
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMacrocyclization via remote meta-selective C–H olefination using a practical indolyl template†
Pengfei
Zhang
 a,
Zhiwei
Jiang
a,
Zhoulong
Fan
b,
Guoshuai
Li
a,
Qingxue
Ma
a,
Jun
Huang
c,
Jinghong
Tang
c,
Xiaohua
Xu
*a,
Jin-Quan
Yu
*b and
Zhong
Jin
*ac
a,
Zhiwei
Jiang
a,
Zhoulong
Fan
b,
Guoshuai
Li
a,
Qingxue
Ma
a,
Jun
Huang
c,
Jinghong
Tang
c,
Xiaohua
Xu
*a,
Jin-Quan
Yu
*b and
Zhong
Jin
*ac
aCollege of Chemistry, State Key Laboratory of Elemento-organic Chemistry, Nankai University, Tianjin 300071, China. E-mail: zjin@nankai.edu.cn
bDepartment of Chemistry, The Scripps Research Institute, La Jolla, CA 92037, United States. E-mail: yu200@scripps.edu
cKey Laboratory of Xinjiang Native Medicinal and Edible Plant Resource Chemistry, College of Chemistry and Environmental Sciences, Kashi University, Kashgar 844007, China
First published on 7th July 2023
Abstract
The synthesis of macrocyclic compounds with different sizes and linkages remains a great challenge via transition metal-catalysed intramolecular C–H activation. Herein, we disclose an efficient macrocyclization strategy via Pd-catalysed remote meta-C–H olefination using a practical indolyl template. This approach was successfully employed to access macrolides and coumarins. In addition, the intermolecular meta-C–H olefination also worked well and was exemplified by the synthesis of antitumor drug belinostat from inexpensive and readily available benzenesulfonyl chloride. Notably, catalytic copper acetate and molecular oxygen were used in place of silver salts as oxidants. Furthermore, for the first time, the formation of a macrocyclophane cyclopalladated intermediate was detected through in situ Fourier-transform infrared monitoring experiments and ESI-MS.
Introduction
Achieving site selectivity in C–H bond functionalization reactions is particularly important for the late-stage diversification of drug molecules.1 With the use of diverse directing groups (DG), the activation of ortho-C–H bonds in arenes has been achieved via a conformationally rigid five- or six-membered ring transition state, i.e. proximity-driven cyclometalation process.2 However, this approach is incapable of accessing remote meta or para C–H bonds, which are spatially and geometrically inaccessible with ortho-directing groups. The development of new strategies for transition metal-catalysed distal C–H bond functionalization of arenes remains a significant challenge.3 Advances4 in directing template (DT) strategies harnessing the understanding of distance and geometry between the target C–H bond and DG have provided a reliable solution to the problem.5 Through tuning of a template's spatial and geometric parameters, distal C–H bonds locating at the meta or para position of the existing functional group could be selectively activated by forming a macrocyclophane (MCP)-like pre-transition state.6Bioactive macrocycles, possessing increased conformational flexibility, larger size, and extended surface area relative to traditional small-molecule drugs, show unique and potentially valuable binding properties towards therapeutic targets (Fig. 1a).7 As a consequence, the efficient synthesis, precise structural modification, and versatile derivatization of macrocycles are of great importance in the drug discovery process. Recently, synthetic methods for expanding the structural diversity of macrocycles, such as the construction of combinatorial libraries incorporating aromatic residue(s) in the macrocyclic backbone, have received intensive interest from the synthetic and medicinal chemistry communities (Fig. 1b).8 Despite this advance, development of macrocyclization strategies for construction of structurally diverse macrocycle libraries remains underexplored.9 The intramolecular meta-selective C–H functionalization to access aromatic macrocycle compounds from a linear precursor would be a highly direct, expedient, and modular method for the preparation of medicinally active macrocycles, yet this strategy has hitherto not been realized.
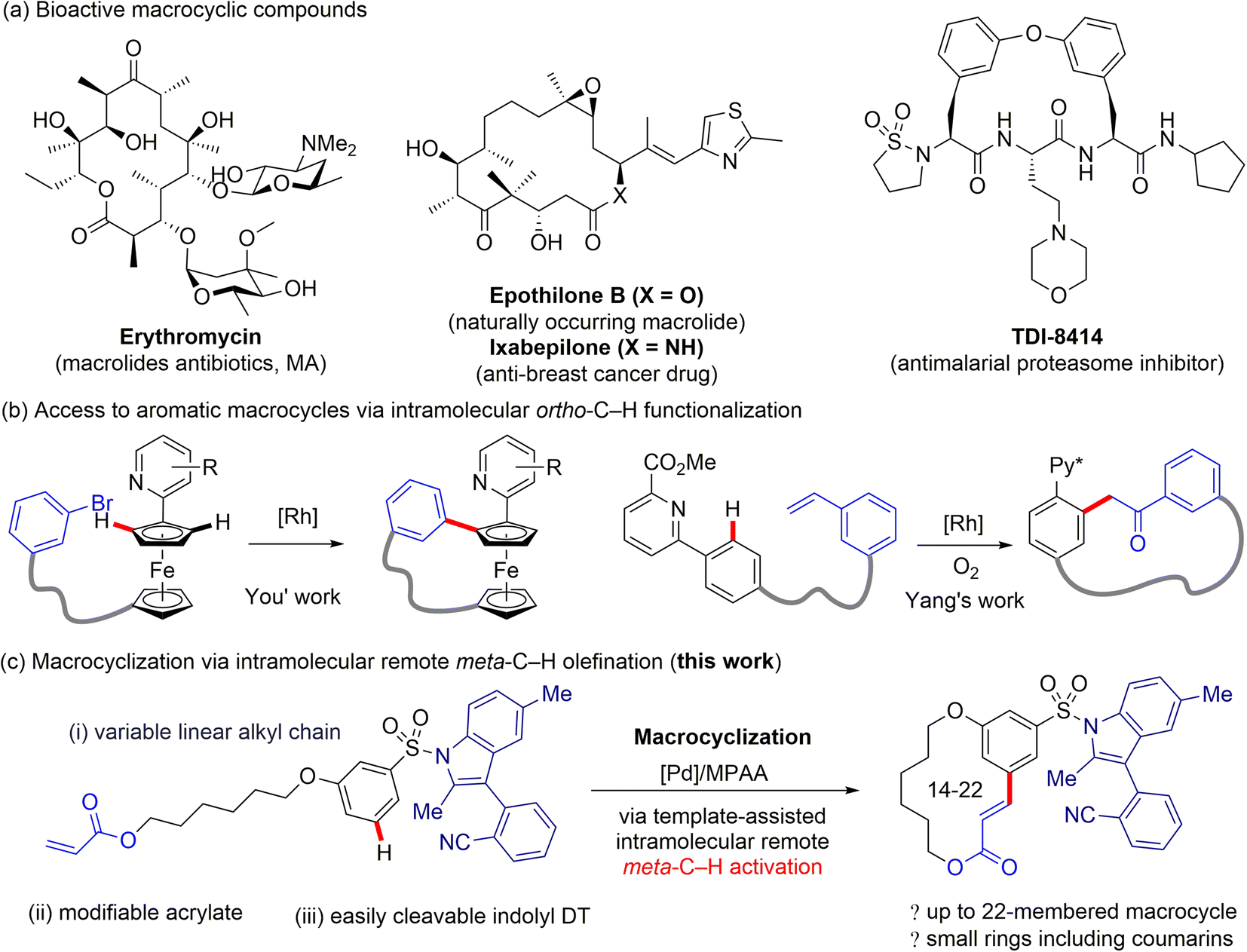 | ||
| Fig. 1 Pharmaceutical importance of macrocyclic compounds and development of strategies for macrocyclization via C–H functionalization. | ||
Inspired by the intriguing biological properties of macrocycle derivatives, we envisioned whether we could achieve a general approach to the synthesis of structurally unique macrocycle libraries through transition metal-catalysed remote meta-selective C–H functionalization. Macrocycles containing an aromatic moiety in their backbones would have planar, relatively rigid conformations, as well as large transannular strain, and thus should display remarkably different properties compared with other macrocycle counterparts.10
Since aryl sulfonic acids and sulfonamides are ubiquitous structural motifs widely present in pharmaceuticals, agrochemicals and other fine chemicals,11 we initially selected aryl sulfonic acids as the substrate for preparing macrocycle derivatives. However, the remote directed C–H bond functionalization of aryl sulfonic acids using reported templates12 has been unsuccessful.5b,13 Competitive ortho-C–H functionalization directed by a secondary sulfonamide severely erodes remote C–H functionalization selectivity, and the inductive electron-withdrawing properties of the sulfonamide further reduces remote C(sp2)–H bond metalation reactivity. To overcome these challenges, a new highly site-selective directing template for intramolecular macrocyclization of aryl sulfonic acids must be developed.
Despite our extensive efforts in template development, we found the reported flexible DT cannot be used for the meta-C–H activation of aryl sulfonic acids. Template-enabled remote C–H functionalization of aryl sulfonate substrates is greatly impeded by the following major problems: the aryl sulfonate ester linkage is unstable; a secondary sulfonamide linkage effectively directs ortho-C–H functionalization;14N-alkyl sulfonamide linkage is difficult to remove. It is well-known that the N-aryl sulfonyl protected indoles can be efficiently formed and cleaved under the relatively mild conditions, therefore, the aryl sulfonyl groups, e.g. p-tosyl, are shown to be a class of superior protecting group for the N–H bond of indoles.15 Given the practicality of N-aryl sulfonyl groups, we questioned whether a substituted indole scaffold can be engineered as a new remote DT for meta- or para-selective C–H functionalization of aryl sulfonates. Our recent statistical analyses suggested that site-selectivity of directed distal C–H functionalization are correlated with the MCP size and rigidity of DT.16 To test our hypothesis, several nitrile-containing 3-arylindole derivatives were prepared and investigated for palladium-catalysed meta-C–H functionalization of aryl sulfonates. We herein disclose a rigid biaryl indole scaffold as the key template motif for intramolecular meta-selective C–H olefination of aryl sulfonate substrates (Fig. 1c). The indolyl template affords a 12-membered pre-transition state, the optimal MCP size for meta site-selection. The N-sulfonamide linkage is readily removable under mild conditions.
Results and discussion
Template design
In our efforts to develop a directing template for the remote C–H functionalization of benzenesulfonic acid derivatives, we initially prepared various nitrile-containing DTs containing 3-aryl indoles, which were then appended to the benzenesulfonic acid substrate via an N-sulfonamide linkage (Scheme 1). With ethyl acrylate as a coupling partner, exploratory studies on the palladium-catalysed remote meta C–H olefination of these benzenesulfonamide substrates were performed. Gratifyingly, with DT1 as a directing template, C–H olefination afforded the desired product in 52% yield with >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 meta selectivity. Furthermore, the directing groups (DT2 and DT3) bearing a C5- or C2-methyl group on the indole moiety also yielded the corresponding meta-C–H olefinated products in 56% and 68% yield, respectively. It is worth noting that, with this type of indole scaffolds as the DT, all C–H olefination reactions provide the target products in high meta-selectivity (meta:others > 20:1). Due to the fact that the starting materials was nearly consumed in all reactions, a thorough investigation on the side products was conducted. It was shown that C–H olefinations at C2 and C5 positions of the indole ring were the main side reactions, likely due to the electrophilic palladation on the electron-rich indole ring.17 Therefore, template (DT4) incorporating two methyl groups into the C2 and C5 positions of indole ring was developed and subjected to the palladium-catalysed C–H olefination reaction. These improved templates delivered the C–H olefination products in 94% yield with exclusive meta selectivity (meta:others > 20:1). In contrast with transition metal-catalysed ortho-C–H functionalization, directed distal C–H bond functionalization commonly involves a conformationally variable macrocyclic transition state.5b The presence of a C2 methyl group in the indole ring, apart from blocking the undesirable C–H palladation, also significantly prevents free rotation of atroposelective axis in the directing template (DT4), likely beneficial to the formation of the conformationally rigid MCP-like pre-transition state.18 A control experiment omitting the nitrile group from the directing template (DT5) results in less than 30% conversion and a complex mixture, validating that the coordinating nitrile is indispensable for meta-selective C–H activation.
:1 meta selectivity. Furthermore, the directing groups (DT2 and DT3) bearing a C5- or C2-methyl group on the indole moiety also yielded the corresponding meta-C–H olefinated products in 56% and 68% yield, respectively. It is worth noting that, with this type of indole scaffolds as the DT, all C–H olefination reactions provide the target products in high meta-selectivity (meta:others > 20:1). Due to the fact that the starting materials was nearly consumed in all reactions, a thorough investigation on the side products was conducted. It was shown that C–H olefinations at C2 and C5 positions of the indole ring were the main side reactions, likely due to the electrophilic palladation on the electron-rich indole ring.17 Therefore, template (DT4) incorporating two methyl groups into the C2 and C5 positions of indole ring was developed and subjected to the palladium-catalysed C–H olefination reaction. These improved templates delivered the C–H olefination products in 94% yield with exclusive meta selectivity (meta:others > 20:1). In contrast with transition metal-catalysed ortho-C–H functionalization, directed distal C–H bond functionalization commonly involves a conformationally variable macrocyclic transition state.5b The presence of a C2 methyl group in the indole ring, apart from blocking the undesirable C–H palladation, also significantly prevents free rotation of atroposelective axis in the directing template (DT4), likely beneficial to the formation of the conformationally rigid MCP-like pre-transition state.18 A control experiment omitting the nitrile group from the directing template (DT5) results in less than 30% conversion and a complex mixture, validating that the coordinating nitrile is indispensable for meta-selective C–H activation.
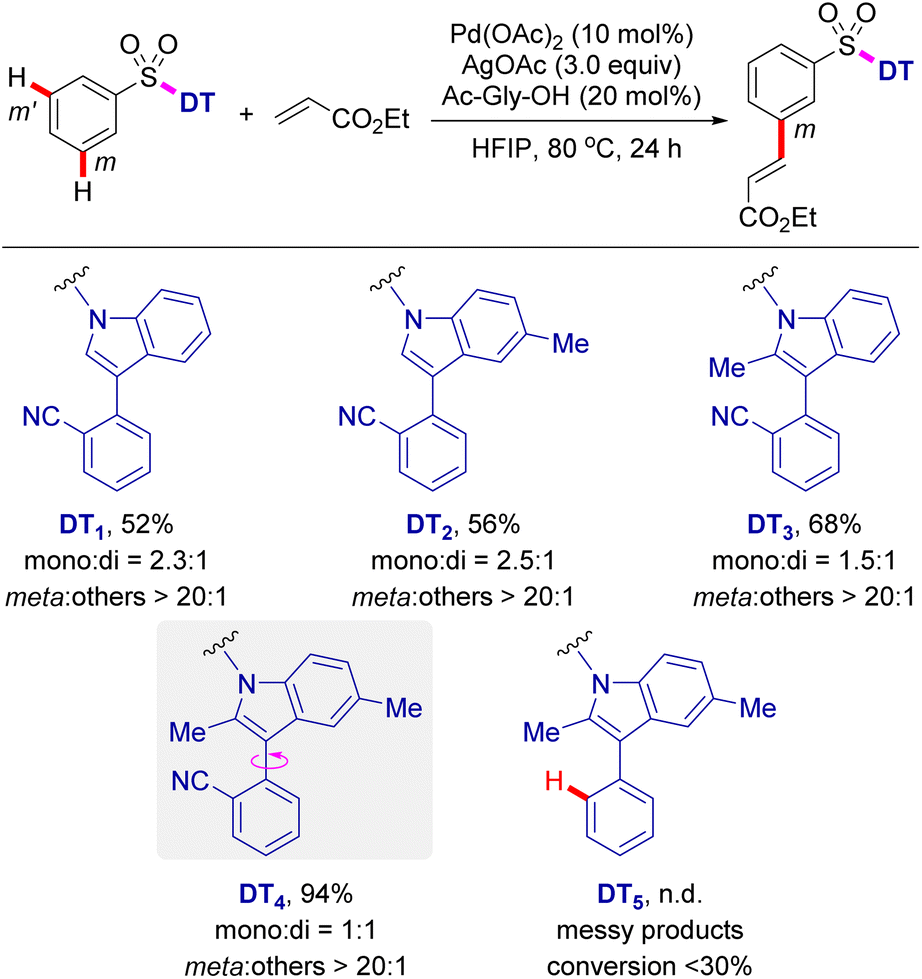 | ||
| Scheme 1 Evaluation of DTs for meta-C–H functionalization of aryl sulfonic acids. Reaction conditions: PhSO2-DTn (0.1 mmol), ethyl acrylate (0.2 mmol), Pd(OAc)2 (10 mol%), Ac-Gly-OH (20 mol%), AgOAc (0.3 mmol), HFIP (1.0 mL), 80 °C, 24 h. Yield, ratio of mono- and di-products and meta selectivity were determined by 1H NMR analysis of the crude products with Cl2CHCHCl2 as an internal standard. | ||
Pd-catalysed meta-C–H olefination of aryl sulfonic acids
With the optimal DT in hand, we subsequently investigated the applicability of palladium-catalysed intermolecular meta-selective C–H olefination (Scheme 2). It was found that substrates bearing the methoxy, methyl, and halo functional groups at meta or para position of benzosulfonates were well tolerated under the present C–H olefination reaction conditions, providing the corresponding meta olefinated products (2c–2e, 2i–2k) in 48–65% yields. Di-olefination occurred prominently with para-substituted benzosulfonates to give the products (2i–2k). The meta selectivity of C–H olefination was comprehensively confirmed by X-ray diffraction analysis of product (2d).‡ Reaction with substrate owning an o-OMe delivered the product (2b) in slightly decreased yield (40%), probably due to the steric repulsion between the methoxy group and tetrahedral sulfonate group, which disfavors the requisite meta-directing conformation. Given the existing electron-withdrawing sulphonamide motif, the incorporation of additional electron-withdrawing substituents might further deactivate the substrate towards C–H palladation. In contrast to this expectation and the reported reactivity trends for the remote meta-C–H functionalization of benzoic acids,12 aryl sulfonamides bearing a diverse set of electron-withdrawing groups, such as trifluoromethoxy, trifluoromethyl, and ester groups at meta or para position, are all compatible, providing the desired products (2f, 2l, 2g, and 2h) in 40–60% yields with exclusive meta selectivity. Di-substituted aryl sulfonates with different substitution patterns are also suitable for the directed meta-C–H functionalization, successfully affording the tetra-substituted aryl sulfonates (2m–2p) in 60–90% yields. Reactions with 3,4-di-halo substituted aryl sulfonates were distinctly sluggish and the desired products (2q and 2r) were isolated only in 26% and 24% yields, respectively. The reason for the stark contrast remains uncertain at this stage.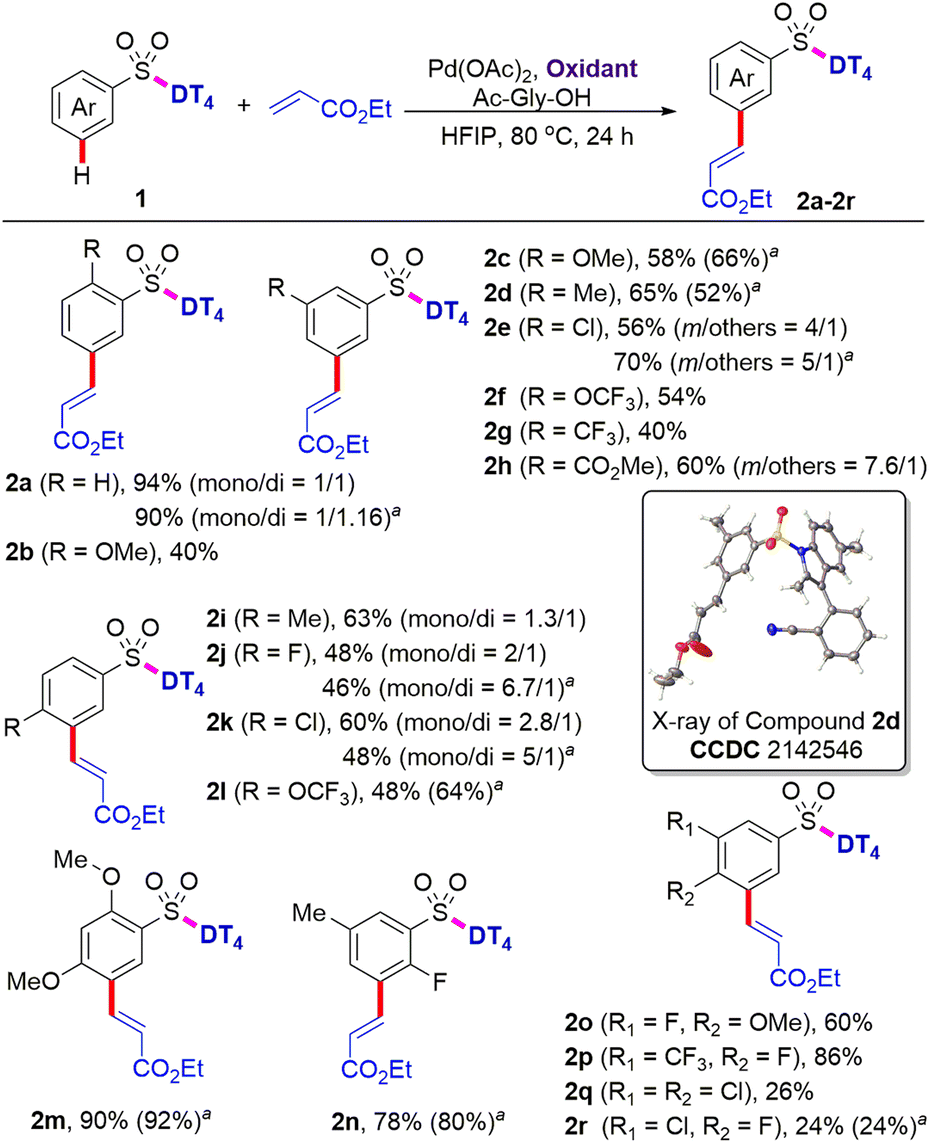 | ||
| Scheme 2 Scope of aryl sulfonic acids. Reaction conditions: ArSO2-DT41 (0.1 mmol), ethyl acrylate (0.2 mmol), Pd(OAc)2 (10 mol%), AgOAc (0.3 mmol), Ac-Gly-OH (20 mol%), HFIP (1.5 mL), 80 °C, 24 h. Isolated yields were reported. Ratio of mono- and di-products were determined by 1H NMR spectra analysis of the crude products with Cl2CHCHCl2 as an internal standard. aIsolated yields when using Cu(OAc)2 (0.05 mmol) and O2 (1 atm) as the terminal oxidant. | ||
Given the high cost of excess silver acetate as the oxidant, the use of greener and more inexpensive oxidants is highly desirable. We found that 0.5 equiv. of copper acetate in combination with O2 (1 atm) as the terminal oxidant (Scheme 2) also successfully provided the corresponding olefinated products (2a, 2c–2e, 2j–2n, and 2r) in comparable yields. These results indicate that Ag salts can be replaced as oxidants in directing template-assisted remote C–H functionalization (vide infra).
After the survey of scope with aryl sulfonates, the established template-directed meta-C–H functionalization was then extended to other olefin partners (Scheme 3). In addition to ethyl acrylate, other alkyl ester (3a) are also suitable substrates for this C–H olefination reaction. Methyl crotonate and methyl cinnamate are compatible under the reaction conditions, giving the desired products (3e and 3f) in 47% and 86% yields, respectively. Likewise, other activated olefins, i.e., vinyl amide and vinyl sulfonates, can also be employed to the present process, providing the corresponding products (3b–3d) in 68–82% yields. Furthermore, C–H olefination with cyclic α,β-unsaturated esters smoothly provided the related products (3g and 3h) in good to excellent yields with exclusive meta selectivity. Electron-deficient perfluorinated olefin is demonstrated to be competent substrate, affording the 3,5-di-substituted perfluoroalkenylated product (3j) in 80% yield. In particular, coupling with olefins containing the biologically significant natural terpenes, e.g. estrone, L-menthol, and stanolone, successfully produced the mono- or di-olefinated products (3i, 3k and 3l) in 56–72% yields, which is highly applicable for synthesis of a diverse range of pharmaceutical conjugates.
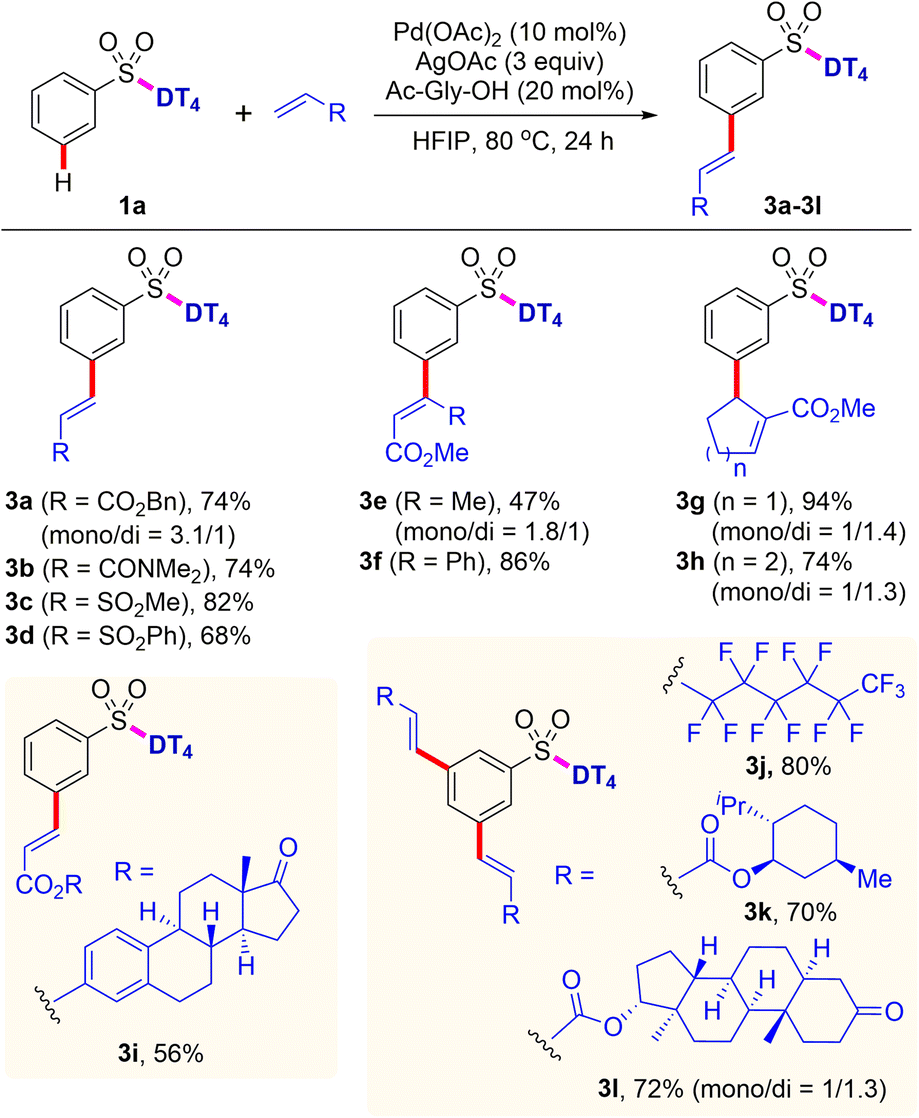 | ||
| Scheme 3 Scope of olefins for intermolecular meta-C–H functionalization. Reaction conditions: PhSO2-DT41a (0.1 mmol), olefin (0.2 mmol), Pd(OAc)2 (10 mol%), AgOAc (0.3 mmol), Ac-Gly-OH (20 mol%), HFIP (1.5 mL), 80 °C, 24 h. Isolated yields were reported. | ||
Macrocyclization via intramolecular meta-C–H olefination
Having successfully realized meta-C–H functionalization of aryl sulfonic acids, we revisited our initial proposal to synthesize aromatic macrocycles using palladium-catalysed intramolecular remote meta-selective C–H olefination. Pleasingly, we found that the macrocyclization of 4c proceeds successfully to generate an 18-membered macrolide product 5c in satisfied yield under the optimized conditions of 10 mol% Pd(OAc)2 catalyst, 20 mol% N-Ac-Gly-OH ligand and 3 equiv. of AgOAc in hexafluoroisopropanol (HFIP) at 60 °C for 1.5 h (see ESI† for detailed reaction conditions optimization), although some competitive intermolecular reaction was observed. It was found that the highest yield (68%) was achieved when macrocyclization reaction was conducted at a 25 mM concentration. With the decrease of Pd catalyst from 10 mol% to 2 mol%, the isolated yield of 5c is reduced to 34%. Extended reaction times did not bring about higher yields. When reaction was carried out over a period of 24 h, no desired macrocyclization product was observed, suggesting macrocycle decomposition under long reaction times. The NMR spectra and HPLC/MS analyses indicate that the hydrolysis of lactone occurred, leading to the formation of the corresponding cinnamic acids in the presence of a small amount of H2O in HFIP.Next, we further investigated the generality of template-enabled macrocyclization methodology (Scheme 4). We were pleased to find the present method demonstrated the considerable tolerance of macrocycle sizes. Under the optimized conditions, by using the different chain lengths of alkyl groups, all of 14- to 22-membered macrolides 5a–5e can be smoothly obtained in 42–68% yields. The macrocyclization protocol was also compatible with the different acrylate tails, for example cinnamates 5h and 5i, albeit in decreased yields. Notably, the use of α-methyl acrylate derivative caused that a macrocycle 5f with exocyclic double bond was isolated as the major product.
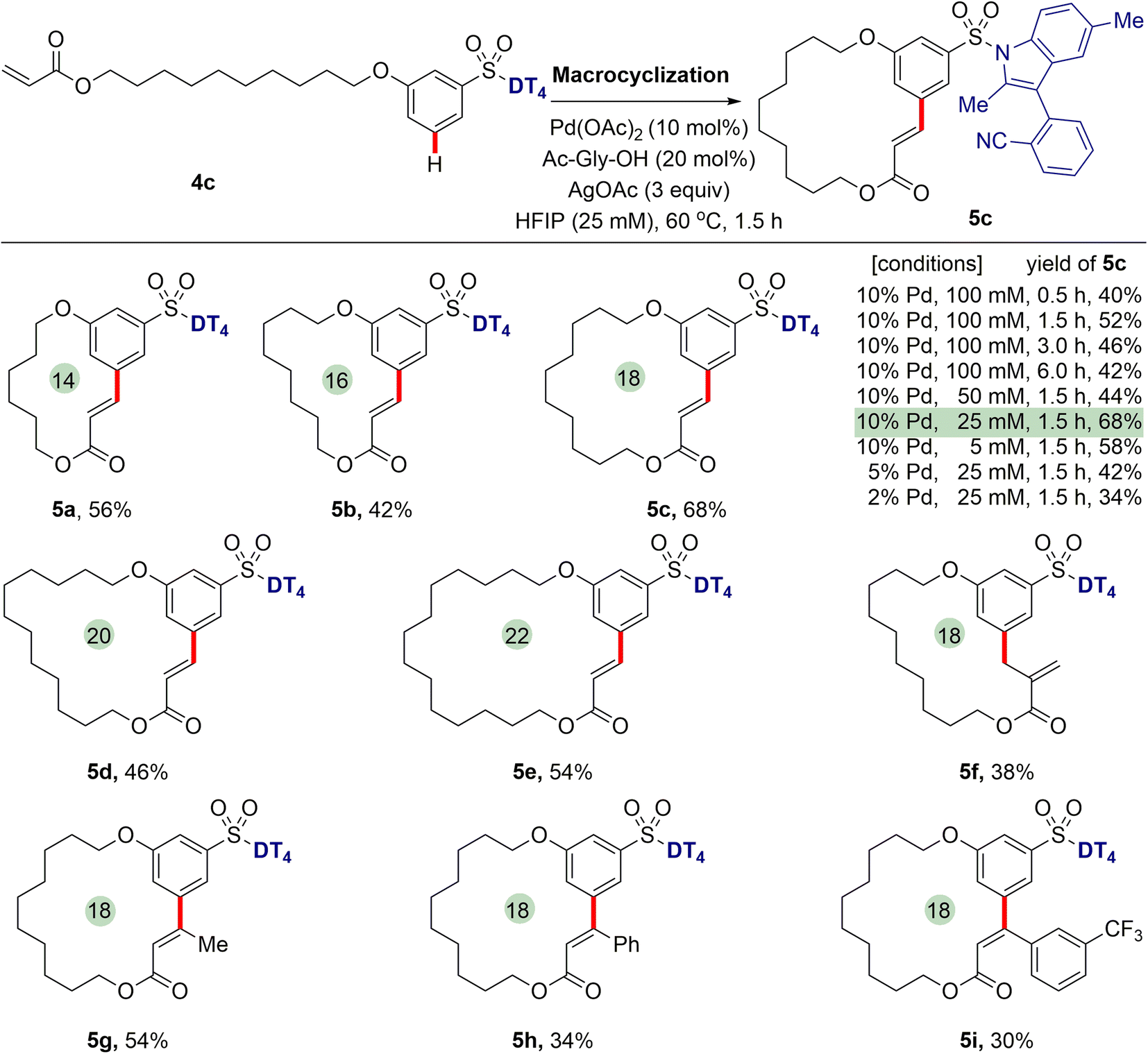 | ||
| Scheme 4 Macrocyclization via template-directed intramolecular remote meta-C–H olefination. Reaction conditions: ArSO2-DT44 (0.1 mmol), Pd(OAc)2 (10 mol%), AgOAc (0.3 mmol), Ac-Gly-OH (20 mol%), HFIP (25 mM), 60 °C, 1.5 h. Isolated yields were reported. | ||
Synthetic utility of meta-C–H functionalization
Encouraged by our success with the Pd-catalysed macrocyclization via intramolecular meta-C–H olefination, we then sought to apply our strategy to the template-directed intramolecular meta-selective C–H functionalization to prepare small ring compounds, such as coumarins, which has yet to be reported.1c Pleasingly, assisted with directing template DT4, Pd-catalysed intramolecular meta-C–H olefination smoothly afforded the 6-sulfonate group-substituted coumarin (7) in 46% yield by using the linear precursor acrylates (6) as the starting substrate (Scheme 5a).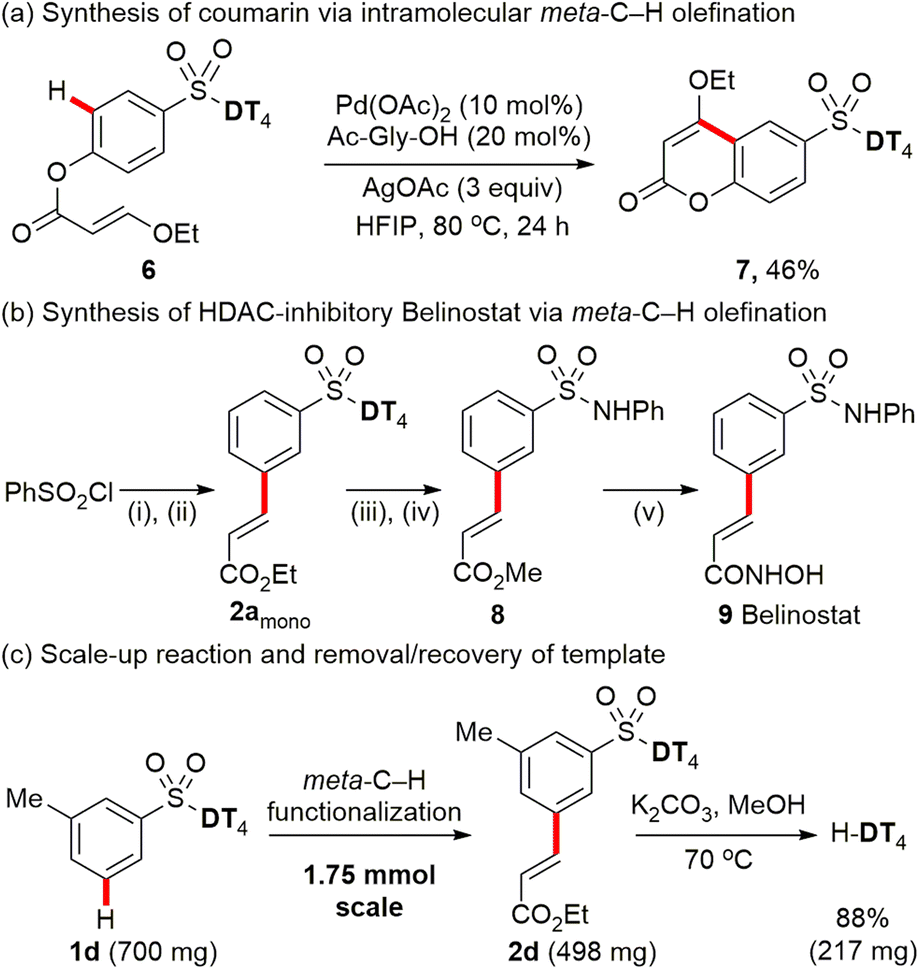 | ||
| Scheme 5 Synthetic application of Pd-catalysed meta-C–H functionalization. Reaction conditions: (i) H-DT4 (1.1 equiv.), NaH, THF, 90%; (ii) PhSO2-DT41a (1 equiv.), ethyl acrylate (1.5 equiv.), Pd(OAc)2 (10 mol%), AgOAc (3 equiv.), Ac-Gly-OH (20 mol%), HFIP (1.5 mL), 80 °C, 6 h, 54% yield for 2amono, 38% yield for 2adi; (iii) K2CO3, MeOH; then aq. HCl; then MeI, K2CO3, DMF, rt. (iv) SOCl2, DMF (cat), toluene; then aniline, pyridine, rt, 70% overall yield from compound 2amono. (v) aq. NaOH, MeOH, rt; then SOCl2, DBU (cat); then NH2OH·HCl, aq. NaHCO3, 65% overall yield from compound 8. | ||
To further test the practicality of template-enabled meta-C–H functionalization of aryl sulfonate derivatives, an antitumor drug Belinostat 9, also known as a pan-histone deacetylase (HDAC) inhibitor, was concisely synthesized through using the established C–H olefination method (Scheme 5b). After further optimization of reaction parameters, a slightly higher ratio (1.4:1) of mono- vs. di-products was achieved when using 1.5 equivalents of ethyl acrylate over 6 h reaction time for preparation of key intermediate 2amono. Finally, belinostat 9 was synthesized in a five-step sequence with an overall yield of 22% from the inexpensive readily available PhSO2Cl.
Furthermore, we also successfully scaled up the reaction to 1.75 mmol (Scheme 5c). Palladium-catalysed meta-C–H olefination of aryl sulfonate 1d provides the target mono-product 2d in 57% isolated yield. The directing template DT4 is readily removed under mild conditions (K2CO3, MeOH) and recovered in 88% isolated yield.
Mechanistic study
For the previously reported Pd-catalysed meta-selective C–H functionalization of arene substrates, such as 3-hydrocinnamic acids4 and distal arene-tethered aliphatic alcohols,6h using nitrile-based DT, silver acetate was often indispensable for the catalytic process. Earlier mechanistic studies indicated Ag salts not only serve as an external oxidant, but also participate the assembly of MCP-like pre-transition state, crucial for the desired positional selectivity. Density functional theory (DFT) studies13 indicated that heterodimeric PdAg(OAc)3 as the active catalytic species is likely involved in the key C–H activation step. In the hypothetical transition state, the DG nitrile coordinates to Ag while the acetate-bridged Pd is located close to the meta C–H bond, thereby accounting for the high site selectivity. To examine the role of silver acetate in the present C–H functionalization, the meta-selective C–H olefination of aryl sulfonates was performed by employing the stoichiometric Pd catalyst, and the comparable site-selectivity and ratio of mono:di products were observed, indicated that AgOAc likely only acts as an external oxidant for this transformation (Scheme 6a). To provide additional support for this proposal, the reaction was also performed when using catalytic Cu(OAc)2 (0.5 equiv.) and O2 (1 atm) as the oxidant system (Scheme 6b) as in our study above (Scheme 2). The yield and meta-selectivity are nearly identical as when using AgOAc as the oxidant (Scheme 2).
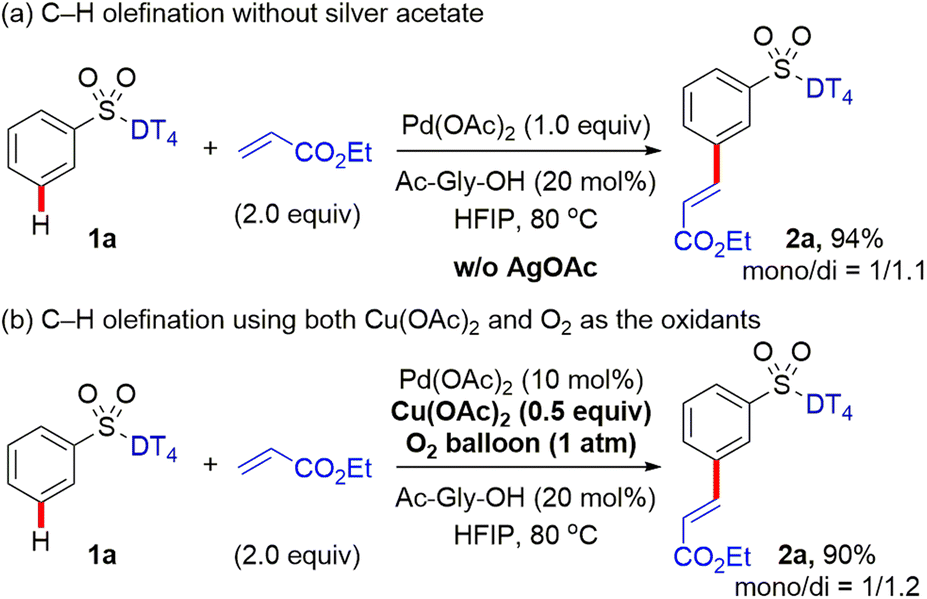 | ||
| Scheme 6 Pd-catalysed meta-C–H olefination under the variable conditions. | ||
Kinetic isotope effect (KIE) studies on the C–H olefination of 1a and its deuterated analog [D]5-1a provided kH/kD value of 3.17 (Scheme 7), implying that the C–H bond cleavage is likely the rate-determining step (RDS) in the catalytic cycle. To gain further mechanistic insights into the catalytic cycle, we also performed a series of control experiments and spectroscopic analyses. In the absence of the Pd catalyst, no desired olefination products were detected. When the N-protected amino acid ligand was omitted (N-Ac-Gly-OH), a sluggish reaction was observed. Subsequently, kinetic studies were carried out to determine the reaction order with respect to the aryl sulfonate substrate and olefin coupling partner (see Fig. S2 and S3 in ESI†). From the initial rate calculations, a first-order rate dependency with respect to aryl sulfonate and zero-order dependency with respect to the olefin were obtained. The observed first order rate dependency on the aryl sulfonate substrate indicates the C–H activation step is the rate-limiting step, consistent with the KIE results.
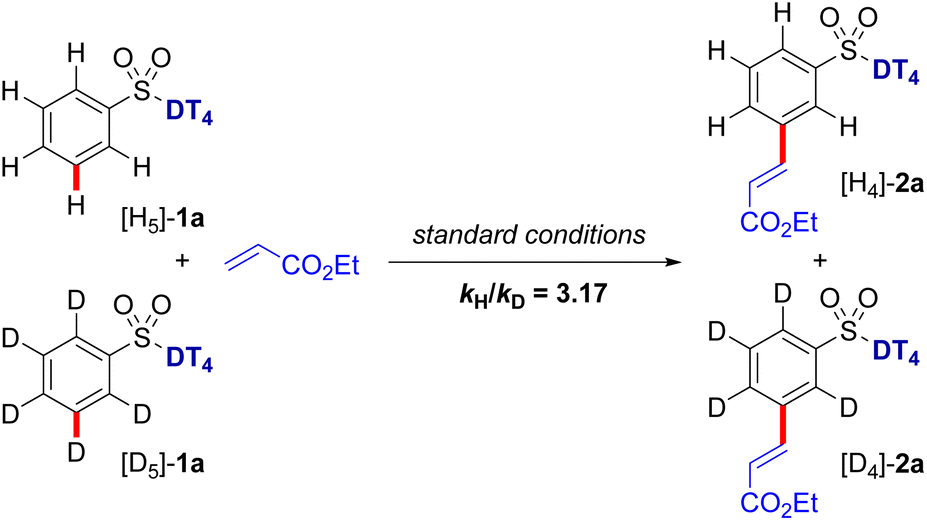 | ||
| Scheme 7 Kinetic isotope effect experiment. | ||
Upon coordination of the nitrile N atom to the Pd catalyst in the directed distal C–H activation, the stretching vibrational frequency (υ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) ) of carbon–nitrogen triple bond on the nitrile group changes dramatically. To detect the possible reaction intermediate in this remote C–H functionalization reaction, in situ Fourier-transform infrared (in situ FTIR) was employed to monitor the reaction with the substrate 1d, stoichiometric Pd catalysts and N-Ac-Gly-OH ligand. From the kinetic profile of the intermediate concentration versus time (ConcIRT), apart from aryl sulfonate substrate and the desired olefinated product (both typical absorbance peak at 2248 cm−1), we observed a new active species (absorbance peak at 2278 cm−1), which probably corresponds to the MCP-like assembly (Fig. 2). When the stoichiometric Pd(OAc)2 and N-Ac-Gly-OH ligand was added into the solution of substrate in hexafluoroisopropanol (HFIP), aryl sulfonate 1d was consumed promptly and a new intermediate (A) was generated (absorbance peak at 2278 cm−1) within 30 minutes. Subsequently, after the introduction of ethyl acrylate, the absorbance peak at 2278 cm−1 subsided instantly, accompanied by the rapid formation of C–H olefination products (also absorbance peak at 2248 cm−1).
) of carbon–nitrogen triple bond on the nitrile group changes dramatically. To detect the possible reaction intermediate in this remote C–H functionalization reaction, in situ Fourier-transform infrared (in situ FTIR) was employed to monitor the reaction with the substrate 1d, stoichiometric Pd catalysts and N-Ac-Gly-OH ligand. From the kinetic profile of the intermediate concentration versus time (ConcIRT), apart from aryl sulfonate substrate and the desired olefinated product (both typical absorbance peak at 2248 cm−1), we observed a new active species (absorbance peak at 2278 cm−1), which probably corresponds to the MCP-like assembly (Fig. 2). When the stoichiometric Pd(OAc)2 and N-Ac-Gly-OH ligand was added into the solution of substrate in hexafluoroisopropanol (HFIP), aryl sulfonate 1d was consumed promptly and a new intermediate (A) was generated (absorbance peak at 2278 cm−1) within 30 minutes. Subsequently, after the introduction of ethyl acrylate, the absorbance peak at 2278 cm−1 subsided instantly, accompanied by the rapid formation of C–H olefination products (also absorbance peak at 2248 cm−1).
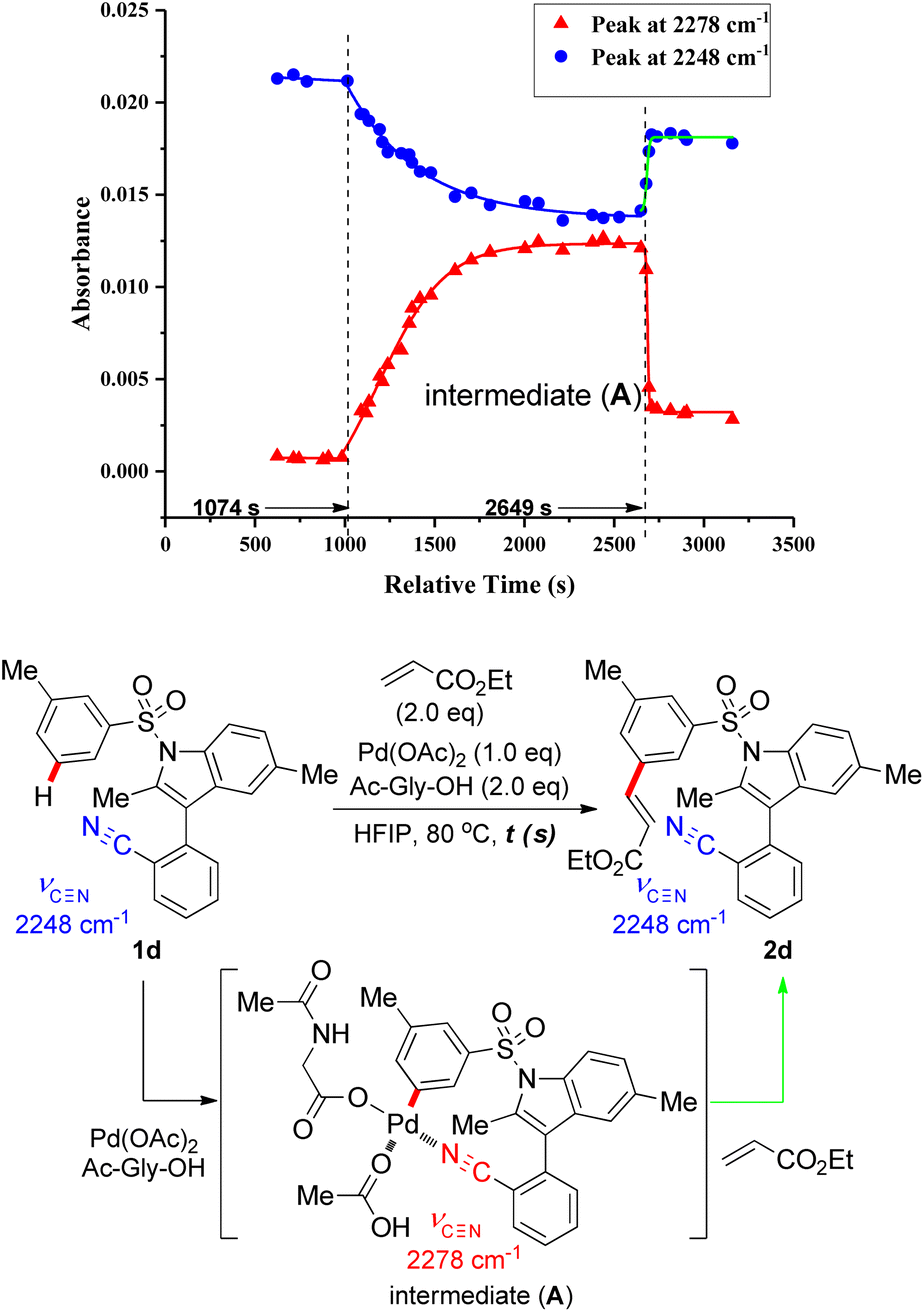 | ||
| Fig. 2 In situ IR reaction kinetic profiles. | ||
ESI-MS studies of the reaction mixture, in the absence of the olefinated reagent, also detected the formation of an MCP-like palladacycle as illustrated in Fig. 3. Identification of this active intermediate, as well as its rate of consumption upon the addition of olefin, represents valuable mechanistic information on template directed C–H activation reactions.
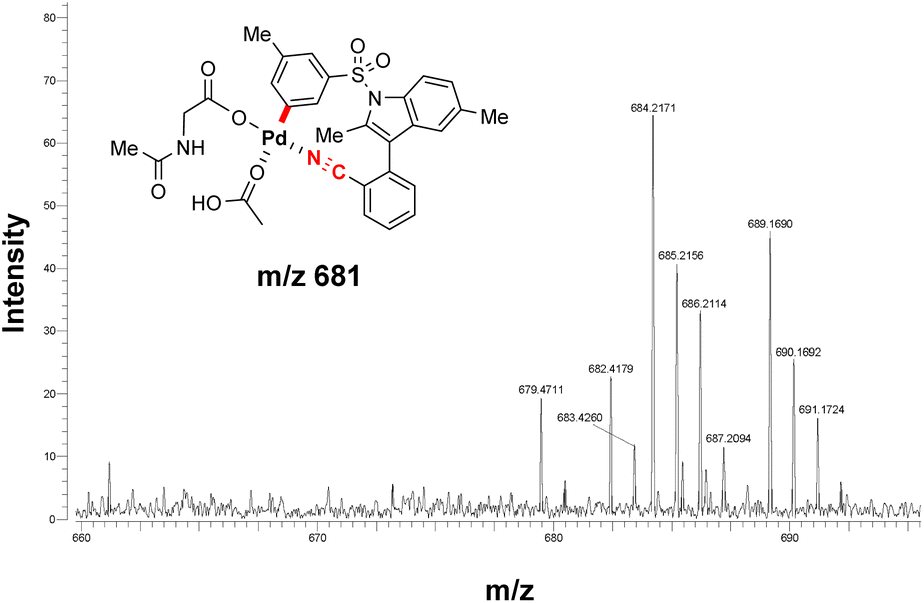 | ||
| Fig. 3 ESI-MS spectra analysis to detect the PdLn-arene complex with substrate 1d. | ||
In combination with in situ IR spectroscopic analysis and earlier reports13,19 on Pd-catalysed meta-C–H activation, a putative catalytic mechanism for the present template-assisted C–H functionalization was therefore proposed (Fig. 4). As shown in the intermediate II, the directing template directs the Pd center to the distal aryl C–H bond with the appropriate distal and geometric arrangement. Next, meta-selective C–H activation, proceeding via a ligand-assisted concerted metalation-deprotonation (CMD) mechanism,20 forms an MCP-like palladated intermediate (A). In the presence of olefin partner, migratory insertion of olefin, followed by sequential β-hydride elimination of the resulting intermediate IV, provides the desired meta-C–H olefinated products.
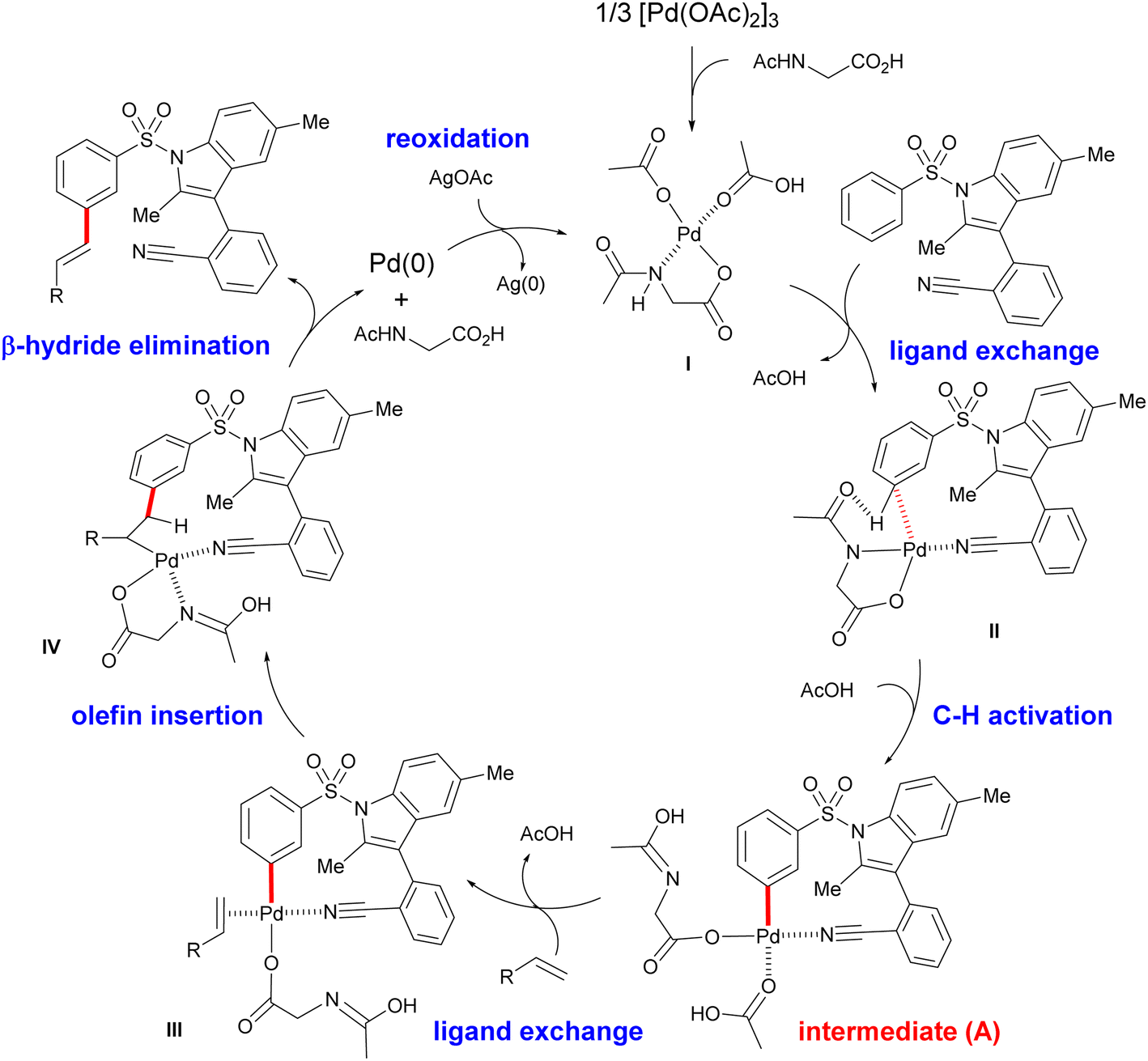 | ||
| Fig. 4 A plausible catalytic cycle. | ||
Conclusions
In summary, Pd-catalysed macrocyclization via remote intramolecular meta-C–H olefination was successfully achieved by utilizing an indolyl DT. The synthetic utility of the strategy was demonstrated in synthesis of a series of macrolides with different scaffold sizes. In addition, application to the concise synthesis of anti-tumor drug Belinostat validates the method's compatibility with intermolecular meta-C–H functionalization as well. Through in situ IR profiling, ESI-MS studies, and kinetic experiments, we provide the first experimental support for the formation of an active macrocyclic palladacycle intermediate from remote C–H activation.Data availability
The datasets supporting this article have been uploaded as part of the ESI† material.Author contributions
X. X., Z. J., and J.-Q. Y. conceptualized the project. P. Z., Z.-W. J., G. L., Q. M., J. H., and J. T. performed the experimental studies. Z. J., Z. F., and J.-Q. Y. prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 22171145 to Z. J. and 32072440 to X. X.) is gratefully acknowledged. The authors thank Dr Kevin Wu for extensive proofreading.Notes and references
- (a) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS PubMed; (b) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 ( Angew. Chem. , 2009 , 121 , 5196 ) CrossRef CAS PubMed; (c) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (d) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS PubMed; (e) J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2 CrossRef CAS.
- (a) O. Daugulis, H. Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (c) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (d) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814 CrossRef CAS PubMed; (e) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 ( Angew. Chem. , 2013 , 125 , 11942 ) CrossRef CAS PubMed; (f) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (g) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed.
- (a) F. Juliá-Hernández, M. Simonetti and I. Larrosa, Angew. Chem., Int. Ed., 2013, 52, 11458 ( Angew. Chem. , 2013 , 125 , 11670 ) CrossRef PubMed; (b) J. Ye and M. Lautens, Nat. Chem., 2015, 7, 863 CrossRef CAS; (c) J. Li, S. De Sarkar and L. Ackermann, Top. Organomet. Chem., 2015, 55, 217 CrossRef; (d) M. T. Mihai, G. R. Genov and R. J. Phipps, Chem. Soc. Rev., 2018, 47, 49 RSC.
- D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518 CrossRef CAS PubMed.
- (a) A. Dey, S. K. Sinha, T. K. Achar and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 10820 ( Angew. Chem. , 2019 , 131 , 10934 ) CrossRef CAS PubMed; (b) G. Meng, N. Y. S. Lam, E. L. Lucas, T. G. Saint-Denis, P. Verma, N. Chekshin and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 10571 CrossRef CAS; (c) W. Ali, G. Prakash and D. Maiti, Chem. Sci., 2021, 12, 2735 RSC; (d) U. Dutta, S. Maiti, T. Bhattacharya and D. Maiti, Science, 2021, 372, eabd5992 CrossRef CAS PubMed; (e) Z. Fan, X. Chen, K. Tanaka, H. S. Park, N. Y. S. Lam, J. J. Wong, K. N. Houk and J.-Q. Yu, Nature, 2022, 610, 87 CrossRef CAS PubMed.
- (a) H.-X. Dai, G. Li, X.-G. Zhang, A. F. Stepan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 7567 CrossRef CAS PubMed; (b) S. Lee, H. Lee and K. L. Tan, J. Am. Chem. Soc., 2013, 135, 18778 CrossRef CAS; (c) R.-Y. Tang, G. Li and J.-Q. Yu, Nature, 2014, 507, 215 CrossRef CAS; (d) G. Yang, P. Lindovska, D. Zhu, J. Kim, P. Wang, R.-Y. Tang, M. Movassaghi and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 10807 CrossRef CAS PubMed; (e) L. Chu, M. Shang, K. Tanaka, Q. Chen, N. Pissarnitski, E. Streckfuss and J.-Q. Yu, ACS Cent. Sci., 2015, 1, 394 CrossRef CAS PubMed; (f) M. Bera, A. Maji, S. K. Sahoo and D. Maiti, Angew. Chem., Int. Ed., 2015, 54, 8515 ( Angew. Chem. , 2015 , 127 , 8635 ) CrossRef CAS; (g) S. Bag, R. Jayarajan, U. Dutta, R. Chowdhury, R. Mondal and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 12538 ( Angew. Chem. , 2017 , 129 , 12712 ) CrossRef CAS PubMed; (h) L. Zhang, C. Zhao, Y. Liu, J. Xu, X. Xu and Z. Jin, Angew. Chem., Int. Ed., 2017, 56, 12245 ( Angew. Chem. , 2017 , 129 , 12413 ) CrossRef CAS; (i) Z. Zhang, K. Tanaka and J.-Q. Yu, Nature, 2017, 543, 538 CrossRef CAS PubMed; (j) S. Li, H. Wang, Y. Weng and G. Li, Angew. Chem., Int. Ed., 2019, 58, 18502 ( Angew. Chem. , 2019 , 131 , 18673 ) CrossRef CAS PubMed; (k) S. Porey, X. Zhang, S. Bhowmick, V. K. Singh, S. Guin, R. S. Paton and D. Maiti, J. Am. Chem. Soc., 2020, 142, 3762 CrossRef CAS PubMed; (l) A. F. Williams, A. J. P. White, A. C. Spivey and C. J. Cordier, Chem. Sci., 2020, 11, 3301 RSC; (m) S. Bag, S. Jana, S. Pradhan, S. Bhowmick, N. Goswami, S. K. Sinha and D. Maiti, Nat. Commun., 2021, 12, 1393 CrossRef CAS; (n) S. Bag, T. Patra, A. Modak, A. Deb, S. Maity, U. Dutta, A. Dey, R. Kancherla, A. Maji, A. Hazra, M. Bera and D. Maiti, J. Am. Chem. Soc., 2015, 137, 11888 CrossRef CAS PubMed; (o) A. Maji, A. Dahiya, G. Lu, T. Bhattacharya, M. Brochetta, G. Zanoni, P. Liu and D. Maiti, Nat. Commun., 2018, 9, 3582 CrossRef; (p) X. Chen, S. Fan, M. Zhang, Y. Gao, S. Li and G. Li, Chem. Sci., 2021, 12, 4126 RSC; (q) P. Ramesh, C. Sreenivasulu, D. R. Kishore, D. Srinivas, K. R. Gorantla, B. S. Malik and G. Satyanarayana, J. Org. Chem., 2022, 87, 2204 CrossRef CAS PubMed; (r) A. Saha, S. Guin, W. Ali, T. Bhattacharya, S. Sasmal, N. Goswami, G. Prakash, S. K. Sinha, H. B. Chandrashekar, S. Panda, S. S. Anjana and D. Maiti, J. Am. Chem. Soc., 2022, 144, 1929 CrossRef CAS PubMed.
- (a) E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608 CrossRef CAS; (b) E. Marsault and M. L. Peterson, J. Med. Chem., 2011, 54, 1961 CrossRef CAS PubMed; (c) T. A. F. Cardote and A. Ciulli, ChemMedChem, 2016, 11, 787 CrossRef CAS PubMed.
- (a) J. R. Frost, C. C. G. Scully and A. K. Yudin, Nat. Chem., 2016, 8, 1105 CrossRef CAS PubMed; (b) X. Zhang, G. Lu, M. Sun, M. Mahankali, Y. Ma, M. Zhang, W. Hua, Y. Hu, Q. Wang, J. Chen, G. He, X. Qi, W. Shen, P. Liu and G. Chen, Nat. Chem., 2018, 10, 540 CrossRef CAS PubMed; (c) C.-X. Liu, P.-P. Xie, F. Zhao, Q. Wang, Z. Feng, H. Wang, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2023, 145, 4765 CrossRef CAS PubMed; (d) B. Song, X. Guo, L. Yang, H. Yu, X. Zong, X. Liu, H. Wang, Z. Xu, Z. Lin and W. Yang, Angew. Chem., Int. Ed., 2023, e202218886 CAS.
- S. Sengupta and G. Mehta, Org. Biomol. Chem., 2020, 18, 1851 RSC.
- K. T. Mortensen, T. J. Osberger, T. A. King, H. F. Sore and D. R. Spring, Chem. Rev., 2019, 119, 10288 CrossRef CAS PubMed.
- U. Lücking, G. Siemeister, M. Schäfer, H. Beiem, M. Krüger, P. Lienau and R. Jautelat, ChemMedChem, 2007, 2, 63 CrossRef.
- (a) S. Li, L. Cai, H. Ji, L. Yang and G. Li, Nat. Commun., 2016, 7, 10443 CrossRef PubMed; (b) L. Fang, T. G. Saint-Denis, B. L. H. Taylor, S. Ahlquist, K. Hong, S. Liu, L. Han, K. N. Houk and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 10702 CrossRef CAS PubMed.
- (a) Y.-F. Yang, G.-J. Cheng, P. Liu, D. Leow, T.-Y. Sun, P. Chen, X. Zhang, J.-Q. Yu and Y.-D. Wu, J. Am. Chem. Soc., 2014, 136, 344 CrossRef CAS; (b) Z. Fan, K. L. Bay, X. Chen, Z. Zhuang, H. S. Park, K.-S. Yeung, K. N. Houk and J.-Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 4770 ( Angew. Chem. , 2020 , 132 , 4800 ) CrossRef CAS PubMed.
- (a) M. V. Pham, B. Ye and N. Cram, Angew. Chem., Int. Ed., 2012, 51, 10610 ( Angew. Chem. , 2012 , 124 , 10762 ) CrossRef CAS PubMed; (b) Y. Ran, Y. Yang, H. You and J. You, ACS Catal., 2018, 8, 1796 CrossRef CAS; (c) Y. Dong, X. Zhang, J. Chen, W. Zou, S. Lin and H. Xu, Chem. Sci., 2019, 10, 8744 RSC; (d) H.-X. Dai, A. F. Stepan, M. S. Plummer, Y.-H. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 7222 CrossRef CAS PubMed; (e) G. Wu, W. Ouyang, Q. Chen, Y. Huo and X. Li, Org. Chem. Front., 2019, 6, 284 RSC; (f) T. Lan, L. Wang and Y. Rao, Org. Lett., 2017, 19, 972 CrossRef CAS; (g) S. Rej and N. Chatani, Chem. Sci., 2020, 11, 389 RSC; (h) W. Liu, D. Wang, Y. Zhao, F. Yi and J. Chen, Adv. Synth. Catal., 2016, 358, 1968 CrossRef CAS; (i) S. Feng, S. Li, J. Li and J. Wei, Org. Chem. Front., 2019, 6, 517 RSC; (j) S. Ojha and N. Panda, Adv. Synth. Catal., 2020, 362, 561 CrossRef CAS.
- P. G. M. Wuts and T. W. Greene, Greene's Protective Groups in Organic Synthesis, Wiley-Interscience, New Jersey, 4th edn, 2007 Search PubMed.
- N. Y. S. Lam, Z. Fan, K. Wu, H. S. Park, S. Y. Shim, D. A. Strassfeld and J.-Q. Yu, J. Am. Chem. Soc., 2022, 144, 2793 CrossRef CAS PubMed.
- A. H. Sandtorv, Adv. Synth. Catal., 2015, 357, 2403 CrossRef CAS.
- J. Xu, J. Chen, F. Gao, S. Xie, X. Xu, Z. Jin and J.-Q. Yu, J. Am. Chem. Soc., 2019, 141, 1903 CrossRef CAS PubMed.
- G.-J. Cheng, Y.-F. Yang, P. Liu, P. Chen, T.-Y. Sun, G. Li, X. Zhang, K. N. Houk, J.-Q. Yu and Y.-D. Wu, J. Am. Chem. Soc., 2014, 136, 894 CrossRef CAS PubMed.
- (a) D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118 CrossRef; (b) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2142546. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01670f |
| ‡ Deposition number 2142546 (for compound 2d) contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre. |
| This journal is © The Royal Society of Chemistry 2023 |