtual screening process. Candidate molecules with higher predicted pIC50 scores exhibit greater inhibitory activity against the target. Higher docking and DTA scores indicate stronger interactions between candidate molecules and target proteins. Candidate molecules with the highest final scores were selected as potential leads pending further in vitro validation.
Finally, we validate the stability of the binding between candidate molecules and targets through MD simulations and in vitro experiments. MD simulations simulate the pose and structure of protein and ligand interactions and observe the generation, number and length of hydrogen bonds in the complex. The greater the number of these hydrogen bonds, the closer their distance, and only fluctuate within a small range, which means that the protein and the ligand have a strong interaction force and a tighter combination.
Further, we verify the accuracy and reliability of the EL model using wet experiments and provide support for further research and applications. The lead compounds interact with the target proteins in vitro, and the interaction between compounds and proteins is evaluated by measuring the results of the reaction (such as enzyme activity, binding affinity, etc.). Cell-based assays are employed to assess the interaction and functionality of drugs with intracellular targets. By observing the effects of drugs on cells, such as cell survival rate, modulation of signal transduction pathways, and other cellular responses, the reliability and accuracy of the prediction results generated by EL models for drug development can be verified.
Colorectal cancer (CRC), a disease in which cells in the colon or rectum grow out of control, is the third most common type of cancer worldwide. Blocking T255 glycosylation on PGK1 decreases colon cancer cell proliferation, suppresses glycolysis, and inhibits tumor growth in xenograft models.77 In order to find potential lead compounds targeting PGK1 to develop drugs for the treatment of CRC, Chen et al. selected flavin adenine dinucleotide (FAD) as a potential lead by applying an EL-based drug discovery framework.19 The docking pose of FAD and PGK1 is shown in Fig. 12A. The results obtained from both wound-healing and transwell wet experiments demonstrated that FAD had a significant inhibitory effect on the migration and invasion of HCT116 cells.
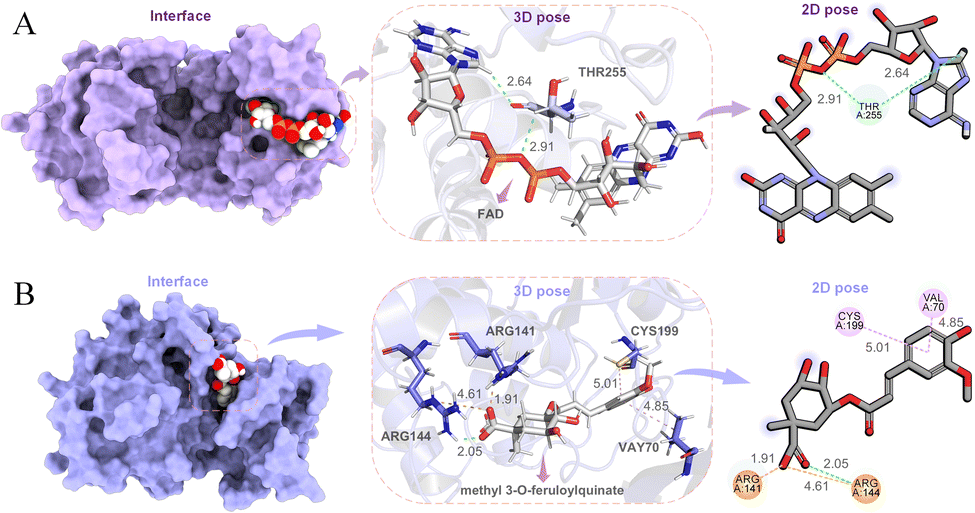 | ||
| Fig. 12 Binding of potential lead compounds to targets. (A) Docking interface, 3D pose, and 2D pose of the PGK1-FAD complex. (B) Interface, 3D pose, and 2D pose for methyl 3-O-feruloylquinate docking with GSK3β. | ||
Alzheimer's disease (AD) is an irreversible impairment of brain function with slow onset and gradual deterioration over time. The accumulation of Aβ peptides formed by continuous cleavage of β-amyloid precursor protein (APP) by β-site amyloid precursor protein cleaving enzyme 1 (BACE1) induce dementia syndrome in patients.78,79 Inhibition of abnormally high phosphorylation activity of glycogen synthase kinase 3β (GSK3β) may prevent an increase in BACE1 production and Aβ generation.80,81 In order to find potential therapeutic drugs for Alzheimer's disease, Chen et al. in our group used support vector machine (SVM), random forests (RF), multiple linear regression, and deep learning methods to propose an ensemble learning for predicting molecular biological activity.17 They first conducted pathway network analysis through the protein–protein interactions to identify an effective target for Alzheimer's disease, and then chose to mine natural ingredients with effective inhibitory functions on potential targets from known herbal medicines. By predicting the biological activities of the compounds in the TCMBank database, they identified that both methyl 3-O-feruloylquinate and cynanogenin A could interact with GSK3β (Fig. 12B). The authors also performed molecular docking and molecular dynamics simulations on them to verify their stability. Methyl 3-O-feruloylquinate is the active ingredient in Phellodendron amurense and Stemona japonica, while cyanoflavin A is the ingredient in Cynanchum atratum. Therefore, it is proposed that the potential lead compounds methyl 3-O-feruloylquinate and cynanogenin A be further developed and applied in the treatment of Alzheimer's disease.
The application of AI in TCM research has facilitated the discovery of new drugs, identified potential drug targets, and revealed novel active ingredients. EL-based drug discovery protocol can be used to identify potential effective clues for drug repurposing, and can also screen components of TCM databases for different diseases according to specific conditions. There are several studies that have used AI to screen potential TCM compounds and investigate their therapeutic effects on various diseases.
Babar et al.82 used MD simulations to virtually screen potential TCM for the treatment of COVID-19, ultimately selecting P1, P5-Di and other five compounds. And Lu et al.83 used CNN and support vector machine (SVM) to screen TCM for the treatment of Alzheimer's disease. Gong et al.84 utilized multiple machine learning methods to screen TCM for the treatment of diabetes, ultimately selecting Hypecoum leptocarpum. Yang et al.85 utilized naïve Bayesian (NB) models and molecular docking to screen FDA-approved drugs for the treatment of metabolic diseases targeting the A-FABP target. He et al.86 used machine learning and graph neural network methods to screen TCM for the treatment of multiple vascular tumors, ultimately selecting Mulberry leaf and Ganoderma lucidum. Wang et al.87 applied CNN, network pharmacology, and MD simulations to obtain multiple-target drugs caffeoyl malic acid for atopic dermatitis. Zhang et al.88 constructed a QSAR model to screen TCM for anti-atopic dermatitis drugs using NB and recursive partitioning (RP) methods. Zhu et al.89 used molecular docking and random forest methods to build a QSAR model to screen 30 potential inhibitors of TLR4 for the treatment of Mycoplasma pneumoniae. Zhang et al.90 used deep learning and MD simulation to screen potential inhibitors for cancer and inflammation, ultimately selecting UM-164 and three other compounds as promising drug candidates.
