
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe proximity-enabled sulfur fluoride exchange reaction in the protein context†
Bingchen
Yu‡
 ,
Li
Cao‡
,
Shanshan
Li
,
Paul C.
Klauser
and
Lei
Wang
*
,
Li
Cao‡
,
Shanshan
Li
,
Paul C.
Klauser
and
Lei
Wang
*
Department of Pharmaceutical Chemistry, The Cardiovascular Research Institute, Helen Diller Family Comprehensive Cancer Center, University of California San Francisco, 555 Mission Bay Blvd. South, San Francisco, California 94158, USA. E-mail: Lei.wang2@ucsf.edu
First published on 20th June 2023
Abstract
The proximity-enabled sulfur(VI) fluoride exchange (SuFEx) reaction generates specific covalent linkages between proteins in cells and in vivo, which opens innovative avenues for studying elusive protein–protein interactions and developing potent covalent protein drugs. To exploit the power and expand the applications of covalent proteins, covalent linkage formation between proteins is the critical step, for which fundamental kinetic and essential properties remain unexplored. Herein, we systematically studied SuFEx kinetics in different proteins and conditions. In contrast to in small molecules, SuFEx in interacting proteins conformed with a two-step mechanism involving noncovalent binding, followed by covalent bond formation, exhibiting nonlinear rate dependence on protein concentration. The protein SuFEx rate consistently changed with protein binding affinity as well as chemical reactivity of the functional group and was impacted by target residue identity and solution pH. In addition, kinetic analyses of nanobody SR4 binding with SARS-CoV-2 spike protein revealed that viral target mutations did not abolish covalent binding but decreased the SuFEx rate with affinity decrease. Moreover, off-target cross-linking of a SuFEx-capable nanobody in human serum was not detected, and the SuFEx-generated protein linkage was stable at cellular acidic pHs, suggesting SuFEx suitability for in vivo usage. These results advanced our understanding of SuFEx reactivity and kinetics in proteins, which is invaluable for ongoing exploration of SuFEx-enabled covalent proteins for basic biological research and creative biotherapeutics.
Introduction
As a new generation of click chemistry,1,2 sulfur(VI) fluoride exchange (SuFEx) has been widely used in small molecules in diverse fields such as chemical synthesis,2,3 medicinal chemistry,4–7 material chemistry8,9 and chemical biology.10,11 Aryl fluorosulfates and aryl sulfonyl fluorides are relatively latent towards biological nucleophiles, yet become reactive upon being placed in proximity to target protein residues, creating stable covalent linkages via SuFEx under cellular conditions.1,11–13 This unique property drives the design and discovery of covalent small molecule inhibitors with therapeutic potentials.5,6,14 Besides applications in small molecules, SuFEx has emerging power in proteins. For instance, it promoted the study of protein–protein interactions through cross-linking interacting protein pairs in vivo,12,13,15 which preserves weak and transient protein–protein interactions for subsequent analysis. Recently, we demonstrated the concept of generating covalent protein drugs via proximity-enabled SuFEx between proteins in vivo.13,15,16 In this strategy, latent bioreactive unnatural amino acids (Uaas) carrying aryl fluorosulfate or sulfonyl fluoride groups are site-specifically incorporated into protein drugs via genetic code expansion. These Uaas include fluorosulfate-L-tyrosine (FSY),13 fluorosulfonyloxybenzoyl-L-lysine (FSK),15meta-fluorosulfate-L-tyrosine (mFSY),17 fluorine-substituted fluorosulfate-L-tyrosine (FFY),18 and o-sulfonyl fluoride-O-methyltyrosine (SFY).19,20 Upon binding with the target protein, these Uaas can crosslink with Lys, His or Tyr on the target via SuFEx to form a stable covalent linkage between the drug and target proteins. Covalent protein drugs were firstly demonstrated in vivo on a programmed cell death protein-1 (PD-1)/PD-L1 pair, where FSY was incorporated into PD-1 to covalently block PD-L1, exhibiting potent antitumor efficacy over the noncovalent wildtype PD-1.21 This strategy has been subsequently used to build covalent antibodies for human rhinovirus 14 (HRV14) 3C protease,22 covalent protein inhibitors for neutralizing SARS-CoV-2,18,23 and covalent protein radiopharmaceuticals to enhance efficacy and safety for targeted radionuclide therapies,24 showing great potential in developing novel biotherapeutics.For various emerging SuFEx applications in proteins, the formation of the protein–protein covalent linkage is a key step. SuFEx kinetics determines the covalent bond formation rate and would thus directly impact the efficacy. Although SuFEx kinetics has been characterized on small molecules,25 it has not been systematically studied in the protein–protein context. Chemical reactivity and kinetics can differ significantly at the level of small molecules from proteins.1,26–28 For instance, target-specific covalent small molecule inhibitors have been shown to inhibit proteins in a generic two-step mechanism involving non-covalent complex formation followed by covalent reaction.29 Here we report a systematic investigation of SuFEx kinetic and essential properties in the protein–protein context. We found that SuFEx kinetics in the protein–protein context conformed with a two-step mechanism, which is different from kinetics on small molecules but analogous to that of covalent small molecules inhibiting their target protein. Protein–protein SuFEx showed nonlinear dependence of protein concentrations, the rate of which was affected by protein binding affinity, chemical reactivity of the functional group, target residue identity, and pH. We further studied the important practical properties of SuFEx in proteins, including the impact of target mutations, off-target reactivity, and linkage stability. These results will be highly valuable in guiding the innovative use of SuFEx for studying and engineering proteins in covalent chemical mode. The general kinetic principles revealed for covalent linkage formation between proteins should further inspire the development of new proximity-enabled chemistry for biocompatible usage in vivo.
Results and discussion
Protein–protein covalent linkage formation via proximity-enabled reactivity
Covalent linkage formation between two interacting proteins through proximity-enabled reactivity generally occurs in two steps (Scheme 1): the two proteins first bind forming a non-covalent complex, which places the latent bioreactive Uaa of one protein close to a target natural residue of the other protein.30 Accelerated by the proximity effects, the latent Uaa and its target residue then undergo reaction, giving rise to a specific covalent linkage. The initial non-covalent binding of the two proteins is governed by their affinity (Ki), and the subsequent covalent bond formation is determined by the covalent reaction rate k2. The reaction under study in this work is SuFEx, for which the latent bioreactive Uaa is FSY carrying the fluorosulfonate group and its target natural residue can be Lys, His, or Tyr. Fluorosulfonate undergoes the SuFEx reaction with the nucleophilic side chains of these natural residues when placed in close proximity. Owing to the two-step mechanism, we expect that the kinetics for such protein–protein covalent linkage formation would be different from the second-order kinetics for two reacting small molecules, in which the reaction rate keeps increasing with reactant concentrations. Instead, we expect that the kinetics would be analogous to that of an irreversible small molecule inhibitor acting on its target protein. We tested this kinetic hypothesis using different binding protein pairs and SuFEx-capable functional groups.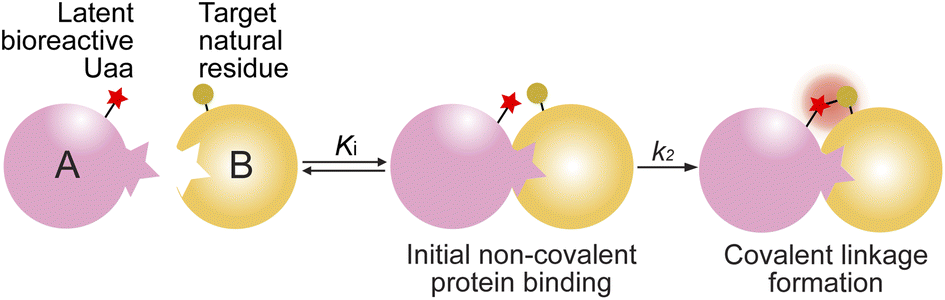 | ||
| Scheme 1 Covalent linkage formation between interacting proteins via proximity-enabled reactivity. | ||
SuFEx kinetics in the Zspa affibody–Z protein pair
We began with an affibody-target protein pair to study the kinetics of SuFEx in the protein context. The Zspa affibody (Afb) binds with its target Z protein with a Kd of ∼6 μM.31 On the basis of the crystal structure of the Afb-Z complex,31 the latent bioreactive Uaa FSY was incorporated at site 24 of the Z protein to covalently target Lys7 of the Afb (Fig. 1A).13 The FSY incorporation and target sites were selected based on their average distance and side chain orientation so that the side chains could contact when the two proteins bind. Maltose binding protein (MBP) was fused to the N-terminus of the Z protein generating the MBP-Z fusion protein to better separate the Z protein from the Afb, which have similar molecular weights. MBP-Z(24FSY) and Afb were expressed in E. coli and affinity purified through the Hisx6-tag appended at their C-termini.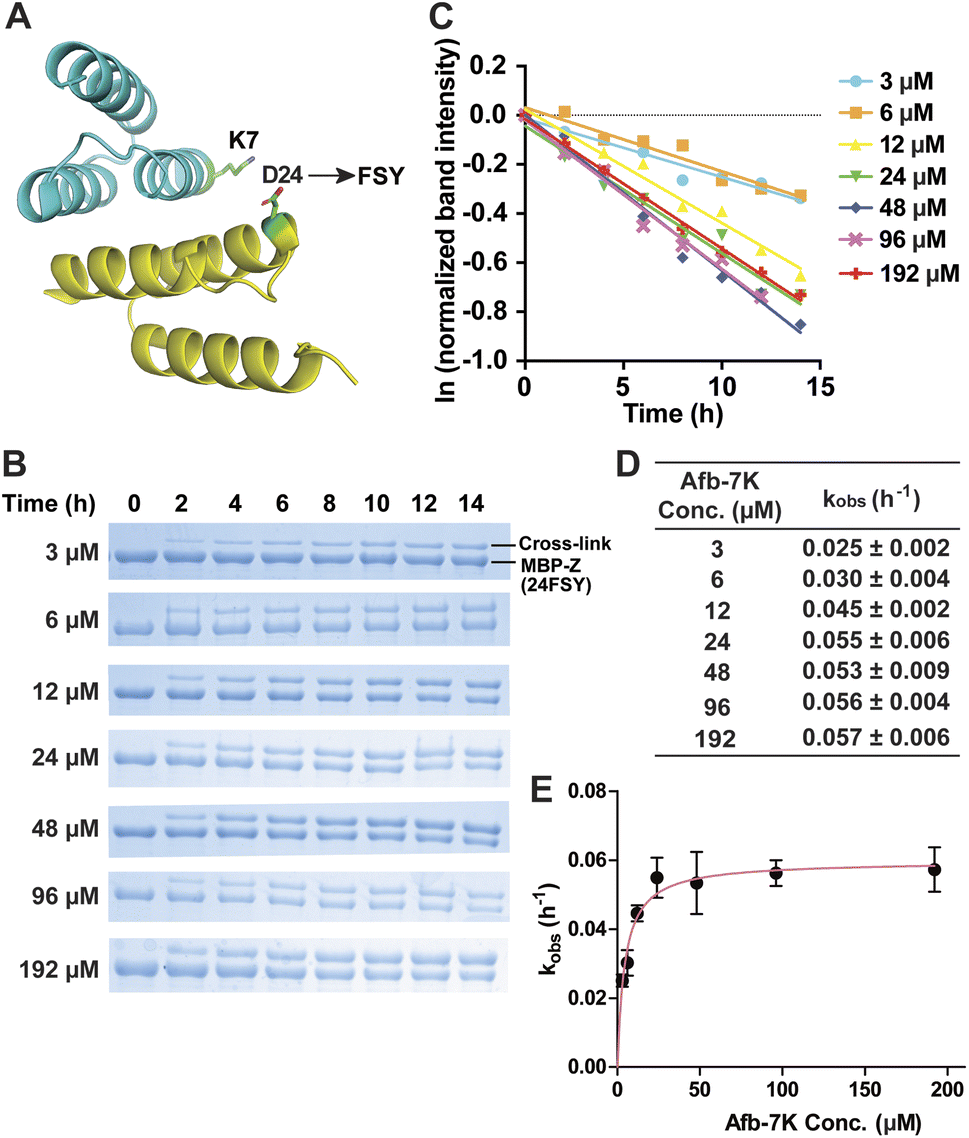 | ||
| Fig. 1 Measurement of SuFEx kinetics in the Zspa affibody–Z protein pair. (A) Crystal structure of the Zspa affibody (Afb, cyan) binding with the Z protein (yellow), with residue Asp24 for FSY incorporation and target Lys7 shown in sticks. (B) SDS-PAGE analysis of cross-linking of 6 μM MBP-Z(24FSY) with different concentrations of Afb in vitro for the indicated duration of incubation time. (C) Determination of the pseudo-first-order rate constant kobs for different concentrations of Afb. The experiments were independently repeated three times, and the data for one time are shown here. (D) Calculated kobs for each Afb concentration. Mean ± s.d., n = 3. (E) Plotting kobs against Afb concentration [Afb] to determine the second-order rate constant k. The data were fitted to equation kobs = kmax[Afb]/(KS + [Afb]). Error bars represent s.d., n = 3. | ||
We first tested the stability of proteins containing FSY in aqueous buffer. MBP-Z(24FSY) was incubated in PBS (pH = 7.4) at 37 °C for 24 h and analyzed with high resolution electrospray ionization time-of-flight mass spectrometry (ESI-TOF MS). The protein's MS peak did not change before and after incubation (Fig. S1†). Similar results were also obtained with another protein, in which FSY was incorporated into site 6 of ubiquitin (Fig. S2†). These results indicate that FSY was stable in proteins under the tested conditions.
Next, 6 μM MBP-Z(24FSY) was incubated with Afb ranging from 3 to 192 μM in phosphate-buffered saline (PBS, pH 7.4) at 37 °C. At different time points, samples were extracted, quenched with 4% SDS, and analyzed with SDS-PAGE under denatured conditions. At all concentrations, we observed a time-dependent decrease of MBP-Z(24FSY) band intensity and a concomitant increase of cross-link band intensity (Fig. 1B).
To determine the kinetics of covalent protein complex formation, we measured the MBP-Z(24FSY) band intensities and normalized the band intensities at different time points relative to the band intensity at 0 h (before cross-linking). The natural logarithm (ln) of the normalized MBP-Z(24FSY) band intensity was plotted against time. For each Afb concentration tested, a linear relationship was observed in the plot with R2 > 0.95 in most cases (Fig. 1C and S3†). The pseudo-first-order rate constant, kobs, was calculated for each concentration (Fig. 1D), and was plotted against Afb concentration in Fig. 1E. The kobs for covalent protein complex formation initially increased with increasing concentration of Afb and then reached a plateau at around 24 μM Afb, exhibiting a hyperbolic dependence that saturated at a maximum rate constant of kmax = 0.0597 ± 0.0019 h−1. The half-saturating concentration KS, the concentration needed to reach half of the maximum reaction rate, of Afb was calculated to be 4.5 μM. Therefore, the second-order rate constant, k = kmax/KS,29,32 for covalent protein complex formation was determined to be (1.32 ± 0.04) × 104 M−1 h−1.
SuFEx kinetics in the nanobody 7D12–EGFR pair
The proximity-enabled SuFEx reaction between proteins is driven by the binding of the two proteins, which places the latent bireactive Uaa in close proximity to its target natural residue. We thus expect that the protein binding affinity could play a critical role in the SuFEx kinetics. To investigate this effect, we further studied the SuFEx kinetics using the 7D12 nanobody, which binds to the human epidermal growth factor receptor (EGFR) with a Kd of 200 nM.33 Its binding affinity is higher than that of the Afb–Z protein pair (Kd = 6 μM).On the basis of the crystal structure of the 7D12–EGFR complex,33 FSY was incorporated at site 109 of the 7D12 nanobody [7D12(109FSY)] to covalently target Lys443 of the EGFR (Fig. 2A).15 The mutant nanobody 7D12(109FSY) was expressed in E. coli and affinity purified through the Hisx6 tag appended at the C-terminus. We then incubated 0.2 μM EGFR with 7D12(109FSY) ranging from 0.2 to 12.8 μM in PBS (pH 7.4) at 37 °C. Samples were extracted, quenched, and analyzed with SDS-PAGE at different incubation time points. The extracellular domain of the EGFR with an Fc tag (total MW = 95 kDa), instead of a full-length EGFR (MW = 200 kDa), was used in these in vitro cross-linking experiments, so that the cross-linked EGFR could be clearly separated from the non-cross-linked EGFR by gel electrophoresis. When 0.2 μM 7D12(109FSY) was used, the cross-linking band was not obvious until 10 h, suggesting that the cross-linking rate was low at this concentration. At other 7D12(109FSY) concentrations, a time-dependent decrease of EGFR band intensities and concomitant increase of cross-link band intensities were detected (Fig. 2B).
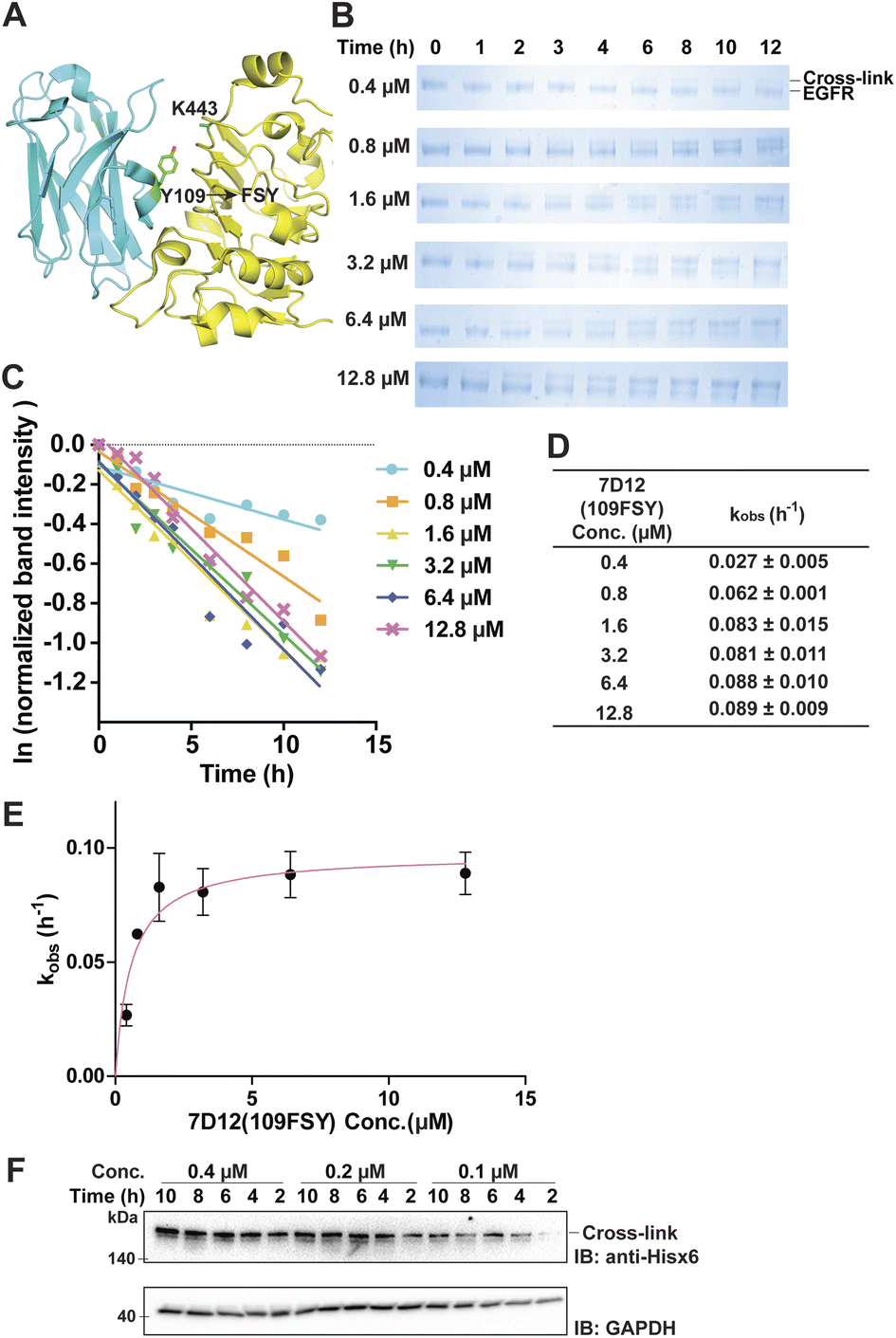 | ||
| Fig. 2 Measurement of SuFEx kinetics in the nanobody 7D12–EGFR pair. (A) Crystal structure of nanobody 7D12 (cyan) binding with the human EGFR (yellow), with residue Tyr109 for FSY incorporation and target Lys443 shown in sticks. (B) SDS-PAGE analysis of cross-linking of 0.2 μM EGFR with different concentrations of 7D12(109FSY) in vitro for the indicated duration of incubation time. (C) Determination of the pseudo-first-order rate constant kobs for different concentrations of 7D12(109FSY). The experiments were independently repeated three times, and the data for one time are shown here. (D) Calculated kobs for each 7D12(109FSY) concentration. Mean ± s.d., n = 3. (E) Plotting kobs against 7D12(109FSY) concentration [7D12] to determine the second-order rate constant k. The data were fitted to equation kobs = kmax[7D12]/(KS + [7D12]). Error bars represent s.d., n = 3. (F) Cross-linking of 7D12(109FSY) with the EGFR on the cell surface. Cells were incubated with varying concentrations of 7D12(109FSY) for the indicated time period, after which cell lysates were analyzed by denatured western blot to detect the cross-linked EGFR–7D12 complex. GAPDH was detected as the loading control. | ||
Plotting the normalized EGFR band intensity against time also showed a linear relationship with R2 > 0.90 in most cases (Fig. 2C and S4†). The pseudo-first-order rate constant, kobs, was calculated for each concentration of 7D12(109FSY) (Fig. 2D) and plotted against 7D12(109FSY) concentration (Fig. 2E). Similar to what was observed in the Afb–Z protein pair, the kobs for covalent 7D12(109FSY)–EGFR complex formation initially increased with increasing concentration of 7D12(109FSY) and then reached a plateau, displaying a hyperbolic dependence. The kmax was calculated to be 0.0972 ± 0.0054 h−1 and KS 0.58 μM, giving a second-order rate constant k of (1.68 ± 0.09) × 105 M−1 h−1.
Protein SuFEx kinetics and effects of binding affinity
In both protein pairs, we observed that the protein SuFEx rate initially increased with protein concentration when protein concentration was low and then reached a maximum rate despite further increase of protein concentration. These results are consistent with the two-step mechanism (Scheme 1) for protein–protein covalent linkage formation via proximity-enabled reactivity and confirmed our kinetics hypothesis. When both protein concentrations are low (≤Kd), a significant amount of protein is not bound, limiting the SuFEx rate; an increase of protein concentration promotes protein association and increases the SuFEx rate. When one protein is supplied in high concentration (>several-fold of Kd), the majority of the other protein has already been bound, and further increase of the former will no longer significantly increase the SuFEx rate, resulting in the observed rate plateau.A comparison of SuFEx kinetics in the above two protein pairs reveals the effect of the binding affinity of the proteins. For the Afb–Z protein pair with a lower binding affinity, the maximum kobs was 0.0597 ± 0.0019 and was reached when MBP-Z(24FSY) and Afb-7K concentrations were 6 μM and 24 μM, respectively. The calculated second-order rate constant was (1.32 ± 0.04) × 104 M−1 h−1. For the 7D12–EGFR pair with a higher binding affinity, the maximum kobs was 0.0972 ± 0.0054 h−1 and was reached when EGFR and 7D12(109FSY) concentrations were 0.2 μM and 1.6 μM, respectively. The calculated second-order rate constant was (1.68 ± 0.09) × 105 M−1 h−1, ten-times higher than that of the Afb–Z protein pair. Therefore, a higher binding affinity resulted in a faster apparent second-order SuFEx rate between proteins.
SuFEx kinetics of the nanobody 7D12 cross-linking endogenous EGFR on the cell surface
Aside from purified proteins binding in vitro, we also evaluated SuFEx kinetics on native proteins expressed on the live cell surface, which represents a general application setting of covalent protein binders or therapeutics. Specifically, EGFR-expressing A431 cells were incubated with varying concentrations of 7D12(109FSY) for the indicated time period, after which cell lysates were analyzed with western blot under denatured conditions to detect the EGFR cross-linked by the nanobody 7D12(109FSY). Ideally, an EGFR-specific antibody should be used to detect both the cross-linked and non-cross-linked EGFRs in order to quantitate the kinetics. However, the full-length native EGFR on cells has a large molecule weight (MW) of 200 kDa, making it difficult to clearly separate it from the EGFR cross-linked by 7D12(109FSY) (MW = 215 kDa). As a result, we used an anti-Hisx6 antibody to detect the Hisx6 tag appended at the C-terminus of 7D12(109FSY) so as to visualize the cross-linked EGFR–7D12(109FSY) complex.As shown in Fig. 2F, the cross-linking bands intensified with the increase of incubation time. On the basis of the band intensities, when 7D12(109FSY) concentration increased from 0.1 μM to 0.2 μM, the cross-linking rates increased; however, no significant rate increase was observed when a higher concentration (0.4 μM) was used. Further increasing concentration of 7D12(109FSY) from 0.4 μM to 3.2 μM did not further increase the cross-link rates (Fig. S5†). These results indicate that the SuFEx rate on the cell surface also increased with increasing concentration of 7D12(109FSY) and plateaued around 0.2 μM. Interestingly, the SuFEx rate for 7D12(109FSY)–EGFR on the cell surface reached the plateau at a lower 7D12(109FSY) concentration (0.2 μM) than that in vitro (1.6 μM). The EGFR is known to cluster on the cell surface, which can enhance the local effective concentration and contribute to this difference.
Effects of chemical reactivity on protein SuFEx kinetics
Besides the fluorosulfonate group, the sulfonyl fluoride group is another latent bioreactive functional group that can react through SuFEx.1,2,34 In the protein–protein context, both fluorosulfonate and sulfonyl fluoride can form stable linkages with side chains of His, Lys, and Tyr, but their resultant linkages with the side chains of Cys, Ser, and Thr are unstable.13,20,25,35,36 From the chemical point of view, sulfonyl fluoride is considered to be more reactive than fluorosulfonate.25,37 Since the second step of the proximity-enabled SuFEx reaction between proteins directly involves reactivity, we thus evaluated how intrinsic chemical reactivity impacts protein SuFEx kinetics. We recently designed and genetically encoded the Uaa o-sulfonyl fluoride-O-methyltyrosine (SFY), which bears a sulfonyl fluoride group. SFY is able to crosslink proteins with different types of biomolecules including proteins, carbohydrates, and RNA.19,20 The sulfonyl fluoride group in SFY is located at the meta position of the phenyl ring, while FSY has the fluorosulfonate group at the para position. We have shown that the orientation of the functional group affects the reactivity in the proximity-enabled SuFEx reaction on proteins.17,34 Therefore, we decided to compare SFY with mFSY, whose fluorosulfonate group is also at the meta position (Fig. 3A).17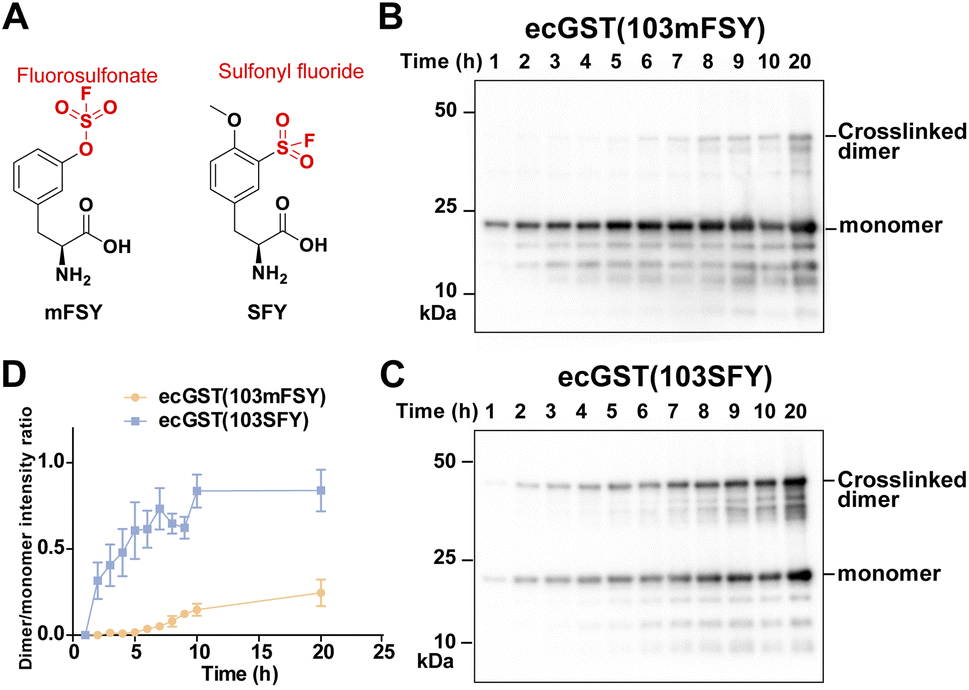 | ||
| Fig. 3 SuFEx kinetics comparison between fluorosulfonate and sulfonyl fluoride in ecGST. (A) Structure of SFY and mFSY bearing sulfonyl fluoride and fluorosulfonate groups, respectively. (B and C) Covalent dimerization of ecGST(103mFSY) (B) and ecGST(103SFY) (C) at different time points in E. coli cells. The experiments were independently repeated three times, and the data for one time are shown here. (D) Band intensity ratio of ecGST crosslinked dimer to monomer at different time points. n = 3 independent repeats; error bars represent s.e.m. | ||
We compared the SuFEx kinetics of mFSY and SFY using E. coli glutathione transferase (ecGST), a homodimeric protein. On the basis of the crystal structure of ecGST,38 mFSY or SFY was incorporated into site 103 of ecGST at the dimer interface to target Lys107 of the other monomer.39 ecGST(103mFSY) and ecGST(103SFY) were separately expressed in E. coli cells. At different time points after the induction of ecGST expression, E. coli cells were extracted, and cell lysates were analyzed by denatured western blot to detect the covalent dimerization of ecGST. As shown in Fig. 3B–D, ecGST(103SFY) exhibited a much faster covalent dimerization rate than ecGST(103mFSY), suggesting that sulfonyl fluoride affords a faster SuFEx reaction rate than fluorosulfonate in this protein context.
Effects of target residue identity and pH on protein SuFEx kinetics
Previously we demonstrated that FSY can crosslink with Lys, His, or Tyr through SuFEx in various proteins. Here we systematically compared the SuFEx kinetics of FSY with these different target residues in the context of the Afb–Z protein pair. We incubated 6 μM MBP-Z(24FSY) with 192 μM Afb-7K, Afb-7H, or Afb-7Y in PBS (pH 7.4) at 37 °C. At different time points, samples were extracted, quenched, and analyzed with SDS-PAGE under denatured conditions. An excess of Afb was used to measure the maximum SuFEx rate. As shown in Fig. 4A, Afb-7K, Afb-7H, and Afb-7Y all cross-linked with MBP-Z(24FSY) in a time-dependent manner. We quantified the MBP-Z(24FSY) band intensities and calculated the kobs using the same method described above (Fig. 4B and S6†). Under these conditions, the maximum SuFEx rate (kobs) for Afb-7H, Afb-7K, and Afb-7Y with FSY was 0.110 ± 0.001 h−1, 0.057 ± 0.006 h−1, and 0.022 ± 0.007 h−1, respectively (Fig. 4C). The binding affinity between WT MBP-Z and Afb-7K, 7H, or 7Y was measured with biolayer interferometry, which showed slight differences and a trend opposite to that of kobs (Fig. S7†), suggesting that the mutation effect on binding affinity was not the main drive of the kobs difference. Afb-7H exhibited the fastest SuFEx rate possibly because the histidine side chain has the lowest pKa and thus the most efficient deprotonation. | ||
| Fig. 4 Effects of the target residue identity and pH on SuFEx kinetics in the Afb–Z protein pair. (A) SDS-PAGE analysis of cross-linking of 6 μM MBP-Z(24FSY) with 192 μM Afb-7K, Afb-7H, or Afb-7Y at pH 7.4 in vitro at different time points. (B) Determination of maximum kobs between MBP-Z(24FSY) and different Afb at pH 7.4. The experiments were independently repeated three times, and the data for one time are shown here. (C) Calculated maximum kobs between MBP-Z(24FSY) and different Afb at pH 7.4 and 8.8 (mean ± s.d., n = 3). (D) SDS-PAGE analysis of cross-linking of 6 μM MBP-Z(24FSY) with 192 μM Afb-7K, Afb-7H, or Afb-7Y at pH 8.8 in vitro at different time points. | ||
We next incubated 192 μM Afb and 6 μM MBP-Z(24FSY) in Tris buffer (pH 8.8) at 37 °C to study the effects of pH on SuFEx kinetics (Fig. 4D and S8†). Afb-7K cross-linked with MBP-Z(24FSY) at pH 8.8 quite fast, so a narrower time range (0 to 5 h) was used to improve the accuracy of measurement. A more basic pH increased the SuFEx rate for all three residues, but to varying extents. Afb-7K cross-linked with MBP-Z(24FSY) about 5-times faster at pH 8.8 than at pH 7.4; Afb-7Y was 3.5-fold faster; Afb-7H had just a slight rate increase (Fig. 4C). Interestingly, while Afb-7H showed the fastest SuFEx kobs rate with FSY at pH 7.4, Afb-7K had the fastest SuFEx kobs rate at pH 8.8.
We selected the Afb–Z protein pair to study the pH effect because the incorporated FSY and its target residue are both at the surface of the protein complex, making them more amenable to pH change of the solution. For Uaa-target residue located in a protein microenvironment that can resist external pH change, adjusting solution pH may or may not impact the SuFEx kinetics.
Effects of target mutation on protein SuFEx kinetics
An emerging application of proximity-enabled reactivity is the development of covalent protein drugs, target of which may undergo mutation during treatment and disease progression. Such mutations may alter the binding affinity of the protein drug and the kinetics of covalent linkage formation between the protein drug and its target, thus affecting the drug efficacy. We therefore evaluated the influence of target protein mutations on SuFEx kinetics in the protein context.Utilizing the proximity-enabled SuFEx reaction, we recently developed covalent nanobodies to neutralize SARS-CoV-2.18 FSY was incorporated into nanobodies and the resultant nanobodies covalently bind with the receptor binding domain (RBD) of the spike protein of SARS-CoV-2, thus blocking the binding of SARS-CoV-2 to the human ACE2 receptor, a critical step for SARS-CoV-2 infection. Various mutated SARS-CoV-2 strains have emerged in the pandemic. Mutations on the spike RBD such as E484K, F490L and N439K have been shown to decrease affinity towards neutralization antibodies;40–42 The B.1.1.7 lineage, which mainly possesses N501Y mutation, shows a stronger interaction with ACE2 and a faster spreading rate;43 The B.1.351 lineage, which has K417N, E484K, and N501Y mutations on the spike RBD, has decreased affinity towards neutralization antibodies and can lower the effectiveness of current vaccine.44–46 It is thus important to study the SuFEx kinetics of the covalent nanobodies with the mutant spike RBD to evaluate the therapeutic potential of covalent nanobodies in fighting against evolving infectious diseases.
On the basis of the structure of the SARS-CoV-2 spike RBD in complex with nanobody SR4,47 we incorporated FSY into site 57 of nanobody SR4 to target Tyr505 of the spike RBD and demonstrated that SR4(57FSY) covalently cross-linked with the wildtype (WT) spike RBD. To assess affinity change caused by SARS-CoV-2 mutation, we then determined the dissociation constant (Kd) between the WT SR4 nanobody and the WT or mutant spike RBD using biolayer interferometry (BLI). As shown in Fig. 5A, all mutated spike RBDs had decreased affinity toward the SR4 nanobody. To measure the cross-linking rates between SR4(57FSY) and various spike RBDs, we next incubated 5 μM SR4(57FSY) with 0.5 μM different spike RBDs at 37 °C in PBS. At different time points, an aliquot was extracted and the cross-linking between SR4(57FSY) and spike RBDs was detected with western blot.
 | ||
| Fig. 5 Effects of target mutation on SuFEx kinetics in nanobody SR4(57FSY) cross-linking SARS-CoV-2 spike RBDs. (A) Biolayer interferometry assay of the dissociation constant Kd between the WT SR4 nanobody and WT or mutant spike RBDs. (B) Western blot analysis of cross-linking of SR4(57FSY) with WT or mutant spike RBDs. An anti-Hisx6 antibody was used to detect the Hisx6 tag appended at the C-terminus of the spike RBD. (C) The binding and kinetic constants measured between nanobody SR4(57FSY) and WT or mutant spike RBDs. | ||
SR4(57FSY) was still able to form covalent adducts with all five mutant spike RBDs efficiently (Fig. 5B). We quantified the spike RBD band intensities and calculated the kobs (Fig. 5C). The results indicate that the SuFEx rate kobs generally decreased when the binding affinity between SR4(57FSY) and spike RBDs decreased. One exception is the N501Y mutant spike RBD, which cross-linked with SR4(57FSY) at a similar rate to the WT spike RBD. It is worth noting that the influence of binding affinity on the SuFEx rate was not necessarily linear. We reason that the cross-linking rate would also be affected by mutation-induced protein conformation and microenvironment change surrounding the FSY-target residue, which warrants further studies.
Covalent specificity of FSY-incorporated protein
A critical concern for covalent drugs is the reaction specificity, as off-target covalent reactions could result in toxicity in cells and in vivo. The covalent reactivity of the proximity-enabled reaction in proteins entails (1) the specific binding of the protein drug toward the target and (2) the proper contact of the latent bioreactive Uaa with a suitable target natural residue of the bound protein. This dual requirement is expected to safeguard the reaction specificity of the protein drug toward its target. To evaluate the covalent specificity of SuFEx capable proteins, we tested whether FSY-incorporated protein would lead to off-target cross-linking of other proteins. Specifically, we incubated 0.5 mg mL−1 7D12(109FSY) in 50% human serum at 37 °C. At different time points, samples were aliquoted and 7D12(109FSY) was pulled down via the C-terminal Hisx6 tag and then analyzed with SDS-PAGE and western blot to examine if any off-target proteins were non-specifically cross-linked. The noncovalent WT 7D12 was used as the control.As shown in Fig. 6A, aside from the 7D12 nanobody band, several other bands with higher molecular weights were also observed. However, these bands also showed up in the WT 7D12 control, which is incapable of forming covalent bonds. In addition, the intensity of these bands did not increase with the incubation time, which further ruled out that these bands were derived from covalent cross-linking of 7D12(109FSY). Moreover, in the western blot in which an anti-Hisx6 antibody was used to detect the Hisx6 tag appended at the C-terminus of 7D12 (Fig. 6B), these bands were not detectable, further indicating that they did not contain 7D12(109FSY). A very faint band was observed in western blot at the position matching the dimer of 7D12, whose intensity was less than 2% of 7D12(109FSY). The 7D12 nanobody has a dimerization tendency, so we suspected that this faint band was from the 7D12(109FSY) covalent dimer, formation of which was facilitated by the high temperature during sample preparation (95 °C and 10 min) for western blot. Because the intensity of this band also did not increase with the incubation time, this band could not be from non-specific cross-linking of 7D12(109FSY) with other serum proteins. In short, these results indicate that 7D12(109FSY) did not result in non-specific cross-linking of off-target proteins in human serum in the time period studied, suggesting high covalent specificity.
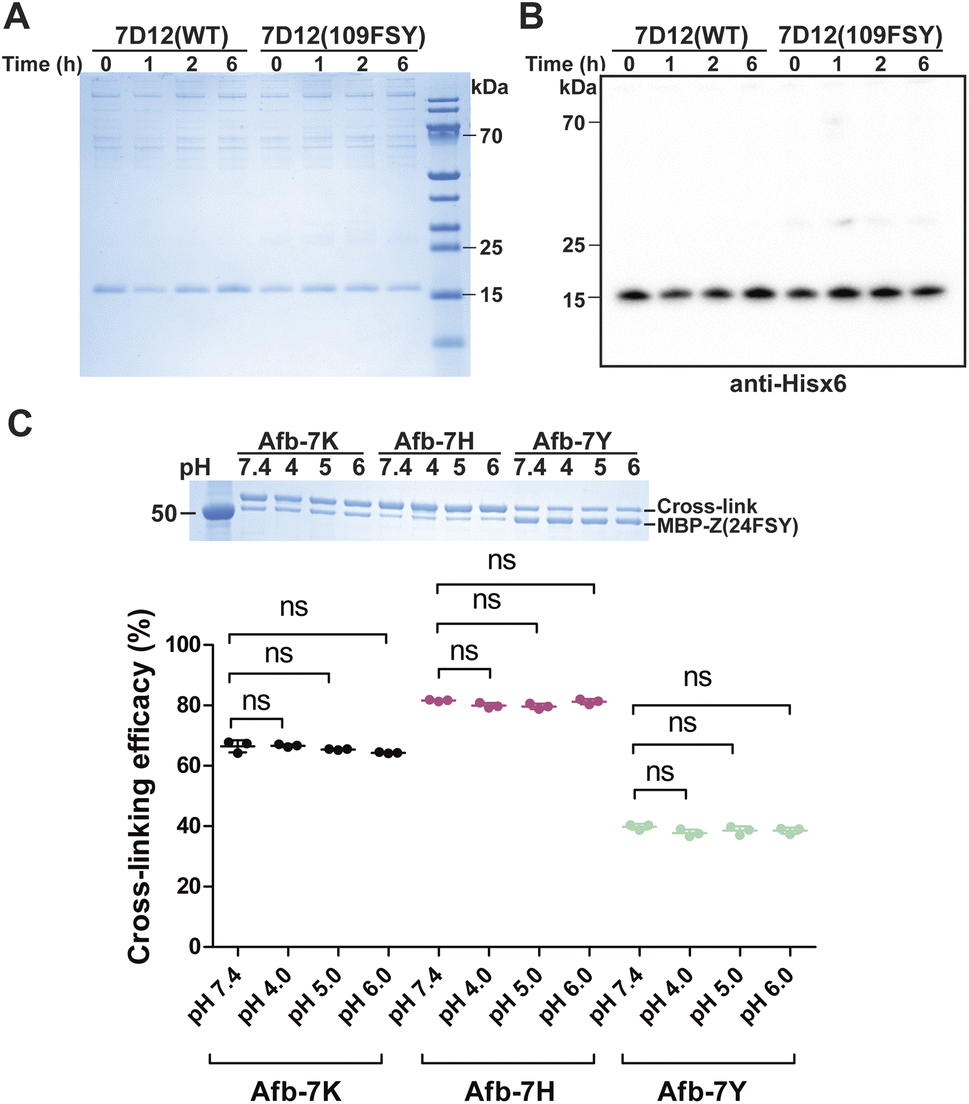 | ||
| Fig. 6 Covalent specificity in human serum and stability of the protein linkage at acidic pH. (A and B) SDS-PAGE (A) and western blot (B) analyses of 7D12(109FSY) and 7D12(WT) after incubation in 50% human serum at 37 °C for the indicated time duration. (C) SDS-PAGE analysis of the stability of the covalent linkage generated between MBP-Z(24FSY) and Afb-7X by FSY reacting with Lys, His, or Tyr. The cross-linked protein samples were incubated at different pH (4, 5, and 6) for 24 h followed by SDS-PAGE analysis. The cross-linking efficacy (%) was quantified as the cross-linking band intensity/[MBP-Z(24FSY) band intensity + cross-link band intensity]. Error bars represent s.d.; n = 3 independent experiments; ns, not significant. | ||
Stability of the protein linkage generated by FSY
Another important practical consideration is the stability of the covalent protein linkage generated by SuFEx, as the covalent protein complex may be internalized inside cells and traffic through the acidic endosomal pathway. We therefore evaluated the stability of the FSY-generated protein covalent linkages at acidic pHs. Specifically, 0.3 mg mL−1 MBP-Z(24FSY) was incubated with 1 mg mL−1 Afb-7X (X = K, Y or H) at 37 °C in PBS (pH 7.4) for 12 h to allow cross-linking. Protein samples were then exchanged into citrate-phosphate buffer with different pH (4, 5, and 6) and incubated for 24 h to test linkage stability, followed by SDS-PAGE analysis. As shown in Fig. 6C, compared with pH 7.4, the cross-linking efficacy of MPB-Z(24FSY) with Afb did not change after incubation at pH 4, 5, or 6, which held for all three linkages generated between FSY and Lys, His, and Tyr, indicating that the FSY-generated covalent protein linkage remained stable at these acidic pHs for the tested duration.Conclusions
In summary, the SuFEx kinetics for protein–protein covalent linkage formation via proximity-enabled reactivity conformed with a two-step mechanism involving protein binding followed by covalent bond formation, showing nonlinear dependence of protein concentration. This kinetics is different from that of the biomolecular reaction of small molecules but analogous to that of covalent small molecules inhibiting target protein. The reaction rate consistently increased with the protein binding affinity and chemical reactivity of the functional group. Target residue identity as well as pH also affected the reaction rate. Mutations in target protein that decreased binding affinity led to decreasing SuFEx rate but in a nonlinear manner. The covalent nanobody based on SuFEx reactivity showed no off-target cross-linking in human serum, and the SuFEx-generated covalent protein linkage remained stable at cellular acidic pHs, suggesting SuFEx suitability for in vivo applications. These results filled the gap of understanding SuFEx reactivity and kinetics in the protein–protein context, which will be invaluable to guide ongoing exploration of SuFEx in proteins in vivo for basic biological studies and innovative covalent biotherapeutics.Data availability
All related data have been included in the paper and the ESI file.†Author contributions
The study was designed and conceptualized by BY, LC, SL and LW. Kinetic and stability studies were performed by BY. Cellular cross-link studies were conducted by LC and SL. mFSY was synthesized by PK. The results were discussed and interpreted by BY, LC, SL, PK and LW. The manuscript was written by BY and LW with input from the other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. W. acknowledges the support of the NIH (R01GM118384 and R01CA258300).Notes and references
- A. S. Barrow, C. J. Smedley, Q. Zheng, S. Li, J. Dong and J. E. Moses, Chem. Soc. Rev., 2019, 48, 4731–4758 RSC.
- J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed.
- B. Gao, S. Li, P. Wu, J. E. Moses and K. B. Sharpless, Angew. Chem., Int. Ed., 2018, 57, 1939–1943 CrossRef CAS PubMed.
- S. Kitamura, Q. Zheng, J. L. Woehl, A. Solania, E. Chen, N. Dillon, M. V. Hull, M. Kotaniguchi, J. R. Cappiello, S. Kitamura, V. Nizet, K. B. Sharpless and D. W. Wolan, J. Am. Chem. Soc., 2020, 142, 10899–10904 CrossRef CAS PubMed.
- Q. Zhao, X. Ouyang, X. Wan, K. S. Gajiwala, J. C. Kath, L. H. Jones, A. L. Burlingame and J. Taunton, J. Am. Chem. Soc., 2017, 139, 680–685 CrossRef CAS PubMed.
- Z. Liu, J. Li, S. Li, G. Li, K. B. Sharpless and P. Wu, J. Am. Chem. Soc., 2018, 140, 2919–2925 CrossRef CAS.
- Y. You, H. S. Kim, I. H. Bae, S. G. Lee, M. H. Jee, G. Keum, S. K. Jang and B. M. Kim, Eur. J. Med. Chem., 2017, 125, 87–100 CrossRef CAS.
- B. Gao, L. Zhang, Q. Zheng, F. Zhou, L. M. Klivansky, J. Lu, Y. Liu, J. Dong, P. Wu and K. B. Sharpless, Nat. Chem., 2017, 9, 1083–1088 CrossRef CAS.
- K. Durie, J. Yatvin, M. Kovaliov, G. H. Crane, J. Horn, S. Averick and J. Locklin, Macromolecules, 2018, 51, 297–305 CrossRef CAS.
- A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650–2659 RSC.
- P. Martín-Gago and C. A. Olsen, Angew. Chem., Int. Ed., 2019, 58, 957–966 CrossRef.
- B. Yang, H. Wu, D. Schnier Paul, Y. Liu, J. Liu, N. Wang, F. DeGrado William and L. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 11162–11167 CrossRef CAS PubMed.
- N. Wang, B. Yang, C. Fu, H. Zhu, F. Zheng, T. Kobayashi, J. Liu, S. Li, C. Ma, P. G. Wang, Q. Wang and L. Wang, J. Am. Chem. Soc., 2018, 140, 4995–4999 CrossRef CAS.
- D. E. Mortenson, G. J. Brighty, L. Plate, G. Bare, W. Chen, S. Li, H. Wang, B. F. Cravatt, S. Forli, E. T. Powers, K. B. Sharpless, I. A. Wilson and J. W. Kelly, J. Am. Chem. Soc., 2018, 140, 200–210 CrossRef CAS.
- J. Liu, L. Cao, P. C. Klauser, R. Cheng, V. Y. Berdan, W. Sun, N. Wang, F. Ghelichkhani, B. Yu, S. Rozovsky and L. Wang, J. Am. Chem. Soc., 2021, 143, 10341–10351 CrossRef CAS PubMed.
- N. Wang and L. Wang, Curr. Opin. Chem. Biol., 2022, 66, 102106 CrossRef CAS PubMed.
- P. C. Klauser, V. Y. Berdan, L. Cao and L. Wang, Chem. Commun., 2022, 58, 6861–6864 RSC.
- B. Yu, S. Li, T. Tabata, N. Wang, L. Cao, G. R. Kumar, W. Sun, J. Liu, M. Ott and L. Wang, Chem, 2022, 8, 2766–2783 CAS.
- S. Li, N. Wang, B. Yu, W. Sun and L. Wang, Nat. Chem., 2023, 15, 33–42 CrossRef CAS PubMed.
- W. Sun, N. Wang, H. Liu, B. Yu, L. Jin, X. Ren, Y. Shen and L. Wang, Nat. Chem., 2023, 15, 21–32 CrossRef CAS PubMed.
- Q. Li, Q. Chen, P. C. Klauser, M. Li, F. Zheng, N. Wang, X. Li, Q. Zhang, X. Fu, Q. Wang, Y. Xu and L. Wang, Cell, 2020, 182, 85–97 CrossRef CAS PubMed.
- Y. Cheng, J. Wu, Y. Han, J. Xu, Y. Da, Q. Zhao, G. Guo, Y. Zhou, Y. Chen, J. Liu, H. Chen, X. Jiang and X. Cai, Bioorg. Med. Chem., 2021, 42, 116219 CrossRef CAS PubMed.
- Y. Han, Z. Yang, H. Hu, H. Zhang, L. Chen, K. Li, L. Kong, Q. Wang, B. Liu, M. Wang, J. Lin and P. R. Chen, J. Am. Chem. Soc., 2022, 144(13), 5702–5707 CrossRef CAS PubMed.
- P. C. Klauser, S. Chopra, L. Cao, K. N. Bobba, B. Yu, Y. Seo, E. Chan, R. R. Flavell, M. J. Evans and L. Wang, ACS Cent. Sci., 2023 DOI:10.1021/acscentsci.3c00288.
- H. Mukherjee, J. Debreczeni, J. Breed, S. Tentarelli, B. Aquila, J. E. Dowling, A. Whitty and N. P. Grimster, Org. Biomol. Chem., 2017, 15, 9685–9695 RSC.
- Z. Xiang, H. Ren, Y. S. Hu, I. Coin, J. Wei, H. Cang and L. Wang, Nat. Methods, 2013, 10, 885–888 CrossRef CAS.
- T. Kobayashi, C. Hoppmann, B. Yang and L. Wang, J. Am. Chem. Soc., 2016, 138, 14832–14835 CrossRef CAS PubMed.
- V. Y. Berdan, P. C. Klauser and L. Wang, Bioorg. Med. Chem., 2021, 29, 115896 CrossRef CAS.
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307–317 CrossRef CAS PubMed.
- L. Cao and L. Wang, Protein Sci., 2022, 31, 312–322 CrossRef CAS PubMed.
- M. Högbom, M. Eklund, P.-A. Nygren and P. Nordlund, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3191–3196 CrossRef.
- J. Singh, E. M. Dobrusin, D. W. Fry, T. Haske, A. Whitty and D. J. McNamara, J. Med. Chem., 1997, 40, 1130–1135 CrossRef CAS PubMed.
- K. R. Schmitz, A. Bagchi, R. C. Roovers, M. Paul, P. van Bergen en Henegouwen and K. M. Ferguson, Structure, 2013, 21, 1214–1224 CrossRef CAS PubMed.
- C. Hoppmann and L. Wang, Chem. Commun., 2016, 52, 5140–5143 RSC.
- O. O. Fadeyi, L. R. Hoth, C. Choi, X. Feng, A. Gopalsamy, E. C. Hett, R. E. Kyne Jr, R. P. Robinson and L. H. Jones, ACS Chem. Biol., 2017, 12, 2015–2020 CrossRef CAS PubMed.
- B. Yang, N. Wang, P. D. Schnier, F. Zheng, H. Zhu, N. F. Polizzi, A. Ittuveetil, V. Saikam, W. F. DeGrado, Q. Wang, P. G. Wang and L. Wang, J. Am. Chem. Soc., 2019, 141, 7698–7703 CrossRef CAS.
- L. H. Jones, ACS Med. Chem. Lett., 2018, 9, 584–586 CrossRef CAS PubMed.
- M. Nishida, S. Harada, S. Noguchi, Y. Satow, H. Inoue and K. Takahashi, J. Mol. Biol., 1998, 281, 135–147 CrossRef CAS PubMed.
- J. Liu, S. Li, N. A. Aslam, F. Zheng, B. Yang, R. Cheng, N. Wang, S. Rozovsky, P. G. Wang, Q. Wang and L. Wang, J. Am. Chem. Soc., 2019, 141, 9458–9462 CrossRef CAS PubMed.
- C. K. V. Nonaka, M. M. Franco, T. Gräf, C. A. de Lorenzo Barcia, R. N. de Ávila Mendonça, K. A. F. de Sousa, L. M. C. Neiva, V. Fosenca, A. V. A. Mendes, R. S. de Aguiar, M. Giovanetti and B. S. de Freitas Souza, Emerging Infect. Dis., 2021, 27, 1522–1524 CrossRef CAS PubMed.
- Q. Li, J. Wu, J. Nie, L. Zhang, H. Hao, S. Liu, C. Zhao, Q. Zhang, H. Liu, L. Nie, H. Qin, M. Wang, Q. Lu, X. Li, Q. Sun, J. Liu, L. Zhang, X. Li, W. Huang and Y. Wang, Cell, 2020, 182, 1284–1294 CrossRef CAS PubMed.
- G. Nelson, O. Buzko, P. Spilman, K. Niazi, S. Rabizadeh and P. Soon-Shiong, bioRxiv, 2021, preprint, DOI:10.1101/2021.01.13.426558.
- F. Tian, B. Tong, L. Sun, S. Shi, B. Zheng, Z. Wang, X. Dong and P. Zheng, eLife, 2021, 10, e69091 CrossRef CAS PubMed.
- D. Planas, T. Bruel, L. Grzelak, F. Guivel-Benhassine, I. Staropoli, F. Porrot, C. Planchais, J. Buchrieser, M. M. Rajah, E. Bishop, M. Albert, F. Donati, M. Prot, S. Behillil, V. Enouf, M. Maquart, M. Smati-Lafarge, E. Varon, F. Schortgen, L. Yahyaoui, M. Gonzalez, J. De Sèze, H. Péré, D. Veyer, A. Sève, E. Simon-Lorière, S. Fafi-Kremer, K. Stefic, H. Mouquet, L. Hocqueloux, S. van der Werf, T. Prazuck and O. Schwartz, Nat. Med., 2021, 27, 917–924 CrossRef CAS.
- P. Wang, M. S. Nair, L. Liu, S. Iketani, Y. Luo, Y. Guo, M. Wang, J. Yu, B. Zhang, P. D. Kwong, B. S. Graham, J. R. Mascola, J. Y. Chang, M. T. Yin, M. Sobieszczyk, C. A. Kyratsous, L. Shapiro, Z. Sheng, Y. Huang and D. D. Ho, Nature, 2021, 593, 130–135 CrossRef CAS PubMed.
- S. A. Madhi, V. Baillie, C. L. Cutland, M. Voysey, A. L. Koen, L. Fairlie, S. D. Padayachee, K. Dheda, S. L. Barnabas, Q. E. Bhorat, C. Briner, G. Kwatra, K. Ahmed, P. Aley, S. Bhikha, J. N. Bhiman, A. a. E. Bhorat, J. du Plessis, A. Esmail, M. Groenewald, E. Horne, S.-H. Hwa, A. Jose, T. Lambe, M. Laubscher, M. Malahleha, M. Masenya, M. Masilela, S. McKenzie, K. Molapo, A. Moultrie, S. Oelofse, F. Patel, S. Pillay, S. Rhead, H. Rodel, L. Rossouw, C. Taoushanis, H. Tegally, A. Thombrayil, S. van Eck, C. K. Wibmer, N. M. Durham, E. J. Kelly, T. L. Villafana, S. Gilbert, A. J. Pollard, T. de Oliveira, P. L. Moore, A. Sigal and A. Izu, N. Engl. J. Med., 2021, 384, 1885–1898 CrossRef CAS PubMed.
- T. Li, H. Cai, H. Yao, B. Zhou, N. Zhang, M. F. van Vlissingen, T. Kuiken, W. Han, C. H. GeurtsvanKessel, Y. Gong, Y. Zhao, Q. Shen, W. Qin, X.-X. Tian, C. Peng, Y. Lai, Y. Wang, C. A. J. Hutter, S.-M. Kuo, J. Bao, C. Liu, Y. Wang, A. S. Richard, H. Raoul, J. Lan, M. A. Seeger, Y. Cong, B. Rockx, G. Wong, Y. Bi, D. Lavillette and D. Li, Nat. Commun., 2021, 12, 4635 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01921g |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |