
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalide solid-state electrolytes for all-solid-state batteries: structural design, synthesis, environmental stability, interface optimization and challenges
Boran
Tao
ab,
Dailin
Zhong
a,
Hongda
Li
 ab,
Guofu
Wang
a and
Haixin
Chang
*b
ab,
Guofu
Wang
a and
Haixin
Chang
*b
aLiuzhou Key Laboratory of New-Energy Vehicle Lithium Battery, School of Electronic Engineering, Guangxi University of Science and Technology, Liuzhou, 545006, China
bQuantum-Nano Matter and Device Lab, State Key Laboratory of Material Processing and Die & Mould Technology, School of Materials Science and Engineering, Huazhong University of Science and Technology, Wuhan, 430074, China. E-mail: hxchang@hust.edu.cn
First published on 10th August 2023
Abstract
Since the huge breakthrough in 2018, research on halide solid-state electrolytes (SSEs) has set off a new craze. In comparison with oxide and sulfide SSEs, halide SSEs have more balanced properties in various aspects, including ionic conductivity, electrochemical stability window, and moisture resistance. Herein, the overall knowledge and deep understanding of halide SSEs and their practical applications in all-solid-state batteries (ASSBs) are introduced. Firstly, the principle of screening halide SSE components is proposed. Among F, Cl, Br and I anions, the Cl anion is excellent owing to its suitable ionic conductivity and electrochemical stability window. The Sc, Y, and lanthanide elements are also more compatible with Cl anions in terms of electronegativity. Secondly, the structural design theory of halide SSEs with high ionic conductivity and the mechanism of Li ion migration are described. A monoclinic structure is more conducive to Li ion migration, compared with trigonal and orthorhombic structures. Additionally, substitution strategies for halide SSEs are discussed, mainly including dual-halogen, isovalent cation substitution, and aliovalent cation substitution. Furthermore, the mechanism of moisture resistance and synthesis method of halide SSEs are analyzed. Compared with the solid-state reaction and mechanochemistry method, wet chemical synthesis is more likely to achieve scale-up production of halide SSEs. Finally, the application prospects and challenges of halide SSEs in ASSBs are outlined.
 Boran Tao | Boran Tao is an assistant professor at the Guangxi University of Science and Technology. He got his PhD in Materials Science and Engineering in 2019 from Chongqing University. He is a visiting scholar at the Huazhong University of Science and Technology for research on new energy materials. His research focuses on solid-state electrolytes, anode materials, and cathode materials for Li ion batteries. |
 Haixin Chang | Haixin Chang is a full professor at the Huazhong University of Science and Technology. He got his PhD in Materials Science in 2007 from the Institute of Metal Research, Chinese Academy of Sciences. Then, he worked at the Department of Chemistry, Tsinghua University, and Nanotechnology Center, ITC, Hong Kong Polytechnic University, before moving to Tohoku University at the beginning of 2011. He joined the faculty of Tohoku University as an assistant professor in 2012. He joined the Huazhong University of Science and Technology as a full professor in 2014. His researches focus on 2D/quantum materials, 2D electronics/optoelectronics, and new energy materials. He has published over 100 papers with a citation over 6000 times. He was also awarded 2020–2022 Elsevier Highly Cited Chinese Researchers. |
1. Introduction
Since the advent of lithium-ion batteries (LIBs), they have been widely considered a research hotspot.1–4 The rapid development of electronics and electric vehicles has put forward higher requirements for rechargeable LIBs, including better safety, higher capacity, higher energy density, and faster charging performance. Faced with these growing demands, conventional LIBs with liquid electrolytes have not performed as expected. Hence, building next-generation “beyond Li-ion” batteries has been key to meet the increasing demands of the energy storage market.5–7 One promising strategy is to assemble all-solid-state batteries (ASSBs) using solid-state electrolytes (SSEs) rather than liquid electrolytes found in conventional LIBs.7–9According to the chemical composition, SSEs are broadly divided into three categories: inorganic electrolytes, polymer electrolytes and organic–inorganic hybrid composites.10–12 Inorganic electrolytes have ionic conductivity comparable to that of liquid electrolytes, exhibiting a greater electrochemical stability window and wider operating temperature window, which provide new opportunities for the development of next-generation “beyond Li-ion” batteries.7,13,14 Based on anion chemistry, inorganic electrolytes can be further divided into oxides, sulfides, and halides.15,16 In contrast, halide SSEs are believed to be the best candidates for ASSB technology due to their intrinsic chemical properties (Fig. 1).17–21 Firstly, the weaker Coulomb force between halogen anions and Li ions and the wider Li ion transport channel formed by relatively larger radius halogen anions (Cl− = 167 pm, Br− = 182 pm, I− = 202 pm, O2− = 126 pm and S2− = 170 pm)22,23 are conducive to ion mobility and guarantee the high ionic conductivity of halide SSEs (e.g., Li3ScCl6,24 3.02 mS cm−1). Secondly, halide SSEs still possess good air stability and recoverability after humidity exposure. Li3InCl6 remains stable after exposure to dry or low humidity air at ambient temperature.25,26 This property can eliminate the need for a rigorous assembly environment for ASSB manufacturing, which is beneficial to significantly reduce production costs. Thirdly, halide anions have a higher electrochemical oxidation stability (such as up to ∼4.21 V for Li3YCl6)27 and can be more compatible with high voltage cathode materials, thus exhibiting good capacities. Finally, halide anions have lower bond strength with polyanions, softer lattice structures, and higher anionic polarizability than oxides and sulfides, which endows halide SSEs with better mechanical deformability and is beneficial for the assembly of ASSBs.27–29
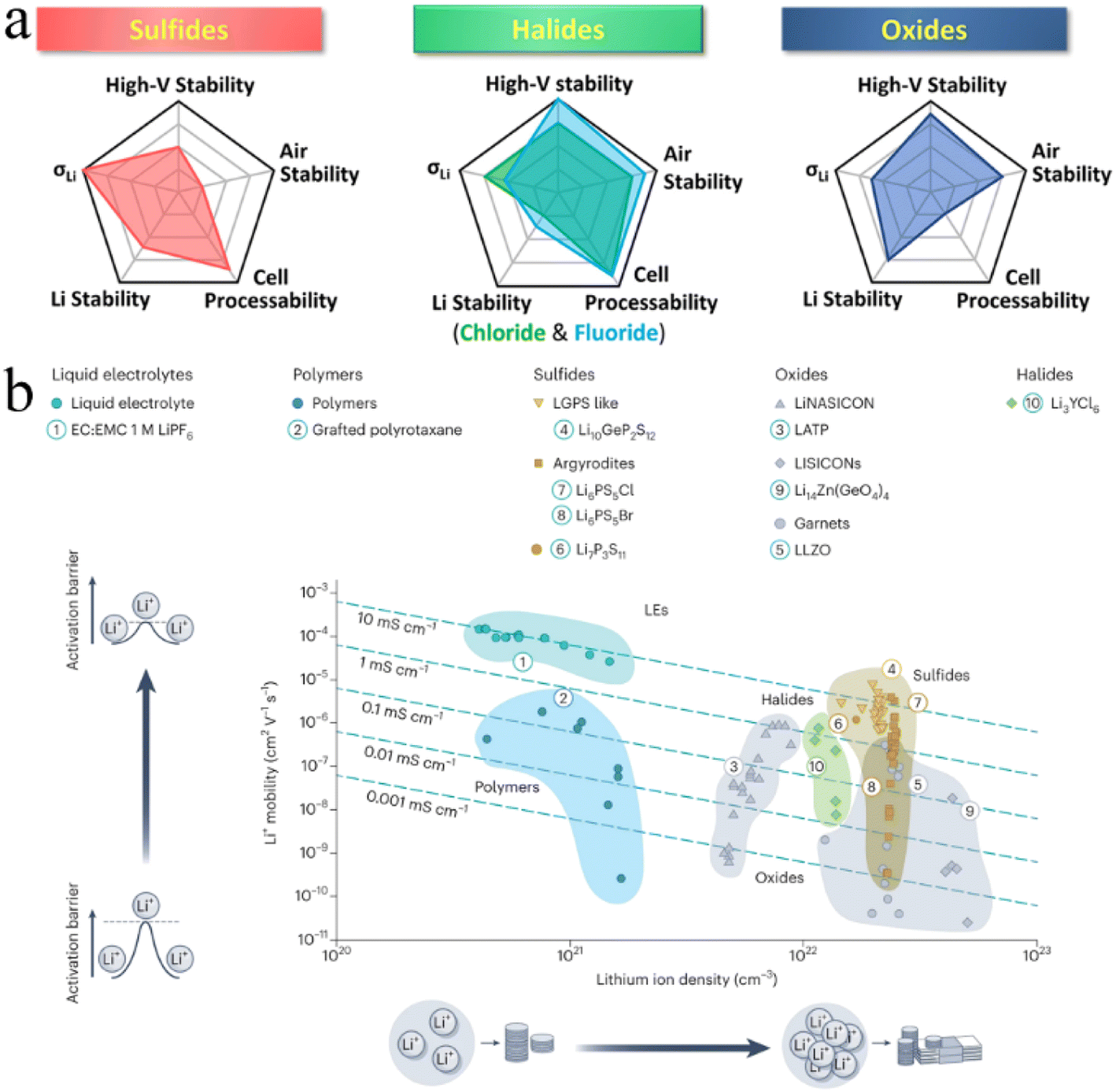 | ||
| Fig. 1 (a) Spider plots exhibiting various properties of three typical inorganic SSEs. Reproduced with permission.19 Copyright 2022, American Chemical Society. (b) Comparison of the ionic conductivity of different electrolytes based on the Li content. Reproduced with permission.18 Copyright 2023, Springer Nature. | ||
In the 1930s, halide electrolytes were first studied and exhibited room-temperature ionic conductivity as low as 10−7 S cm−1, so they didn't attract wide publicity.30–32 Until 2018, Asano et al. significantly increased the room-temperature ionic conductivity of halide SSEs to 1.7 mS cm−1, which triggered a research hotspot in halide SSEs.19,23,33–37 In terms of composition, fast Li-ion conductors based on ternary halides can be roughly divided into three categories: (i) Li3MX6 halide electrolytes with group 3 elements (M = Sc, Y, and lanthanides); (ii) Li3MX6 halide electrolytes with group 13 elements (M = Al, Ga, In); and (iii) Li2MX4 or Li6MX8 halide electrolytes with divalent metal elements (M = Ti, Zr, Hf, V, Cr, Mn, Fe, Zn, Mg).34 In addition, there are quaternary halide compounds formed by chemical substitutions to improve ionic conductivity and humidity tolerance, e.g., Li3Y1−xInxCl6,38 Li3−xEr1−xZrxCl6.39 From another viewpoint, halide fast Li-ion conductors can be generally divided into four categories according to crystalline structures, including (i) trigonal structures (space groups: P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1), such as Li3YCl6,33 and Li3ErCl6;40,41 (ii) monoclinic structures (space groups: C2/m), such as Li3YBr6,33 Li3InCl6,42 and Li3ScCl6;24 (iii) orthorhombic structures (space groups: Pnma), such as Li3YbCl643 and Li2.5Y0.5Zr0.5Cl6;44 (iv) spinel structures (space groups: Fdm), such as Li2Sc2/3Cl4,45 Li2Sc2/3−xErxCl4,46 and Li2FeCl4.47 In parallel, halide fast Na-ion conductors, mainly including Na2ZrCl6,48 NaAlCl4,49 Na3MCl6 (M = Y, Er, In, Sc, and Yb),50–52 Na3MBr6,51,53 Na3MI6 (M = Sc, Y, La, and In),54,55 Na3−xY1−xZrxCl6,56,57 Na3−xEr1−xZrxCl6,58 Na2InxSc0.666−xCl4,59 and Na3In1−xScxCl6 (ref. 59) have also been investigated. However, there is still a large gap between the limited ionic conductivity (<0.1 mS cm−1) of halide fast Na-ion conductors and the demand for practical applications. Besides, halide SSEs have the obvious advantage of abundant synthesis pathways, such as mechanochemical synthesis,41,60 co-melting synthesis,45 and wet chemical synthesis,61–63 which can avoid the high-temperature sintering process used in sulfide and oxide SSE synthesis and greatly reduce energy consumption and manufacturing cost.
m1), such as Li3YCl6,33 and Li3ErCl6;40,41 (ii) monoclinic structures (space groups: C2/m), such as Li3YBr6,33 Li3InCl6,42 and Li3ScCl6;24 (iii) orthorhombic structures (space groups: Pnma), such as Li3YbCl643 and Li2.5Y0.5Zr0.5Cl6;44 (iv) spinel structures (space groups: Fdm), such as Li2Sc2/3Cl4,45 Li2Sc2/3−xErxCl4,46 and Li2FeCl4.47 In parallel, halide fast Na-ion conductors, mainly including Na2ZrCl6,48 NaAlCl4,49 Na3MCl6 (M = Y, Er, In, Sc, and Yb),50–52 Na3MBr6,51,53 Na3MI6 (M = Sc, Y, La, and In),54,55 Na3−xY1−xZrxCl6,56,57 Na3−xEr1−xZrxCl6,58 Na2InxSc0.666−xCl4,59 and Na3In1−xScxCl6 (ref. 59) have also been investigated. However, there is still a large gap between the limited ionic conductivity (<0.1 mS cm−1) of halide fast Na-ion conductors and the demand for practical applications. Besides, halide SSEs have the obvious advantage of abundant synthesis pathways, such as mechanochemical synthesis,41,60 co-melting synthesis,45 and wet chemical synthesis,61–63 which can avoid the high-temperature sintering process used in sulfide and oxide SSE synthesis and greatly reduce energy consumption and manufacturing cost.
In this review, the present development of halide SSEs and their practical applications in ASSBs are introduced. Firstly, the principle of screening halide SSE components is proposed. Secondly, the structural design theory of halide SSEs with high ionic conductivity and the mechanism of Li ion migration are described. Additionally, substitution strategies for halide SSEs are discussed, including dual-halogen, isovalent cation substitution, and aliovalent cation substitution. Furthermore, the mechanism of moisture resistance and the synthesis of halide electrolytes are presented. Next, the application prospects and challenges of halide SSEs in ASSBs are outlined. Finally, the summary and outlook of future development and challenges for halide SSEs are also presented along with the discussions for their commercial applications in ASSBs.
2. Composition, structure, and ion migration mechanism of halide SSEs
The composition and structure of materials determine their intrinsic chemical properties to some extent. In the past few years, the discovery of new materials has seriously relied on the researcher's experience and intuition and then been further validated by synthesis and characterization in a laboratory, which is considered an inefficient, resource-consuming, and expensive process. There are more than 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 entries in the Inorganic Crystal Structure Database (ICSD).64 If traditional trial-and-error approaches are used to screen them one by one, it can't meet the urgent demand for the development of new materials and energy technologies. Luckily, with the rapid development of material informatics, numerous advanced technologies such as artificial intelligence,65,66 machine learning,67,68 high-throughput screening,40,69ab initio molecular dynamics (AIMD),70,71 density functional theory (DFT),72 and first-principles calculations,73–75 are used to guide component screening, structural design, and ion diffusion prediction of crystalline inorganic SSEs.
000 entries in the Inorganic Crystal Structure Database (ICSD).64 If traditional trial-and-error approaches are used to screen them one by one, it can't meet the urgent demand for the development of new materials and energy technologies. Luckily, with the rapid development of material informatics, numerous advanced technologies such as artificial intelligence,65,66 machine learning,67,68 high-throughput screening,40,69ab initio molecular dynamics (AIMD),70,71 density functional theory (DFT),72 and first-principles calculations,73–75 are used to guide component screening, structural design, and ion diffusion prediction of crystalline inorganic SSEs.
2.1 Component screening of halide SSEs
In halide SSEs, the elemental composition of the phase field determines the range of chemical structure and bonding, thus affecting the related properties. The common method of designing SSEs is to select suitable primary elements to construct a non-rigid frame for fast Li ion conduction and compact solid–solid contact. The elements that are applicable to halide SSEs are shown in Fig. 2.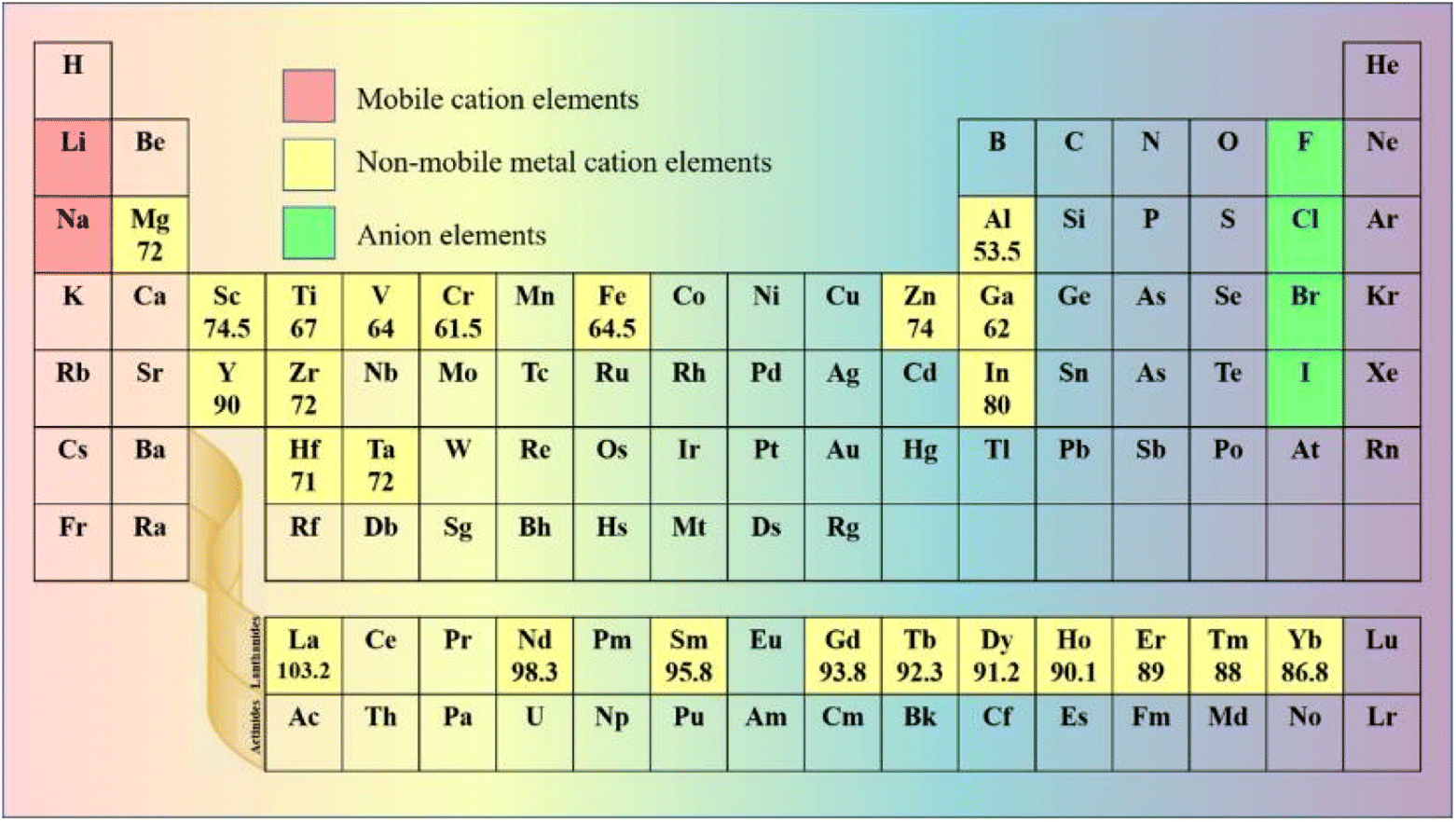 | ||
| Fig. 2 Elements in the periodic table that are applied to halide SSEs. The unit of ionic radius is pm, and the data come from ref. 76. | ||
Halides have become a better choice for SSE materials than oxides and sulfides. Muy et al.40 used lattice-dynamics descriptors to screen more than 14000 compounds, where 18 components were identified as the most promising fast Li-ion conductors according to computed Li-phonon band centers and electrochemical stability windows, including 4 fluorides, 9 chlorides, and 2 bromides. Among them, Li3ErCl6 was successfully synthesized and showed an ionic conductivity of 0.05–0.3 mS cm−1, which well proved the rationality of prediction results. Based on a data-driven approach, 6600 materials containing O, S, F, Cl, Br, I, N, P, Ge, or Si monatomic anions were screened, among which only chlorides and bromides could exhibit both fast ionic conduction and high oxidation potential.77 Rational selection of a halogen anion can also further improve the comprehensive performance of SSEs. Based on the DFT method, chlorides in trigonal halides Li3MX6 (X = Cl, Br, and I) exhibited faster ionic conductivity, a wider electrochemical stability window and a higher elastic modulus, which were more suitable for high-voltage cathodes.78 The first-principles calculations of Li2Sc2/3X4 (X = Cl, Br, and I) showed that the Cl anion was the best choice for superior performance in ASSB applications, due to its excellent ionic conductivity, electrochemical stability, and interfacial compatibility.79 This was mainly attributed to the formation of an ion pair between Cl and Sc, leading to high ionic conductivity (2.07 mS cm−1) and a wide electrochemical stability window (0.91–4.25 V). Consistent with this, the AIMD study for Li3YX6 (X = F, Cl, Br, and I) series showed that Li3YCl6 possessed the lowest activation energy and the highest ionic conductivity.71 The octahedron–octahedron (Oct–Oct) Li ion diffusion pathway and weak coulombic force between Li and Cl ions were the main factors that made it outstanding. Unfortunately, Li3YCl6 was most energetically favorable to form an antisite defect and transform into a lower ionic conductivity lattice, which made the experimentally measured ionic conductivity (0.51 mS cm−1) of Li3YCl6 far away from the predicted value (10.4 mS cm−1).33,71
Except for halogen anions, Li/cation configurations also have an important influence on ionic conductivity. The atomic size and valence electron configuration of non-mobile cationic elements determine their coordination environment and lattice volume and ultimately affect the corresponding ionic conductivity.73 The 202 Li-containing chlorides in the ICSD were screened using first-principles calculations,80 where 19 of them were considered potential Li superionic conductors (σLi > 1 mS cm−1 at room temperature), and their cations mainly included In, Mg, Zn, Zr, Er, Al, Sc, and Y elements. The low activation energy barrier for Li ion migration could be achieved in a target crystalline structure frame by adjusting the electronegativity difference between anionic and nonmobile cationic elements. Non-mobile cationic elements with high electronegativity were preferred for Li superionic conductors with a tetrahedral substructure. In contrast, non-mobile cationic elements with low electronegativity were preferred for Li superionic conductors with an octahedral substructure.73,81 DFT calculations for Li3MI6 (M = Sc, Y, and La) showed that the key event to the extremely fast diffusion of Li ions in Li superionic conductors with an octahedral substructure lied in the large electronegativity difference between anion elements and non-mobile metal cation elemenst.82 The high ionic conductivity of Li3YBr6, Li3LaI6, and Li3ErI6 was consistent with the octahedral principle.33,82,83
In general, Li3MCl6 (M = Sc, Y, and lanthanides) are the most potential halide SSEs, which can reconcile ionic conductivity with the electrochemical stability window.
2.2 Structural design of halide SSEs
The crystal structure of SSEs directly determines the diffusion pathway of Li ions and affects their ionic conductivity. The calculation result of sulfide indicated that body-centered cubic (bcc) allowed Li ions to hop directly from adjacent tetrahedral sites, with the lowest energy barrier and the highest ionic conductivity.84 However, most halide SSEs had anion sublattices with a hexagonal close-packed (hcp) or face centered cubic (fcc) structure, which were beneficial for the higher electronegativity and lower polarizability of halogen anions compared with O2− and S2− anions.27,80 For halide SSEs, the anion sublattice formed the framework of the crystal structure, where interstitial sites were occupied by Li ions and other cations. The anion sublattice was commonly hcp or cubic close-packed (ccp), while cation arrangement mainly depended on the component and synthetic method.41,85–87 The crystallographic structure of halide SSEs was dependent on the ionic radius (r) of the central metal element. When r was less than or equal to 80 pm, halide SSEs tended to have a monoclinic structure. When r was between 80 pm and 85 pm, they exhibited a trigonal structure. When r was greater than or equal to 85 pm, they seemed to have an orthorhombic structure.23,34,88 In general, ternary halides had five crystal structures, the monoclinic structure (space group C2/m, C2/c), halospinel structure (space group Fdm) with a ccp anion framework, orthorhombic structure (space group Pnma) and trigonal structure (Pm1) with an hcp anion framework.
Li3YCl6 and Li3YBr6 were the earliest reported halide SSEs with high ionic conductivity.33 Li3YCl6 had a trigonal structure and hcp anion framework with the octahedral coordination of all Cl atoms forming YCl63− and LiCl65− octahedra and the six LiCl66− octahedra surrounding every YCl63− octahedron (Fig. 3a).27,33 Y atoms occupied two distinct sites. One was a 1a site where the YCl63− octahedra were isolated from each other and were completely occupied by Y. Another one was a 2d site where the YCl63− octahedron shared a face along the c-axis direction and exhibited a noteworthy Y disorder. Li occupied 6g and 6h sites in whole or in part, forming chains with face-sharing along the c-axis direction and edge-sharing within the ab-plane, thus creating tetrahedral interstitial sites for Li+ diffusion. Li3ErCl6 and Li3HoCl6 were isostructural to Li3YBr6.41,89 Li3BrCl6 had a monoclinic structure and ccp anion framework. In the ccp anion framework, all the Br atoms were octahedrally coordinated and formed the edge-sharing YBr63− and LiBr65− octahedra (Fig. 3b).27,33 Y mostly occupied the 2a site and occupied the 4h site together with Li in the meantime, thus resulting in Li/Y-site disorder. Li was octahedrally coordinated (4g and 4h) and the edge-sharing octahedral arrangement led to multiple tetrahedral voids, where only one was occupied and the others were vacant. Another Li with tetrahedral coordination (8j) was reported later,90,91 in which LiBr43− tetrahedra shared an edge and connected two LiBr65− octahedra via a face and simultaneously connected the LiBr65− and YBr63− octahedra along the (001) plane (Fig. 3c). The chlorides Li3ScCl6, Li3TiCl6, and Li3InCl6 were isostructural to Li3YBr6.24,25,92
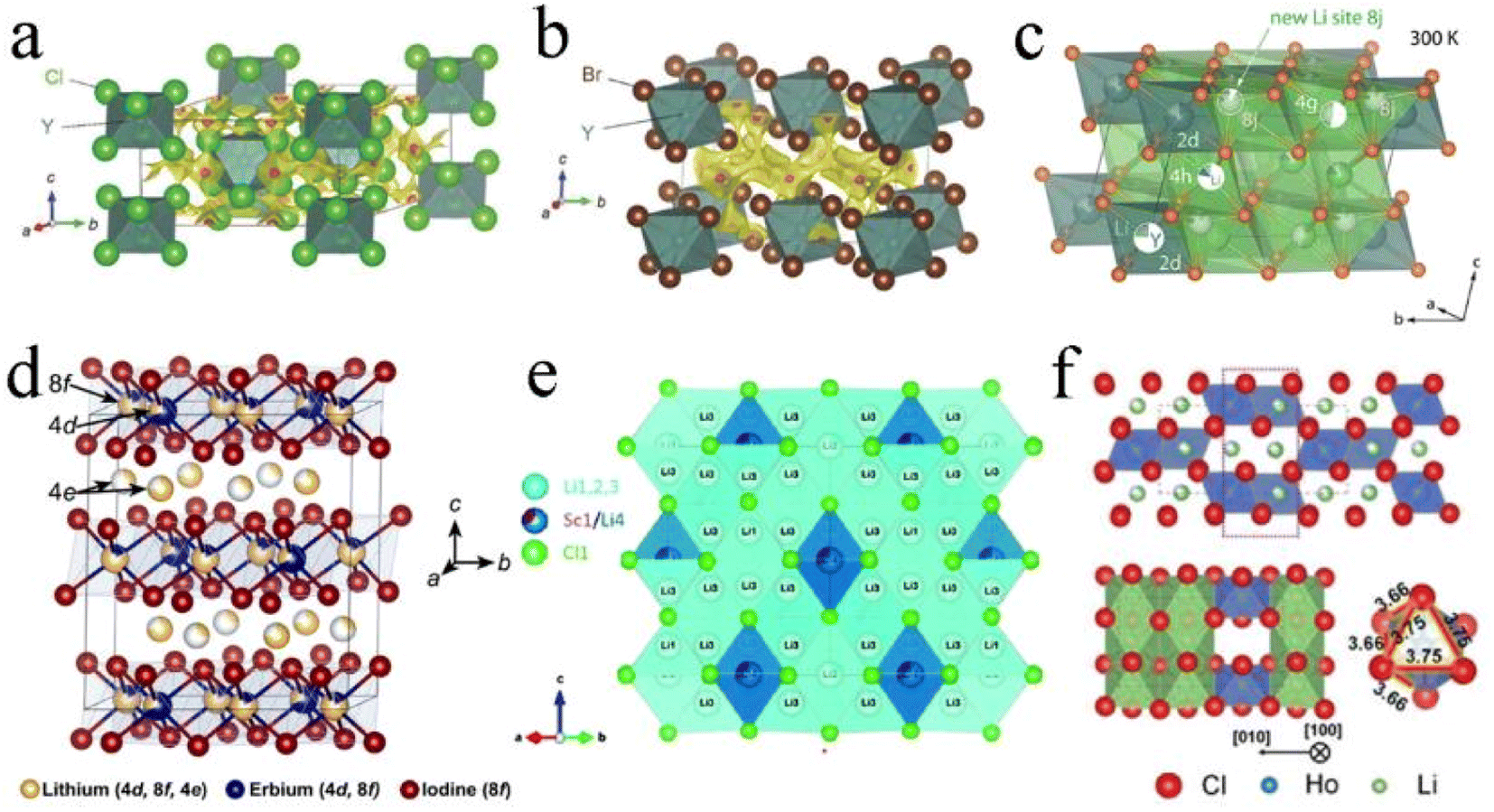 | ||
| Fig. 3 The crystal structures of (a) Li3YCl6 and (b) Li3YBr6 refined by Rietveld analysis and superimposed with a Li ion potential map. Reproduced with permission.33 Copyright 2018, Wiley-VCH. (c) The additional Li ion occupying interstitial 8j sites that connect 4g and 4h sites by face sharing in Li3YBr6. Reproduced with permission.91 Copyright 2020. American Chemical Society. (d) The crystal structure of Li3ErI6, where the Li occupies partially vacant sites, resulting in a cation-exchange defect. Reproduced with permission.83 Copyright 2020. American Chemical Society. (e) The crystal structure of disordered spinel Li2Sc2/3Cl4 from neutron diffraction. Reproduced with permission.45 Copyright 2020. American Chemical Society. (f) Orthorhombic structure and the HoCl63− octahedron of the Li3HoCl6 phase. Reproduced with permission.96 Copyright 2020. American Chemical Society. | ||
Another monoclinic structure with a C2/c space group was only reported in iodide SSEs.82,83 The structural framework of Li3ErI6 was constructed by edge-sharing ErI63− octahedra, in which Er partially occupied 4d and 8f sites. Li+ was in partially vacant sites and formed the Li/Er-site disorder within layers. Moreover, another Li+ site (4e) existed in interlayers between the edge-sharing ErI63− octahedra and formed an edge-sharing LiI65− octahedra layer. These edge-sharing octahedra created tetrahedral vacancies that could provide transitional sites for Li+ migration (Fig. 3d). In addition, the lithium tetrahaloaluminates LiAlX4 (X = Cl, Br, I) also had a monoclinic structure, but this series of SSEs had limited ionic conductivity.60,93,94
Moreover, Li2Sc2/3Cl4 with an Fdm space group also possessed the ccp anion framework. The halospinel Li2Sc2/3Cl4 was constructed from edge-sharing (Sc1/Li4)Cl6 octahedra with the remaining Li occupying the face-sharing octahedral (16c) and tetrahedral sites (8a and 48f) (Fig. 3e).45,79,95 The rigid connectivity of face-sharing octahedra and tetrahedra was more conducive to structural integrity. Both halospinel and monoclinic structures possessed a ccp anion sublattice, and the most important difference was that the Li/metal sites in the halospinel structure were shared, whereas all Li and metal sites were crystallographically ordered in the monoclinic structure.46 This disordered cation (Li+ and Sc3+) structure was conducive to Li ion migration.
The orthorhombic structure with a Pnma space group was reported only in Li3YbCl6 and Li3HoCl6, and the unit cell had 4c, 8d1, and 8d2 octahedral sites. Among them, the Li portion occupied 8d1 and 8d2 sites and M fully occupied the 4c site. Each MX63− octahedron was surrounded by three edge-sharing LiM65− octahedra (Fig. 3f).96,97
Currently, the reported quadrivalent metal chlorides only include Li2ZrCl6 and Li2HfCl6.68,86 Li2ZrCl6 had two different structures. The one was as-milled Li2ZrCl6 and exhibited a trigonal structure, which could be transformed into a monoclinic structure after annealing at 350 °C.86 The trigonal Li2ZrCl6 was isostructural to Li3YCl6, and the Li atom preferentially stayed at the 6h site. Another one was the monoclinic Li2ZrCl6, which was isostructural to Li3InCl6, and had lower occupancy at 2a and 4g sites. Unlike previous cases, the Li2ZrCl6 with a trigonal structure and hcp anion sublattice had a higher ionic conductivity than that with a monoclinic structure and ccp anion sublattice (0.81 mS cm−1vs. 5.81 × 10−3 mS cm−1). Amounts of nonperiodic features formed in the process of ball milling promoted Li+ migration in the trigonal structure. For the monoclinic structure, although Li+ migration in the ab plane had an energy barrier, the Zr sites effectively hindered Li+ migration between the neighboring ab planes, which made it possess two-dimensional (2D) diffusion characteristics. Based on the stochastic surface walking-neural network potential method, Li2ZrCl6 and Li2HfCl6 phases with novel layered structures were designed.68 This layered Li2ZrCl6 phase was composed of LiCl65− and ZrCl62− octahedra, which shared the common edges and formed layer-by-layer configuration along the c-axis direction. Based on six-coordinating halogen anions with Oct-sites occupied by Zr4+ and Li+ cations, the Li2ZrCl6 layered structure contained ideal and Zr-deficient layers with sufficient intrinsic vacancies. This layered structure was quite different from other reports,85,86 containing more vacancies and defects and giving Li2ZrCl6 more substantial ionic conductivity (∼1 mS cm−1).
In addition to hcp and ccp anion sublattices, a high ionic conductivity halide SSE with a non-close-packed anion lattice has recently been reported.17 This halide SSE was based on LaCl3 with a P63/m space group and doped by Ta5+ to form a three-dimensional (3D) Li+ diffusion channel to obtain considerable ionic conductivity. In Li0.388Ta0.238La0.475Cl3, Li+ has two sites: a 2b site in channels (Li1) and 6h1 site near La sites (Li2). The former coordinates with 6Cl− to form a compressed [LiCl6] octahedron. The latter is a metastable intermediate site conducive to the mobility of Li+ and coordinates with adjacent 5Cl− to form a rectangular pyramid. La3+ and Ta5+ occupy part of the 2c site together. The key factor for the high ionic conductivity of Li0.388Ta0.238La0.475Cl3 is the abundance of La vacancies formed by Ta5+ doping, which connects the one-dimensional (1D) channels in the raw structure into three dimensions.
In both Pm1 Li3YCl6 and C2/m Li3YBr6, the cations (Y3+ and Li+) occupied six-coordinated octahedral sites with the halogen anions. Vacancies and Li ion disorders occupied one third of Oct sites, which could facilitate Li ion migration and enable the conductivity of Li3YCl6 and Li3YBr6 to be 0.51 mS cm−1 and 1.7 mS cm−1, respectively. It seemed that the ccp sublattice was more favorable for Li ion migration. By adjusting the lattice chemistry of hexagonal Li3YCl6 into a spinel-like cubic structure, a three-dimensional (3D) channel for efficient Li+ transportation could be formed, thus achieving higher ionic conductivity.98 Park et al. calculated 51 structures of 17 chloride electrolytes (Li3MCl6), clearly showing that a monoclinic structure had a lower migration energy barrier and activation energy than orthorhombic and trigonal structures.99 In orthorhombic and trigonal structures, the sluggish migration of Li+ along the 2D pathway was the main factor for its mediocre ionic conductivity. Among these three space groups of ternary chlorides, the trigonal structure exhibited the highest energy barriers.99 Besides, a tremendous amount of experimental results also confirmed that the monoclinic structure was more favorable for Li ion diffusion than the orthorhombic structure of halides with the same composition.38,43,99 This was primarily benefited from the existence of tetrahedral Li+ sites and reasonable 3D ionic migration pathways in monoclinic structures. The existence of Li ions in tetrahedral sites was favorable for Li ion diffusion.43 However, Wan and co-workers took the opposite viewpoint, arguing that the ionic conductivity of the trigonal Pm1 Li3YCl6 phase was tens of times higher than that of the monoclinic Li3YCl6 phase C2/c Li3YCl6 phase (10.4 vs. 0.69 mS cm−1 at 300 K).71 The results of DFT showed that there were two types of 2D ab-plane diffusion paths (Oct–Tet–Oct) with migration barriers of 0.23 and 0.20 eV in the Pm1 phase, while there were also other one-dimensional (1D) diffusion paths (Oct–Oct) with a lower barrier (0.16 eV) in the C2/c phase.
However, the trigonal Li2ZrCl6 exhibits higher ionic conductivity than the monoclinic phase.85,86 The monoclinic Li2ZrCl6 has a similar crystal structure to Li3InCl6, but the concentration of mobile charge carriers is lower. Furthermore, the ionic radius of Zr4+ is smaller than that of In3+, and the stronger coulombic repulsion between Zr4+ and Li+ and narrower ion diffusion channel further raise the activation energy barrier for Li+ migration. Therefore, the ionic conductivity of monoclinic Li2ZrCl6 is not as expected. The trigonal Li2ZrCl6 phase is a low-temperature metastable phase with a low crystallinity. The trigonal Li2ZrCl6 has a similar crystal structure to Li3YCl6, and significant amount of nonperiodic features in the crystal structure are key factors for maintaining considerable ionic conductivity. This means the factors affecting the ionic conductivity of halide electrolytes are complex and the crystal structure, carrier concentration, cation radius and defects jointly determine the Li+ migration energy barrier.
Studies on a range of Li3−3xM1+xCl6 (M = Tb, Dy, Ho, Y, Er, Tm) with hcp anion stacking showed that the ionic conductivity of the orthorhombic phase was approximately one order of magnitude higher than that of the trigonal phase.96 According to the AIMD results, the orthorhombic phase had a clear long-range migration pathway along the z-axis direction and the hop along the z-axis direction had a lower energy barrier than the cross-plane hop. However, the hop was mostly around the hexagon rather than connecting the hexagon in the trigonal phase. The first-principles calculation result of Li3YbCl6 also considered that the orthorhombic structure was superior to the trigonal structure for ionic conductivity.100
In brief, halides with a ccp sublattice are more likely to obtain high ionic conductivity, and an orthorhombic structure with a hcp sublattice is more favorable to Li ion migration than a trigonal structure.
2.3 Ionic migration mechanism
Ionic conductivity is easily affected by the ionic diffusion channel and carrier concentration, which were mainly dependent on the crystalline structure, ion spatial array, crystal defect and ion migration mechanism.84,101,102 The summary of halide SSEs with an ionic conductivity of >0.1 mS cm−1 since 2018 is shown in Table 1.| Composition | Structure | Synthesis method | σ [mS cm−1] | Ref. |
|---|---|---|---|---|
| a BM: ball milling; SSR: solid state reaction; WCS: wet-chemistry synthesis. | ||||
| Li3YCl6 | Trigonal, Pm1 |
BM | 0.51 | 33 |
| Li3YCl6 | Trigonal, Pm1 |
WCS | 0.345 | 63 |
| Li2.5Y0.5In0.5Cl6 | Monoclinic, C2/m | BM + annealing (260 °C) | 1.42 | 38 |
| Li2.5Y0.5Zr0.5Cl6 | Orthorhombic, Pnma | SSR (450 °C) | 1.4 | 44 |
| Li3YBr1.5Cl4.5 | Trigonal, Pm1 |
SSR (650 °C) | 2.1 | 103 |
| Li3YBr3Cl3 | Monoclinic, C2/m | BM + annealing (200 °C) | 7.2 | 104 |
| Li3YBr4.5Cl1.5 | Monoclinic, C2/m | SSR (650 °C) | 5.36 | 103 |
| Li3InCl6 | Monoclinic, C2/m | WCS | 0.79 | 62 |
| Li3InCl6 | Monoclinic, C2/m | BM + annealing (260 °C) | 1.49 | 25 |
| Li3InCl6 | Monoclinic, C2/m | WCS | 2.04 | 61 |
| Li2.7In0.7Zr0.3Cl6 | Monoclinic, C2/m | BM + annealing (550 °C) | 2.1 | 105 |
| Li2.7In0.7Zr0.3Cl6 | Monoclinic, C2/m | SSR (450 °C) | 2.02 | 106 |
| Li2.6In0.6Zr0.4Cl6 | Monoclinic, C2/m | BM + annealing (260 °C) | 1.25 | 107 |
| Li2.75In0.75Zr0.25Cl6 | Monoclinic, C2/m | BM + annealing (450 °C) | 5.82 | 108 |
| Li2.9In0.9Zr0.1Cl6 | Monoclinic, C2/m | BM + annealing (260 °C) | 1.54 | 109 |
| Li2.7In0.7Hf0.3Cl6 | Monoclinic, C2/m | BM + annealing (350 °C) | 1.28 | 88 |
| Li2In0.444Sc0.222Cl4 | Spine, Fdm |
BM + annealing (450 °C) | 2.03 | 46 |
| Li3InCl4.8F1.2 | Monoclinic, C2/m | BM + annealing (260 °C) | 0.51 | 110 |
| Li3InCl5.6F0.4 | Monoclinic, C2/m | BM + annealing (300 °C) | 1.37 | 111 |
| Li3ErCl6 | Trigonal, Pm1 |
BM + annealing (550 °C) | 0.3 | 40 |
| Li3ErCl6 | Trigonal, Pm1 |
WCS | 0.407 | 63 |
| Li3ErCl6 | Trigonal, Pm1 |
BM | 0.33 | 41 |
| Li3ErCl6 | Trigonal, Pm1 |
BM + annealing (550 °C) | 0.1 | 41 |
| Li2.6Er0.6Zr0.4Cl6 | Trigonal, Pm1 |
BM | 1.38 | 39 |
| Li2.633Er0.633Zr0.367Cl6 | Orthorhombic, Pnma | SSR (450 °C) | 1.1 | 44 |
| Li3YbCl6 | Trigonal, Pm1 |
BM + annealing (400 °C) | 0.19 | 43 |
| Li3YbCl6 | Orthorhombic, Pnma | BM + annealing (500 °C) | 0.14 | 43 |
| Li2.556Yb0.492Zr0.492Cl6 | Orthorhombic, Pnma | SSR (450 °C) | 1.58 | 100 |
| Li2.7Yb0.7Hf0.3Cl6 | Orthorhombic, Pnma | BM + annealing (350 °C) | 1.1 | 97 |
| Li2.6Yb0.6Hf0.4Cl6 | Monoclinic, C2/m | BM + annealing (400 °C) | 1.5 | 43 |
| Li2.6Yb0.6Hf0.4Cl6 | Orthorhombic, Pnma | BM + annealing (500 °C) | 1.2 | 43 |
| Li3TiCl6 | Monoclinic, C2/m | BM | 0.115 | 92 |
| Li3TiCl6 | Monoclinic, C2/m | BM + annealing (300 °C) | 1.04 | 92 |
| Li2ZrCl6 | Trigonal, Pm1 |
BM | 0.4 | 85 |
| Li2.25Zr0.75Fe0.25Cl6 | Trigonal, Pm1 |
BM | 0.98 | 85 |
| Li2.25Zr0.75V0.25Cl6 | Trigonal, Pm1 |
BM | ∼0.9 | 85 |
| Li2.1Zr0.9Cr0.1Cl6 | Trigonal, Pm1 |
BM | ∼0.85 | 85 |
| Li2.25Zr0.75In0.25Cl6 | Trigonal, Pm1 |
BM | 1.08 | 112 |
| Li2.1Zr0.95Mg0.05Cl6 | Trigonal, Pm1 |
BM | 0.62 | 113 |
| ZrO2–2LiCl–Li2ZrCl6 | Trigonal, Pm1 |
BM + annealing (550 °C) | 1.3 | 114 |
| Li2ZrCl6 | Layered structure | BM | ∼1.0 | 68 |
| Li2HfCl6 | Layered structure | BM | ∼0.5 | 68 |
| Li3HoCl6 | Trigonal, Pm1 |
SSR | 1.05 | 89 |
| Li2.73Ho1.09Cl6 | Trigonal, Pm1 |
SSR (650 °C) | 1.3 | 96 |
| Li2Sc2/3Cl4 | Spine, Fdm |
SSR (680 °C) | 1.5 | 45 |
| Li3ScCl6 | Monoclinic, C2/m | SSR (650 °C) | 3.02 | 24 |
| Li3ScCl6 | Monoclinic, C2/m | WCS | 1.25 | 63 |
| Li2.5Sc0.5Zr0.5Cl6 | Monoclinic, C2/m | SSR (500 °C) | 2.23 | 115 |
| Li2.6Sc0.6Zr0.4Cl6 | Monoclinic, C2/m | BM + annealing (450 °C) | 1.61 | 116 |
| Li2.6Sc0.6Hf0.4Cl6 | Monoclinic, C2/m | BM + annealing (450 °C) | 1.33 | 116 |
| LiTaCl6 | Glass-phase | BM + annealing (120 °C) | 10.95 | 117 |
| Li3YBr6 | Monoclinic, C2/m | BM + annealing (550 °C) | 1.7 | 33 |
| Li3YBr6 | Monoclinic, C2/m | WCS | 1.09 | 63 |
| Li3YBr5.7F0.3 | Monoclinic, C2/m | SSR (950 °C) | 1.8 | 118 |
| Li3HoBr6 | Monoclinic, C12/m1 | SSR (450 °C) | 1.1 | 119 |
| Li3HoBr6 | Monoclinic, C12/m1 | WCS | 1.25 | 120 |
| Li3HoBr2.9I3.1 | Monoclinic, C2/m | SSR (1000 °C) | 2.7 | 121 |
| Li3ErI6 | Monoclinic, C2/c | BM | 0.65 | 83 |
| Li3ErI6 | Monoclinic, C2/c | BM + annealing (550 °C) | 0.39 | 83 |
For Li3YCl6 with an hcp-like anion arrangement, there existed two different hop possibilities for transport among connected polyhedra:27,33,71 (1) the hop along the c-axis direction, where the pathway was directly connected between adjacent octahedral sites (Oct–Oct) and had a low energy barrier of 0.25 eV and (2) the hop in the ab-plane, where the pathway was via tetrahedral interstitial sites (Oct–Tet–Oct) and exhibited an energy barrier of 0.29 eV (Fig. 4a). The ionic conductivities along the c-, a-, and b-axis in Li3YCl6 were 85.4, 0.3, and 0.7 mS cm−1, respectively.122 It could be seen that Li+ diffusion in Li3YCl6 was highly anisotropic and had a fast c-axis 1D diffusion channel. Such anisotropic diffusion made the conductivity extremely susceptible to channel-blocking defects, including anti-site defects, impurities, and grain boundaries, which caused ionic conductivity to deviate significantly from theoretical prediction. For Li3YBr6 with a ccp anion arrangement, the Li+ conducting pathway showed a 3D isotropic network and Li+ hopped to the adjacent octahedral site through a tetrahedral interstitial site (Oct–Tet–Oct pathway), thereby exhibiting a barrier of 0.28 eV (Fig. 4b).27 Further AIMD results showed that the 3D isotropic diffusion pathway was based on two channels: the hopping between different Li-1 sites in the (001) plane, and the activation energy was between 0.11 and 0.19 eV; the hopping between Li-1 and Li-2 sites along the [001] direction, and the activation energy was 0.39 eV.123
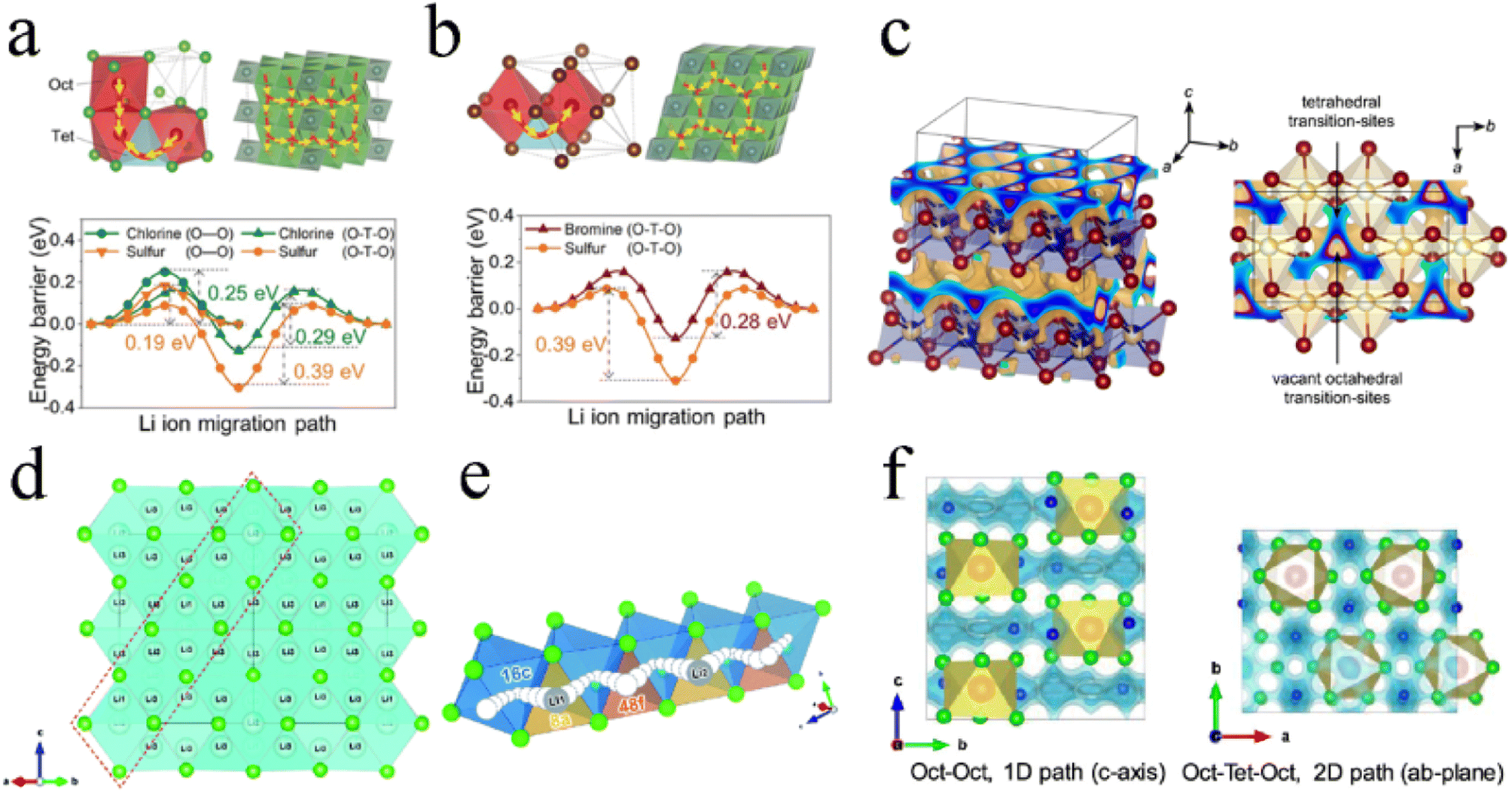 | ||
| Fig. 4 The Li+ migration pathways and corresponding energy landscape in (a) Li3YCl6 and (b) Li3YBr6. Reproduced with permission.27 Copyright 2019, Wiley-VCH. (c) Possible Li+ diffusion pathways of Li3ErI6 obtained by a bond valence sum calculation. Reproduced with permission.83 Copyright 2020. American Chemical Society. (d) Structure and 3D Li+ diffusion pathway of disordered spinel Li2Sc2/3Cl4. Reproduced with permission.45 Copyright 2020. The Royal Society of Chemistry. (e) Schematic diagram of collective Li+ motion. Reproduced with permission.79 Copyright 2020. The Royal Society of Chemistry. (f) Li+ migration pathway in orthorhombic Li3MCl6 obtained by the BVSE method. Reproduced with permission.99 Copyright 2020. American Chemical Society. | ||
The ion migration of Li3ErI6 with a monoclinic structure was mainly 2D in the ab-plane (Fig. 4c). In the ab-plane, Li sites were strongly interconnected with each other, and the tetrahedral voids left by the edge-sharing of octahedra were used as transition sites for Li+ diffusion. Of course, the strong disorder between Li+ and Er3+ indicated the possibility of Li+ migration along the c-axis direction. Based on DFT calculations, monoclinic Li3HoBr6 had four possible diffusion pathways.119 The vacancies in the lattice provided different pathways for Li ion diffusion. The energy barriers of these two in-plane pathways were 0.22 eV and 0.46 eV for Oct–VOct and Oct–Oct pathways, respectively. In addition, the energy barriers for two out-of-plane paths were 0.12 eV and 0.24 eV for Oct–Tet–VOct and Oct–Tet–Oct pathways, respectively. In contrast, Li ions showed a lower energy barrier to migrate through vacant octahedral sites. However, the number of such natural vacant octahedral sites was limited and couldn't fully supply Li ion migration. The Oct–Tet–Oct pathway in the out-of-plane could be used as a complementary contribution to Li ion migration. The synergistic effect of multiple diffusion pathways with low activation barriers in Li3HoBr6 well guaranteed the high ionic conductivity.
The high ionic conductivity of Li2Sc2/3Cl4 was mainly due to multiple Li sites in the spinel lattice. The rigid framework formed by the Li/Sc shared site and the presence of Li+ throughout the face-sharing octahedral and tetrahedral sites, provided the conditions for infinite 3D Li+ diffusion pathways (Fig. 4d). In addition, the large number of vacancies formed by the Li deficiency strategy was beneficial in eliminating the defect formation step for Li+ diffusion.45 Li2Sc2/3Cl4 prepared by a facile synthesis process exhibited an ionic conductivity of 1.5 mS cm−1. The results of DFT showed that halospinel Li2Sc2/3Cl4 had the potential for ionic conductivity of up to ∼2.07 mS cm−1 under a collective ionic motion mechanism.79 Before the first Li (Li1) at the 8a site hopped to the 48f site, the strong Coulomb repulsion between Li1 and Li2 drove Li2 to jump to the next 48f site, followed by Li1 hopping to the 8a site, where Li2 was initially located. Hence, rapid Li+ migration was achieved repeatedly (Fig. 4e).
The orthorhombic structure was similar to the trigonal structure and exhibited the anisotropic diffusion pathways of Li ions (Fig. 4f): 1D diffusion pathway along the c-axis direction between the octahedral sites (Oct–Oct pathway) with lower activation energy and 2D diffusion pathway in the ab-plane between octahedral sites and interstitial tetrahedral sites (Oct–Tet–Oct pathway) with higher activation energy.
The site disorder in the crystal structure had a direct influence on the ion diffusion pathway of halide SSEs. At present, researchers don't have a unified understanding of the relevant mechanism. Experimental studies on Li3YCl6 and Li3ErCl6 showed that higher cationic site disorder was favorable to ion transport.33,40,41 The disordered and distorted structure expanded the bottleneck of Li+ diffusion and significantly affected the transport mechanism. Consistent with this result, the stacking faults in Li3YCl6 could reduce the Li+ migration barrier and generate more interlayer channels, thereby promoting Li+ conduction in the structure.124 In contrast, theoretical work suggested that antisite disorder blocked the Li+ diffusion channel and reduced conductivity by about one order of magnitude.25 Calculation results by Wan et al. showed that antisite defects formed in Li3YCl6 were favorable for converting the Pm1 lattice to the C2/c lattice with low ionic conductivity and limiting Li+ transportation.71
3. Substitution in halide SSEs
Chemical substitution in known ionic conductors was often used to develop new electrolytes with improved ionic conductivity, electrochemical/chemical stability, and environmental stability.35,125,1263.1 Haloanion substitution
Fluoride exhibited a wider electrochemical stability window, while chloride and bromide had more prominent ionic conductivity. Therefore, dual-halogen SSEs were formed by haloanion substitution, which could balance oxidative stability and ionic conductivity and obtain better comprehensive performance. The introduction of Cl− into Li2ZrF6−xClx could maintain the excellent Li interface stability, meanwhile improving ionic conductivity by two orders of magnitude.127 Compared with other halide anions, F− had the smallest ionic radius. Therefore, F− substitution usually led to lattice shrinkage.110,111,118,128 F− had shorter and stronger bonds with Li+ in comparison with Cl−, increasing the barrier for Li+ migration. And the conductivity of chloride decreased slightly after F− doping.71 In F-doped Li3InCl4.8F1.2, Cl occupied symmetrical 4i and 8j sites and F occupied partial 8j sites. Cl and F were stacked layer-by-layer to form an edge-share six-coordinated octahedron, and the cation and vacancy occupied octahedral interstitial sites (Fig. 5a).110 The experimental results showed that the ionic conductivity of Li3InCl6−xFx gradually decreased with the F content increasing (Fig. 5b).111 However, according to the first-principles theoretical method, the influence of F-doping on Li+ migration in Li3InCl6 was not unilateral.128 On the one hand, F-doping reduced the energy barrier of site-to-site hops, which was conducive to the migration of some Li ions to the empty space of the In layer and generating Li vacancies and triggering the diffusion of other Li ions. On the other hand, the strong electronegativity of F− led to high electrostatic interactions between Li+ and F−, which limited the long-term travel range of nearby Li+ under a high correlation effect. Therefore, it was necessary to balance the positive and negative effects by controlling the concentration of F-doping for achieving desired ionic conductivity. Interestingly, no matter whether the F-doping increased or decreased ionic conductivity, F-doping improved the crystal structure rigidity of halides, expanded the electrochemical window, and enhanced the stability of the cathode interface.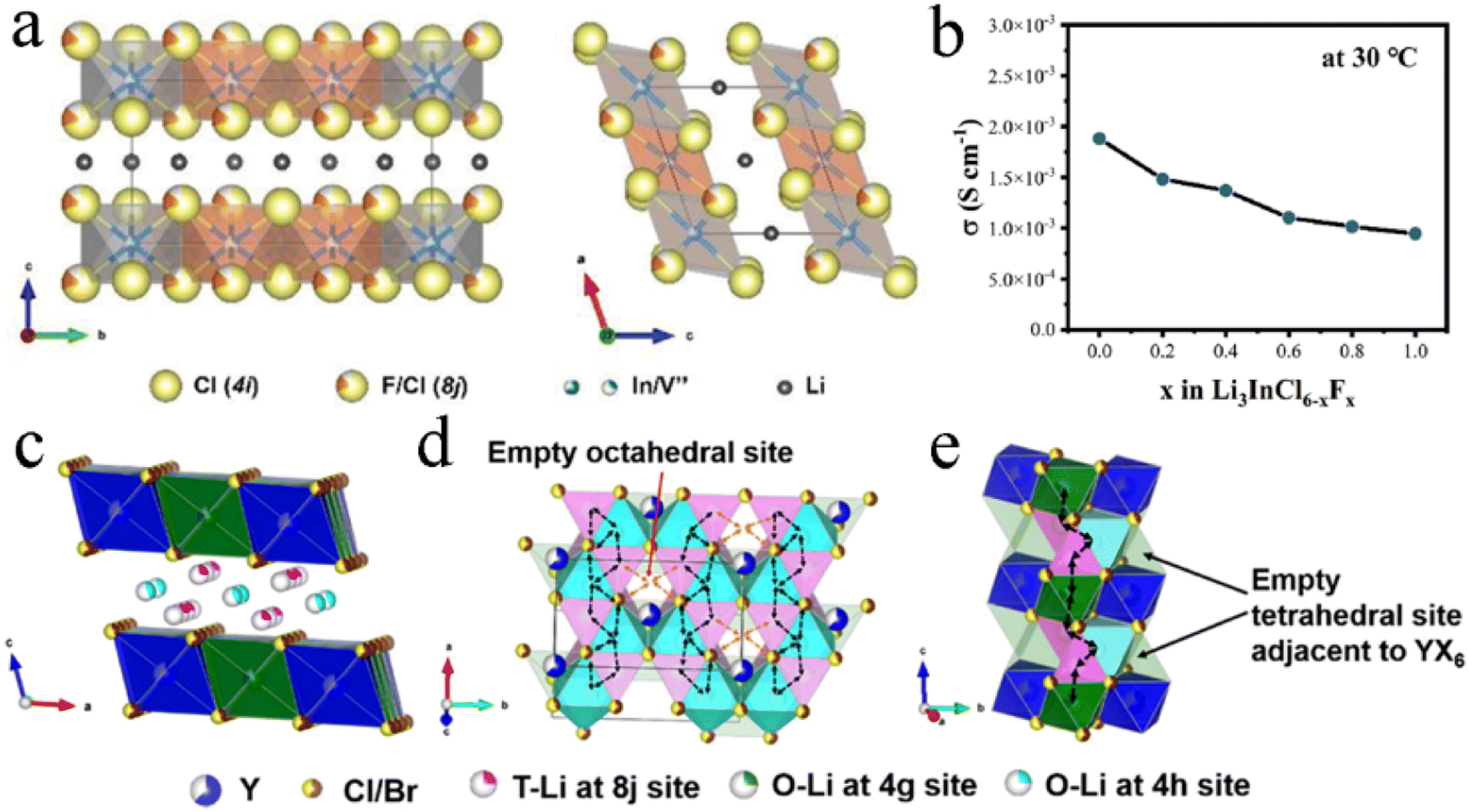 | ||
| Fig. 5 (a) Crystal structure of Li3InCl4.8F1.2 viewed from different orientations. Reproduced with permission.110 Copyright 2021, Wiley-VCH. (b) Ionic conductivity of Li3InCl6−xFx with different F-contents at room temperature. Reproduced with permission.111 Copyright 2022. Elsevier B.V. (c) Crystal structure of Li3Y(Br3Cl3) viewed from parallel to the a-axis. Reproduced with permission.104 Copyright 2020. American Chemical Society. Migration routes of Li ions along the (d) ab-plane and (e) c-axis direction. Reproduced with permission.104 Copyright 2020. American Chemical Society. | ||
Introducing Br into Li3YCl6 could tune the ionic conductivity and oxidative stability.103 As the Br content increased, the structure of Li3YBrxCl6−x changed from trigonal to monoclinic when x was greater than 1.5. The ionic conductivity of Li3YBrxCl6−x increased from 2.1 to 5.36 mS cm−1, with the Br content increasing until the formation of Li3YBr6. This was mainly attributed to the larger ionic radius, smaller electronegativity, and larger lattice polarizability of Br− than Cl− and also the change in the crystal structure. However, Br-enriched materials showed lower oxidative stability.103 In other studies, Li3Y(Br3Cl3) with a mixed haloanion exhibited ionic conductivity up to 7.2 mS cm−1 in practice and was estimated to reach 22.3 mS cm−1 in theory.104,122 This performance was probably due to the unique Li ion site and interlayer concerted diffusion mechanism. Unlike the crystal structure of C2/m Li3YBr6, a considerable amount of Li+ occupied the multiple tetrahedral sites (8j) in addition to octahedral sites (4h and 4g) (Fig. 5c).104 Octahedral Li occupied about one third of the total, while tetrahedral Li occupied the remaining two thirds. The existence of tetrahedral Li produced more octahedral vacancies and expanded the diversity of Li+ diffusion pathways. In the ab plane, two tetrahedral Li ions surround one neighboring octahedral Li ion, forming an Oct–Tet–Oct chain along the a-axis direction for Li hopping. These parallel chains were connected by empty octahedral sites, forming a 2D diffusion network (Fig. 5d). Along the c-axis direction, zigzag “Oct–Ter–Oct–Ter” routes near the Y sites were provided for Li ions to hop in adjacent ab-planes and construct 3D diffusion pathways (Fig. 5e). In addition to the 3D diffusion pathways, the favorable grain boundaries were also contributors to the high ionic conductivity of Li3Y(Br3Cl3).104 The first-principles study revealed that the high ionic conductivity of Li3Y(Br3Cl3) was due to an interlayer concerted diffusion mechanism.122 In Li3Y(Br3Cl3), the intralayer vacancy diffusion in the b-axis direction promoted the interlayer concerted diffusion in the diagonal direction between the a- and c-axis, with two Li ions moving simultaneously across Li, halide, and Y layers, thereby collectively contributing to the ultra-high ionic conductivity.
It is also a method to tune the ionic conductivity of halide SSEs by doping halide anions to construct the structure of intralayer cation disorder and stacking faults.121 Introducing I− into Li3HoBr6 does not change its monoclinic structure, but results in an increase in the a- and b-lattice parameters and the interlayer distance. Meanwhile, Li3HoBr6−xIx exhibits complete randomization in the staggered stacking direction. In addition, substitution causes an increase in the volumes of the different coordination polyhedra, Li enters the Ho sites, and the cation disorder in the layer increases greatly. The doping of I− on the one hand softens the lattice and promotes the diffusion of Li ions, and on the other hand increases the cation disorder and hinders the movement of Li ions. The two factors compete and jointly affect the ionic conductivity of Li3HoBr6−xIx. The degree of stacking faults does not seem to have a decisive effect on ionic conductivity.121 However, another study showed that stacking faults in Li3YCl6 can reduce Li+ migration barriers, generate more interlayer channels for Li+ transport, and promote long-range Li+ conduction.124
In addition, O2− can be used to replace the haloanion in the halide SSE to improve its ionic conductivity.114,117 The introduction of O2− into Li2ZrCl6 by one-pot mechanochemical synthesis to form halide nanocomposite SSE (ZrO2–2Li2ZrCl6) can increase the ionic conductivity of the electrolyte by more than three times, from 0.40 to 1.3 mS cm−1.114 This enhancement is due to the widening of Li+ transport channels and the increase in the Li+ concentration caused by the local anion substitution at the interface of the nanocomposite SSE. At the same time, the halide nanocomposite SSE has higher compatibility with sulfide Li6PS5Cl at elevated temperature. Moreover, this strategy is universal and can be applied to Al2O3–3Li2ZrCl6, SnO2–2Li2ZrCl6, 0.75ZrO2–Li2.25Zr0.75Fe0.25Cl6, and ZrO2–2Li2ZrCl5F.114 The ionic conductivity of LiTaCl6-based polyanionic glass-phase LiTaCl5X1/nn− (Xn− = F−, Cl−, Br−, I−, O2−, OH−, O2−, S2−) can even reach 10 mS cm−1.117
3.2 Isovalent cation substitution
The effect of isovalent cation substitution on halide SSEs was mainly due to the otherness of ionic sizes. Replacing Y3+ in Li3YCl6 with a larger La3+ could expand the size of the Li ion diffusion channel and thus reduce activation energy.71 In addition, substitution also caused a change in the crystal structure. In Li3Y1−xInxCl6, the anion substructure changed from hcp to ccp with the In content increasing.38 After substitution, Y and In atoms occupied the 4g position together, forming (Y/In)Cl63− octahedra. The increase in In3+ content led to a shrinkage of the unit cell volume, mainly due to the smaller ionic radius of In3+ (80 pm) than that of Y3+ (90 pm). The ionic conductivity of Li3Y1−xInxCl6 increased abruptly with the structural change from pristine hcp to the ccp anion sublattice (Fig. 6a), which further demonstrated the superiority of the ccp anion sublattice structure in Li ion transport. The ionic conductivity of monoclinic Li3Y1−xInxCl6 (x ≥ 0.2) remained at a high level and was not affected by lattice shrinkage. With the increase in In content, the activation energy continued to decrease, but the ionic conductivity didn't linearly increase. This was because the ionic conductivity was affected not only by activation energy, but also by the concentration of mobile ions, activation entropy and other factors.38 With the increase in In content, the crystal structure of Li2InxSc0.666−xCl4 (0 < x < 0.666) changed from halospinel to monoclinic.46 The spinel Li2In1/3Sc1/3Cl4 was constructed from the edge-sharing (In1/Sc1/Li4)Cl6 octahedral rigid framework, while the additional Li+ spread throughout face-sharing octahedral and tetrahedral sites (Fig. 6b). The low occupancy of face-sharing octahedral and tetrahedral sites provided a considerable number of vacancies for Li+ mobility, which could form 3D ion diffusion channels with relatively low activation energy and obtain an ionic conductivity of more than 2 mS cm−1 (Fig. 6c).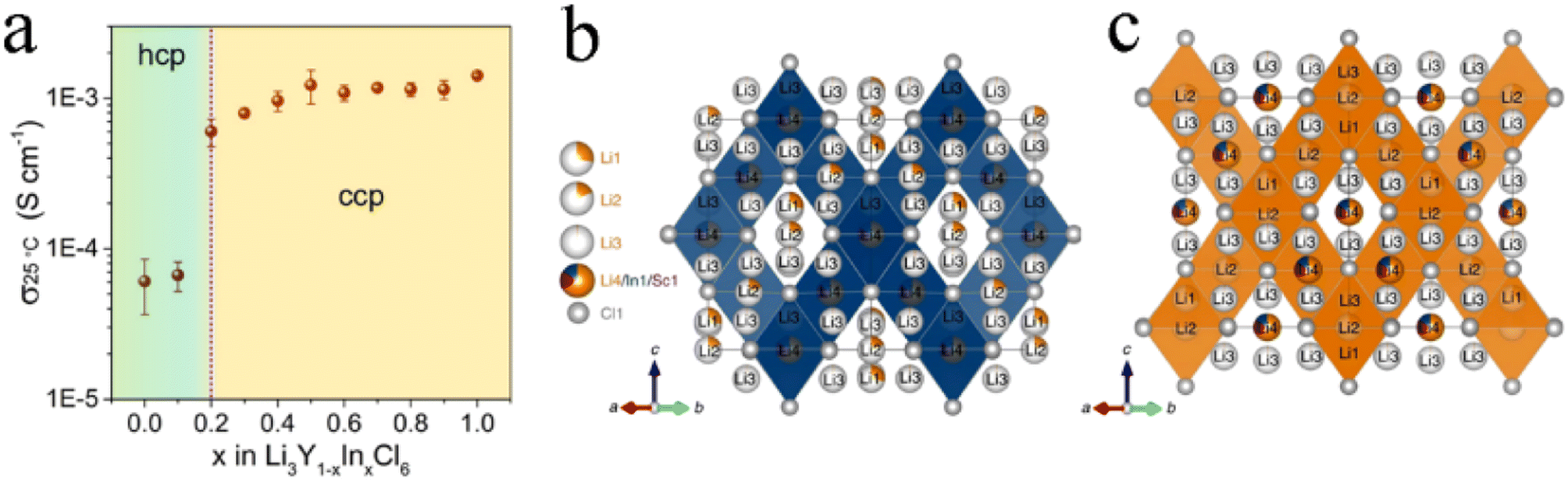 | ||
| Fig. 6 (a) Ionic conductivity of Li3Y1−xInxCl6 with different In contents at 25 °C. Reproduced with permission.38 Copyright 2020, American Chemical Society. Structure (b) and the main 3D Li ion diffusion pathway (c) of Li2In1/3Sc1/3Cl4. Reproduced with permission.46 Copyright 2022, Springer Nature. | ||
3.3 Aliovalent cation substitution
An aliovalent substitution could significantly affect the ionic conductivity and activation energy, due to providing more free interstitial sites, increasing the number of charge carriers, and broadening diffusion pathways.Tetravalent Zr4+ and Hf4+ were usually doped into trivalent metal halides as aliovalent ions.39,44,88,97,106–108 The introduction of Zr4+ into Li3YCl6 and Li3ErCl6 by a solid-state reaction at 450 °C resulted in crystal structure changes.44 As Zr content increased, their crystal structure changed from the trigonal structure, first to an orthorhombic-I structure and then to an orthorhombic-II structure (Fig. 7a). The former phase transformation process involved the rearrangement of metal ions, and the latter one only led to the occurrence of the tilt for (Er/Zr)Cl6 octahedra and created an additional tetrahedral Li site. Compared with the other two crystal structures, the orthorhombic-II structure had obvious advantages in ionic conductivity. The ionic conductivities of Li2.633Er0.633Zr0.367Cl6 and Li2.5Y0.5Zr0.5Cl6 with the orthorhombic-II structure were 1.1 and 1.4 mS cm−1, respectively. However, another report was against this viewpoint that Zr-doping caused phase transitions.39 Li3−xEr1−xZrxCl6 prepared by ball-milling had the same trigonal structure as Li3ErCl6 regardless of the Zr content (Fig. 7b). And this structure didn't change during annealing. The ionic conductivity of ball-milled Li3−xEr1−xZrxCl6 first increased and then decreased with an increase in Zr content, where the maximum value reached 1.38 mS cm−1 at x = 0.6 (Fig. 7c). On the one hand, the appropriate Zr4+ substitution increased the concentration of Li vacancies and Er/Zr site disorder, facilitating the increase in ionic conductivity. On the other hand, excessive Zr4+ substitution reduced the concentration of Li+ and then caused lattice shrinkage and narrowed the transport channel of Li+, thus impeding the migration of Li+. The AIMD results also showed that replacing Y3+ with Zr4+ in Li3YCl6 could not only increase the Li vacancy concentration through charge compensation, but also increase the Coulomb force between Li and immobile ions, thus limiting the migration of Li+.71
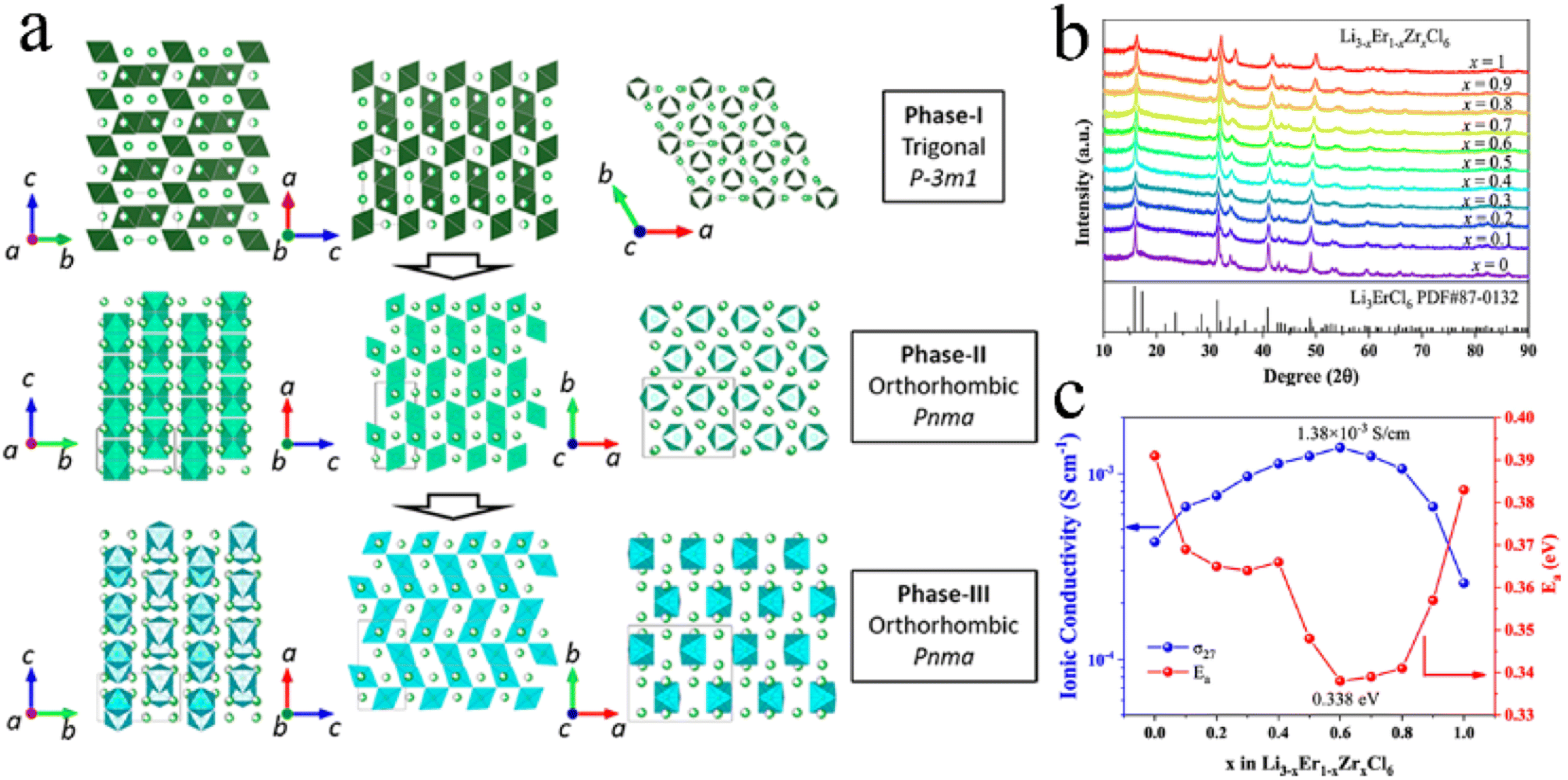 | ||
| Fig. 7 (a) Phase evolution of Li3M1−xZrxCl6 (M = Er, Y) upon Zr substitution. Reproduced with permission.44 Copyright 2020, American Chemical Society. (b) XRD patterns of Li3−xEr1−xZrxCl (x = 0–1) obtained from ball-milling. Reproduced with permission.39 Copyright 2022. American Chemical Society. (c) Ionic conductivity and activation energy for Li3−xEr1−xZrxCl as a function of x. Reproduced with permission.39 Copyright 2022. American Chemical Society. | ||
In addition to the size of doped metal ions, the heat treatment scheme was also a key factor in phase transformation. For Li3YbCl6, the orthorhombic phase was stable, while the metastable trigonal phase was formed when the solid state reaction or annealing was performed at low temperature.43,97 The appropriate aliovalent cation substitution (Zr4+ or Hf4+) for Yb3+ in Li3YbCl6 triggered the phase transition, whether it was a metastable trigonal phase or stable orthorhombic phase. With the increase in Hf content, the crystal structure of Li3−xYb1−xHfxCl6 prepared by mechanochemical synthesis could change from a trigonal structure to a monoclinic structure when annealed at 400 °C (Fig. 8a), and from the original orthorhombic-I structure to an orthorhombic-II structure when annealed at 500 °C (Fig. 8b). Their ionic conductivity also showed a parabolic trend of reaching a peak value of 1.5 and 1.2 mS cm−1 at x = 0.4 with the increase in Hf content, respectively.43 The effect of substitution on ionic conductivity derived from the combination of phase transition and the concentration of charge carriers of ions or Li vacancies.43 With the increase in Zr content, the crystal structure of Li3−xYb1−xZrxCl6 synthesized by a solid state reaction at 350 °C changed from a trigonal structure to an orthorhombic structure (Fig. 8c).97 The phase transition triggered metal ion rearrangement and then generated an interstitial tetrahedral site between Li1 and Li2 sites, which provided an intermediate “stepping stone” for Li+ migration. The 1D Li+ migration pathway through the face sharing Li1 and Li2 octahedral sites along the c-axis path was expanded to a 3D network. The ionic conductivity of orthorhombic Li2.7Yb0.7Zr0.3Cl6 was improved by nearly ten times compared with that of trigonal Li3YbCl6, reaching 1.1 mS cm−1. After further optimization, the ionic conductivity of Li2.556Yb0.492Zr0.492Cl6 reached up to 1.58 mS cm−1.100
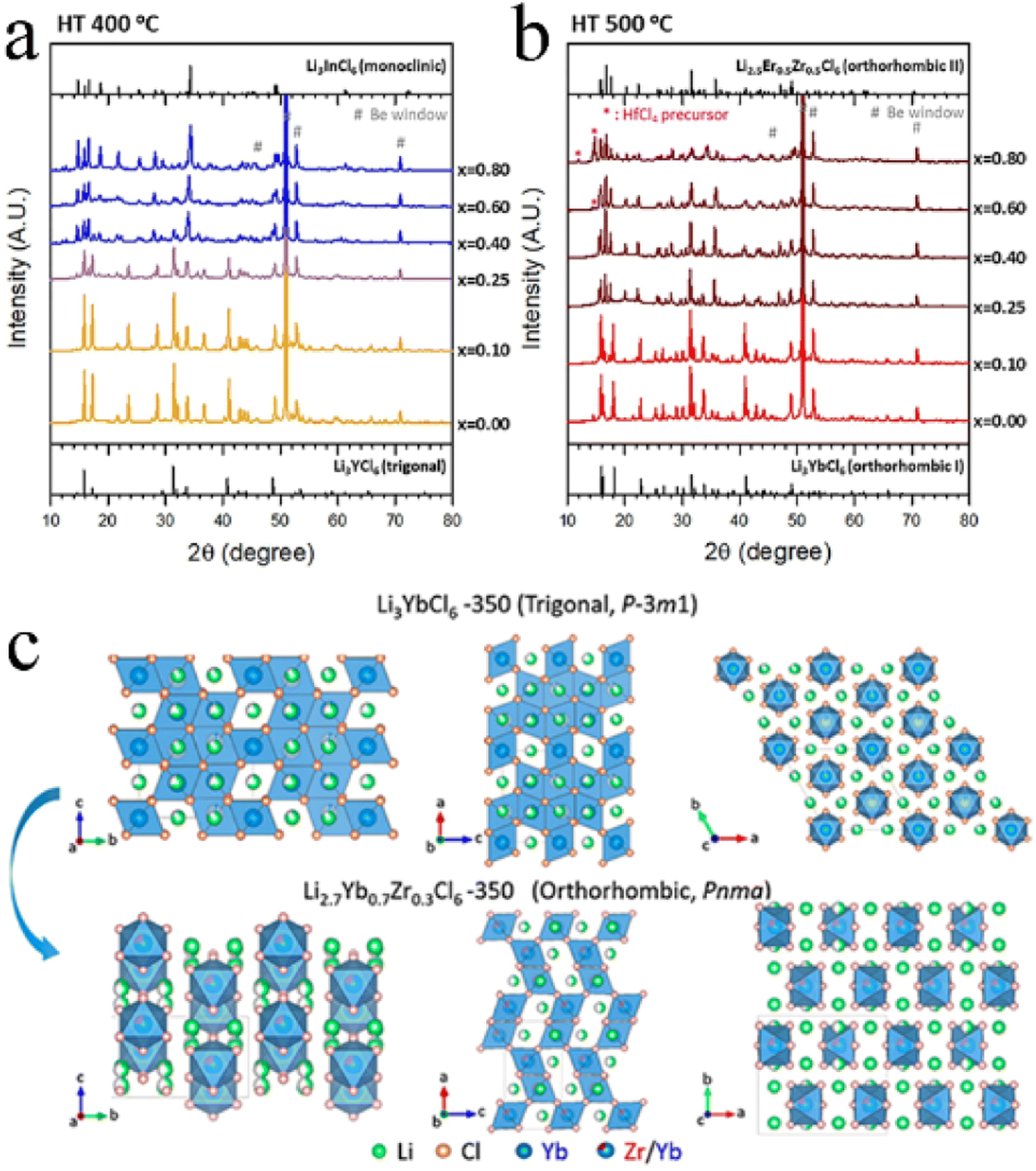 | ||
| Fig. 8 XRD patterns of Li3−xYb1−xHfxCl6 annealed at (a) 400 °C and (b) 500 °C. Reproduced with permission.43 Copyright 2021. Elsevier B.V. (c) Structural evolution of Li3YbCl6-350 to Li2.7Yb0.7Zr0.3Cl6-350 by Zr substitution. Reproduced with permission.97 Copyright 2021. American Chemical Society. | ||
Replacing In3+ with different amounts of Zr4+ or Hf4+ in Li3InCl6 could still maintain a monoclinic structure.88,106–109 In Li3−xIn1−xZrxCl6, the Zr4+ only occupied the 2b site (Fig. 9a) and the occupancy of In at the 4g site gradually decreased with the Zr content increasing. It was noteworthy that the occupancy of tetrahedral Li3 (8j) sites almost linearly decreased with the increase in Zr content, until complete disappearance.106,107 Li3InCl6 had two possible Li+ diffusion pathways: one was along the c-direction, involving the octahedral 2c site, tetrahedral 8j site, and shared octahedral 4g site. The other was along the Li-layer in the ab-plane, involving both the octahedral Li-sites in the layer and the vacant tetrahedra. The contribution ratio of the former and the latter to ionic conductivity was about 1:10. After the introduction of Zr4+, In3+ and Zr4+ occupied the octahedral site in large quantities, hindered the diffusion of Li+ in the mixed cation layer, and then reduced the possibility of long-range diffusion. The Oct–Tet–Oct pathway along the c-axis was also blocked by the high cumulative occupancy of the In2/Li4 site. At the same time, Zr4+ substitution led to a decrease in In occupation at the 4g site, and Li was removed from the tetrahedral site under charge compensation, thus opening up a new diffusion pathway along the c-axis (Fig. 9b). Moreover, Zr4+ substitution changed the preferred orientation of Li3InCl6 from the (001) plane to the (131) plane, which might be conducive to the construction of 3D Li+ migration channels. The Zr4+ substitution strategy made the ionic conductivity of Li2.9In0.9Zr0.1Cl6 significantly increase to 1.54 mS cm−1, which was nearly double that of Li3InCl6.109 The research of van der Maas et al. on a series of Li3−xIn1−xZrxCl6 showed that the conductivity reached a maximum value of 2.02 mS cm−1, when x = 0.3.106 However, Helm recognized that conductivity peaks at 1.2 mS cm−1 when x = 0.4.107 This remarkable difference might be due to the different methods of preparing Li3−xIn1−xZrxCl6. Based on the AIMD modeling results, the ionic conductivity of Li2.75In0.75Zr0.25Cl6 synthesized by Fu et al. could reach as high as 5.82 mS cm−1.108 The mechanism of tetravalent Hf4+ substitution on Li3−xIn1−xHfxCl6 was analogous to that of Zr4+, which improved ionic conductivity by forming interstitial tetrahedral sites and Li vacancies.88 At x = 0.3, the ionic conductivity of Li2.7In0.7Hf0.3Cl6 reached a maximum of 1.54 mS cm−1.
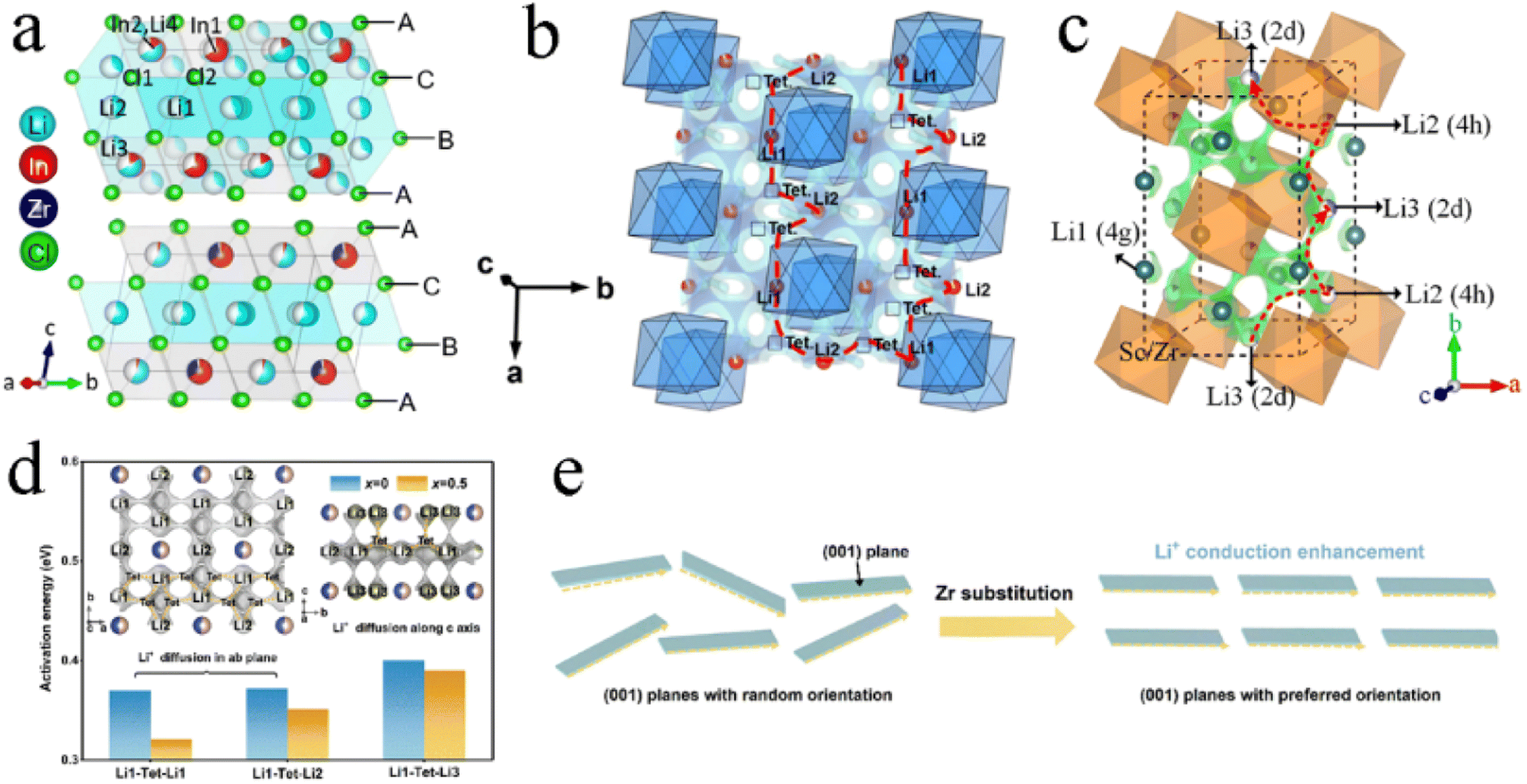 | ||
| Fig. 9 (a) The structure of Li3InCl6 (upper) and Li2.7In0.7Zr0.3Cl6 (below). Reproduced with permission.106 Copyright 2023, The Royal Society of Chemistry. (b) Migration pathways of Li3−xIn1−xHfxCl6 along the a-axis direction. Reproduced with permission.88 Copyright 2023, American Chemical Society. (c) The Li+ migration pathways of Li3−xSc1−xZrxCl6 marked with the green isosurfaces, and one of the migration pathways is highlighted with a red dotted line with arrows. Reproduced with permission.116 Copyright 2023, Wiley-VCH. (d) Comparison of the energy barrier values for Li+ migration through different pathways in Li3ScCl6 and Li2.5Sc0.5Zr0.5Cl6. Reproduced with permission.115 Copyright 2022, Elsevier B.V. (e) Schematic illustration of Li+ conduction in (001) planes with a randomly distributed orientation and preferred orientation. Reproduced with permission.115 Copyright 2022, Elsevier B.V. | ||
Similarly, introducing Zr4+ or Hf4+ into Li3ScCl6 didn't change the monoclinic structure.115,116 Zr4+/Hf4+ was randomly located at the Sc3+ 2a site, resulting in local structural distortion due to the difference in ion radii. The partially occupied Li2- and Li3-centered octahedra expanded, while the fully occupied Li1-centered octahedra shrank. The former could weaken the restriction of surrounding Cl−, thus promoting the migration of Li ions. This substitution couldn't change the 2D diffusion pathway of Li+ in the Li2/Li3-plane, which used tetrahedral interspaces as an intermediary site (Fig. 9c). Since Zr4+ and Hf4+ had a higher valence than Sc3+, they exhibited a stronger coulombic repulsion with Li+, thus well stabilizing the 2D diffusion path in the Li2/Li3-plane by limiting the random diffusion of Li ions. The resulting ionic conductivity was 1.61 and 1.33 mS cm−1 for Li2.6Sc0.6Zr0.4Cl6 and Li2.6Sc0.6Hf0.4Cl6 respectively, which were much higher than that of Li3ScCl6 (0.6 mS cm−1).116 Li et al. thought that both Li3ScCl6 and Li3−xSc1−xZrxCl6 had a 3D Li+ diffusive channel.115 The migration energy barriers of pathways in the ab plane were lower than those along the c-axis, and the migration of Li+ was still dominated by the ab plane pathway (Fig. 9d). The substitution of Sc3+ by Zr4+ reduced the migration energy barriers in this pathway. In addition, the Zr4+ substitution increased the concentration of Li vacancies. Most surprisingly, the Zr4+ substitution could improve the degree of preferred orientation in (001) planes (Fig. 9e). This structure reduced Li+ migration resistance along each ab plane and between contiguous ab planes and enabled Li+ to hop rapidly along the parallelly aligned ab planes with small misorientation, thus improving the ionic conductivity. Li2.5Sc0.5Zr0.5Cl6 exhibited an ionic conductivity of up to 2.23 mS cm−1, which was 3.28-fold higher than that of pristine Li3ScCl6.115
Li2ZrCl6 had a metastable trigonal structure and stable monoclinic structure, and the trivalent metal ions (In3+, Sc3+, and Fe3+) exhibited different substitution effects on these two structures. All Li2+xZr1−xInxCl6 (0 ≤ x ≤ 1.0) annealed at 260 °C after ball milling were of monoclinic phase with a space group of C2/m, and doping In3+ didn't change the crystal structure.105 As the content of In3+ increased, the lattice of Li2+xZr1−xInxCl6 expanded asymmetrically and the ZrCl62− octahedra distorted. The introduced In3+ initially occupied the 2a site and gradually occupied the 4g site when x exceeded 0.6, resulting in the disappearance of Zr4+ at that site. In unsubstituted Li2ZrCl6, Li+ preferred to present at the M2/Li3 site in the (001) plane rather than Li1, Li2, and Li4 sites in the (002) plane. The introduction of In3+ enabled the occupancy of the Li3 site to be decreased and enabled the occupancy of L1 and L2 sites to be increased upon substitution. Even when x was greater than 0.4, the Li4 site was occupied. This played a key role in improving ionic conductivity. The Li4 site was conducive not only to the intra-layer Li+ diffusion in the ab-plane, but also to the interlayer Li+ diffusion along the c-axis, which was the basis of the 3D diffusion pathway (Fig. 10a–c). In addition, the substitution of In3+ made the concentration of Li+ more abundant and expanded the anisotropic lattice, and the Li+ redistribution in the lattice also made the energy landscape more favorable for Li+ migration. Under the action of these comprehensive factors, the ionic conductivity of Li2+xZr1−xInxCl6 increased by several orders of magnitude, and the ionic conductivity of Li2.7Zr0.3In0.7Cl6 reached up to 2.1 mS cm−1. When synthesizing Li2+xZr1−xInxCl6 by mechanical milling, the crystal structure changed from trigonal to monoclinic with an increase in In3+ content (x ≥ 0.4) (Fig. 10d).112 In the series of Li2+xZr1−xInxCl6 prepared by this method, the ionic conductivity of Li2.25Zr0.75In0.25Cl6 was the highest (1.08 mS cm−1) and decreased in the subsequent annealing process. The ionic conductivity was doubled to 0.98 mS cm−1 by appropriate trivalent metal ion (Fe3+, V3+, and Cr3+) doping in hcp-Li2ZrCl6 (Fig. 10e).85 On the one hand, the Fe3+ substitution to Zr4+ by trivalent metal ions relieved the coulombic repulsion between Li+ and other metal cations, thus reducing the activation barrier for Li+ transport. On the other hand, the trivalent metal ion substitution modulated the overall potential energy landscape and facilitated Li+ migration. In addition, the Fe3+ substitution increased the number of Li+ and raised the concentration of effective charge carriers. The ionic conductivity of Li2.25Zr0.75In0.25Cl6 prepared by the mechanochemical method decreased sharply during annealing with an increase in crystallinity (Fig. 10f).129
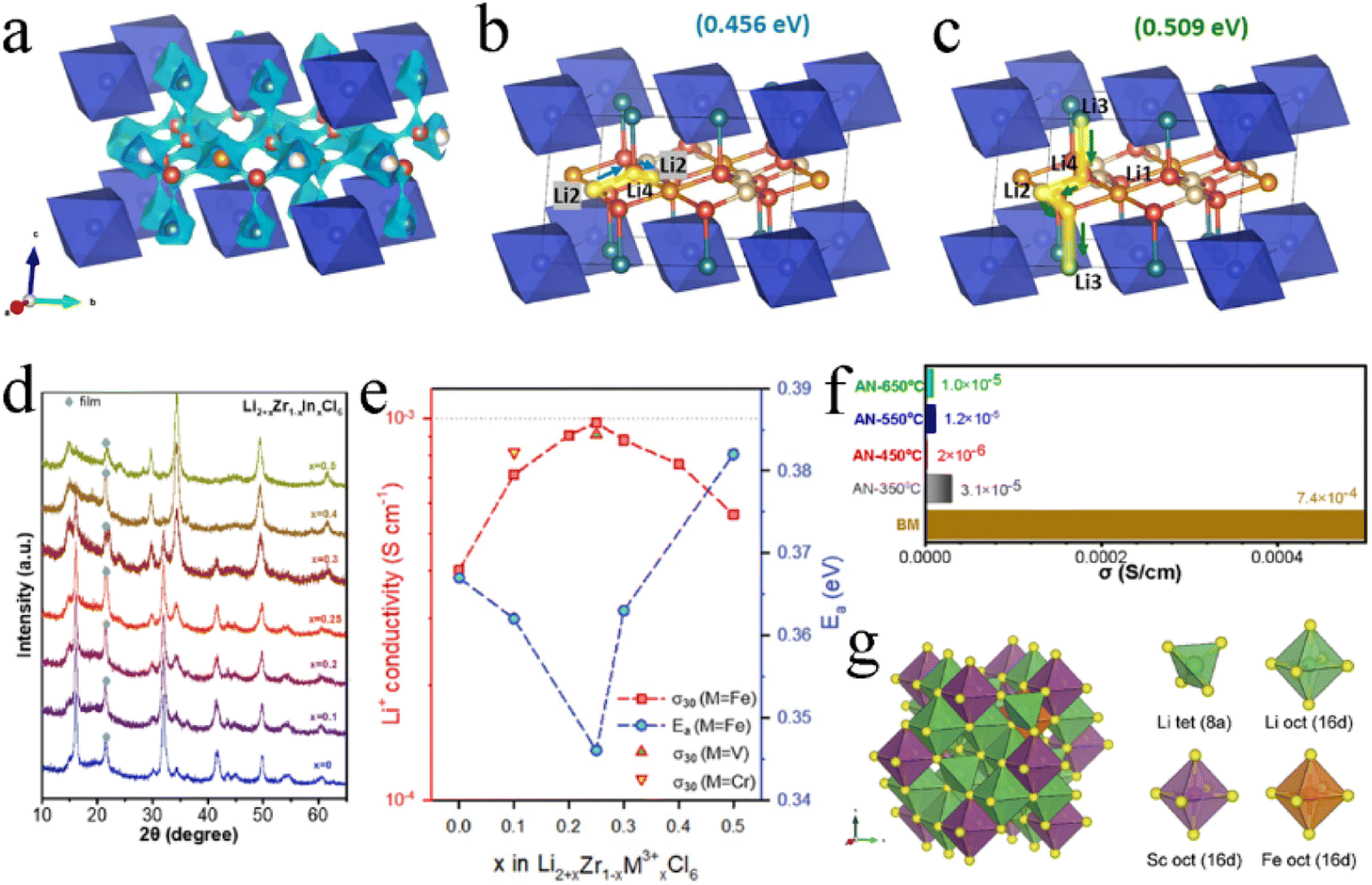 | ||
| Fig. 10 (a) Li+ diffusion pathways of Li2+xZr1−xInxCl6 with an iso-surface value of ±0.5 v.u. Reproduced with permission.105 Copyright 2022, Elsevier B.V. Possible Li+ conduction pathways in the ab-plane (b) and along the c-axis (c). Reproduced with permission.105 Copyright 2022, Elsevier B.V. (d) XRD patterns of Li2+xZr1−xInxCl6. Reproduced with permission.112 Copyright 2022, Elsevier B.V. (e) Ionic conductivity at 30 °C and activation energy for Fe3+/V3+/Cr3+-substituted Li2ZrCl6. Reproduced with permission.85 Copyright 2021, Wiley-VCH. (f) Comparison of ionic conductivity between ball-milled and annealed Li2.25Zr0.75Fe0.25Cl6 electrolytes. Reproduced with permission.129 Copyright 2022, Elsevier B.V. (g) The most thermodynamically stable structure of Li2S2/3Cl4–0.2Fe and its components. Reproduced with permission.95 Copyright 2023, The Royal Society of Chemistry. | ||
Replacing Sc3+ in Li2Sc2/3Cl4 with the divalent Fe2+ also effectively improved its ionic conductivity.95 Due to the ionic properties, Fe2+ tended to replace Sc3+ to occupy the 16d site rather than a tetrahedral site, forming a stable octahedral framework. At the same time, the additional Li+ occupied the 8a vacant site to maintain charge balance (Fig. 10g). In halospinel Li2Sc2/3Cl4, the Li+ diffused only through octahedral sites and 3D diffusion pathways were easily blocked by other cations located at the same site. Compared with Sc3+, Fe2+ had a larger ionic radius and a lower oxidation number. The doped Fe2+ and the extra Li+ formed new links, allowing the Oct–Oct diffusion to occur more frequently. Doped Fe2+ contributed to the formation of bonding networks between Li octahedra and then formed multi-diffusion channels with firm topological connectivity between the octahedra along Li diffusion pathways, thus promoting Li+ diffusion and obtaining a high ionic conductivity of 2.72 mS cm−1.
4. Synthesis methods of halide SSEs
In order to commercialize halide SSEs, the development of a stable and efficient large-scale synthesis method was key. At present, the synthesis of halide SSEs is divided into three categories: solid state reaction methods, mechanochemical synthesis, and wet chemistry synthesis (Fig. 11). The synthesis approach affected the local structure, local cationic ordering, and ion diffusion pathways of halide SSEs.41,90,91 | ||
| Fig. 11 Synthesis methods of halide SSEs, including (a) solid-state reaction, (b) mechanochemical synthesis and (c) wet-chemistry synthesis. | ||
4.1 Solid-state reaction
The intrinsic nature of conventional solid-state reactions required good solid–solid particle contact and enhanced the reaction kinetics by high-temperature co-melting.130 From the viewpoint of thermodynamics, a high temperature solid-state reaction was the most likely to achieve a phase that was close to the thermodynamic equilibrium state. Due to the sensitivity of halide SSEs to moist air, the reaction was usually carried out in vacuum quartz tubes, which limited the scale-up production of electrolytes. Moreover, the solid-state reaction often needs maintaining high temperature for a long time. For example, the synthesis of Li3YBr6−xFx required heating to 950 °C for 15 h,118 the synthesis of Li3YBrxF6−x required heating to 680 °C for 24 h,103 and the synthesis of Li4−3xScxCl4 required heating to 650 °C for 48 h.45 The huge energy consumption further limited its practical applications.4.2 Mechanochemical synthesis
Generally, the mechanochemical method allowed for the synthesis of nonequilibrium compounds. Moreover, there were large amounts of structurally disordered interfacial regions, local distortions, and defect structures in the electrolytes by mechanochemical synthesis. The presence of these defects was advantageous for some electrolytes. For ternary trigonal chloride electrolytes, high M2/M3 site disorder played a key role in their high ionic conductivity. As shown in Fig. 12a, the ball milled Li3YCl6 had the lowest activation energy and the highest ionic conductivity. By the annealing treatment at different temperatures, crystallinity increased but the ionic conductivity decreased.124 The trigonal Li3ErCl6, Li2.6Er0.6Zr0.4Cl6 and monoclinic Li3ErI6 were also consistent with this law.39,41,83 In contrast, the ionic conductivity of Li3YBr6 and Li3InCl6 increased, with crystallinity increasing (Fig. 12b and c).25,33,91,123 Annealing treatment could not only improve crystallinity, but also change the crystal structure of electrolyte in some cases. Li2ZrCl6 prepared by ball milling has a trigonal structure, which could transform into a monoclinic structure after annealing at 350 °C (Fig. 12d).86 Although crystallinity was improved, the ionic conductivity decreased sharply. Annealing temperature was also an important parameter affecting the ion conductivity and phase structure of electrolyte (Fig. 12e).43 After mechanochemical milling, Li3YbCl6 prepared at 400 °C crystallized into a trigonal structure, while the material prepared at 500 °C formed an orthorhombic structure. There was a slight difference in ionic conductivity between them. In order to obtain the as-expected ionic conductivity, it was necessary to consider the necessity of annealing and rationally select annealing temperature for the synthesis of halide SSEs by mechanochemical synthesis.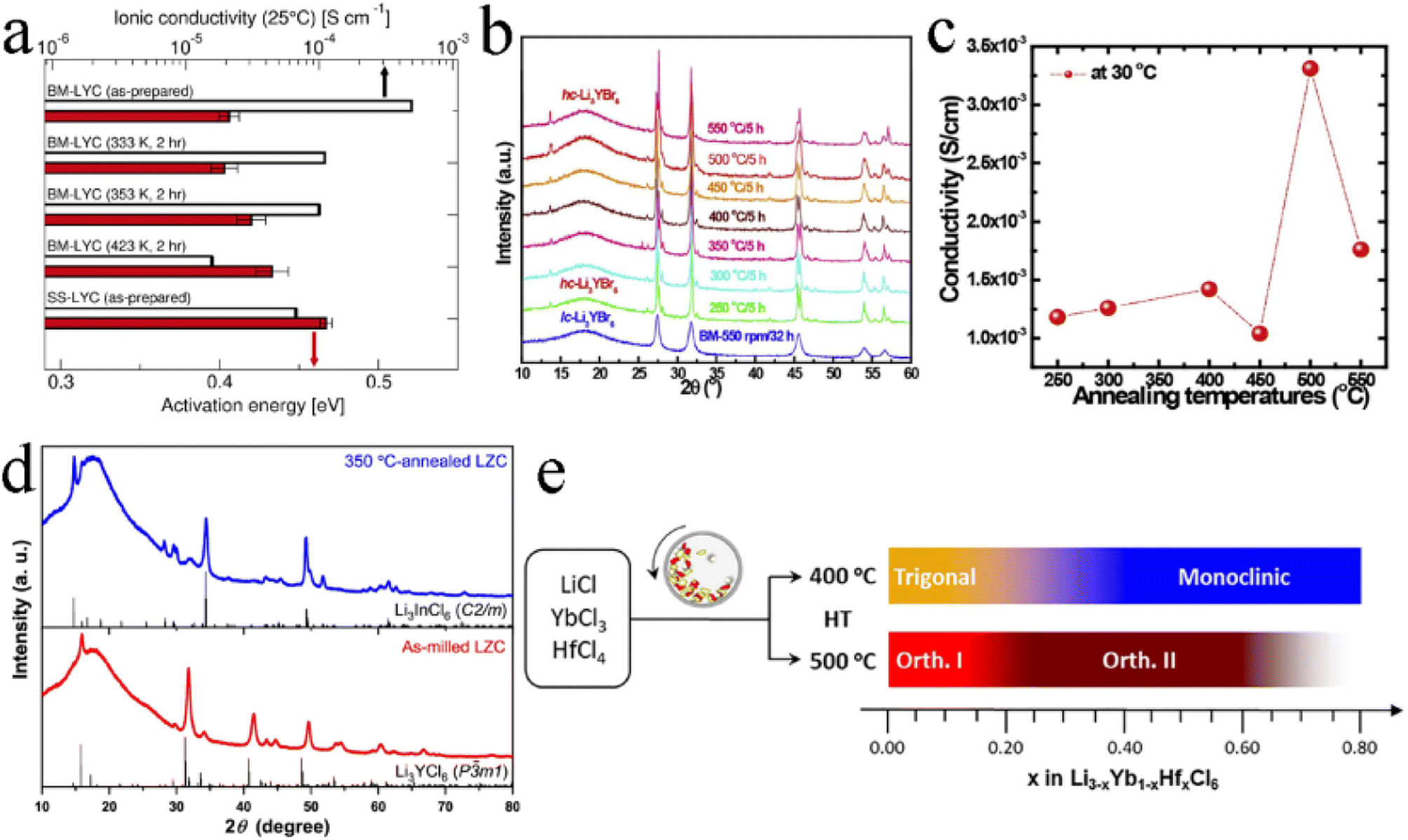 | ||
| Fig. 12 (a) Room temperature ionic conductivity and activation energy for Li+ long-range diffusion among various Li3YCl6 samples. Reproduced with permission.122 Copyright 2022, American Chemical Society. (b) The XRD patterns of Li3YBr6 annealed at various temperatures. Reproduced with permission.123 Copyright 2020, Elsevier Ltd. (c) Ionic conductivities of annealed Li3YBr6 changed with annealing temperatures. Reproduced with permission.123 Copyright 2020, Elsevier Ltd. (d) The XRD patterns of the as-milled and annealed Li2ZrCl6 at 350 °C. Reproduced with permission.86 Copyright 2020, Springer Nature. (e) Schematic illustrating the phase evolution of Li3−xYb1−xHfxCl6 at 400 or 500 °C. Reproduced with permission.43 Copyright 2021, Elsevier B.V. | ||
4.3 Wet chemistry synthesis
Compared with the mechanochemical and co-melting synthesis, wet chemistry synthesis could avoid long periods of high-energy ball-milling or high-temperature heating treatment and was more efficient and time-saving. Wet chemistry synthesis was the most potential route for the large-scale manufacturing of halide SSEs.Because the dehydration/hydration process between Li3InCl6·2H2O and Li3InCl6 was reversible and obtained high crystallinity in dehydrated Li3InCl6, it was feasible to use deionized water as solvent to synthesize Li3InCl6 on a large scale. Sun's research group was the first to synthesize Li3InCl6 with ionic conductivity up to 2.04 mS cm−1 using distilled water as the medium (Fig. 13a).61 Through simple dissolution and vacuum heating, the high purity and crystallinity of Li3InCl6 could be obtained. Vacuum drying was conducive to the formation of small-size particles, which could completely remove trace water and avoid oxygen pollution due to a shorter diffusion length and larger surface area.131 The particle size of Li3InCl6 was greatly reduced by introducing freeze drying technology into wet chemistry synthesis (Fig. 13b–d).132 The effective removal of free water by freeze drying significantly alleviated the increase in particle size caused by particle collision during thermal evaporation of solution. The uniform particles of electrolyte greatly reduced the porosity of ASSBs to obtain better interfacial contact and excellent cycling performance. As shown in Fig. 13e, the ASSB based on freeze-dried Li3InCl6 had little capacity attenuation after 150 cycles at 10C.
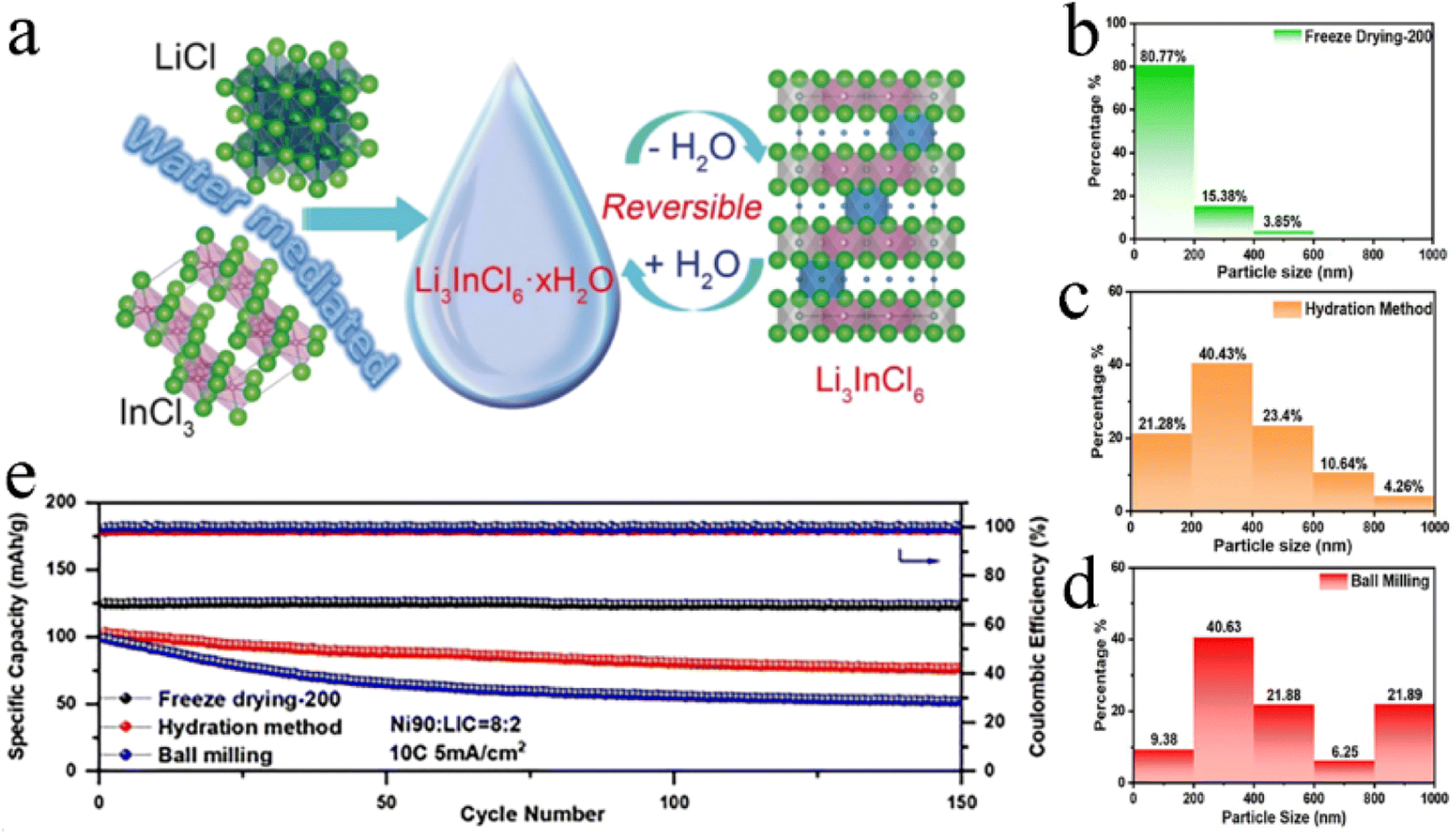 | ||
| Fig. 13 (a) Illustration of a water-mediated synthesis route for Li3InCl6 and the reversible interconversion between hydrated Li3InCl6·xH2O and dehydrated Li3InCl6. Reproduced with permission.61 Copyright 2019, Wiley-VCH. The histograms of the particle size distribution of Li3InCl6 synthesized by different methods: (b) freeze drying, (c) hydration method and (d) ball milling. Reproduced with permission.132 Copyright 2023, The Royal Society of Chemistry. (e) Cycling performance at 10C of NCM90-Li3InCl6/Li3InCl6/Li6PS5Cl/Li cells with Li3InCl6 prepared by different methods. Reproduced with permission.132 Copyright 2023, The Royal Society of Chemistry. | ||
In addition, ethanol was also used as solvent for the synthesis of Li3InCl6 electrolyte.62 The advantage of this method was that it eliminated the adverse impact of trace water on the battery performance. At the same time, this method only needed heating at 200 °C for 3 h to make the intermediate phase completely decomposed and obtain high crystallinity Li3InCl6 electrolyte. The raw materials (LiCl and InCl3) had a very low solubility in ethanol, meaning more solvents were needed for the same amount of production. And the price of ethanol was much higher than that of deionized water, so the ethanol-mediated route showed a disadvantage in manufacturing cost.
The ammonia-assisted wet chemical synthesis was more universal, which could lift the restriction on reversible hydration/dehydration of electrolyte and extend the wet chemical route to the preparation of Li3MX6 (M = Y, Sc, and Er; X = Cl and Br).63 The relevant equation is as follows:
| MCl3·nH2O + 3NH4Cl → (NH4)3[MCl6] | (1) |
| (NH4)3[MCl6] +3LiCl → Li3MCl6 + 3NH3 + 3HCl | (2) |
Firstly, the NH4+ and MX6 were dissolved in deionized water to form an intermediate phase. Then, the intermediate ammonium was completely decomposed after heating and the halide electrolytes with good crystallinity were obtained. The halide electrolyte synthesized by this method had nanoscale size and formed a localized microstrain in the material under the small size effect. The microstrain-induced local structural change might be favorable for Li+ transport along the ab plane in an hcp anion framework, but not in a ccp anion sublattice.
Inspired by ammonia-assisted wet chemical synthesis, the vacuum evaporation-assisted synthesis was developed for the scale-up synthesis of Li3HoBr6 using the following equation:120
| 3Li2CO3 + Ho2O3 +12NH4Br → 2Li3HoBr6 + 3CO2 + 12NH4 + 6H2O | (3) |
This pathway used relatively inexpensive precursors such as rare earth oxides, lithium carbonate, and ammonium halide. The ionic conductivity of synthesized Li3HoBr6 was equivalent to or even better than the ionic conductivity of that synthesized by the solid-state reaction method.119
By comparison, the wet chemistry synthesis had lower equipment requirements, cheaper raw materials, less energy consumption, and produced halide SSEs with both high ionic conductivity and electrochemical stability. In summary, wet chemistry synthesis was the most promising method for large-scale preparation of halide ASSBs.
5. Air environmental stability of halide SSEs
Air environment stability of SSEs was always a hard-to-overcome difficulty.133–135 It was directly related to the manufacturing cost, transportation cost, and application cost. Oxide-based SSEs had relatively good air stability, which slowly reacted with moisture and CO2 through Li+/H+ exchange, formed LiOH, Li2CO3 and Li2O on the surface and increased the interface resistance.136 Sulfide-based SSEs were extremely unstable in the air, where the S2− tended to bond with H− in moist air to form toxic H2S gas.133 In contrast, the hydrolysis energy of ternary chloride was positive, so the reaction with water was basically stable.1375.1 Degradation mechanism
Li3InCl6 and Li3YCl6 were very sensitive to moisture in the air and absorbed water quickly when exposed to air and then are completely liquefied into transparent solutions after 2 h and 8 h, respectively (Fig. 14a).138 Li3InCl6 absorbed water faster than Li3YCl6, while Li3YCl6 absorbed more water than Li3InCl6. Their absorption rate was proportional to the contact area with moisture air. The schematic diagram of the degradation mechanism of Li3InCl6 is shown in Fig. 14b. Li3InCl6 absorbed water rapidly after being exposed to the air, to form Li3InCl6·2H2O crystalline hydrate in the initial stage. With the progress of the hydrolysis process, part of Li3InCl6 was decomposed into InCl3 and LiCl, and InCl3 could further hydrolyze to produce the In(OH)3 intermediate phase and finally dehydrated to form In2O3 impurities.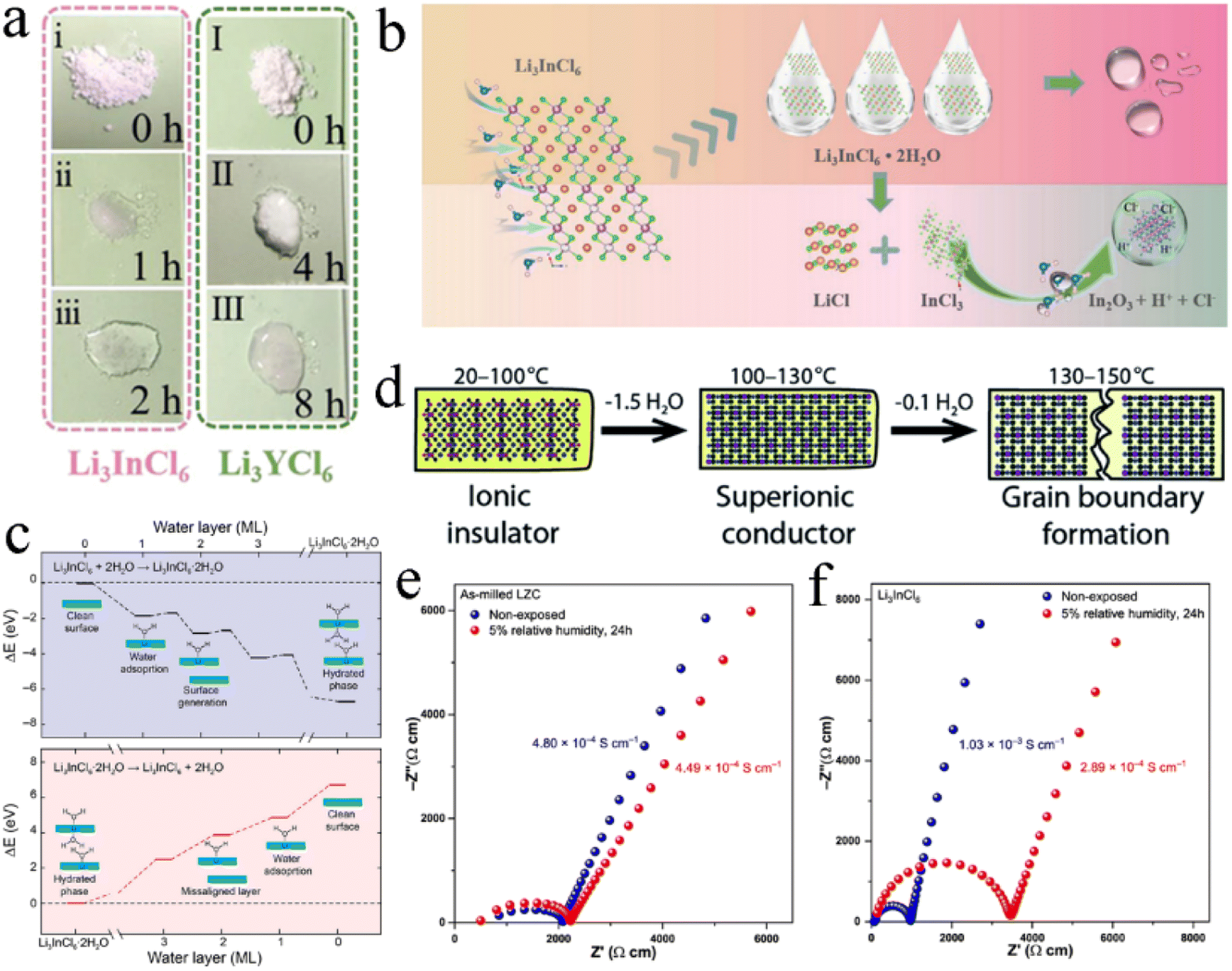 | ||
| Fig. 14 (a) Images of water absorption morphology evolution of Li3InCl6 and Li3YCl6 in an air environment. Reproduced with permission.138 Copyright 2021, Wiley-VCH. (b) Schematic diagram exhibiting the hydrolysis mechanism of Li3InCl6 in an air environment. Reproduced with permission.138 Copyright 2021, Wiley-VCH. (c) Profiles of the relative energy in hydration (upper) and dehydration (below). Reproduced with permission.139 Copyright 2021, Elsevier B.V. (d) Illustration of Li3InCl6 hydrate evolution during heating. Reproduced with permission.140 Copyright 2021, The Royal Society of Chemistry. Nyquist plots of (e) as-milled Li2ZrCl6 and (f) Li3InCl6 before and after being exposed to the atmosphere with 5% relative humidity. Reproduced with permission.86 Copyright 2021, Springer Nature. | ||
In combination with types of advanced characterization methods, Li et al. revealed the degradation process of Li3InCl6 when exposed to humid air.26 Li3InCl6 remained stable in dry air and exhibited a certain tolerance to low humidity air (3–5%), but quickly decomposed in high humidity air (30%). In general, the hydrolysis of Li3InCl6 proceeded according to the following two reactions:
| 2Li3InCl6 + 3H2O → In2O3 (s) + 6HCl (g) + 6LiCl | (4) |
| Li3InCl6 + xH2O → Li3InCl6·xH2O | (5) |
In moist air, hydrophilic Li3InCl6 first absorbed water and part of Li3InCl6 reacted with H2O to form In2O3 as a precipitate, as well as LiCl and HCl (eqn (4)). Besides, the remaining Li3InCl6 absorbed H2O to form Li3InCl6·xH2O hydrate (eqn (5)).
The hydration reaction of Li3InCl6 was a completely reversible process, and the ionic conductivity of Li3InCl6·xH2O could recover over 92% after removing H2O by vacuum heating. However, the ionic conductivity of Li3YCl6 could only retain 0.8% after the same treatment.25,38,61 According to the DFT, the surface adsorption energy of Li3InCl6 was only −0.60 eV, while the hydrolysis reaction energy barrier was high enough to inhibit the spontaneous hydrolysis reaction.139Fig. 14c exhibits the schematic and energy profiles of hydration and dehydration reactions of Li3InCl6. During hydration, Li3InCl6 stabilized by adsorbing H2O on the surface. The adsorption of H2O could reduce the surface energy below 0 J m−2, making it easier to form a new surface. The newly formed surface further promoted a hydration layer on the surface of Li3InCl6. In the case of dehydration, the reaction was non-spontaneous and required tremendous energies to remove H2O from the hydration phase. The monotonous reaction pathways of hydration and dehydration and their energies enabled the reversible phase evolution of Li3InCl6.139 The H2O in the Li3InCl6·xH2O hydrate reduced the mobility of Li by extending the required jumping distance and blocking facile migration pathways. With the removal of H2O, the lattice shrank and the ionic conductivity was restored.140 When the unit formula amount of H2O was less than 0.5, the ionic conductivity of the sub-hydrate phase improved significantly, until the superionic conducting phase was formed after complete dehydration. However, the evaporation of the last trace of water contributed to the formation of stress cracks and grain boundaries (Fig. 14d).
The moisture resistance of Li3InCl6 came from its good recoverability after hydrolysis. By contrast, Li2ZrCl6 was indeed moisture resistant at relative humidity even higher than 1%.86 Both Li2ZrCl6 and Li3InCl6 were exposed to N2 with 5% relative humidity at the same time, and the crystal structure and ionic conductivity of Li2ZrCl6 remained unchanged after 24 h, whereas the ionic conductivity of Li3InCl6 was largely decreased by nearly an order of magnitude (Fig. 14e and f).
5.2 Strategies to improve moisture resistance
In order to improve the air stability of halide SSEs, the coating strategy is considered one of the possible solutions. Coating the surface of Li3InCl6 with Al2O3 through powder atomic layer deposition could isolate the contact with humid air, which effectively improved its moisture resistance (Fig. 15a).138 For pristine Li3InCl6 with different particle sizes, the air stability time of Li3InCl6@Al2O3 after coating was prolonged by 4 or even 7 times. However, compared with the original Li3InCl6, the ionic conductivity of Li3InCl6@Al2O3 was slightly decreased. Also, the surface coating obviously increased manufacturing cost, which was far from a perfect solution.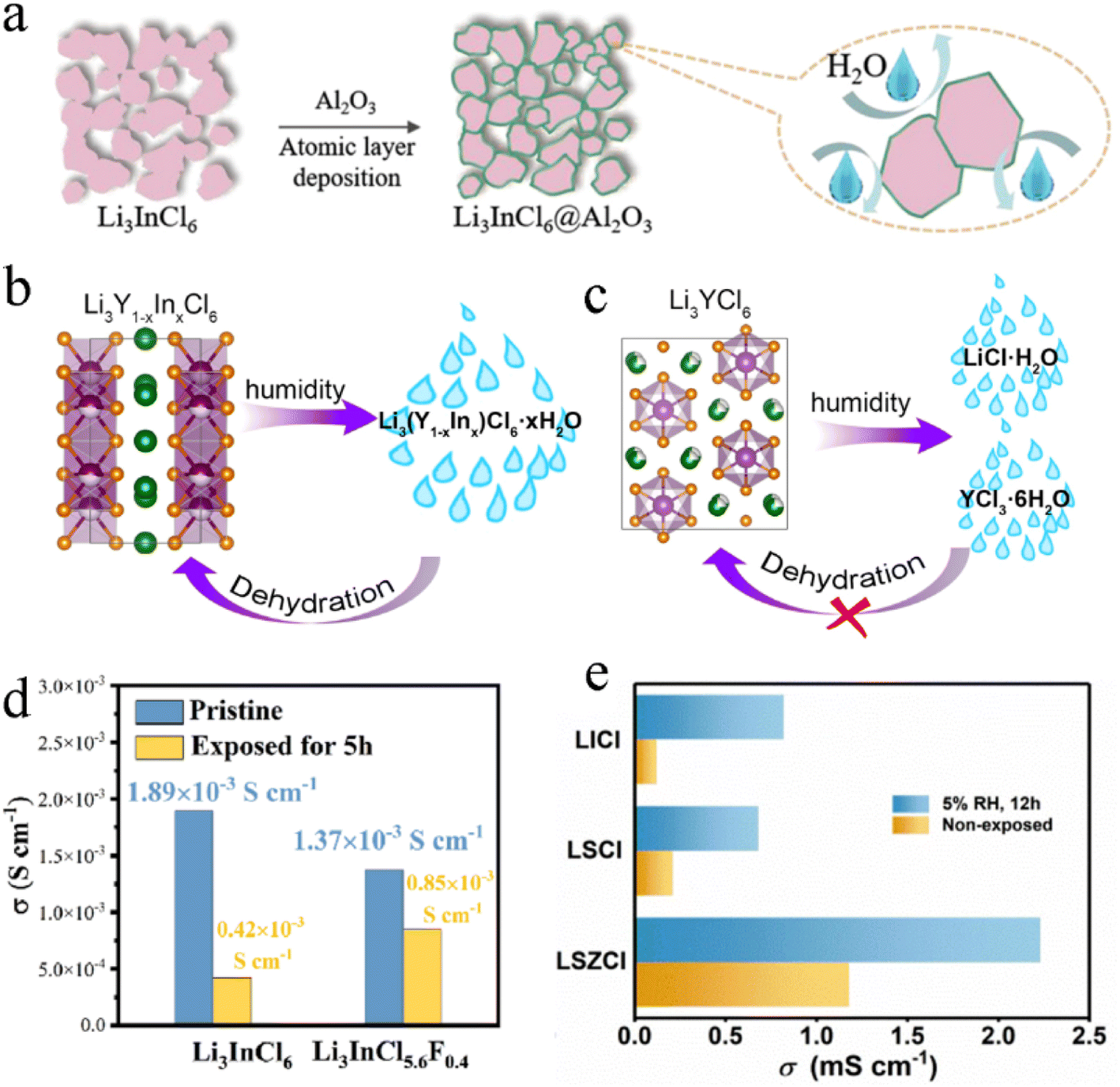 | ||
| Fig. 15 (a) Schematic diagram showing that the Al2O3 coating enhanced Li3InCl6 air stability via powder atomic layer deposition. Reproduced with permission.138 Copyright 2021, Wiley-VCH. Schematic illustration of the humidity stabilities of (b) Li3Y1−xInxCl6 and (c) Li3YCl6, respectively. Reproduced with permission.38 Copyright 2020, American Chemical Society. (d) Ionic conductivity evolution of Li3InCl6 and Li3InCl5.6F0.4 after being exposed to a dew-point dry room (−20 ± 3 °C) for 5 h. Reproduced with permission.115 Copyright 2022, Elsevier B.V. (e) Ionic conductivity of Li3InCl6, Li3ScCl6, and Li2.5Sc0.5Zr0.5Cl6 before and after exposure to an Ar atmosphere with 5% relative humidity. Reproduced with permission.115 Copyright 2022, Elsevier B.V. | ||
The doping strategy could improve the ionic conductivity of halide SSEs and enhance their moisture resistance.38,115 Li3YCl6 was unstable in air and easily changed to YCl3·6H2O and LiCl·H2O upon contact with moisture, which was irreversible and couldn't be recovered by vacuum heating. After doping through the In3+, hydration intermediates were formed when Li3Y1−xInxCl6 was exposed to moist air, rather than separated phases (Fig. 15b and c). The recovery of Li3Y1−xInxCl6 increased with an increase in In3+ content. When x ≥ 0.5, the ionic conductivity of Li3Y1−xInxCl6 reheated after humidity exposure reached more than 85% that of the original material.38 F− doping could greatly reduce the water absorption rate of Li3InCl6.111 After exposure to a dew-point dry room (−20 ± 3 °C) for 5 h, Li3InCl6 retained only 22.2% of ionic conductivity, while Li3InCl5.6F0.4 retained 62% of ionic conductivity (Fig. 15d). The increasing moisture stability might be related to the formation of robust Li–F and In–F bonds by F− doping.111 Inspired by the high moisture resistance of Li2ZrCl6, Zr4+ was introduced into Li3ScCl6 to form a robust Zr–Cl bond and improve the moisture resistance.115 The moisture resistance of Li2.5Sc0.5Zr0.5Cl6 was obviously improved by the Zr4+ replacement. After being exposed to an Ar atmosphere with 5% relative humidity for 12 h, Li2.5Sc0.5Zr0.5Cl6 retained the original crystal structure and decreased its ionic conductivity by only 50%. In contrast, the ionic conductivities of Li3ScCl6 and Li3InCl6 decreased by 70% and 85%, respectively, under the same conditions (Fig. 15e). When exposed to high humidity (30% of relative humidity), Li2.5Sc0.5Zr0.5Cl6 absorbed water and further reacted with it to form a ZrOCl2(H2O)8 phase, which could avoid the subsequent water absorption and enhanced moisture resistance.
6. Interface optimization and application challenges of halide SSEs in ASSBs
With the rapid development of halide SSEs, a series of halides with high room temperature ionic conductivity, even exceeding mS cm−1 have been fabricated.33,104 The overall reaction kinetics of ASSBs were determined by ionic conductivity and also depended on interfacial resistance.141 These halides SSEs, especially chlorides, typically had a wider electrochemical stability window than sulfides and oxides SSEs, which provided ASSBs the potential for high energy density.96,100 However, as with other inorganic SSEs, halide electrolytes also need to overcome the interface problem with electrode materials.20,142,1436.1 Interfacial stability of halide SSEs and cathodes
According to Wang et al.'s calculations, halide SSEs had a wide thermodynamic intrinsic electrochemical window, while sulfides and oxides couldn't match it (Fig. 16a).27 This made halides extremely compatible with high-voltage cathodes without the need for any protective coating.44,46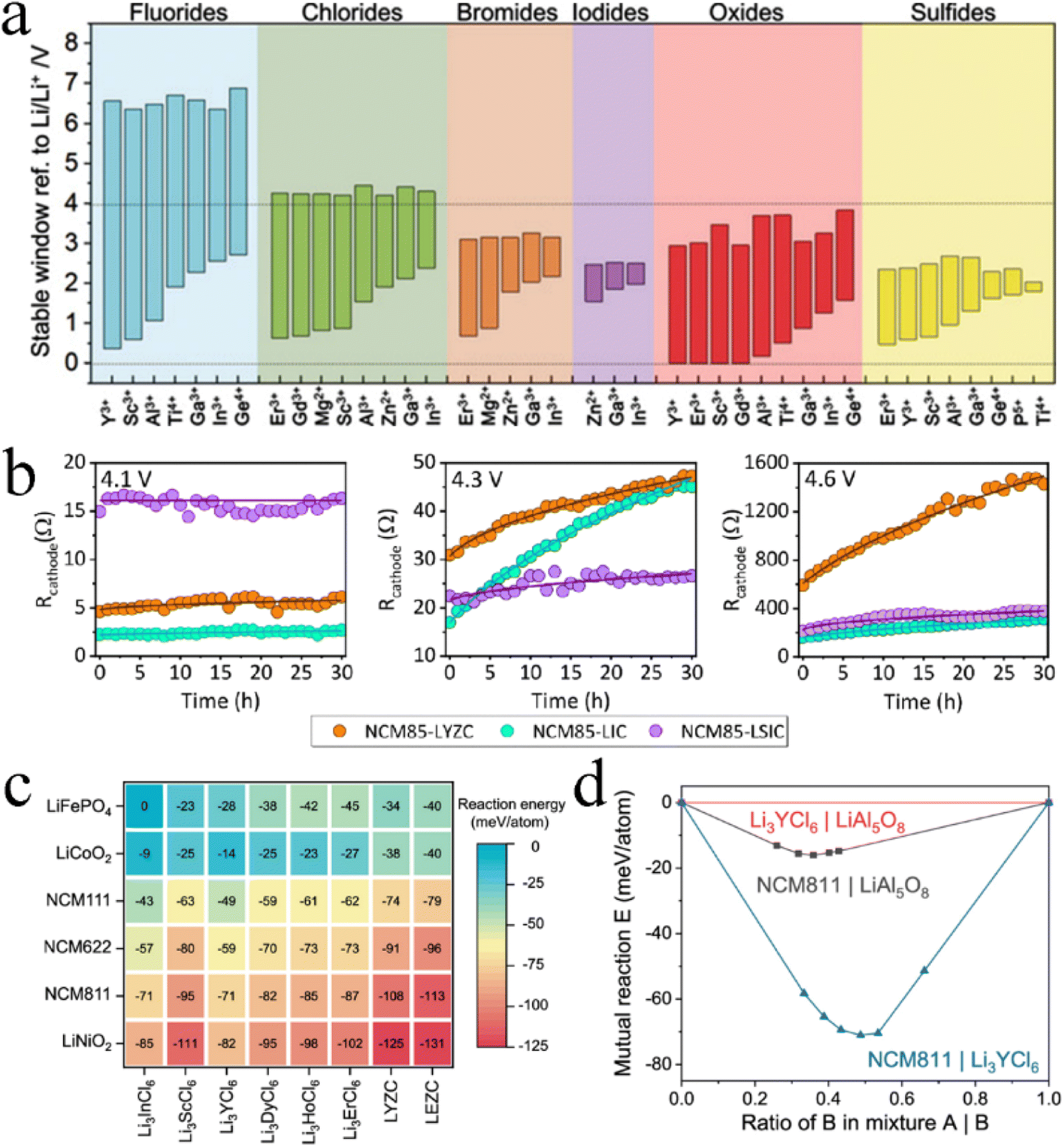 | ||
| Fig. 16 (a) Comparison of thermodynamic intrinsic electrochemical windows of ternary halides, oxides, and sulfides. Reproduced with permission.27 Copyright 2019, Wiley-VCH. (b) The cathode resistance of the charged In/InLi‖NCM85 cells during aging at 4.1–4.6 V vs. Li+/Li. Reproduced with permission.144 Copyright 2022, The Royal Society of Chemistry. (c) Heatmap of the mutual reaction energy between chloride SSEs and cathode materials. Reproduced with permission.145 Copyright 2021, American Chemical Society. (d) Mutual reaction energy between Li3YCl6 and NCM811, between LiAl5O8 and NCM811, and between LiAl5O8 and Li3YCl6, as a function of the mixing ratio. Reproduced with permission.145 Copyright 2021, American Chemical Society. | ||
Almost all chloride electrolytes exhibited high oxidation potentials of ∼4.3 V, which could fully cover the typical working potential of cathode materials. However, chloride electrolytes and cathodes weren't stable enough against chemical decomposition, and the stability of the interface between them depended on the central metal cation and the type of cathode materials.99,144,145 By comparing the electrochemical properties of Li3InCl6, Li2Sc1/3In1/3Cl4, and Li2.5Y0.5Zr0.5Cl6, chlorides with similar ionic conductivity, it was found that In3+ and Sc3+ central cations were beneficial for the kinetically stable interface between chloride electrolyte and the NCM85 cathode. However, some serious side reactions occurred at the interface of Li2.5Y0.5Zr0.5Cl6 and NCM85 at or above 4.3 V vs. Li+/Li. The interfacial decomposition products (YOCl or ZrO2) were conducive to the mass transfer of oxygen-containing components and promote continuous side reactions at the interface and lead to an appreciable increase of cathode impedance and a significant decay of capacity (Fig. 16b).144 The first-principles calculation results showed that Zr4+ doping significantly increased the mutual reaction energy between chloride (Li3YCl6 and Li3ErCl6) and cathode materials.145 As shown in Fig. 16c, the mutual reaction energies between all chlorides and LiFePO4 and LiCoO2 (LCO) were below 50 meV per atom, indicating good chemical stability and good compatibility between chloride electrolytes and cathode materials. Similarly, LiMn2O4 also had low chemical reaction energy with chloride electrolytes.99 By contrast, the Li(NiMnCo)1/3O2 (NCM111) situation wasn't so optimistic. The chemical reaction energy between NCM111 and chlorides was much higher than that of the other three anode materials, which could increase with an increase in Ni content in NCM. And therefore, NCM cathode materials weren't a good choice for chloride electrolytes. Coating was considered a common strategy to solve SSE interface problems. From the aspects of phase stability, electrochemical stability, and chemical stability with cathode materials and chloride electrolytes, it was found that 54 Li-containing compounds were noteworthy by the high-throughput computational screening of 20237 Li-containing compounds.145 In Fig. 16d, the LiAl5O8 coating material exhibited much lower reaction energy on Li3YCl6 and NCM811, meaning that it could stabilize the interface between chloride electrolyte and high-voltage cathode materials. But this effect has not been confirmed by the experimental results. It was more convenient and feasible to improve the stability of the interface between chloride electrolyte and the LCO cathode by doping F− into dual-halogen SSE.110 The F-doping generated F-enriched passivating interphases in situ on the cathode interface, which protected the electrolyte from further decomposition and was beneficial for the promising cycling stability of ASSBs. The Li3InCl4.8F1.2-based ASSB in the voltage range of 2.6–4.47 V retained a capacity of 102 mA h g−1 after 70 cycles. Notably, the average coulombic efficiency was up to 99.5% during the cycling process, implying the highly reversible Li+ de-/intercalation behavior and interfacial stability between Li3InCl4.8F1.2 and the cathode material.
6.2 Interfacial stability of halide SSEs and anodes
Li metal is considered the “holy grail” of next-generation LIB anode materials because of its extremely high theoretical capacity of 3860 mA h g−1 and the lowest redox potential (−3.04 V vs. standard hydrogen electrode).146,147 However, due to the low electronegativity of Li, almost all SSEs containing transition metal components were reduced upon contact with a bare Li anode.148,149 Although a more stable interface could be obtained by replacing Li metal with a Li–In alloy, it came at the expense of capacity.Depending on the type of central element, the reduction potential of ternary chloride electrolyte ranged from 0.7 to 2.6 V.145 The reduction potential of chlorides with group 3 elements was slightly lower and that of group 13 elements was higher. In addition, Zr4+ substitution significantly increased the reduction potential of chloride electrolyte, which was an aspect to be considered when reaping the increased ionic conductivity. First-principles calculations showed that Li3YCl6 and Li3ErCl6 possessed a relatively lower decomposition energy than Li3InCl6 and Li2.5Y0.5Zr0.5Cl6, but they all exceeded 200 meV per atom, indicating good chemical instability with the Li metal anode. In situ X-ray photoelectron spectroscopy analysis of Li3MCl6/Li interfacial decomposition products showed that the high-valence metal cations (M3+) in the chlorides were easily reduced to M0 when they encountered Li metal, according to the following balanced chemical equation:150
| Li3MCl6 + 3Li → 6LiCl + M0 (M0 = In, Y) |
The reaction product LiCl was a Li+ conductor and M was an electron conductor, so the interphase was a mixed ionic and electronic conductor (MIEC). The MIEC interphase encouraged the continuation of thermodynamically favorable decomposition reaction and and inhibited the formation of a passivation layer.151 Both Li+ and electrons migrated through MIEC interphases, and the adverse side reaction continued during the Li+ plating/stripping process until halide electrolyte or Li metal was depleted (Fig. 17a).63,152
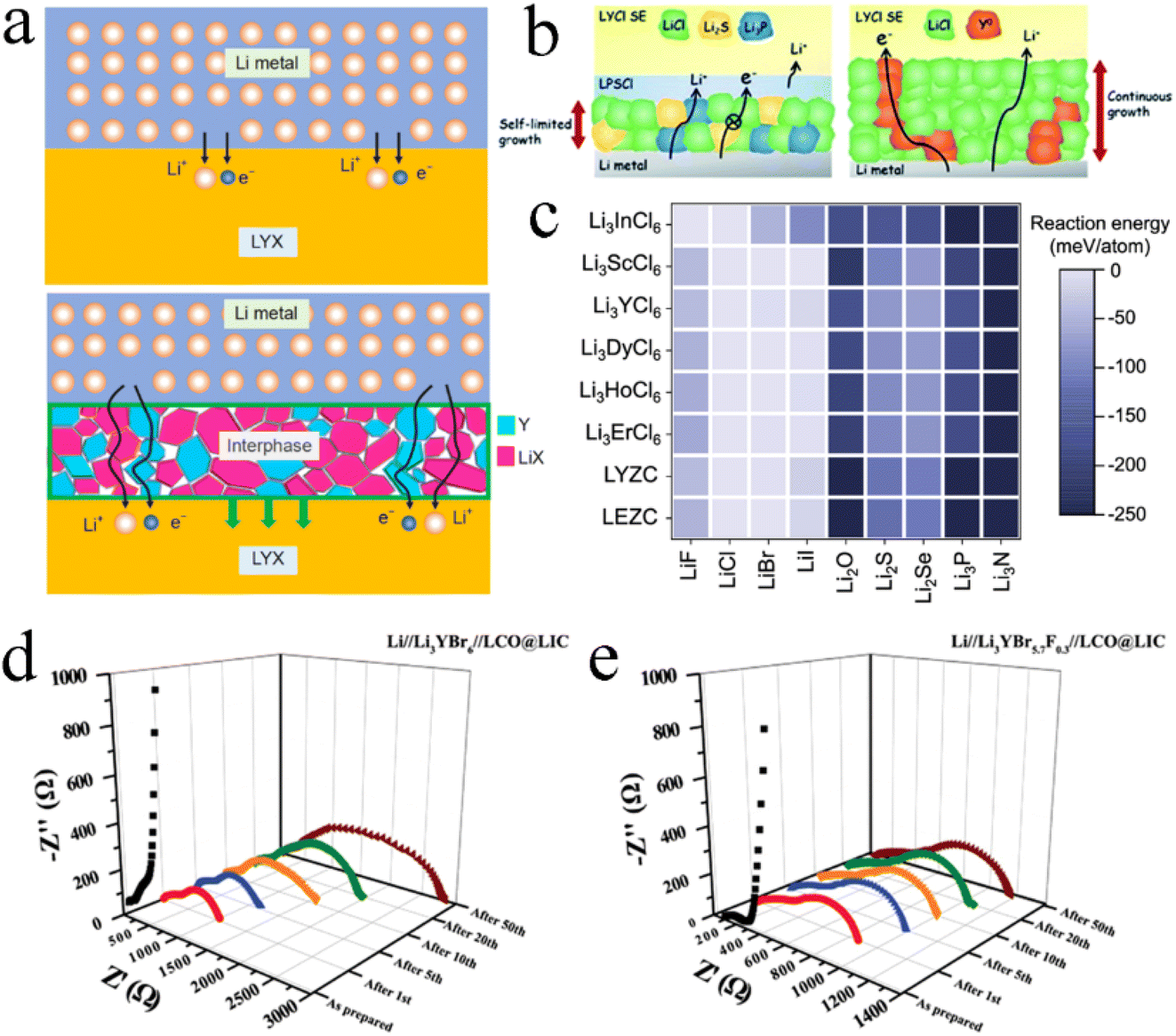 | ||
| Fig. 17 (a) Schematic illustration of the reaction of LYX electrolyte and the Li anode. Once LYX electrolyte was in contact with Li metal, it was reduced to form a Y and LiX interphase that could conduct both Li+ and electrons, allowing the adverse side reaction to continue. Reproduced with permission.152 Copyright 2021, Science China Press and Springer. (b) Schematic illustration showing the mechanism of LPSCl action on the interface stability. Reproduced with permission.153 Copyright 2021, The Royal Society of Chemistry. (c) Heatmap of the reaction energy between binary coating materials and lithium chloride electrolyte. Reproduced with permission.145 Copyright 2021, American Chemical Society. The EIS evolution at different cycles of (d) Li//Li3YBr6//LCO@LIC and (e) Li//Li3YBr5.7F0.3//LCO@LIC cells. Reproduced with permission.116 Copyright 2021, Wiley-VCH. | ||
Li6PS5Cl was the ideal protection layer to prevent direct physical contact between halide electrolyte and Li metal anode.63,150,153 On the one hand, the Li6PS5Cl/halide interface had high chemical compatibility, which was conducive to the charge transfer process. On the other hand, the primary ionic conducting nature of the Li/Li6PS5Cl interface formed a stable self-limiting SEI layer (Fig. 17b). In addition, the good ductility of Li6PS5Cl ensured that cracks couldn't form during the charge–discharge cycle. The NCM-811/Li3YCl6/Li6PS5Cl/Li full cell displayed excellent recycling ability with a capacity retention of 91% and a high coulombic efficiency of 99.7% after 100 cycles.153 The NCM/Li2ZrCl6/Li6PS5Cl/Li–In full cell also exhibited a stable capacity of ∼150 mA h g−1 at 200 mA g−1 after 200 cycles.86 The Li/L6PS5Cl–Li2ZrCl6/LCO cell showed good cycling stability over 70 cycles at a 0.1C rate, with a capacity retention of 80.5% and coulombic efficiency of up to 100%. By contrast, the Li/Li2ZrCl6/LCO cell lost more than 70% of its capacity after only 3 cycles, due to severe side reactions between Li2ZrCl6 and the Li metal anode.154 However, the other experimental results showed that Li6PS5Cl and Li3InCl6 were chemically incompatible.155–157 The parasitic reaction occurred when these two came into direct contact and formed the indium sulfide-like compound in the interfacial region, which resulted in interfacial deterioration and an increase in interfacial resistance. These weren't conducive to the cycle performance of ASSBs. Moreover, Li6PS5Cl possessed intrinsic chemical incompatibility with high voltage cathode materials.158,159 Li6PS5Cl was oxidatively decomposed in the reaction voltage range of NCM811 to produce Li2S, P2S5, Li2Sn, and other phosphorus species. The molar volume of these decomposition products was smaller than that of original Li6PS5Cl, forming large numbers of voids between Li6PS5Cl and the NCM cathode material. This was greatly detrimental to the cycle stability of ASSBs.
Due to the limitations of Li6PS5Cl, it was necessary to screen out the coating materials that were compatible with halide electrolytes and cathode materials to improve the stability of the interface.145 According to calculations, the oxidation potential of binary halides (LiF, LiCl, LiBr, and LiI) and oxides (Li2O) was higher than the reduction potential of Li3InCl6, which could be used as efficient coating materials. And Li2S, Li2Se, and Li3P with a narrow electrochemical stability window were only suitable for Li3MCl6 with group 3 elements. As shown in Fig. 17c, the reaction energy between Li3N and chloride exceeded 100 meV per atom, with the risk of a chemical decomposition reaction. However, it was proved that β-Li3N could be used as a coating material to improve the interfacial stability of Li2ZrCl6 and the Li metal anode.160 β-LiN3 was an excellent ionic conductor and electrical insulator and was fully compatible with Li metal. β-Li3N could prevent direct physical contact between the halide SSE and Li metal to avoid interface side reactions. Furthermore, the high ionic conductivity of β-Li3N didn't hinder the rapid ion migration of Li+ and was conducive to the plating/stripping homogenization of Li metal. The β-Li3N layer dramatically reduced the interfacial impedance between the halide SSE and Li anode and effectively heightened the interfacial stability. It was reported that the Li6PS5Cl and Li3N mixture effectively inhibited the growth of Li dendrites, thus improving the rate capability and cycle stability of ASSBs.159 Amorphous LiNbO3 reduced the oxidation decomposition of Li3YCl6 at a high voltage of ∼4.5 V in the cycle process, so as to ensure the chemical and electrochemical compatibility between electrolyte and electrode materials.158
In addition, F− doping could also greatly improve the cycle stability of ASSBs.110,111,118 Compared with Cl− and Br−, the F− in halide SSEs had shorter and stronger bonds with Li, thus causing local distortion in the local Li coordination environment, increasing the barrier for Li+ migration and slightly reducing the ionic conductivity. Although the F− doped Li3YBr5.7F0.3 was still unstable to Li metal, a consecutive and homogeneous fluoride (LiF and YFx) layer was formed at the interface during the charging/discharging process, which could effectively inhibit interface side reactions and guarantee long cycling durability. The interface resistance between Li3YBr5.7F0.3 and the Li metal anode was relatively stable and was increased by only 300 Ω after 50 cycles (Fig. 17e). Using Li3YBr5.7F0.3 as electrolyte, the Li plating/stripping maintained over 1000 h at 0.75 mA cm−2, and the Li//Li3YBr5.7F0.3//LCO@LIC cell could still retain 60% of discharge capacity and 99% of Coulomb efficiency after 70 cycles. In contrast, the side reactions between undoped Li3YBr6 and the Li metal interface continue to occur, contributing to the non-homogeneous deposition of Li+, increased interface polarization, interface structure decomposition, and contact failure. The interface resistance between Li3YBr6 and the Li metal anode increased significantly and reached about 3000 Ω in the 50th cycle (Fig. 17d). The plating/stripping potential of the cell with Li3YBr6 increased gradually after 50 h and short-circuit failure occurred after 500 h. The ASSB assembled based on Li3YBr6 could only retain 12% of its discharge capacity after 70 cycles. By introducing Zr4+ into Li3InCl6, the system formation energy was reduced and the stability between Li3−xIn1−xZrxCl6 and the Li metal anode was improved. No chemical reaction occurred after the direct contact between Li3−xIn1−xZrxCl6 and Li metal for 24 h, while the side reaction occurred and formed a visible black spot on the Li metal sheet surface when Li3InCl6 came into contact with Li metal.108
6.3 Halide SSEs for high voltage ASSBs
Halide SSEs could combine the advantages of oxides and sulfides and exhibited good mechanical formability, considerable ionic conductivity, and excellent electrochemical oxidation stability and were outstanding candidates for next generation LIBs. After solving the interface problem with the Li metal anode, halide-based ASSBs always showed excellent electrochemical performance.156,161,162 The satisfactory capacity and rate performance of ASSLBs with halide SSEs and high voltage Li-enriched oxide cathode materials are summarized in Table 2.| Cathode | Solid electrolyte | Separator | Anode | Voltage range vs. Li+/Li [V] | Cell performance [mA h g−1] | Ref. | |
|---|---|---|---|---|---|---|---|
| First cycle CE [%]/capacity | Capacity/current density/cycle | ||||||
| a SC: super C; VGCF: vapour-grown carbon fibre. | |||||||
| LCO@Li3YCl6 | Li3YCl6 | — | Li–In | 2.5–4.2 | 94.8/119 | 111/0.1C/100 | 31 |
| NCM811@Li3YCl6@C | Li3YCl6 | Li6PS5Cl | Li | 2.9–4.3 | 87/181 | 164.7/0.1 mA cm−2/100 | 153 |
| LiNi0.88Co0.11Al0.01O2/Li3YCl6@SC | Li3YCl6 | — | Li–In | 3.0–4.3 | 89.6/199 | 192.6/0.1/200 | 28 |
| LCO@Li3InCl6 | Li3InCl6 | Li10GeP2S12 | Li–In | 3.1–4.2 | 92.7/132 | 90.3/0.5C/200 | 163 |
| NCM811@Li3InCl6@C | Li3InCl6 | Li10GeP2S12 | Li–In | 1.9–3.8 | 80.44/174.8 | 165.7/0.1C/200 | 62 |
| NCM811@Li3InCl6 | Li3InCl6 | Li10GeP2S12 | Li–In | 1.9–3.8 | 84.2/154 | 150/0.13 mA cm−2/70 | 61 |
| LCO@Li3InCl6 | Li3InCl6 | Li6PS5Cl | Li | 2.5–4.2 | —/125 | 124/10C/150 | 132 |
| LCO@Li3InCl6 | Li3InCl6 | Li10GeP2S12 | Li | 2.5–4.2 | 92/127 | 95/0.1/100 | 25 |
| LCO@Li3InCl4.8F1.2 | Li3InCl4.8F1.2/Li3InCl6 | Li6PS5Cl | In | 2.6–4.47 | 92/160.6 | 102/0.125 mA cm−2/70 | 110 |
| LCO@Li2.7In0.7Hf0.3Cl6 | Li2.7In0.7Hf0.3Cl6 | Li6PS5Cl | Li–In | 3.0–4.2 | 92.2/104.4 | 76.3/0.1C/50 | 88 |
| LCO@Li2ZrCl6 | Li2ZrCl6 | Li6PS5Cl | Li–In | 1.9–3.6 | 97.9/137 | 114/0.5C/100 | 86 |
| NCM811@Li2ZrCl6 | Li2ZrCl6 | Li6PS5Cl | Li–In | 2.2–3.8 | 90.3/181 | 181/0.1C/200 | 86 |
| NCM622@Li2ZrCl6 | Li2ZrCl6 | Li6PS5Cl | Li | 3.0–4.3 | 96.1/158.8 | 138.3/0.3C/70 | 154 |
| LCO@Li2ZrCl6 | Li2ZrCl6 | — | Li–In | 3.0–4.3 | 91.4/156 | 142.1/0.1C/100 | 85 |
| LiNi0.88Co0.11Al0.01O2/Li2.25Zr0.75Fe0.25Cl6/SC | Li2.25Zr0.75Fe0.25Cl6 | — | Li–In | 3.0–4.3 | 85.8/206 | 188.1/0.5C/100 | 85 |
| LiNi0.6Co0.2Mn0.2O2@Li2.25Zr0.75Fe0.25 | Li2.25Zr0.75Fe0.25Cl6 | Li5.5PS4.5Cl1.5 | In–Li | 3.0–4.3 | 86.99/153.1 | 105.5/0.2C/90 | 129 |
| NCM88@Li2.5Zr0.5In0.5Cl6@SC | Li2.5Zr0.5In0.5Cl6 | Li6PS5Cl | Li–In | 3.0–4.3 | 87.2/202 | 174.5/0.5C/100 | 105 |
| LCO@Li3ScCl6 | Li3ScCl6 | — | In | 2.5–4.2 | 90.3/126 | 104.5/0.1/160 | 24 |
| NCM@Li3ScCl6 | Li3ScCl6 | Li6PS5Cl | Li | 2.8–4.4 | 85.6/166.9 | 85.9/0.2C/100 | 63 |
| NMC622@Li2Sc2/3Cl4 | Li2Sc2/3Cl4 | Li6.7Si0.7Sb0.3S5I | Li–In | 2.8–4.5 | 93.9/180 | 170/0.1C/110 | 45 |
| LCO@Li2Sc2/3Cl4 | Li2Sc2/3Cl4 | Li6.7Si0.7Sb0.3S5I | Li–In | 3.0–4.3 | 93.7/135 | 120/1C/70 | 45 |
| NCM622@Li2In1/3Sc1/3Cl4 | Li2In1/3Sc1/3Cl4 | Li6.7Si0.7Sb0.3S5I | In/In–Li | 2.8–4.6 | —/194 | 180/0.2C/320 | 46 |
| NCM85@Li2In1/3Sc1/3Cl4 | Li2In1/3Sc1/3Cl4 | Li6.7Si0.7Sb0.3S5I | In/In–Li | 2.8–4.3 | —/200 | 180/0.2/600 | 46 |
| NCM85@Li2In1/3Sc1/3Cl4 | Li2In1/3Sc1/3Cl4 | Li6.7Si0.7Sb0.3S5I | In/In–Li | 2.8–4.3 | —/90 | 72/3C/3000 | 46 |
| NCM811@Li2.5Sc0.5Zr0.5Cl6 | Li2.5Sc0.5Zr0.5Cl6 | Li6PS5Cl | Li–In | 2.8–4.3 | 89.6/203.6 | 174.5/0.2C/200 | 115 |
| LCO@Li2.6Er0.6Zr0.4Cl6@VGCF | Li2.6Er0.6Zr0.4Cl6 | Li6PS5Cl | Li–In | 3.0–4.2 | 97.4/140 | 106.4/0.1C/500 | 39 |
| LCO@Li2.633Er0.633Zr0.367Cl6 | Li2.633Er0.633Zr0.367Cl6 | Li3PS4 | Li11Sn6 | 3.0–4.3 | 96.4/110 | 80/0.5C/200 | 44 |
| NCA88@Li2.6Yb0.6Hf0.4Cl6@SC | Li2.6Yb0.6Hf0.4Cl6 | Li6PS5Cl0.5Br0.5 | Li–In | 3.0–4.3 | 84.8/188 | 157.2/0.5C/1000 | 43 |
| LCO@Li2.556Yb0.492Zr0.492Cl6 | Li2.556Yb0.492Zr0.492Cl6 | Li10GeP2S12 | In–Li | 2.5–4.5 | 93.3/193.9 | 159.2/0.3C/50 | 100 |
| LCO@Li3YBr6 | Li3YBr6 | — | Li–In | 2.5–4.2 | 94.2/120 | 117/0.1C/100 | 33 |
| NMC811@Li3YBr6 | Li3YBr6 | Li5.7PS4.7Cl1.3 | In | 2.5–4.4 | —/180.2 | 67.8/0.127 mA cm−2/90 | 121 |
| LCO@Li3InCl6@Li3YBr5.7F0.3 | Li3YBr5.7F0.3 | — | Li | 2.5–4.2 | 89/126.7 | 85.1/0.1 mA cm−2/70 | 118 |
| NCM523@Li0.388Ta0.238La0.475Cl3@VGCF | Li0.388Ta0.238La0.475Cl3 | — | Li | 2.2–4.35 | 84.96/163 | 138.5/0.44C/100 | 17 |
| NCM91@LiTaCl6 | LiTaCl6 | Li5.4PS4.4Cl1.6 | Li–In | 2.5–4.8 | 91.17/232.39 | 207.6/0.3C/200 | 117 |
| LCO@ZrO2–2Li2ZrCl6@C65 | ZrO2–2Li2ZrCl6 | Li6PS5Cl | Li–In | 3.0–4.3 | 95.4/156 | 134.1/82 mA g−1/100 | 114 |
Zr was more abundant in the Earth's crust than other rare earth elements, which gave Li2ZrCl6-based ASSBs an advantage in terms of raw material cost (Fig. 18a). In addition, Li2ZrCl6 had excellent moisture resistance and further reduced storage cost and manufacturing cost. Therefore, Li2ZrCl6 exhibited broad application prospects. Li2ZrCl6 with the as-expected ionic conductivity could be prepared by ball milling, and then Li2ZrCl6-based ASSBs with excellent performance could be fabricated by the facile cold pressing method.86 The LCO/Li2ZrCl6/Li6PS5Cl/Li–In cell exhibited an initial discharge capacity of 137 mA h g−1 and a Coulomb efficiency of 97.9% at 0.1C between 1.9 and 3.6 V. After 100 cycles at 0.5C, the capacity was 114 mA h g−1 and the Coulomb efficiency reached 99.9%. By replacing the cathode material with NMC811, the performance of ASSBs could be further improved. The NMC811/Li2ZrCl6/Li6PS5Cl/Li–In cell could deliver an initial discharge capacity of 181 mA h g−1 and a Coulomb efficiency of 90.3% at 0.1C between 2.2 and 3.8 V. After 200 cycles at 1C, the capacity was 149 mA h g−1 and the Coulomb efficiency was 99.9%. After the Fe doping, the rate performance of Li2ZrCl6 was further improved.85
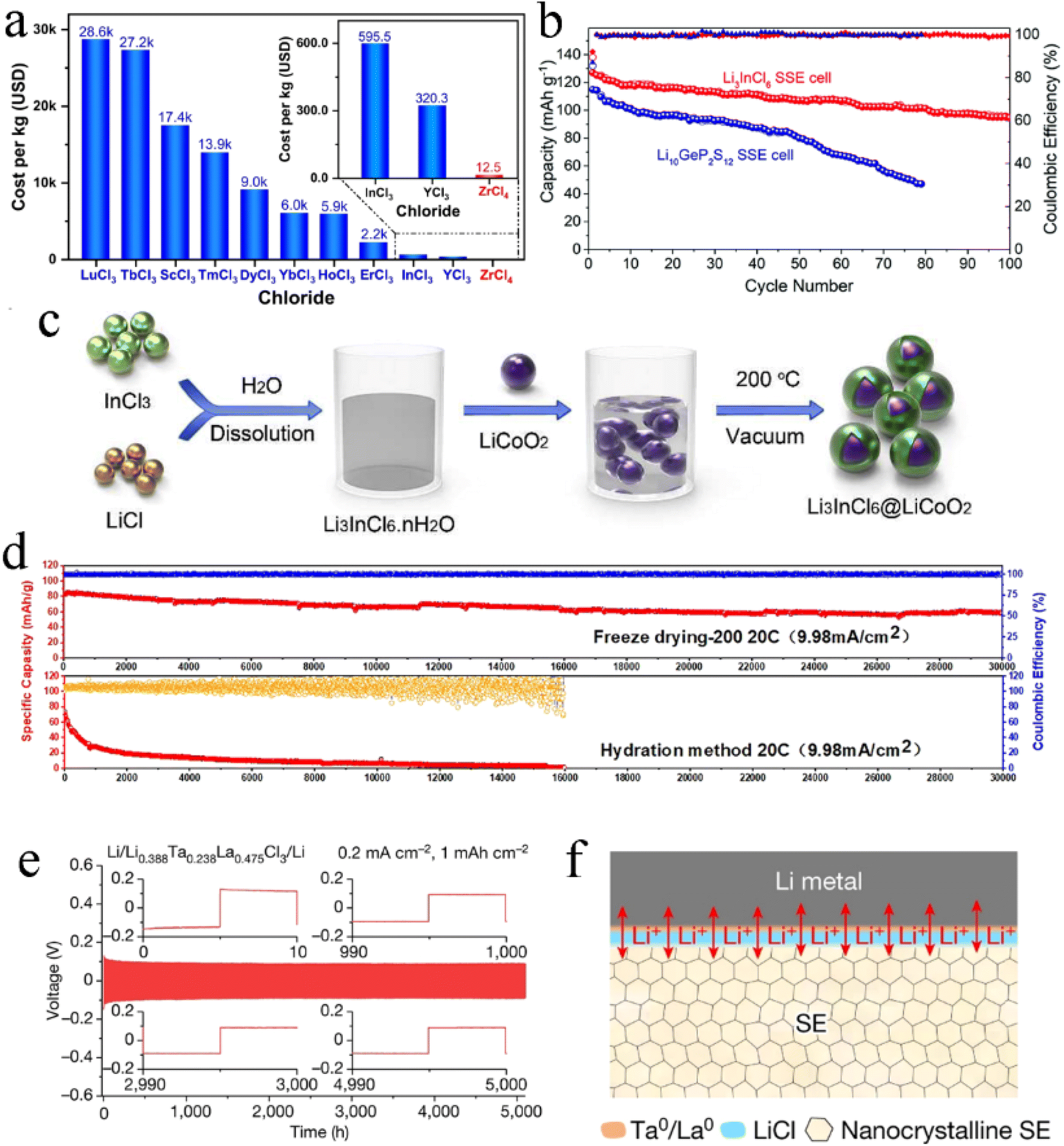 | ||
| Fig. 18 (a) The price per unit of raw materials required for the synthesis of different chloride SSEs. Reproduced with permission.86 Copyright 2021, Springer Nature. (b) The cycling performance and coulombic efficiency of LiCoO2@Li3InCl6/Li3InCl6/In and LiCoO2@Li10GeP2S12/Li10GeP2S12/In cells at 0.1C. Reproduced with permission.25 Copyright 2019, The Royal Society of Chemistry. (c) Illustration of the in situ synthesis of Li3InCl6 on the LCO surface. Reproduced with permission.163 Copyright 2020, Elsevier Ltd. (d) Cycling performance of the LCO/Li3InCl6/Li6PS5Cl/Li cell at 20C. Reproduced with permission.132 Copyright 2023, The Royal Society of Chemistry. (e) Voltage profile of the Li/Li0.388Ta0.238La0.475Cl3/Li symmetric cell cycled at a current density of 0.2 mA cm−2 at 30 °C. Reproduced with permission.17 Copyright 2023, Springer Nature. (f) Schematic of the gradient structural interphase layer generated at the Li/Li0.388Ta0.238La0.475Cl3 interface. Reproduced with permission.17 Copyright 2023, Springer Nature. | ||
Li3InCl6 could be synthesized through a H2O-mediated synthesis route, which reduced the requirement for a manufacturing facility and had the potential for scale-up production. Li3InCl6 synthesized by mechanochemistry had better cycling stability and reversible capacity than Li10GeP2S12 (Fig. 18b). The LCO/Li3InCl6/Li–In cell still had a specific capacity of 95 mA h g−1 after 100 cycles at 0.1C.25 Li3InCl6 synthesized by the wet-chemistry method also showed a stable cycling performance. The LCO/Li3InCl6/Li10GeP2S12/In cell exhibited an initial reversible specific capacity of 154 mA h g−1 and retained up to ∼150 mA h g−1 after 70 cycles.61 Li3InCl6 could be grown in situ on the surface of the LCO cathode for intimate solid–solid contact and ultra-small interfacial resistance (Fig. 18c).163 The LCO@Li3InCl6 composite cathode delivered an initial discharge capacity of 131.7 mA h g−1 and a coulombic efficiency of 92.7% at 0.1C. After 200 cycles, the capacity retention rate was 68.6%. By refining the Li3InCl6 particles, the performance of the ASSB was further improved.132 Small electrolyte particles were conducive to interfacial contact and ion transportation, which were the basis of achieving excellent rate performance and cycle performance. Most of the Li3InCl6 particles prepared by freeze-drying technology were less than 200 nm in diameter. The LCO/Li3InCl6/Li6PS5Cl/Li cell reached an initial capacity of 201 mA h g−1 at 0.5C or even 125 mA h g−1 at a high rate of 10C and then released a capacity of 124 mA h g−1 after 150 cycles at 10C. Even up to 49C, the Li3InCl6-based ASSB could still be charged and discharged normally, with a capacity of 17 mA h g−1. At 20C, the LCO/Li3InCl6/Li6PS5Cl/Li cell had an ultra-long cycle life, and the capacity retention rate reached 70 after 30000 cycles (Fig. 18d). The ASSB fabricated with the large-particle Li3InCl6 failed completely after 16, 000 cycles.
Yin et al. reported a novel chloride electrolyte Li0.388Ta0.238La0.475Cl3, with a room temperature ionic conductivity of 3.02 mS cm−1 and stability with a Li metal electrode.17 In the Li/Li0.388Ta0.238La0.475Cl3/Li symmetric cell, the interphase impedance only increased slightly during the first 20 h and then stabilized at 5000 h (Fig. 18e), which was better than that of the inorganic SSEs previously reported. This was due to the formation of a dense gradient interface passivation layer during the Li stripping/plating (Fig. 18f). The passivation layer isolated the direct contact between electrolyte and Li metal, relieved the interfacial strain and inhibited the growth of Li dendrites. Thus, Li0.388Ta0.238La0.475Cl3-based ASSBs used bare Li metal as an anode without the need for an extra buffer layer. The Li/Li0.388Ta0.238La0.475Cl3/NCM523 full cell delivered a specific capacity of 163 mA h g−1 at a 0.44 C rate and an initial coulombic efficiency of 84.96%. After 100 cycles in the cut-off voltage range of 2.2–4.35 V, the capacity retention was 81.6%.
Halide SSEs could also be used in other ASSBs. The Se@Li3HoCl6@C/Li3HoCl6/Li cell exhibited a reversible capacity of 402 mA h g−1 at 0.1C after 750 cycles.89 The Li/Li7P3S11/Li3HoBr6/S cell could maintain high coulombic efficiency (close to 100%) at 0.2C after 400 cycles.119 Li3InCl6 could be used as an interlayer to improve the stability of the Li10SnP2S12-based ASSB's cathode interface.164 Li3InCl6 could also be used to modify the interface for high-performance solid-state Li–O2 batteries.165 Li3TiCl6 could be used as a positive electrode active material for Li3TiCl6/Li2ZrCl6/Li6PS5Cl/Li–In cells with an initial capacity of 92.5 mA h g−1 at 0.1C.92 After 2500 cycles, the capacity retention was 62.3% and the final coulombic efficiency was as high as 99.7%. The halide–sulfide hybrid SSEs formed by the combination of Li3YCl6 and Li6PS5Cl showed excellent electrochemical performance in terms of discharge capacity, rate capability and cycling performance.166
7. Conclusion and outlook
In summary, this review presented the cognition and understanding of halide SSEs and their applications in ASSBs. Firstly, the screening principle of halide SSE composition was proposed. With the assistance of computational simulation, Cl− was considered to be the most suitable halogen anion because of chloride's ability to well balance ionic conductivity and the electrochemical stability window. Group 3 elements (Sc, Y, and lanthanides) were the most promising metal cations because they matched the electronegativity of halogen anions. Secondly, the theory of structural design of halide electrolytes with high ionic conductivity and the mechanism of Li ion migration were described. Compared with trigonal and orthorhombic structures, the monoclinic structure-based electrolyte had a 3D diffusion pathway with a low energy barrier and obtained higher ionic conductivity. Additionally, strategies for halide SSEs were discussed, including dual-halogen, isovalent cation substitution, and aliovalent cation substitution. Reasonable substitution could improve the ionic conductivity of halide SSEs, broaden the electrochemical stability window, and enhance moisture resistance. Furthermore, the mechanism of moisture resistance and synthesis of halide electrolytes were analyzed. Wet chemical synthesis was the most potential method, which had the advantages of convenience and high efficiency and was beneficial for scale-up preparation of halide SSEs. Finally, the applications of halide SSEs in ASSBs were outlined. Li2ZrCl6 had more advantages in terms of cost, while Li3InCl6 was outstanding in terms of electrochemical performance. A Li3InCl6-based ASSB could cycle normally up to 30000 times at a high rate of 20C.
Although halide SSEs ushered in their second spring since 2018 and made breakthrough progress in recent years, to achieve commercial applications as soon as possible, there are still urgent issues to be solved in the following aspects.
(1) The upper limit of ionic conductivity for halide SSEs is still an open question. Although the formation of dual-halogen SSEs by haloanion substitution makes the ionic conductivity of Li3Y(Br3Cl3) reach up to 7.2 mS cm−1, enough to be comparable to that of sulfide SSEs known for their high ionic conductivity, there is still a significant gap from theoretical prediction. Some strategies should be used to try to narrow this gap, such as defect design and grain boundary enhancement.
(2) At present, halide SSEs with high ionic conductivity mainly rely on rare earth metals as central elements, resulting in high raw material cost. Li2ZrCl6 can greatly reduce the manufacturing cost, but its ionic conductivity is only 0.81 mS cm−1. Through partial substitution of Sc, the ionic conductivity of Li2.5Sc0.5Zr0.5Cl6 reaches 2.23 mS cm−1, which is still not outstanding. Other substitution strategies are needed to further improve the ionic conductivity of Li2ZrCl6 under the premise of controlling the cost of raw materials.
(3) Wet chemistry synthesis is the most convenient and efficient preparation method, which can realize the large-scale manufacturing of halide SSEs. The H2O-mediated synthesis route is only applicable to Li3InCl6. Although wet chemistry synthesis is extended to the preparation of other electrolytes by ammonium-assisted methods, it is only successful for ternary electrolytes. Developing a quaternary electrolyte through substitution is an effective way to improve the comprehensive properties of halide SSEs. Cationic substitution by the wet chemistry synthesis should be attempted.
(4) The compatibility of halide SSEs with high voltage cathodes has reached a satisfactory level, but the interface instability with Li metal anodes is still a difficult problem. Introducing an additional separator mitigates this dilemma, but the manufacturing process increases. The report of Li0.388Ta0.238La0.475Cl3 inspires researchers to improve the interfacial stability between halide SSEs and Li metal by designing electrolytes to in situ generate a gradient interfacial passivation layer.
Author contributions
All authors contributed to the writing and revision of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Guangxi Scientific Base and Talent Special Project (No. AD20297134), National Key Research and Development Program (No. 2022YFEO134600 and 2021YFA0715404), Guangxi Key Research and Development Program (No. 2021AB05083) and National Natural Science Foundation of China (No. 52272152).References
- Y. Bi, J. Tao, Y. Wu, L. Li, Y. Xu, E. Hu, B. Wu, J. Hu, C. Wang, J.-G. Zhang, Y. Qi and J. Xiao, Science, 2020, 370, 1313–1317 CrossRef CAS.
- X. Fan and C. Wang, Chem. Soc. Rev., 2021, 50, 10486–10566 RSC.
- T. Liu, J. Liu, L. Li, L. Yu, J. Diao, T. Zhou, S. Li, A. Dai, W. Zhao, S. Xu, Y. Ren, L. Wang, T. Wu, R. Qi, Y. Xiao, J. Zheng, W. Cha, R. Harder, I. Robinson, J. Wen, J. Lu, F. Pan and K. Amine, Nature, 2022, 606, 305–312 CrossRef CAS.
- L. Wang, T. Liu, T. Wu and J. Lu, Nature, 2022, 611, 61–67 CrossRef CAS.
- F. Wu, J. Maier and Y. Yu, Chem. Soc. Rev., 2020, 49, 1569–1614 RSC.
- Y. Gao, Z. Pan, J. Sun, Z. Liu and J. Wang, Nano-Micro Lett., 2022, 14, 94 CrossRef CAS.
- Y. Tian, G. Zeng, A. Rutt, T. Shi, H. Kim, J. Wang, J. Koettgen, Y. Sun, B. Ouyang, T. Chen, Z. Lun, Z. Rong, K. Persson and G. Ceder, Chem. Rev., 2021, 121, 1623–1669 CrossRef CAS PubMed.
- X. Fan, C. Zhong, J. Liu, J. Ding, Y. Deng, X. Han, L. Zhang, W. Hu, D. P. Wilkinson and J. Zhang, Chem. Rev., 2022, 122, 17155–17239 CrossRef CAS PubMed.
- B. S. Vishnugopi, E. Kazyak, J. A. Lewis, J. Nanda, M. T. McDowell, N. P. Dasgupta and P. P. Mukherjee, ACS Energy Lett., 2021, 6, 3734–3749 CrossRef CAS.
- C. Sun, J. Liu, Y. Gong, D. P. Wilkinson and J. Zhang, Nano Energy, 2017, 33, 363–386 CrossRef CAS.
- Y. Zheng, Y. Yao, J. Ou, M. Li, D. Luo, H. Dou, Z. Li, K. Amine, A. Yu and Z. Chen, Chem. Soc. Rev., 2020, 49, 8790–8839 RSC.
- B. Tao, C. Ren, H. Li, B. Liu, X. Jia, X. Dong, S. Zhang and H. Chang, Adv. Funct. Mater., 2022, 32, 2203551 CrossRef CAS.
- R. Chen, Q. Li, X. Yu, L. Chen and H. Li, Chem. Rev., 2020, 120, 6820–6877 CrossRef CAS PubMed.
- K. B. Hatzell, Matter, 2021, 3, 2533–2535 Search PubMed.
- T. Yu, X. Yang, R. Yang, X. Bai, G. Xu, S. Zhao, Y. Duan, Y. Wu and J. Wang, J. Alloys Compd., 2021, 885, 161013 CrossRef CAS.
- D. Wu, L. Chen, H. Li and F. Wu, Appl. Phys. Lett., 2022, 121, 120502 CrossRef CAS.
- Y. C. Yin, J. T. Yang, J. D. Luo, G. X. Lu, Z. Huang, J. P. Wang, P. Li, F. Li, Y. C. Wu, T. Tian, Y. F. Meng, H. S. Mo, Y. H. Song, J. N. Yang, L. Z. Feng, T. Ma, W. Wen, K. Gong, L. J. Wang, H. X. Ju, Y. Xiao, Z. Li, X. Tao and H. B. Yao, Nature, 2023, 616, 77–83 CrossRef CAS.
- J. Janek and W. G. Zeier, Nat. Energy, 2023, 8, 230–240 CrossRef.
- H. Kwak, S. Wang, J. Park, Y. Liu, K. T. Kim, Y. Choi, Y. Mo and Y. S. Jung, ACS Energy Lett., 2022, 7, 1776–1805 CrossRef CAS.
- Y. Nikodimos, W. N. Su and B. J. Hwang, Adv. Energy Mater., 2023, 13, 2202854 CrossRef CAS.
- H. Wu, H. Han, Z. Yan, Q. Zhao and J. Chen, J. Solid State Electrochem., 2022, 26, 1791–1808 CrossRef CAS.
- J. Liang, X. Li, K. R. Adair and X. Sun, Acc. Chem. Res., 2021, 54, 1023–1033 CrossRef CAS.
- C. Wang, J. Liang, J. T. Kim and X. Sun, Sci. Adv., 2022, 8, eadc9516 CrossRef CAS.
- J. Liang, X. Li, S. Wang, K. R. Adair, W. Li, Y. Zhao, C. Wang, Y. Hu, L. Zhang, S. Zhao, S. Lu, H. Huang, R. Li, Y. Mo and X. Sun, J. Am. Chem. Soc., 2020, 142, 7012–7022 CrossRef CAS PubMed.
- X. Li, J. Liang, J. Luo, M. Norouzi Banis, C. Wang, W. Li, S. Deng, C. Yu, F. Zhao, Y. Hu, T.-K. Sham, L. Zhang, S. Zhao, S. Lu, H. Huang, R. Li, K. R. Adair and X. Sun, Energy Environ. Sci., 2019, 12, 2665–2671 RSC.
- W. Li, J. Liang, M. Li, K. R. Adair, X. Li, Y. Hu, Q. Xiao, R. Feng, R. Li, L. Zhang, S. Lu, H. Huang, S. Zhao, T.-K. Sham and X. Sun, Chem. Mater., 2020, 32, 7019–7027 CrossRef CAS.
- S. Wang, Q. Bai, A. M. Nolan, Y. Liu, S. Gong, Q. Sun and Y. Mo, Angew. Chem., Int. Ed., 2019, 58, 8039–8043 CrossRef CAS.
- Y. Han, S. H. Jung, H. Kwak, S. Jun, H. H. Kwak, J. H. Lee, S. T. Hong and Y. S. Jung, Adv. Energy Mater., 2021, 11, 2100126 CrossRef CAS.
- M. Jiang, S. Mukherjee, Z. W. Chen, L. X. Chen, M. L. Li, H. Y. Xiao, C. Gao and C. V. Singh, Phys. Chem. Chem. Phys., 2020, 22, 22758–22767 RSC.
- D. C. Ginnings and T. E. Phipps, J. Am. Chem. Soc., 1930, 52, 1340–1345 CrossRef CAS.
- K. Ryoji, T. Yasuo, M. Masashi and Y. Osamu, Chem. Lett., 1989, 18, 223–226 CrossRef.
- C. Li, L. Gu and J. Maier, Adv. Funct. Mater., 2012, 22, 1145–1149 CrossRef CAS.
- T. Asano, A. Sakai, S. Ouchi, M. Sakaida, A. Miyazaki and S. Hasegawa, Adv. Mater., 2018, 30, 1803075 CrossRef.
- X. Li, J. Liang, X. Yang, K. R. Adair, C. Wang, F. Zhao and X. Sun, Energy Environ. Sci., 2020, 13, 1429–1461 RSC.
- S. R. Combs, P. K. Todd, P. Gorai and A. E. Maughan, J. Electrochem. Soc., 2022, 169, 040551 CrossRef CAS.
- J. Liang, X. Li, J. T. Kim, X. Hao, H. Duan, R. Li and X. Sun, Angew. Chem., Int. Ed., 2023, 62, e202217081 CAS.
- J. Y. Huang, K. Iputera, A. Jena, Z. Tong, D. H. Wei, S. F. Hu and R. S. Liu, J. Chin. Chem. Soc., 2022, 69, 1233–1241 CrossRef CAS.
- X. Li, J. Liang, K. R. Adair, J. Li, W. Li, F. Zhao, Y. Hu, T. K. Sham, L. Zhang, S. Zhao, S. Lu, H. Huang, R. Li, N. Chen and X. Sun, Nano Lett., 2020, 20, 4384–4392 CrossRef CAS PubMed.
- Q. Shao, C. Yan, M. Gao, W. Du, J. Chen, Y. Yang, J. Gan, Z. Wu, W. Sun, Y. Jiang, Y. Liu, M. Gao and H. Pan, ACS Appl. Mater. Interfaces, 2022, 14, 8095–8105 CrossRef CAS.
- S. Muy, J. Voss, R. Schlem, R. Koerver, S. J. Sedlmaier, F. Maglia, P. Lamp, W. G. Zeier and Y. Shao-Horn, iScience, 2019, 16, 270–282 CrossRef CAS.
- R. Schlem, S. Muy, N. Prinz, A. Banik, Y. Shao-Horn, M. Zobel and W. G. Zeier, Adv. Energy Mater., 2020, 10, 1903719 CrossRef CAS.
- P. Molaiyan, S. E. Mailhiot, K. Voges, A. M. Kantola, T. Hu, P. Michalowski, A. Kwade, V.-V. Telkki and U. Lassi, Mater. Des., 2023, 227, 111690 CrossRef CAS.
- J. Park, D. Han, H. Kwak, Y. Han, Y. J. Choi, K.-W. Nam and Y. S. Jung, Chem. Eng. J., 2021, 425, 130630 CrossRef CAS.
- K.-H. Park, K. Kaup, A. Assoud, Q. Zhang, X. Wu and L. F. Nazar, ACS Energy Lett., 2020, 5, 533–539 CrossRef CAS.
- L. Zhou, C. Y. Kwok, A. Shyamsunder, Q. Zhang, X. Wu and L. F. Nazar, Energy Environ. Sci., 2020, 13, 2056–2063 RSC.
- L. Zhou, T.-T. Zuo, C. Y. Kwok, S. Y. Kim, A. Assoud, Q. Zhang, J. Janek and L. F. Nazar, Nat. Energy, 2022, 7, 83–93 CrossRef CAS.
- N. Tanibata, M. Kato, S. Takimoto, H. Takeda, M. Nakayama and H. Sumi, Adv. Energy Sustainability Res., 2020, 1, 2000025 CrossRef.
- H. Kwak, J. Lyoo, J. Park, Y. Han, R. Asakura, A. Remhof, C. Battaglia, H. Kim, S.-T. Hong and Y. S. Jung, Energy Storage Mater., 2021, 37, 47–54 CrossRef.
- J. Park, J. P. Son, W. Ko, J.-S. Kim, Y. Choi, H. Kim, H. Kwak, D.-H. Seo, J. Kim and Y. S. Jung, ACS Energy Lett., 2022, 7, 3293–3301 CrossRef CAS.
- D. Park, K. Kim, G. H. Chun, B. C. Wood, J. H. Shim and S. Yu, J. Mater. Chem. A, 2021, 9, 23037–23045 RSC.
- Y. Qie, S. Wang, S. Fu, H. Xie, Q. Sun and P. Jena, J. Phys. Chem. Lett., 2020, 11, 3376–3383 CrossRef PubMed.
- Y. Lian, M. Wu, B. Xu, B. He, G. Liu, J. Shi, Q. Kuang, H. Wang and C. Ouyang, J. Mater. Chem. A, 2023, 11, 1906–1919 RSC.
- R. Li, K. Xu, K. Liu, R. Si and Z. Zhang, Chem. Mater., 2022, 34, 8356–8365 CrossRef CAS.
- H. Huang, C. Chi, J. Zhang, X. Zheng, Y. Wu, J. Shen, X. Wang and S. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 36864–36874 CrossRef CAS PubMed.
- H. Huang, H.-H. Wu, C. Chi, Y. Yang, J. Zheng, B. Huang and S. Wang, J. Mater. Chem. A, 2021, 9, 26256–26265 RSC.
- E. A. Wu, S. Banerjee, H. Tang, P. M. Richardson, J.-M. Doux, J. Qi, Z. Zhu, A. Grenier, Y. Li, E. Zhao, G. Deysher, E. Sebti, H. Nguyen, R. Stephens, G. Verbist, K. W. Chapman, R. J. Clément, A. Banerjee, Y. S. Meng and S. P. Ong, Nat. Commun., 2021, 12, 1256 CrossRef CAS PubMed.
- E. Sebti, J. Qi, P. M. Richardson, P. Ridley, E. A. Wu, S. Banerjee, R. Giovine, A. Cronk, S.-Y. Ham, Y. S. Meng, S. P. Ong and R. J. Clément, J. Mater. Chem. A, 2022, 10, 21565–21578 RSC.
- R. Schlem, A. Banik, M. Eckardt, M. Zobel and W. G. Zeier, ACS Appl. Energy Mater., 2020, 3, 10164–10173 CrossRef CAS.
- F. Hussain, P. Yu, J. Zhu, H. Xia, Y. Zhao and W. Xia, Adv. Theory Simul., 2023, 6, 2200569 CrossRef CAS.
- N. Flores-Gonzalez, N. Minafra, G. Dewald, H. Reardon, R. I. Smith, S. Adams, W. G. Zeier and D. H. Gregory, ACS Mater. Lett., 2021, 3, 652–657 CrossRef CAS.
- X. Li, J. Liang, N. Chen, J. Luo, K. R. Adair, C. Wang, M. N. Banis, T. K. Sham, L. Zhang, S. Zhao, S. Lu, H. Huang, R. Li and X. Sun, Angew. Chem., Int. Ed., 2019, 58, 16427–16432 CrossRef CAS.
- X. Luo, D. Cai, X. Wang, X. Xia, C. Gu and J. Tu, ACS Appl. Mater. Interfaces, 2022, 14, 29844–29855 CrossRef CAS.
- C. Wang, J. Liang, J. Luo, J. Liu, X. Li, F. Zhao, R. Li, H. Huang, S. Zhao, L. Zhang, J. Wang and X. Sun, Sci. Adv., 2021, 7, eabh1896 CrossRef CAS PubMed.
- D. Zagorac, H. Muller, S. Ruehl, J. Zagorac and S. Rehme, J. Appl. Crystallogr., 2019, 52, 918–925 CrossRef CAS PubMed.
- Z. Wang, X. Lin, Y. Han, J. Cai, S. Wu, X. Yu and J. Li, Nano Energy, 2021, 89, 106337 CrossRef CAS.
- W. Qiu, Y. Wang and J. Liu, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1592 CAS.
- A. Vasylenko, J. Gamon, B. B. Duff, V. V. Gusev, L. M. Daniels, M. Zanella, J. F. Shin, P. M. Sharp, A. Morscher, R. Chen, A. R. Neale, L. J. Hardwick, J. B. Claridge, F. Blanc, M. W. Gaultois, M. S. Dyer and M. J. Rosseinsky, Nat. Commun., 2021, 12, 5561 CrossRef PubMed.
- F. Li, X. Cheng, L. L. Lu, Y. C. Yin, J. D. Luo, G. Lu, Y. F. Meng, H. Mo, T. Tian, J. T. Yang, W. Wen, Z. P. Liu, G. Zhang, C. Shang and H. B. Yao, Nano Lett., 2022, 22, 2461–2469 CrossRef CAS PubMed.
- L. Kahle, A. Marcolongo and N. Marzari, Energy Environ. Sci., 2020, 13, 928–948 RSC.
- J. Qi, S. Banerjee, Y. Zuo, C. Chen, Z. Zhu, M. L. Holekevi Chandrappa, X. Li and S. P. Ong, Mater. Today Phys., 2021, 21, 100463 CrossRef CAS.
- T. H. Wan and F. Ciucci, ACS Appl. Energy Mater., 2021, 4, 7930–7941 CrossRef CAS.
- W. Chen, Y. Li, D. Feng, C. Lv, H. Li, S. Zhou, Q. Jiang, J. Yang, Z. Gao, Y. He and J. Luo, J. Power Sources, 2023, 561, 232720 CrossRef CAS.
- Z. Xu, X. Chen, R. Chen, X. Li and H. Zhu, npj Comput. Mater., 2020, 6, 47 CrossRef CAS.
- Y. Yu, Z. Wang and G. Shao, J. Mater. Chem. A, 2021, 9, 25585–25594 RSC.
- S. Zhang, J. Ma, S. Dong and G. Cui, Electrochem. Energy Rev., 2023, 6, 4 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A: Found. Adv., 1976, 32, 751–767 CrossRef.
- A. D. Sendek, G. Cheon, M. Pasta and E. J. Reed, J. Phys. Chem. C, 2020, 124, 8067–8079 CrossRef CAS.
- K. Kim, D. Park, H.-G. Jung, K. Y. Chung, J. H. Shim, B. C. Wood and S. Yu, Chem. Mater., 2021, 33, 3669–3677 CrossRef CAS.
- H. Chun, K. Nam, S. J. Hong, J. Kang and B. Han, J. Mater. Chem. A, 2021, 9, 15605–15612 RSC.
- Y. Liu, S. Wang, A. M. Nolan, C. Ling and Y. Mo, Adv. Energy Mater., 2020, 10, 2002356 CrossRef CAS.
- Z. Xu and H. Zhu, Chem. Mater., 2020, 32, 4618–4626 CrossRef CAS.
- Z. Xu, X. Chen, K. Liu, R. Chen, X. Zeng and H. Zhu, Chem. Mater., 2019, 31, 7425–7433 CrossRef CAS.
- R. Schlem, T. Bernges, C. Li, M. A. Kraft, N. Minafra and W. G. Zeier, ACS Appl. Energy Mater., 2020, 3, 3684–3691 CrossRef CAS.
- Y. Wang, W. D. Richards, S. P. Ong, L. J. Miara, J. C. Kim, Y. Mo and G. Ceder, Nat. Mater., 2015, 14, 1026–1031 CrossRef CAS PubMed.
- H. Kwak, D. Han, J. Lyoo, J. Park, S. H. Jung, Y. Han, G. Kwon, H. Kim, S. T. Hong, K. W. Nam and Y. S. Jung, Adv. Energy Mater., 2021, 11, 2003190 CrossRef CAS.
- K. Wang, Q. Ren, Z. Gu, C. Duan, J. Wang, F. Zhu, Y. Fu, J. Hao, J. Zhu, L. He, C. W. Wang, Y. Lu, J. Ma and C. Ma, Nat. Commun., 2021, 12, 4410 CrossRef CAS PubMed.
- H. Ito, K. Shitara, Y. Wang, K. Fujii, M. Yashima, Y. Goto, C. Moriyoshi, N. C. Rosero-Navarro, A. Miura and K. Tadanaga, Adv. Sci., 2021, 8, 2101413 CrossRef CAS.
- H. Wang, Y. Li, Y. Tang, D. Ye, T. He, H. Zhao and J. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 5504–5511 CrossRef CAS PubMed.
- X. Li, J. Liang, J. T. Kim, J. Fu, H. Duan, N. Chen, R. Li, S. Zhao, J. Wang, H. Huang and X. Sun, Adv. Mater., 2022, 34, 2200856 CrossRef CAS PubMed.
- R. Schlem, A. Banik, S. Ohno, E. Suard and W. G. Zeier, Chem. Mater., 2021, 33, 327–337 CrossRef CAS.
- M. Gombotz and H. M. R. Wilkening, ACS Sustainable Chem. Eng., 2021, 9, 743–755 CrossRef CAS.
- K. Wang, Z. Gu, Z. Xi, L. Hu and C. Ma, Nat. Commun., 2023, 14, 1396 CrossRef CAS PubMed.
- N. Flores-González, M. López, N. Minafra, J. Bohnenberger, F. Viñes, S. Rudić, I. Krossing, W. G. Zeier, F. Illas and D. H. Gregory, J. Mater. Chem. A, 2022, 10, 13467–13475 RSC.
- F. Hussain, J. Zhu, H. Xia, Y. Zhao and W. Xia, J. Phys. Chem. C, 2022, 126, 13105–13113 CrossRef CAS.
- S. Hyun, H. Chun, M. Hong, J. Kang and B. Han, J. Mater. Chem. A, 2023, 11, 4272–4279 RSC.
- J. Liang, E. Maas, J. Luo, X. Li, N. Chen, K. R. Adair, W. Li, J. Li, Y. Hu, J. Liu, L. Zhang, S. Zhao, S. Lu, J. Wang, H. Huang, W. Zhao, S. Parnell, R. I. Smith, S. Ganapathy, M. Wagemaker and X. Sun, Adv. Energy Mater., 2022, 12, 2103921 CrossRef CAS.
- S. Y. Kim, K. Kaup, K.-H. Park, A. Assoud, L. Zhou, J. Liu, X. Wu and L. F. Nazar, ACS Mater. Lett., 2021, 3, 930–938 CrossRef CAS.
- Y. Huang, Y. Yu, H. Xu, X. Zhang, Z. Wang and G. Shao, J. Mater. Chem. A, 2021, 9, 14969–14976 RSC.
- D. Park, H. Park, Y. Lee, S. O. Kim, H. G. Jung, K. Y. Chung, J. H. Shim and S. Yu, ACS Appl. Mater. Interfaces, 2020, 12, 34806–34814 CrossRef CAS PubMed.
- G. Xu, L. Luo, J. Liang, S. Zhao, R. Yang, C. Wang, T. Yu, L. Wang, W. Xiao, J. Wang, J. Yu and X. Sun, Nano Energy, 2022, 92, 106674 CrossRef CAS.
- B. He, A. Ye, S. Chi, P. Mi, Y. Ran, L. Zhang, X. Zou, B. Pu, Q. Zhao, Z. Zou, D. Wang, W. Zhang, J. Zhao, M. Avdeev and S. Shi, Sci. Data, 2020, 7, 153 CrossRef CAS PubMed.
- B. He, P. Mi, A. Ye, S. Chi, Y. Jiao, L. Zhang, B. Pu, Z. Zou, W. Zhang, M. Avdeev, S. Adams, J. Zhao and S. Shi, Acta Mater., 2021, 203, 116490 CrossRef CAS.
- E. van der Maas, W. Zhao, Z. Cheng, T. Famprikis, M. Thijs, S. R. Parnell, S. Ganapathy and M. Wagemaker, J. Phys. Chem. C, 2023, 127, 125–132 CrossRef CAS.
- Z. Liu, S. Ma, J. Liu, S. Xiong, Y. Ma and H. Chen, ACS Energy Lett., 2021, 6, 298–304 CrossRef CAS.
- H. Kwak, D. Han, J. P. Son, J. S. Kim, J. Park, K.-W. Nam, H. Kim and Y. S. Jung, Chem. Eng. J., 2022, 437, 135413 CrossRef CAS.
- E. van der Maas, T. Famprikis, S. Pieters, J. P. Dijkstra, Z. Li, S. R. Parnell, R. I. Smith, E. R. H. van Eck, S. Ganapathy and M. Wagemaker, J. Mater. Chem. A, 2023, 11, 4559–4571 RSC.
- B. Helm, R. Schlem, B. Wankmiller, A. Banik, A. Gautam, J. Ruhl, C. Li, M. R. Hansen and W. G. Zeier, Chem. Mater., 2021, 33, 4773–4782 CrossRef CAS.
- J. Fu, S. Yang, J. Hou, L. Azhari, Z. Yao, X. Ma, Y. Liu, P. Vanaphuti, Z. Meng, Z. Yang, Y. Zhong and Y. Wang, J. Power Sources, 2023, 556, 232465 CrossRef CAS.
- X. Luo, X. Wu, J. Xiang, D. Cai, M. Li, X. Wang, X. Xia, C. Gu and J. Tu, ACS Appl. Mater. Interfaces, 2021, 13, 47610–47618 CrossRef CAS PubMed.
- S. Zhang, F. Zhao, S. Wang, J. Liang, J. Wang, C. Wang, H. Zhang, K. Adair, W. Li, M. Li, H. Duan, Y. Zhao, R. Yu, R. Li, H. Huang, L. Zhang, S. Zhao, S. Lu, T. K. Sham, Y. Mo and X. Sun, Adv. Energy Mater., 2021, 11, 2100836 CrossRef CAS.
- X. Chen, Z. Jia, H. Lv, C. Wang, N. Zhao and X. Guo, J. Power Sources, 2022, 545, 231939 CrossRef CAS.
- S. Chen, C. Yu, S. Chen, L. Peng, C. Liao, C. Wei, Z. Wu, S. Cheng and J. Xie, Chin. Chem. Lett., 2022, 33, 4635–4639 CrossRef CAS.
- H. Zhang, Z. Yu, H. Chen, Y. Zhou, X. Huang and B. Tian, J. Energy Chem., 2023, 79, 348–356 CrossRef CAS.
- H. Kwak, J. S. Kim, D. Han, J. S. Kim, J. Park, G. Kwon, S. M. Bak, U. Heo, C. Park, H. W. Lee, K. W. Nam, D. H. Seo and Y. S. Jung, Nat. Commun., 2023, 14, 2459 CrossRef CAS.
- W. Li, Z. Chen, Y. Chen, W. Duan, G. Liu, Y. Lv, H. Yang and L. Yao, Chem. Eng. J., 2023, 455, 140509 CrossRef CAS.
- H. Zhang, Z. Zeng, X. Shi, C. H. Wang and Y. Du, EcoMat, 2023, 5, e12315 CAS.
- Y. Ishiguro, K. Ueno, S. Nishimura, G. Iida and Y. Igarashib, Chem. Lett., 2023, 52, 237–241 CrossRef CAS.
- T. Yu, J. Liang, L. Luo, L. Wang, F. Zhao, G. Xu, X. Bai, R. Yang, S. Zhao, J. Wang, J. Yu and X. Sun, Adv. Energy Mater., 2021, 11, 2101915 CrossRef CAS.
- X. Shi, Z. Zeng, M. Sun, B. Huang, H. Zhang, W. Luo, Y. Huang, Y. Du and C. Yan, Nano Lett., 2021, 21, 9325–9331 CrossRef CAS PubMed.
- X. Shi, Z. Zeng, H. Zhang, B. Huang, M. Sun, H. H. Wong, Q. Lu, W. Luo, Y. Huang, Y. Du and C. H. Yan, Small Methods, 2021, 5, 2101002 CrossRef CAS.
- M. A. Plass, S. Bette, R. E. Dinnebier and B. V. Lotsch, Chem. Mater., 2022, 34, 3227–3235 CrossRef CAS.
- T. Jeon and S. C. Jung, J. Mater. Chem. A, 2023, 11, 4334–4344 RSC.
- C. Yu, Y. Li, K. R. Adair, W. Li, K. Goubitz, Y. Zhao, M. J. Willans, M. A. Thijs, C. Wang, F. Zhao, Q. Sun, S. Deng, J. Liang, X. Li, R. Li, T.-K. Sham, H. Huang, S. Lu, S. Zhao, L. Zhang, L. van Eijck, Y. Huang and X. Sun, Nano Energy, 2020, 77, 105097 CrossRef CAS.
- E. Sebti, H. A. Evans, H. Chen, P. M. Richardson, K. M. White, R. Giovine, K. P. Koirala, Y. Xu, E. Gonzalez-Correa, C. Wang, C. M. Brown, A. K. Cheetham, P. Canepa and R. J. Clement, J. Am. Chem. Soc., 2022, 144, 5795–5811 CrossRef CAS PubMed.
- Y. Wang, Y. Wu, Z. Wang, L. Chen, H. Li and F. Wu, J. Mater. Chem. A, 2022, 10, 4517–4532 RSC.
- Y. Ni, C. Huang, H. Liu, Y. Liang and L. Z. Fan, Adv. Funct. Mater., 2022, 32, 2205998 CrossRef CAS.
- E. Umeshbabu, S. Maddukuri, Y. Hu, M. Fichtner and A. R. Munnangi, ACS Appl. Mater. Interfaces, 2022, 14, 25448–25456 CrossRef CAS PubMed.
- Y. Kim and S. Choi, J. Power Sources, 2023, 567, 232962 CrossRef CAS.
- S. Chen, C. Yu, C. Wei, L. Peng, S. Cheng and J. Xie, Chin. Chem. Lett., 2023, 34, 107544 CrossRef CAS.
- Ö. U. Kudu, T. Famprikis, B. Fleutot, M.-D. Braida, T. Le Mercier, M. S. Islam and C. Masquelier, J. Power Sources, 2018, 407, 31–43 CrossRef.
- H.-W. Liu, C.-C. Lin, P.-Y. Chang, S.-C. Haw, H.-S. Sheu, J.-M. Chen, C.-C. Chen, R.-J. Jeng and N.-L. Wu, J. Solid State Electrochem., 2022, 26, 2089–2096 CrossRef CAS.
- T. Ma, Z. Wang, D. Wu, P. Lu, X. Zhu, M. Yang, J. Peng, L. Chen, H. Li and F. Wu, Energy Environ. Sci., 2023, 16, 2142–2152 RSC.
- M. Yang, L. Chen, H. Li and F. Wu, Energy Mater. Adv., 2022, 2022, 9842651 Search PubMed.
- L. Zhu, Y. Wang, J. Chen, W. Li, T. Wang, J. Wu, S. Han, Y. Xia, Y. Wu, M. Wu, F. Wang, Y. Zheng, L. Peng, J. Liu, L. Chen and W. Tang, Sci. Adv., 2022, 8, eabj7698 CrossRef CAS PubMed.
- P. Lu, D. Wu, L. Chen, H. Li and F. Wu, Electrochem. Energy Rev., 2022, 5, 3 CrossRef CAS.
- A. Sharafi, E. Kazyak, A. L. Davis, S. Yu, T. Thompson, D. J. Siegel, N. P. Dasgupta and J. Sakamoto, Chem. Mater., 2017, 29, 7961–7968 CrossRef CAS.
- Y. Zhu and Y. Mo, Angew. Chem., Int. Ed., 2020, 59, 17472–17476 CrossRef CAS PubMed.
- S. Wang, X. Xu, C. Cui, C. Zeng, J. Liang, J. Fu, R. Zhang, T. Zhai and H. Li, Adv. Funct. Mater., 2022, 32, 2108805 CrossRef CAS.
- J.-S. Kim, S. Soo Shin, J.-H. Lee, B.-K. Kim and H. Kim, Appl. Surf. Sci., 2022, 574, 151621 CrossRef CAS.
- R. L. Sacci, T. H. Bennett, A. R. Drews, V. Anandan, M. J. Kirkham, L. L. Daemen and J. Nanda, J. Mater. Chem. A, 2021, 9, 990–996 RSC.
- E. McCalla, M. T. Sougrati, G. Rousse, E. J. Berg, A. Abakumov, N. Recham, K. Ramesha, M. Sathiya, R. Dominko, G. Van Tendeloo, P. Novak and J. M. Tarascon, J. Am. Chem. Soc., 2015, 137, 4804–4814 CrossRef CAS PubMed.
- B. Zahiri, A. Patra, C. Kiggins, A. X. B. Yong, E. Ertekin, J. B. Cook and P. V. Braun, Nat. Mater., 2021, 20, 1392–1400 CrossRef CAS PubMed.
- X. Miao, S. Guan, C. Ma, L. Li and C. W. Nan, Adv. Mater., 2023 DOI:10.1002/adma.202206402.
- I. Kochetkov, T.-T. Zuo, R. Ruess, B. Singh, L. Zhou, K. Kaup, J. Janek and L. Nazar, Energy Environ. Sci., 2022, 15, 3933–3944 RSC.
- G. H. Chun, J. H. Shim and S. Yu, ACS Appl. Mater. Interfaces, 2022, 14, 1241–1248 CrossRef CAS PubMed.
- P. Bonnick and J. Muldoon, Energy Environ. Sci., 2022, 15, 1840–1860 RSC.
- G. Wang, M. Zhu, Y. Zhang, C. Song, X. Zhu, Z. Huang, Y. Zhang, F. Yu, G. Xu, M. Wu, H. K. Liu, S. X. Dou and C. Wu, InfoMat, 2022, 4, e12293 CAS.
- K. B. Hatzell, X. C. Chen, C. L. Cobb, N. P. Dasgupta, M. B. Dixit, L. E. Marbella, M. T. McDowell, P. P. Mukherjee, A. Verma, V. Viswanathan, A. S. Westover and W. G. Zeier, ACS Energy Lett., 2020, 5, 922–934 CrossRef CAS.
- D. K. Singh, T. Fuchs, C. Krempaszky, B. Mogwitz, S. Burkhardt, F. H. Richter and J. Janek, Adv. Funct. Mater., 2022, 33, 2211067 CrossRef.
- L. M. Riegger, R. Schlem, J. Sann, W. G. Zeier and J. Janek, Angew. Chem., Int. Ed., 2021, 60, 6718–6723 CrossRef CAS.
- Y. Zhu, X. He and Y. Mo, J. Mater. Chem. A, 2016, 4, 3253–3266 RSC.
- Y. Fu and C. Ma, Sci. China Mater., 2021, 64, 1378–1385 CrossRef CAS.
- W. Ji, D. Zheng, X. Zhang, T. Ding and D. Qu, J. Mater. Chem. A, 2021, 9, 15012–15018 RSC.
- H. Zhang, Z. Yu, J. Cheng, H. Chen, X. Huang and B. Tian, Chin. Chem. Lett., 2023 DOI:10.1016/j.cclet.2023.108228.
- T. Koç, M. Hallot, E. Quemin, B. Hennequart, R. Dugas, A. M. Abakumov, C. Lethien and J.-M. Tarascon, ACS Energy Lett., 2022, 7, 2979–2987 CrossRef.
- T. Koç, F. Marchini, G. Rousse, R. Dugas and J.-M. Tarascon, ACS Appl. Energy Mater., 2021, 4, 13575–13585 CrossRef.
- C. Rosenbach, F. Walther, J. Ruhl, M. Hartmann, T. A. Hendriks, S. Ohno, J. Janek and W. G. Zeier, Adv. Energy Mater., 2023, 13, 2203673 CrossRef CAS.
- J. Jang, Y.-T. Chen, G. Deysher, D. Cheng, S.-Y. Ham, A. Cronk, P. Ridley, H. Yang, B. Sayahpour, B. Han, W. Li, W. Yao, E. A. Wu, J.-M. Doux, L. H. B. Nguyen, J. A. S. Oh, D. H. S. Tan and Y. S. Meng, ACS Energy Lett., 2022, 7, 2531–2539 CrossRef CAS.
- Y. Subramanian, R. Rajagopal and K.-S. Ryu, J. Alloys Compd., 2023, 940, 168867 CrossRef CAS.
- X. Xu, G. Du, C. Cui, J. Liang, C. Zeng, S. Wang, Y. Ma and H. Li, ACS Appl. Mater. Interfaces, 2022, 14, 39951–39958 CrossRef CAS PubMed.
- R. Yu, C. Wang, H. Duan, M. Jiang, A. Zhang, A. Fraser, J. Zuo, Y. Wu, Y. Sun, Y. Zhao, J. Liang, J. Fu, S. Deng, Z. Ren, G. Li, H. Huang, R. Li, N. Chen, J. Wang, X. Li, C. V. Singh and X. Sun, Adv. Mater., 2023, 35, 2207234 CrossRef CAS.
- T. A. Hendriks, M. A. Lange, E. M. Kiens, C. Baeumer and W. G. Zeier, Batteries Supercaps, 2023, 6, e202200544 CrossRef CAS.
- C. Wang, J. Liang, M. Jiang, X. Li, S. Mukherjee, K. Adair, M. Zheng, Y. Zhao, F. Zhao, S. Zhang, R. Li, H. Huang, S. Zhao, L. Zhang, S. Lu, C. V. Singh and X. Sun, Nano Energy, 2020, 76, 105015 CrossRef CAS.
- Q. Luo, C. Yu, C. Wei, S. Chen, S. Chen, Z. Jiang, L. Peng, S. Cheng and J. Xie, Ceram. Int., 2023, 49, 11485–11493 CrossRef CAS.
- C. Zhao, J. Liang, X. Li, N. Holmes, C. Wang, J. Wang, F. Zhao, S. Li, Q. Sun, X. Yang, J. Liang, X. Lin, W. Li, R. Li, S. Zhao, H. Huang, L. Zhang, S. Lu and X. Sun, Nano Energy, 2020, 75, 105036 CrossRef CAS.
- J. S. Kim, S. Jung, H. Kwak, Y. Han, S. Kim, J. Lim, Y. M. Lee and Y. S. Jung, Energy Storage Mater., 2023, 55, 193–204 CrossRef.
| This journal is © The Royal Society of Chemistry 2023 |