With the release of TCMBank and the continued development of AI, it is expected that more efficient and effective TCM research will be conducted in the future, leading to the discovery of new drugs for various diseases.
3.5 Case study of IDIM
We take literature ref. 91 and 92 as examples to illustrate the performance of two tasks, automatic summarization and keyword extraction. For the automatic summarization task, because the whole article is long, only the extraction results of key sentences in the summary section are posted here. For the keyword extraction task, we extracted 10 keywords in order.For ref. 91, the extracted key sentences include: “Urolithin-A (mostly present in UM-A) was positively correlated with apolipoprotein A-I (P ≤ 0.05) and intermediate-HDL-cholesterol (P ≤ 0.05) while urolithin-B and isourolithin-A (characteristic from UM-B) were positively correlated with total-cholesterol, LDL-cholesterol (P ≤ 0.001), apolipoprotein B (P ≤ 0.01), VLDL-cholesterol, IDL-cholesterol, oxidized-LDL and apolipoprotein B:apolipoprotein A-I ratio (P ≤ 0.05)”, “Urolithins are microbial metabolites produced after consumption of ellagitannin-containing foods such as pomegranates and walnuts”, “Overweight-obese individuals with UM-B are at increased risk of cardiometabolic disease, whereas urolithin-A production could protect against CMR factors”. The keywords extracted from ref. 91 include: “cholesterol”, “cardiometabolic risk factors”, “urolithin metabotypes A”, “healthy normoweight individuals”, “MetS individuals”, “overweight-obese individuals”, “ellagitannins”, “gut microbial metabolism”, “urolithin metabotype B”, “LDL-cholesterol”.
The key sentences extracted from ref. 92 include: “Microarray analyses were performed to determine whether standard diet ELVs (SD-ELVs) and high palmitate diet ELVs (HPD-ELVs) induced specific transcriptional signatures in MIN6B1 cells”, “To validate this, we demonstrated that miR-16, which is overexpressed in HPD-ELVs, was transferred to MIN6B1 cells and regulated Ptch1, involved in pancreas development”, “In vivo, islets from HPD mice showed increased size and altered expression of genes involved in the development, including Ptch1, suggesting that the effect of palm oil on islet size in vivo was reproduced in vitro by treating beta cells with HPD-ELVs”. The keywords include: “MIN6B1”, “pancreatic”, “exosome-like vesicles”, “miRNA”, “Ptch1”, “diabetes”, “insulin”, “mice”, “palmitate”, “high palmitate diet”.
These results indicate that IDIM can recognize TCM-related information. Due to the limited ability to infer associations and different expressions of the same thing, IDIM cannot directly validate TCM-related information. It is encouraging that with the assistance of IDIM, the efficiency of researchers in processing TCM-related information has been significantly improved.
3.6 Case study of TCMBank
Herbs are often used in the treatment of traditional Chinese medicine, but they generally contain a variety of ingredients and are associated with a variety of diseases and targets, resulting in many difficulties in the quantitative study of herbs. However, the TCMBank database can obtain various ingredients, targets, and diseases associated with the specified herbs, which will provide extensive support for relevant scientific research.Take pomegranate (POM, Chinese pinyin name: shi liu) as an example, it has moderate calories and is an excellent source of dietary fiber. It has been widely used in TCM to relieve burns, treat sore throat, cough, diarrhea, overweight, Inflammation caused by binge drinking, cardiovascular diseases, and stimulate the contraction of the uterus during childbirth. Through TCMBank, we can not only view the relevant properties of pomegranate but also obtain related 1 ingredient, 7 diseases, and 6 targets. Moreover, TCMBank also provides a visual relationship map of pomegranates and its associated items. The generated herb-ingredient-diseases-target network suggests that the diseases overweight, cardiovascular diseases, binge drinking, and the target proteins Tight Junction Protein 1 (TJP1), Zonula occludens-1 (ZO-1), and claudin-1 are crucial constituents with noteworthy functions. For instance, pre-treatment with POM substantially improved the levels of tight junction (TJ) proteins in the intestines, including ZO-1, occludin, claudin-1, and claudin-3, which were significantly reduced after exposure to alcohol. The pre-treatment with POM markedly prevented nitration and ubiquitination of claudin-1 protein in the intestines. POM prevents alcohol-induced gut permeability and inflammatory liver injury by inhibiting oxidation and nitration.93 In addition, urolithin-A (UA) or urolithin-B (UB) from a gut microbiota-derived metabolite of POM may be promising biomarkers for assessing cardiometabolic risk.91 The UA is a metabolite with effective anti-inflammatory properties and alleviates adiposity and metabolic disorders for mice without side effects.94
Turpentine Oil (Chinese pinyin name: song jie you) is a natural product extracted from pine trees, and the main components are terpene compounds. It has warming properties that improve blood circulation and soothe pain. In traditional Chinese medicine, turpentine oil is widely used to treat arthritis, sprains and muscle pain. It can also be used as an osmotic agent, which has a dispersing effect and can help to dissipate congestion and blood stasis. There are 5 components, 7 diseases, and 6 genes associated with turpentine oil in TCMBank. The herb-ingredient-diseases-target network in the Turpentine Oil details page contains ingredients such as tannins, hydroxybenzenes, amino acids, etc., and is associated with diseases such as liver diseases, anemia, hepatitis C, hepatitis B, etc. Turpentine is an established inducer of IL-6.95 Both K8 and K18 were overexpressed in subjects with moderate and mild liver inflammation and were upregulated in patients with advanced liver fibrosis. The expression of K8 and K18 was significantly increased after treatment of HepG2/Hep3B cells with IL-6.96 It has been proved that long-term Turpentine treatment can lead to small-cell anemia.97 Bone morphogenetic protein (BMP) and IL-6 work together to regulate iron homeostasis, and inhibition of BMP signaling may be an effective strategy for the treatment of inflammatory anemia. These results indicate that the disease and gene data associated with Turpentine Oil are correct and abundant.
3.7 Unique advantages of TCMBank
Several different TCM databases have been published, which have greatly contributed to TCM modernization. However, these databases may have several disadvantages. HIT focuses on the association of herbal ingredients and their corresponding targets. It has limited ingredient data and is no longer maintained. TCM-ID has no linkage information between ingredients and targets. Its website is currently accessible, but it is no longer updated. TCMID gathered mass spectrometry data of 3895 ingredients/compounds in herbs for reliability assurance purposes, but it has no herb classification and is no longer maintained. According to the HIT and TCMID publications contained more than 25000 entries, they are now not accessible and may have been lost. TCMSP is a database focused on TCM systems pharmacology containing herbs, ingredients, targets, and diseases, but it lacks TCM categories and reliability assurance information. ETCM is an encyclopedia of traditional Chinese medicine that contains various types of data, maintained but no longer updated, and each type of data has only a few. SymMap is also maintained but no longer updated. HERB is a high-throughput TCM resource with TCM experiments and reference guidance. However, its coverage is limited as it only provides transcriptome data for 20 herbs and 152 components. In addition, variations in experimental conditions and methods may render gene expression analysis results unconvincing.98
To evaluate the completeness of TCMBank, we took the top 6 serial numbers of herbs in TCMBank as an example and compared them with other TCM databases. They were turpentine oil (song jie you), pomegranate fruit (shi liu), fish liver oil (yu gan you), fortune windmillpalm petiole (zong lv), spirulina major kuetz (luo xuan zao), and oil of oriental sesame (zhi ma you). The Chinese pinyin name is in brackets. HIT and TCMID cannot be accessed, so it is not included in the comparison. We used each herb to search in other databases, and the results showed that only 1, pomegranate fruit, out of 6 herbs could be searched by hits from TCMSP. There is no classification of herbal medicines in TCM-ID. 2 out of 6 herbs, pomegranate fruit and fortune windmillpalm petiole, could be searched by hits from SymMap, and ETCM yielded the same results as SymMap. Only HERB hits all of them. This situation is very consistent with the statistical intuition that the herb entries in TCMBank and HERB are comprehensive, while the number in other databases is small. Further, we analyze the comparison of ingredients, genes and diseases in TCMBank with other databases. For the herb pomegranate fruit, 5 ingredients, 250 gene targets and corresponding diseases were retrieved in TCMSP. 11 ingredients, 191 gene targets and corresponding diseases were matched in ETCM. We keep skeptical about the results in TCMSP and ETCM. Only 1 ingredient was obtained in the TCM-ID, Punica granatum, which has very little information available and may be missing or inaccurate. Only one ingredient, ellagic acid, and 17 gene targets were found in SymMap. Both only found 1 ingredient, gallic acid, in TCMBank and HERB, and the number of gene targets and corresponding diseases obtained were 7 and 5, respectively. For the herb fortune windmillpalm petiole, 5 ingredients, 70 gene targets and corresponding diseases were retrieved in ETCM. TCMBank, SymMap and HERB share the same results for ingredient search, palmitic acid, stearic acid, and myristic acid, while 24 targets and 26 diseases were obtained in SymMap, and there are 2 gene targets and their corresponding diseases in both TCMBank and HERB. For the other 4 herbs, TCMBank provided a total of 47 ingredients, 9 gene targets and corresponding 13 diseases. HERB provides a total of 40 components, 8 gene targets and corresponding 11 diseases. TCMBank is relatively more complete, showing unique advantages.
In general, due to the time-consuming and labor-intensive work of data collation, these databases inevitably have several shortcomings, such as lack of link information, or limited data volume. TCMBank presents the most systematic list, detailed information and their relationships of 9192 herbs, 61966 unduplicated ingredients with 3D structures, 15179 targets and 32529 diseases by intelligently identifying documents and offers jump links to public data sources. In addition, most of these databases are not being updated due to the infamous “publish or perish”. TCM Database@Taiwan was established in 2011 and contains more than 20000 ingredient entries, and after updating in 2016 it contains more than 58000 entries. TCMBank is updated from TCM Database@Taiwan. The IDIM in TCMBank can intelligently identify published references and books, so as to continuously provide the latest TCM-related information. Furthermore, the chemical structure of ingredients in these TCM databases is not easy to retrieve, while TCMBank provides a structure-based ingredient compound search function and simple batch download of molecular 3D structures, which is the uniquely valuable and exceptional features of the database. Each of the above TCM databases has advantages and disadvantages, and they complement each other, but TCMBank stands out by incorporating their advantages.
3.8 The analogy of TCMBank and natural product databases
Natural product (NP) has been used as a source of traditional medicines to treat diseases for centuries. In recent years, various databases and collections of natural product databases (NPs) have increased rapidly. TCM is determined to be used as medicinal materials in human practice and is a part of natural products, so TCMBank and NPs have a huge number of overlapping compounds. Since the utilization of TCM dates back thousands of years ago in Asia, people may think that TCM is more “human body-friendly” than Western drugs, but this is actually a misconception.We compared TCMBank with a combination of several natural product databases (Table 1) from ZINC.37 Discovery Studio (DS) is a life science molecular simulation software that allows analysis and visualization of molecular data on a personal laptop. We perform extensive molecular property prediction on 3D molecular data in TCMBank by various computational models in DS software. As shown in Fig. 13A and B, the statistical trend of molecular property predicted results in TCMBank is very similar to that of NPs. They all reach the highest point at a certain point on the abscissa and then decrease rapidly. However, the proportion of extreme chemical substances in TCMBank is higher (i.e. excessive molecular weight and overdose of rotational bonds). The ratios of TCMBank in Lipinski's rule of five, absorption, distribution, metabolism, excretion, toxicity (ADMET), drug-like (DL), lead-like (LL), and fragment-like (FL) are 63%, 20%, 6%, 2%, 1%, respectively, while the ratios of NP database are 25%, 53%, 17%, 4%, and 2%, respectively. There are significant differences in the ratios between the two databases (Fig. 13C). In terms of categorization, the total ratio of 23% of overall NPs in DL, LL, and FL categories is more than 2 times higher than the 9% of TCMBank. Whereas from a database perspective, the ratios of TCMBank databases in DL and LL are 41.2%, and 7.8% respectively, reaching the lowest ratio (Fig. 14C). The ADMET distributions in TCMBank are significantly more divisible compared to NPs, whereas NPs are able to concentrate near the 95% and 99% ellipse range of absorption and blood–brain barriers (BBB) (Fig. 13D). Although a portion of chemical compounds fail both the rule of five and ADMET, a higher proportion 63% of TCMBank compounds fail the rule of five, while a higher proportion 53% of natural product compounds fail ADMET.
| Database | Actual no. of compounds | No. of zinc entries | Release time | Weblink | Commercial |
|---|---|---|---|---|---|
| Ambinter Natural Products | 32998 |
49234 |
2011-12-07 | https://www.ambinter.com/ | Yes |
| AnalytiCon Discovery NP | 5154 | 31482 |
2013-02-17 | https://ac-discovery.com/ | Yes |
| IBScreen NP | 49596 |
91785 |
2013-02-26 | https://www.ibscreen.com/ | Yes |
| Indofine Natural Products | 64 | 64 | 2013-02-18 | http://www.indofinechemical.com/ | Yes |
| Molecular Diversity Preservation International | 22181 |
32393 |
2013-08-14 | http://www.molmall.net/ | No |
| Nubbe Natural Products | 643 | 712 | 2013-01-28 | http://nubbe.iq.unesp.br/ | No |
| Princeton NP | 13284 |
18423 |
2013-02-15 | https://princetonbio.com/ | Yes |
| SelleckBioChemicals NP | 130 | 200 | 2012-04-17 | https://www.selleckchem.com/ | Yes |
| Specs Natural Products | 456 | 745 | 2011-12-06 | http://www.specs.net/ | Yes |
| UEFS Natural Products | 503 | 590 | 2011-11-29 | http://www.uefs.br/ | No |
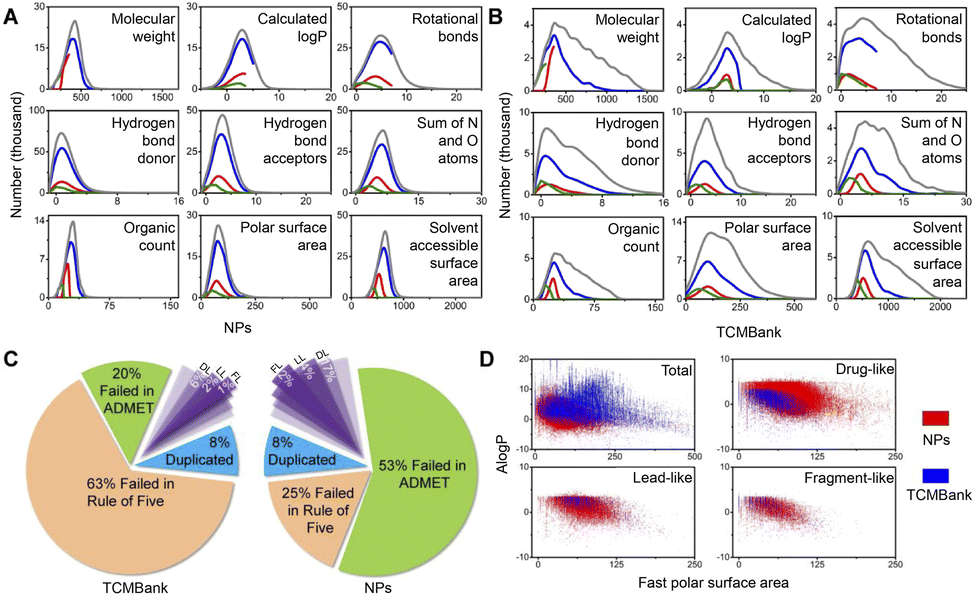 | ||
| Fig. 13 Comparison with natural product databases. (A) In NPs’ overall physical properties and (B) TCMBank overall physical properties, grey, blue, red, and green respectively represent all chemical compounds, drug-like, lead-like, and fragment-like. (C) Results of TCMBank and NPs going through ADMET and Rule of five selection. (D) ADMET distributions for TCMBank and NPs. NPs refer to natural product databases. | ||
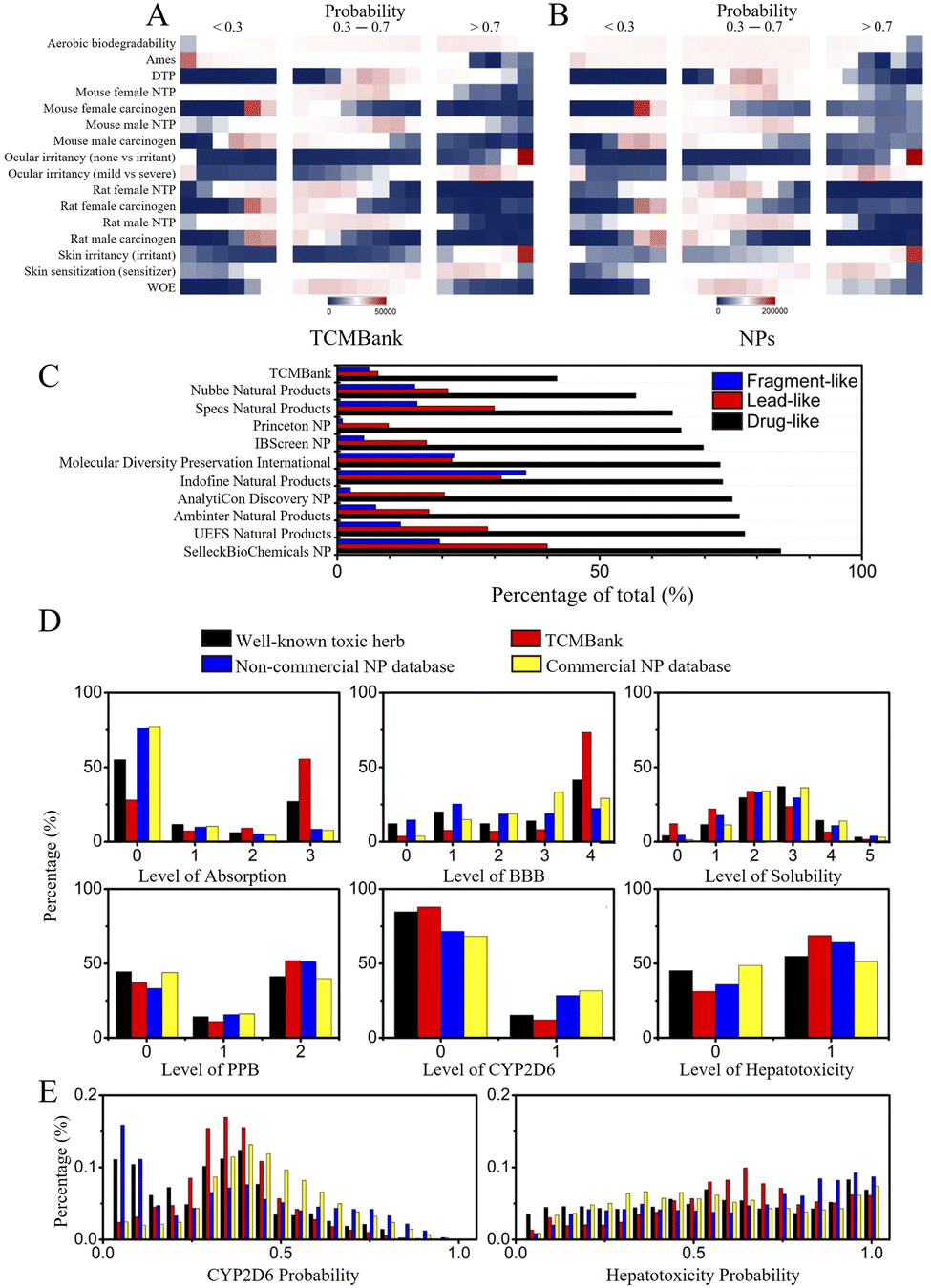 | ||
| Fig. 14 Comparative analysis of ADMET of TCMBank and NPs. (A) Toxicity prediction results of TCMBank. (B) Toxicity prediction results of NPs. (C) Comparison results of DL, LL, and FL between TCMBank and other databases. (D) Prediction results of ADMET from TCMBank. (E) Prediction of the probability of CYP2D6 and hepatotoxicity properties. DTP, developmental toxicity potential; NTP, national toxicology program; WOE, weight of evidence; UEFS, universidade Estadual de Feira de Santana database; BBB, blood–brain barrier penetration; MDPI, molecular diversity preservation international database; PPB, plasma protein binding; CYP2D6, cytochrome P450 2D6 inhibition. | ||
In addition, we also explored the toxicity of TCM. The toxicity prediction results of TCMBank and NPs are very similar (Fig. 14), with both only having ocular irritancy, skin irritancy, and skin sensitization with high toxicity risks (probability > 0.7) (Fig. 14A and B). However, 55% of TCMBank compounds show bad absorption, at a polar surface area (PSA) 2D ≥ 150.0 or partition coefficient (AlogP) ≤ 2.0 or AlogP ≥ 7.0, whereas only 8% of NPs show bad absorption (absorption sub-figure in Fig. 14D). A total of 36% of TCMs are extremely low solubility (log(Sw) ≤ 6.0), whereas only 17% of NPs are extremely low solubility (solubility sub-figure in Fig. 14D). 69% of TCMBank results in dose-dependent liver injuries, whereas only 53% of NPs result in dose-dependent liver injuries (hepatotoxicity sub-figure in Fig. 14D).
Although TCM is part of natural products, these results show that TCM is not necessarily friendly to the human body, and the use of TCM must be careful. It is particularly important to use modern analytical methods to study the active ingredients in herbal medicine. Decoding the mechanisms of active components and gene targets, and deeply exploring the pharmacodynamics and toxicological effects of TCM in the human body based on clinical reality. Fully understanding the mechanism of action of these compounds at the molecular level makes the application of TCM more effective, safe, and reliable.
Furthermore, the origins and structure of most NPs are vague or lacking. Researchers usually need expertise and more inquiries to determine their source or to obtain it experimentally. TCMBank provides a simple batch download of molecular 3D structure, further facilitating the use of local tools for virtual screening. TCMBank reduces the extra effort of users and alleviates the problem of structure lack in NPs.
Finally, although there are many existing sources for NPs, more than 20% of NPs’ sources are no longer maintained or accessed intermittently after publication time.99 TCMBank is an updated version of TCM Database@Taiwan released in 2011, and we have continued to update it for more than ten years. Now, TCMBank has the IDIM module, which can intelligently identify TCM-related information in newly published literature. In the future, we will stick to the maintenance work and data management work of TCMBank.
3.9 The future of AI-based TCM
The future of TCM holds great promise with the integration of AI technologies. AI has the potential to revolutionize TCM practices by enhancing diagnosis, treatment, and research. This section will introduce the future work of TCMBank and explore the future prospects and potential applications of AI-based TCM.The future work of TCMBank mainly involves increasing the amount of data and exploring the associations between herbs, ingredients, targets, and diseases. At present, the herbs, ingredients, targets, and diseases in TCMBank have covered most of the items recognized by humans. Therefore, the future expansion of data in TCMBank will primarily rely on the integration of other public databases, such as OMIM, DrugBank, DisGeNET, etc. Compared with other TCM databases, the amount of gene data in TCMBank is not the largest. In the next step, we plan to integrate gene-related public databases and other TCM databases to enhance gene richness in TCMBank.
Furthermore, to make the data in TCMBank really useful, it is more important to increase the links between the data. This may mainly rely on the developed IDIM module for literature recognition. The IDIM model regularly retrieves and recognizes information in published articles every day, and saves all information such as original PDF files, parsed texts, graphs, tables, keywords, summaries, SMILES, SD, and time stamps into the MySQL v5.7.36 database. We plan to organize volunteers to update the data in TCMBank every year and update the TCM-related information contained in the articles during this year. An undergraduate student majoring in chemistry or pharmacy spends only 5 minutes on average reviewing the data in an article using the IDIM model, but the publication of the article is relatively slow. Such an update method is completely achievable, without relying heavily on manual labor.
Additionally, we plan to provide user interaction and feedback mechanisms where users can provide suggestions and new data. This increases user engagement, improves database quality, and yields valuable insights.
Fig. 15 shows five future prospects of artificial intelligence in traditional Chinese medicine.
 | ||
| Fig. 15 Future applications of AI in TCM include diagnosis, treatment recommendation, selection of herbal remedies, research, and patient monitoring. AI enhances accuracy, optimizes treatment, accelerates research, and enables proactive care. | ||
(1) AI-driven diagnostic systems: AI can play a vital role in improving diagnostic accuracy and efficiency in TCM.100 By leveraging machine learning algorithms, AI can analyze patient data, including symptoms, medical history, and diagnostic indicators, to assist TCM Practitioners in making more precise and timely diagnoses. These AI-driven diagnostic systems can aid in identifying patterns and relationships in complex TCM data, leading to improved diagnostic outcomes.
(2) Personalized treatment recommendations: AI can facilitate personalized treatment recommendations in TCM.101 By analyzing individual patient characteristics, such as constitution, lifestyle, and genetic factors, AI algorithms can generate tailored treatment plans that optimize therapeutic outcomes. This personalized approach ensures that TCM treatments are customized to the unique needs of each patient, improving treatment efficacy.
(3) Intelligent herb recommendation systems: AI can assist in the selection of herbal remedies in TCM.102 By analyzing the properties and therapeutic effects of various herbs, as well as considering individual patient characteristics, AI algorithms can recommend specific herbal formulations for different health conditions. These intelligent herb recommendation systems can enhance the precision and effectiveness of TCM herbal treatments.
(4) AI-enabled TCM research and drug development: AI technologies offer opportunities for accelerated TCM research and drug development.103 Through data mining and analysis, AI can uncover hidden patterns in large-scale TCM databases, facilitating the identification of potential therapeutic t targets and the discovery of new herbal formulations. AI can also aid in predicting the efficacy and safety profiles of TCM compounds, expediting the development of novel TCM drugs.
(5) Patient monitoring and prognostic tools: AI-based systems can continuously monitor patient health parameters, providing real-time feedback to both patients and TCM practitioners.104 By analyzing data from wearable devices, AI algorithms can detect subtle changes in health status and predict disease progression. This enables early intervention and proactive management of health conditions, leading to improved patient outcomes.
In conclusion, the future of AI-based TCM is poised to bring significant advancements to the field. The integration of AI technologies can enhance diagnostic accuracy, enable personalized treatment recommendations, facilitate intelligent herb selection, accelerate TCM research, and empower patient monitoring. However, further research, collaboration, and validation are essential to fully unlock the potential of AI in TCM.
4 Conclusions
Traditional Chinese Medicine boasts millennia of clinical experience and serves as a crucial source of modern drug development. Modern drug development requires the separation of active compounds from herbs and further analysis of the potential therapeutic mechanisms of herb preparations at the molecular level. Research and development of TCM requires a systematic method to explore rational modern drug discovery efforts. The rise of AI technology fits perfectly with this need.We developed TCMBank (https://TCMBank.CN/), which is the largest comprehensive systematic and high-quality TCM information database. The database organizes herbal active ingredients, target and diseases information from books, published references, and public databases in a standardized format. The intelligent document identification module in TCMBank is used to aggregate TCM research scattered in various forms of sources. The module assisted volunteers to identify TCM-related information by extracting SMILES, chemical formulae, abstracts, keywords and other information from published documents. TCMBank can be continuously updated every year without relying too much on manpower. TCMBank has free access, and the data can be easily downloaded. TCMBank provides strong support for the development of new drug molecules, and the research on the action mechanism of active ingredients and targets.
Furthermore, we proposed an EL-based drug discovery protocol for identifying potentially effective leads and drug repurposing. Wet experiments allow the study of living cells under controlled conditions and infer cell behavior in vivo, making the EL-based framework for drug development more convincing. We take colorectal cancer and Alzheimer's disease as examples to demonstrate how to accelerate drug discovery by artificial intelligence.
Data availability
All data are available at https://TCMBank.CN/.Author contributions
Calvin Yu-Chian Chen designed research. Qiujie Lv, Guanxing Chen, Lu Zhao, and Haohuai He worked together to complete the experiment. Qiujie Lv and Ziduo Yang contributed to analytic tools. Lu Zhao, Ziduo Yang, and Hsin-Yi Chen analyzed the data. Qiujie Lv, Guanxing Chen, Lu Zhao, Haohuai He and Calvin Yu-Chian Chen wrote the manuscript together.Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.Acknowledgements
We acknowledge the help of Yuteng Xu, Qiongwen Liu, Yaohao Wang, Shiyue Cheng, Zhicheng Yang, Liwei Yang, Rui Huang, Hao Liu, Hui Peng, Binghao Cheng, and Minqing Lin, et al. for their hard work in data processing and website construction. The authors thank the anonymous reviewers for their valuable suggestions. This work was supported by the National Natural Science Foundation of China (Grant No. 62176272), the Research and Development Program of Guangzhou Science and Technology Bureau (Grant No. 2023B01J1016), Key-Area Research and Development Program of Guangdong Province (Grant No. 2020B1111100001).Notes and references
- J. Qiu, Nature, 2007, 448, 126–129 CrossRef CAS PubMed.
- F. Cheung, Nature, 2011, 480, S82–S83 CrossRef CAS.
- D. Normile, Science, 2003, 299, 188–190 CrossRef CAS PubMed.
- J.-F. Wang, D.-Q. Wei and K.-C. Chou, Curr. Top. Med. Chem., 2008, 8, 1656–1665 CrossRef CAS.
- W. R. Strohl, Drug discovery today, 2000, 5, 39–41 CrossRef.
- J.-F. Wang and D.-Q. Wei, Pharmacogenomics, 2009, 10, 1213–1215 CrossRef CAS PubMed.
- Y. Tu, Nat. Med., 2011, 17, 1217–1220 CrossRef CAS PubMed.
- K. Chen, J. Am. Pharmaceut. Assoc., 1925, 14, 189–194 Search PubMed.
- A. L. Harvey, R. Edrada-Ebel and R. J. Quinn, Nat. Rev. Drug Discov., 2015, 14, 111–129 CrossRef CAS.
- F. Saldívar-González, V. Aldas-Bulos, J. Medina-Franco and F. Plisson, Chem. Sci., 2022, 13, 1526–1546 RSC.
- Q.-J. Lv, H.-Y. Chen, W.-B. Zhong, Y.-Y. Wang, J.-Y. Song, S.-D. Guo, L.-X. Qi and C. Y.-C. Chen, IEEE J. Transl. Eng. Health. Med., 2019, 8, 1–11 Search PubMed.
- S. Guo, L. Xu, C. Feng, H. Xiong, Z. Gao and H. Zhang, Med. Image Anal., 2021, 73, 102170 CrossRef PubMed.
- Q. Lv, G. Chen, Z. Yang, W. Zhong and C. Y.-C. Chen, IEEE Transact. Neural Networks Learn. Syst., 2023, 1–13 Search PubMed.
- S. Guo, H. Zhang, Y. Gao, H. Wang, L. Xu, Z. Gao, A. Guzzo and G. Fortino, Computer Methods and Programs in Biomedicine, 2023, 107547 Search PubMed.
- Q. Lv, G. Chen, L. Zhao, W. Zhong and C. Y.-C. Chen, Briefings Bioinf., 2021, 22, bbab317 CrossRef PubMed.
- Z. Yang, W. Zhong, Q. Lv and C. Y.-C. Chen, Chem. Sci., 2022, 13, 8693–8703 RSC.
- H.-Y. Chen, J.-Q. Chen, J.-Y. Li, H.-J. Huang, X. Chen, H.-Y. Zhang and C. Y.-C. Chen, J. Chem. Inf. Model., 2019, 59, 1605–1623 CrossRef CAS PubMed.
- Q. Lv, J. Zhou, Z. Yang, H. He and C. Y.-C. Chen, Neural Network., 2023, 165, 94–105 CrossRef.
- G. Chen, X. Jiang, Q. Lv, X. Tan, Z. Yang and C. Y.-C. Chen, Knowl. Base Syst., 2022, 257, 109925 CrossRef.
- M. Zhao, Q. Zhou, W. Ma and D.-Q. Wei, et al. , Evid. base Compl. Alternative Med., 2013, 1–15 Search PubMed.
- D. Melchart, S. Hager, J. Dai and W. Weidenhammer, Complement. Med. Res., 2016, 23, 21–28 CrossRef PubMed.
- H. Gao, Z. Wang, Y. Li and Z. Qian, Front. Med., 2011, 5, 195–202 CrossRef PubMed.
- H. Ye, L. Ye, H. Kang, D. Zhang, L. Tao, K. Tang, X. Liu, R. Zhu, Q. Liu and Y. Z. Chen, et al. , Nucleic Acids Res., 2010, 39, D1055–D1059 CrossRef PubMed.
- C. Y.-C. Chen, PLoS One, 2011, 6, e15939 CrossRef CAS PubMed.
- Y. Wu, F. Zhang, K. Yang, S. Fang, D. Bu, H. Li, L. Sun, H. Hu, K. Gao and W. Wang, et al. , Nucleic Acids Res., 2019, 47, D1110–D1117 CrossRef PubMed.
- R. Xue, Z. Fang, M. Zhang, Z. Yi, C. Wen and T. Shi, Nucleic Acids Res., 2012, 41, D1089–D1095 CrossRef PubMed.
- L. Huang, D. Xie, Y. Yu, H. Liu, Y. Shi, T. Shi and C. Wen, Nucleic Acids Res., 2018, 46, D1117–D1120 CrossRef CAS PubMed.
- J. Ru, P. Li, J. Wang, W. Zhou, B. Li, C. Huang, P. Li, Z. Guo, W. Tao and Y. Yang, et al. , J. Cheminf., 2014, 6, 1–6 Search PubMed.
- H.-Y. Xu, Y.-Q. Zhang, Z.-M. Liu, T. Chen, C.-Y. Lv, S.-H. Tang, X.-B. Zhang, W. Zhang, Z.-Y. Li and R.-R. Zhou, et al. , Nucleic Acids Res., 2019, 47, D976–D982 CrossRef CAS PubMed.
- X. Chen, H. Zhou, Y. Liu, J. Wang, H. Li, C. Ung, L. Han, Z. Cao and Y. Chen, Br. J. Pharmacol., 2006, 149, 1092–1103 CrossRef CAS.
- S. Fang, L. Dong, L. Liu, J. Guo, L. Zhao, J. Zhang, D. Bu, X. Liu, P. Huo and W. Cao, et al. , Nucleic Acids Res., 2021, 49, D1197–D1206 CrossRef CAS PubMed.
- X. Yang, Y. Wang, R. Byrne, G. Schneider and S. Yang, Chem. Rev., 2019, 119, 10520–10594 CrossRef CAS.
- R. P. Sheridan, J. C. Culberson, E. Joshi, M. Tudor and P. Karnachi, J. Chem. Inf. Model., 2022, 62, 3275–3280 CrossRef CAS PubMed.
- J.-Y. Li, H.-Y. Chen, W.-j. Dai, Q.-J. Lv and C. Y.-C. Chen, J. Phys. Chem. Lett., 2019, 10, 4947–4961 CrossRef CAS PubMed.
- J.-Q. Chen, H.-Y. Chen, W.-j. Dai, Q.-J. Lv and C. Y.-C. Chen, J. Phys. Chem. Lett., 2019, 10, 4382–4400 CrossRef CAS PubMed.
- P. Nietert and L. Thabane, Nat. Med., 2011, 17, 1531 Search PubMed.
- J. J. Irwin, K. G. Tang, J. Young, C. Dandarchuluun, B. R. Wong, M. Khurelbaatar, Y. S. Moroz, J. Mayfield and R. A. Sayle, J. Chem. Inf. Model., 2020, 60, 6065–6073 CrossRef CAS PubMed.
- T.-Y. Tsai, K.-W. Chang and C. Y.-C. Chen, J. Comput. Aided Mol. Des., 2011, 25, 525–531 CrossRef CAS.
- Selenium is a suite of tools for automating web browsers, https://www.selenium.dev/, accessed July 12, 2020 Search PubMed.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen and B. Yu, et al. , Nucleic Acids Res., 2019, 47, D1102–D1109 CrossRef.
- Plumb a PDF for detailed information about each char, rectangle, and line, https://pypi.org/project/pdfplumber/, accessed October 18, 2020 Search PubMed.
- Python-tesseract is an optical character recognition tool for python, https://pypi.org/project/pytesseract/, accessed July 12, 2020 Search PubMed.
- A. T. Schutz, et al., Keyphrase Extraction from Single Documents in the Open Domain Exploiting Linguistic and Statistical Methods, M. App. Sc thesis, Citeseer, 2008 Search PubMed.
- F. Barrios, F. López, L. Argerich and R. Wachenchauzer, arXiv, preprint, arXiv:1602.03606, 2016, DOI:10.48550/arXiv.1602.03606.
- I. V. Filippov and M. C. Nicklaus, J. Chem. Inf. Model., 2009, 49, 740–743 CrossRef CAS.
- Y.-C. Chen, Trends Pharmacol. Sci., 2015, 36, 78–95 CrossRef PubMed.
- J. Zhou, G. Xie and X. Yan, Isolat Compound AB, 2011, 1, 455 Search PubMed.
- G. Chen and S. Li, Ben cao gang mu tong shi, Xue Yuan Publishing House, 1992 Search PubMed.
- N. Zhong, G. Zhao, S. Dai and R. Chen, Zhong yao da ci dian, Shanghai Scientific & Technical Publishers, 2006 Search PubMed.
- X. Miao and J. Zheng, Shennong ben cao jing shu, Chinese Medicine Ancient Books Publishing House, 2002 Search PubMed.
- L. X., Z. YaoJian bie da quan, Hunan Science and Technology Press, 2002 Search PubMed.
- Y. Fang, Z. Zhang and X. Miao, Shang han lun tiao bian, Shanghai Classics Publishing House, 1991 Search PubMed.
- L. Shen and S. Li, Ben cao gang mu cai se tu pu, Huaxia Publishing House, 1998 Search PubMed.
- S. Yang, The divine farmer's materia medica: a translation of the Shen Nong Ben Cao Jing, Blue Poppy Enterprises, Inc., 1998 Search PubMed.
- B. Borate and A. D. Baxevanis, Curr. Protoc. Bioinformatics, 2009, 27, 1–2 Search PubMed.
- S. Povey, R. Lovering, E. Bruford, M. Wright, M. Lush and H. Wain, Hum. Genet., 2001, 109, 678–680 CrossRef CAS.
- F. Minguet, T. M. Salgado, L. Van Den Boogerd and F. Fernandez-Llimos, Res. Soc. Adm. Pharm., 2015, 11, 686–695 CrossRef PubMed.
- A. D. Yates, P. Achuthan, W. Akanni, J. Allen, J. Allen, J. Alvarez-Jarreta, M. R. Amode, I. M. Armean, A. G. Azov and R. Bennett, et al. , Nucleic Acids Res., 2020, 48, D682–D688 CrossRef CAS PubMed.
- L. M. Schriml, C. Arze, S. Nadendla, Y.-W. W. Chang, M. Mazaitis, V. Felix, G. Feng and W. A. Kibbe, Nucleic Acids Res., 2012, 40, D940–D946 CrossRef CAS.
- S. Köhler, L. Carmody, N. Vasilevsky, J. O. B. Jacobsen, D. Danis, J.-P. Gourdine, M. Gargano, N. L. Harris, N. Matentzoglu and J. A. McMurry, et al. , Nucleic Acids Res., 2019, 47, D1018–D1027 CrossRef PubMed.
- J. Piñero, J. M. Ramírez-Anguita, J. Saüch-Pitarch, F. Ronzano, E. Centeno, F. Sanz and L. I. Furlong, Nucleic Acids Res., 2020, 48, D845–D855 Search PubMed.
- D. S. Wishart, Y. D. Feunang, A. C. Guo, E. J. Lo, A. Marcu, J. R. Grant, T. Sajed, D. Johnson, C. Li and Z. Sayeeda, et al. , Nucleic Acids Res., 2018, 46, D1074–D1082 CrossRef CAS.
- Q. Lv, G. Chen, H. He, Z. Yang, L. Zhao, K. Zhang and C. Y.-C. Chen, Signal Transduct. Targeted Ther., 2023, 8, 127 CrossRef PubMed.
- G. Erkan and D. R. Radev, J. Artif. Intell. Res., 2004, 22, 457–479 CrossRef.
- A. R. Aronson, O. Bodenreider, H. F. Chang, S. M. Humphrey, J. G. Mork, S. J. Nelson, T. C. Rindflesch and W. J. Wilbur, Proceedings of the AMIA Symposium, 2000, p. 17 Search PubMed.
- K. T. Dibia, P. K. Igbokwe, G. I. Ezemagu and C. O. Asadu, Results Chem., 2022, 4, 100272 CrossRef CAS.
- C.-H. Chen, K. Tanaka, M. Kotera and K. Funatsu, J. Cheminf., 2020, 12, 1–16 Search PubMed.
- Z. Yang, W. Zhong, L. Zhao and C. Y.-C. Chen, Chem. Sci., 2022, 13, 816–833 RSC.
- M. Karimi, D. Wu, Z. Wang and Y. Shen, Bioinformatics, 2019, 35, 3329–3338 CrossRef CAS PubMed.
- B. Bruno and E. Peter, JSME: a free molecule editor in JavaScript, J. Cheminf., 2013, 5(1), 1–6 Search PubMed.
- B. Bienfait and P. Ertl, J. Cheminf., 2013, 5, 1–6 Search PubMed.
- M. Brunn, Y. Chali and C. J. Pinchak, Proc. of Document Understanding Conference, 2001, p. 29 Search PubMed.
- P. Over and W. Liggett, Document Understanding Conference, 2002 Search PubMed.
- Y. Gong and X. Liu, Proceedings of the 24th annual international ACM SIGIR conference on Research and development in information retrieval, 2001, pp. 19–25 Search PubMed.
- S. N. Kim, O. Medelyan, M.-Y. Kan and T. Baldwin, Proceedings of the 5th International Workshop on Semantic Evaluation, USA, 2010, p. 21–26 Search PubMed.
- I. Augenstein, M. Das, S. Riedel, L. Vikraman and A. McCallum, Proceedings of the 11th International Workshop on Semantic Evaluation (SemEval-2017), Vancouver, Canada, 2017, pp. 546–555 Search PubMed.
- H. Nie, H. Ju, J. Fan, X. Shi, Y. Cheng, X. Cang, Z. Zheng, X. Duan and W. Yi, Nat. Commun., 2020, 11, 36 CrossRef CAS PubMed.
- R. E. Tanzi and L. Bertram, Cell, 2005, 120, 545–555 CrossRef CAS PubMed.
- Y. Li, W. Zhou, Y. Tong, G. He and W. Song, Faseb. J., 2006, 20, 285–292 CrossRef PubMed.
- H. Eldar-Finkelman, Trends Mol. Med., 2002, 8, 126–132 CrossRef CAS PubMed.
- P. H. Reddy, Biochim. Biophys. Acta, 2013, 1832, 1913–1921 CrossRef CAS PubMed.
- Z. Babar, M. Khan, M. Zahra, M. Anwar, K. Noor, H. F. Hashmi, M. Suleman, M. Waseem, A. Shah and S. Ali, et al. , J. Biomol. Struct. Dyn., 2022, 40, 523–537 CrossRef CAS PubMed.
- J. Lu, P. Tang, W. Qiu, H. Wang and J. Guo, Security and Privacy in Social Networks and Big Data: 6th International Symposium, SocialSec 2020, Tianjin, China, September 26–27, 2020, Proceedings 6, 2020, pp. 203–214 Search PubMed.
- J.-N. Gong, L. Zhao, G. Chen, X. Chen, Z.-D. Chen and C. Y.-C. Chen, Mol. Diversity, 2021, 25, 1375–1393 CrossRef CAS PubMed.
- S. Yang, S. Li and J. Chang, RSC Adv., 2022, 12, 13500–13510 RSC.
- H. He, G. Chen and C. Y.-C. Chen, New J. Chem., 2022, 46, 5188–5200 RSC.
- Y. Wang, D. Qin, L. Jin and G. Liang, Comput. Biol. Med., 2022, 145, 105410 CrossRef CAS PubMed.
- B. Zhang, J. Zhao, Z. Wang, P. Guo, A. Liu and G. Du, Front. Pharmacol., 2021, 12, 709607 CrossRef CAS PubMed.
- Z. Zhu, Z. Rahman, M. Aamir, S. Z. A. Shah, S. Hamid, A. Bilawal, S. Li and M. Ishfaq, RSC Adv., 2023, 13, 2057–2069 RSC.
- H. Zhang, J. Li, K. M. Saravanan, H. Wu, Z. Wang, D. Wu, Y. Wei, Z. Lu, Y. H. Chen and X. Wan, et al. , Front. Pharmacol., 2021, 3297 Search PubMed.
- M. V. Selma, A. González-Sarrías, J. Salas-Salvadó, C. Andrés-Lacueva, C. Alasalvar, A. Örem, F. A. Tomás-Barberán and J. C. Espín, Clin. Nutr., 2018, 37, 897–905 CrossRef CAS PubMed.
- A. Jalabert, G. Vial, C. Guay, O. P. Wiklander, J. Z. Nordin, H. Aswad, A. Forterre, E. Meugnier, S. Pesenti and R. Regazzi, et al. , Diabetologia, 2016, 59, 1049–1058 CrossRef CAS PubMed.
- Y.-E. Cho and B.-J. Song, Redox Biol., 2018, 18, 266–278 CrossRef CAS PubMed.
- B. Xia, X. C. Shi, B. C. Xie, M. Q. Zhu, Y. Chen, X. Y. Chu, G. H. Cai, M. Liu, S. Z. Yang and G. A. Mitchell, et al. , PLoS Biol., 2020, 18, e3000688 CrossRef CAS PubMed.
- W. Kozak, M. J. Kluger, D. Soszynski, C. A. Conn, K. Rudolph, L. R. Leon and H. Zheng, Ann. N. Y. Acad. Sci., 1998, 856, 33–47 CrossRef CAS PubMed.
- N. Guldiken, V. Usachov, K. Levada, C. Trautwein, M. Ziol, P. Nahon and P. Strnad, Liver Int., 2015, 35, 1203–1212 CrossRef CAS PubMed.
- A. U. Steinbicker, C. Sachidanandan, A. J. Vonner, R. Z. Yusuf, D. Y. Deng, C. S. Lai, K. M. Rauwerdink, J. C. Winn, B. Saez, C. M. Cook, B. A. Szekely, C. N. Roy, J. S. Seehra, G. D. Cuny, D. T. Scadden, R. T. Peterson, K. D. Bloch and P. B. Yu, Blood, 2011, 117, 4915–4923 CrossRef CAS PubMed.
- S. Tian, J. Zhang, S. Yuan, Q. Wang, C. Lv, J. Wang, J. Fang, L. Fu, J. Yang and X. Zu, et al. , Briefings Bioinf., 2023, bbad027 CrossRef.
- M. Sorokina and C. Steinbeck, J. Cheminf., 2020, 12, 20 CAS.
- C. Feng, Y. Shao, B. Wang, Y. Qu, Q. Wang, Y. Li and T. Yang, Evid. base Compl. Alternative Med., 2021, 2021, 1–8 Search PubMed.
- X. Chu, B. Sun, Q. Huang, S. Peng, Y. Zhou and Y. Zhang, Artif. Intell. Med., 2020, 103, 101810 CrossRef PubMed.
- H. Bao, R. Wen, X. Li, C. Zhao and Z. Chen, TMR Mod. Herb. Med., 2021, 4, 13 CrossRef.
- G. Tian, K. Qian, X. Li, M. Sun, H. Jiang, W. Qiu, X. Xie, Z. Zhao, L. Huang and S. Luo, et al. , IEEE Trans. Comput. Soc. Syst., 2023, 10(2), 700–713 Search PubMed.
- Y. Zheng, N. Tang, R. Omar, Z. Hu, T. Duong, J. Wang, W. Wu and H. Haick, Adv. Funct. Mater., 2021, 31, 2105482 CrossRef CAS.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |