
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsertion of CO2 and CS2 into Bi–N bonds enables catalyzed CH-activation and light-induced bismuthinidene transfer†
Kai
Oberdorf
a,
Anna
Hanft
a,
Xiulan
Xie
a,
F. Matthias
Bickelhaupt
 bcd,
Jordi
Poater
*e and
Crispin
Lichtenberg
*a
bcd,
Jordi
Poater
*e and
Crispin
Lichtenberg
*a
aFachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Str. 4, 35043 Marburg, Germany. E-mail: crispin.lichtenberg@chemie.uni-marburg.de
bTheoretical Chemistry, Department of Chemistry and Pharmaceutical Sciences, Vrije Universiteit Amsterdam, The Netherlands
cInstitute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands
dDepartment of Chemical Sciences, University of Johannesburg, Auckland Park, Johannesburg 2006, South Africa
eDepartament de Química Inorgànica i Orgànica, IQTCUB, Universitat de Barcelona, ICREA, Pg. Lluís Companys 23, 08010 Barcelona, Spain. E-mail: jordi.poater@ub.edu
First published on 28th April 2023
Abstract
The uptake and release of small molecules continue to be challenging tasks of utmost importance in synthetic chemistry. The combination of such small molecule activation with subsequent transformations to generate unusual reactivity patterns opens up new prospects for this field of research. Here, we report the reaction of CO2 and CS2 with cationic bismuth(III) amides. CO2-uptake gives isolable, but metastable compounds, which upon release of CO2 undergo CH activation. These transformations could be transferred to the catalytic regime, which formally corresponds to a CO2-catalyzed CH activation. The CS2-insertion products are thermally stable, but undergo a highly selective reductive elimination under photochemical conditions to give benzothiazolethiones. The low-valent inorganic product of this reaction, Bi(I)OTf, could be trapped, showcasing the first example of light-induced bismuthinidene transfer.
Introduction
The activation of small molecules such as H2, CO, CO2, P4, and ethylene used to be considered a domain of transition-metal complexes, but recent advances in main group chemistry have set the stage for s- and p-block compounds to become a unique part of this field of research.1–8 Strategies that have successfully been applied for small molecule activation by main group compounds include the exploitation of frustrated Lewis pairs,6 low valent species,1–4 (bi)radicals,1–4,9 compounds featuring element–element multiple bonds,1–4 ring-strain,2,10 shuttling between unusual oxidation states,11,12 and element-ligand cooperativity.8Among the main group compounds, species based on heavy p-block elements with a main quantum number ≥5 stand out due to large covalent radii, flexible coordination environments, soft Lewis acidity, relativistic effects becoming more relevant, and low bond dissociation energies.13–17 These properties have paved the ground for fundamentally important findings, including the reversible addition of H2 and ethylene to distannynes,18 the reversible fixation of CO2 and CS2 by complexes with hyper-coordinate antimony or bismuth centers,19,20 CO insertion into a Bi–N bond,10 and the reversible P4 activation by a bismuth radical.21 The valuable insights gained from research in this field have so far mostly been focused on the activation, release, and degradation of small molecules, as well as their utilization as building blocks (for highlights on the reversible uptake and release of small molecules see: Scheme 1, top). In contrast, the concept of incorporating a small molecule into a heavy main group complex in order to grant access to new reactivity patterns in subsequent reactions is far less explored. This requires the generation of reactive sites that can be addressed by an external stimulus. Along these lines, the insertion of CO2 and CS2 into well-defined bismuth complexes is a promising approach, since Bi–O and Bi–S bonds can readily be cleaved via heterolytic or homolytic pathways22–24 and even chelating bonding modes of carboxylates and dithiocarboxylates do not generate deep thermodynamic sinks due to the coordination chemical flexibility of the large central atom (Scheme 1, bottom).
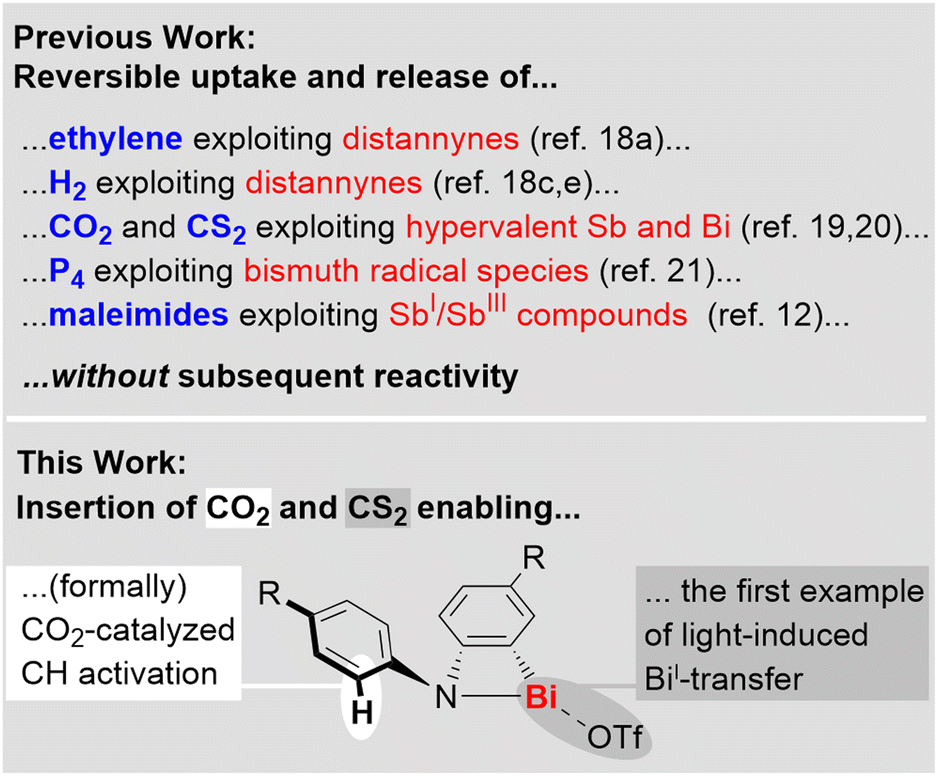 | ||
| Scheme 1 Activation of small molecules with compounds of heavy p-block elements. Top: reversible activation without subsequent novel reactivity (literature). Bottom: activation of CO2 and CS2 followed by CH activation and light-induced bismuthinidene transfer (this work). | ||
Here we report the selective insertion of CO2 and CS2 into the Bi–N bonds of cationic bismuth amides and the unusual reactivity patterns of the resulting products, which cover CH activation and an unprecedented light-induced bismuthinidene transfer.
Results and discussion
Previously reported ring-strained cationic bismuth compounds 1-R were selected as starting materials since they exhibit a sufficiently high reactivity and one reactive site per bismuth atom (Scheme 2a).25–27 Reactions of 1-R with CO2 and CS2 gave compounds 2-R and 3-R in 72–89% yield, resulting from the selective insertion of the heterocumulenes into the Bi–N bond (Scheme 2a). These are rare examples of compounds based on heavier p-block elements, for which well-defined insertion reactions with both, CO2 and CS2 could be realized.28 NMR spectra of 2-R and 3-R revealed the expected signal patterns, albeit signal broadening was observed for 3-R (and for 2-R in pyridine), suggesting the accessibility of higher aggregates in equilibrium reactions. This is supported by a combination of DOSY NMR spectroscopy, high-resolution mass spectrometry, and DFT calculations. DOSY NMR spectroscopy indicated the presence of aggregated species, where the formally determined degrees of aggregation represent averaged values when involving equilibrium reactions that proceed beyond the time scale of the experiment.29 For 2-H, high-resolution ESI-MS revealed a mononuclear species resulting from loss of CO2 and the [OTf]− counteranion, suggesting the possibility of de-aggregation and giving a first hint at the facile liberation of CO2 from this molecule (vide infra). For 3-H mono- and dinuclear monocations (obtained by loss of [OTf]−) were detected. DFT calculations at the ZORA-BLYP-D3(BJ)/TZ2P level30 with a pyridine solvent model, suggest that the aggregation to form a dinuclear species is energetically disfavored for 2-H (ΔG = +10.9 kcal mol−1), but feasible for 3-H (ΔG = −8.8 kcal mol−1).29 Overall, these results suggest that the solution structures of compounds 2-R and 3-R involve aggregation phenomena with mononuclear species being accessible. The mononuclear representation of these compounds in Scheme 2 is chosen for simplicity.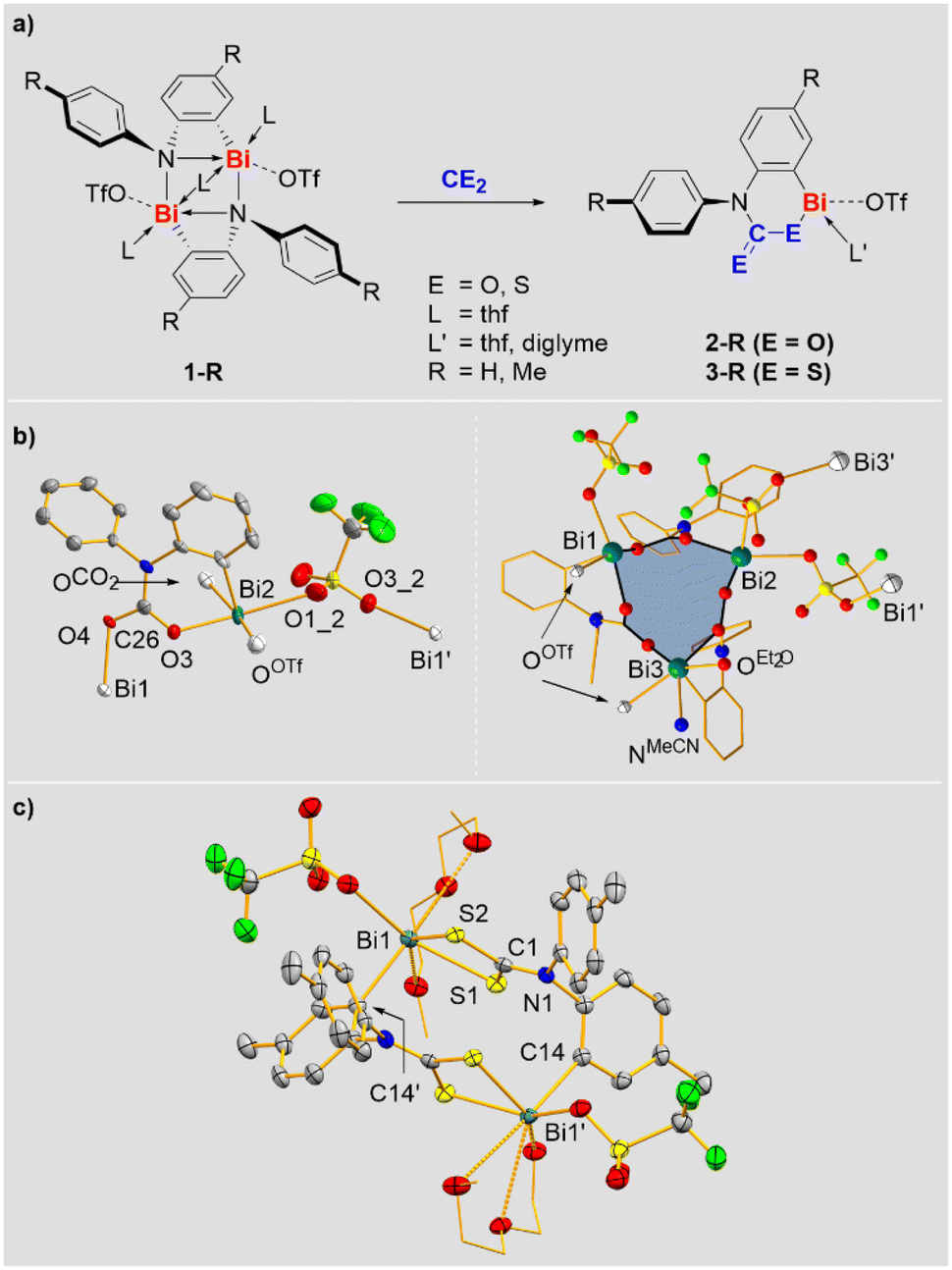 | ||
| Scheme 2 (a) Insertion of CO2 and CS2 into Bi–N bonds to give 2-R and 3-R. For a discussion of the CE2-bonding mode and the aggregation behavior of 2-R and 3-R see main text. A mononuclear representation of 2-H and 3-H is chosen for simplicity. (b and c) Molecular structures of 2-H (b) and 3-Me (c) in the solid state. Displacement ellipsoids are shown at the 50% probability level. Hydrogen atoms are omitted for clarity and C atoms are shown as wireframe in the right part of (b); atoms exceeding one subunit are drawn as white ellipsoids. Selected bond lengths [Å] for 2-H: Bi1–O4, 2.232(5), Bi2–O3, 2.222(5); Bi2–C14, 2.206(8). Selected bond lengths [Å] for 3-Me: Bi1–S1, 2.6260(8); Bi1–S2, 2.6354(7), Bi1–C14′, 2.305(3); S1–C1, 1.716(3); S2–C1, 1.720(3); C1–N1, 1.332(4). | ||
The molecular structures of 2-R and 3-R in the solid state were elucidated by single-crystal X-ray diffraction analyses and evidence the coordination chemical flexibility of cationic bismuth species (the quality of the X-ray data on 2-Me and 3-H does not allow the discussion of bonding parameters, but serves as a proof of connectivity). Single-crystals of 2-H were obtained by layering a saturated solution of 2-H in acetonitrile with diethyl ether, leading to partial exchange of the solvent ligands (vide infra). The CO2 moieties in 2-H adopt bridging μ2-η1,η1-coordination modes to form a trinuclear species (one mononuclear subunit and the trimer are depicted in Scheme 2b). Two of the three triflate anions also adopt a bridging coordination mode, resulting in the interconnection of trinuclear subunits to form a double-stranded coordination polymer along the crystallographic b-axis. The bismuth atoms Bi1 and Bi2 adopt a square pyramidal coordination geometry due to contacts with one aryl, two triflate and two CO2 units, while Bi3 shows a very unusual pentagonal pyramidal coordination geometry due to contacts with one aryl, one triflate, one CO2 moiety and two neutral ligands (Et2O and MeCN). The Bi–O bonds (2.22–2.30 Å) and the C–O bonds (1.27–1.30 Å) involving CO2 moieties are similar in length, suggesting a delocalization of electron density across the CO2 unit. The analysis of 2-Me confirmed the μ2-η1:η1-coordination mode of the CO2 group and the tendency towards aggregation in the solid state (to form a dinuclear species in this case).29
The structural analysis of 3-R gives further evidence of the rich coordination chemistry of bismuth cations: 3-H forms a tetranuclear species with the CS2 moiety in a bridging μ2-η1,η1-coordination mode, while 3-Me crystallized as a dinuclear aggregate with the CS2 unit showing a non-bridging, chelating η1,η1-coordination (Scheme 2c).29 The bismuth atoms in 3-Me show a coordination number of seven with an irregular coordination geometry around Bi1. The Bi1–S1/2 bond lengths (2.63–2.64 Å) do not differ significantly. Together with the C1–S1/2 distances (1.72 Å), which are identical within limits of error, they indicate effective delocalization of the electron density in the CS2 unit.
While only a few bismuth compounds have been reported to selectively react with CO2 or CS2, even less is known about the reactivity of the products resulting from these reactions.19,28,31 As a prominent example, the shallow potential energy landscape related to the insertion of CO2 into a Bi–O bond of (Ar2Bi)2O has been exploited to reversibly bind and release this heterocumulene.19 Inspired by these pivotal contributions, we set out to map the reactivity of 2-R and 3-R centered around the stimulus-induced release of CO2 and CS2.
Indeed, compounds 2-R eliminate CO2 at ambient temperature in pyridine or at 60 °C in THF, irrespective of the presence or absence of ambient light. The decay of 2-R and the formation of CO2 was monitored by 1H and 13C NMR spectroscopy, respectively. Remarkably, the CO2 release goes along with selective formation of the previously reported25–27ortho CH-activation products 4-R in quantitative spectroscopic yield (Scheme 3a). The formation of 4-Me was also confirmed by single-crystal X-ray analysis.29 Access to the ortho CH-activated species 4-R has previously been achieved by heating 1-R in pyridine at 80 °C (Scheme 3a, bottom).25,27 When THF is used as the solvent, 1-H in particular can be kept at 60 °C over an extended period of time without any signs of decomposition or transformation.32 In the presence of 10 mol% of 2-H, however, compound 1-H was readily transformed into 4-H with >90% spectroscopic yield over 4 days in THF (Scheme 3a, bottom). Lowering the amount of 2-H in the reaction mixture to 1 mol% still resulted in the selective formation of 4-H (84% spectroscopic yield), albeit longer reaction times of 17 d were necessary. These observations formally correspond to a CO2-catalyzed CH-activation reaction.33 In contrast to the CO2-insertion products 2-R, the sulfur analogs 3-R proved to be stable in THF at 60 °C over days, when ambient light was excluded.
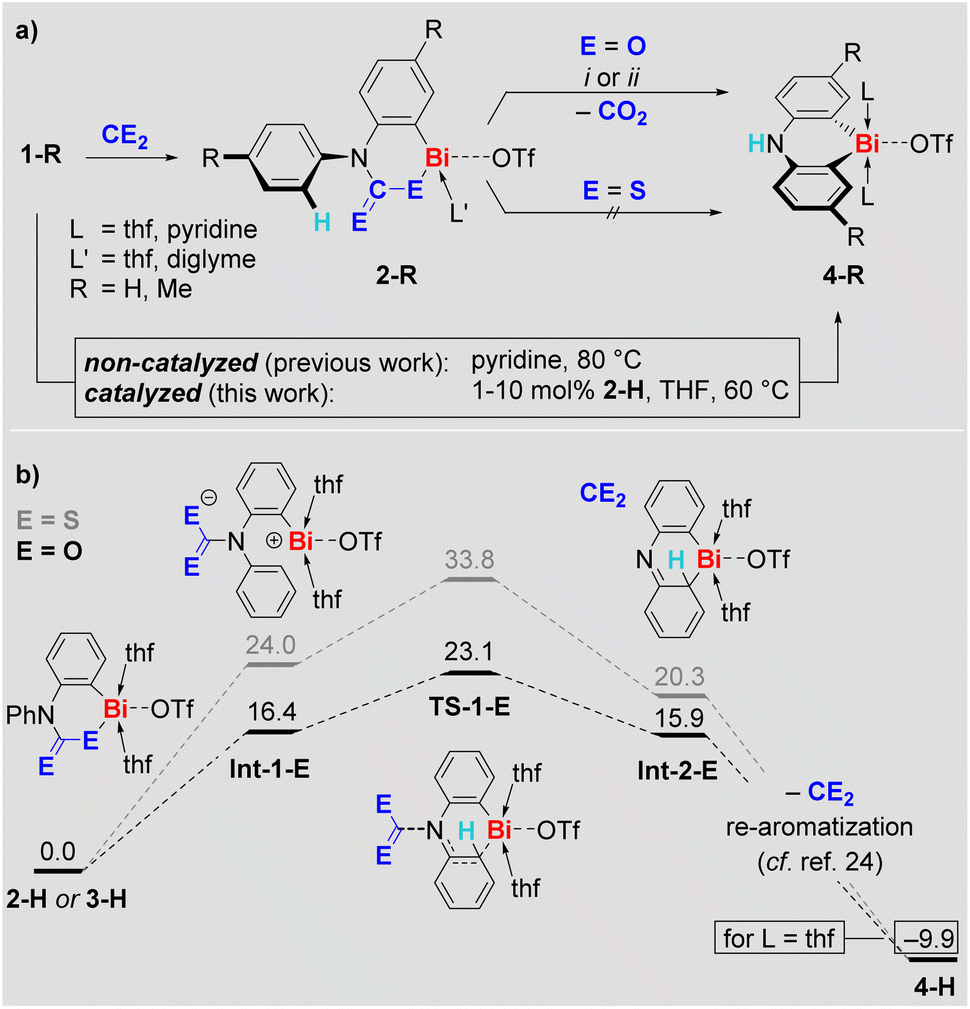 | ||
| Scheme 3 (a) Reactivity of compounds 2-R in the context of CH activation of 1-R to give 4-R ((i) THF, 60 °C, (ii) pyridine, r.t.). (b) Proposed mechanism for the extrusion of CE2 from 2-H (black) and 3-H (dark grey) to give 4-H. Gibbs energies are given in kcal mol−1.29 | ||
Mechanistic aspects of the CE2 extrusion from 2-H and 3-H were investigated by DFT calculations (Scheme 3b). Mononuclear model systems were chosen based on the investigations of their aggregation behavior (vide supra), to keep the computational approach feasible at a sufficiently high level of theory, and because a higher reactivity for mononuclear species has been proposed in related systems.34 We suggest a heterolytic cleavage of the Bi–E bond as the initiating step of the reaction to give the charge-separated intermediate Int-1-E. This intermediate is energetically more favorable for E = O than for E = S due to stabilization of the negative charge by the electronegative oxygen atoms in the (R2NCO2)− moiety. Release of CE2 with concomitant addition of the cationic bismuth center to the phenyl group via transition state TS-1-E gives Int-2-E. Again, the oxygen-based species is lower in energy, which is due to the energy gain resulting from the formation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) E double bond in CE2 being higher for E = O than for E = S.35 Re-aromatization of Int-2-E gives the final product 4-H – a sequence of reactions that has previously been investigated in detail during the CH activation of 1-H and is thus suggested to proceed in an analogous manner here.25 Overall, the results of this mechanistic study indicate release of CO2 at early stages of the reaction, rationalize the feasibility vs. reluctance of CE2 release for 2-R and 3-R, and demonstrate the accessibility of previously identified reaction pathways for bismuth-mediated CH activation upon CE2 elimination. The sensitivity of the CH activation 1-H → 4-H towards the polarity of the reaction medium32 motivated us to investigate the effect of the CO2 insertion on the polarity of the bismuth species by DFT methods. Indeed, the CO2-insertion-product 2-H shows a high dipole moment of 6.1 D, exceeding those of pyridine (2.3 D) or 1,2-difluorobnezene (2.6 D), which also facilitate the CH activation of 1-H when used as solvents.29 These findings indicate that an increase of the polarity of the solvent medium as a result of CO2 insertion is an important factor facilitating the CO2-catalyzed CH activation 1-H–(cat. 2-H) → 4-H.
E double bond in CE2 being higher for E = O than for E = S.35 Re-aromatization of Int-2-E gives the final product 4-H – a sequence of reactions that has previously been investigated in detail during the CH activation of 1-H and is thus suggested to proceed in an analogous manner here.25 Overall, the results of this mechanistic study indicate release of CO2 at early stages of the reaction, rationalize the feasibility vs. reluctance of CE2 release for 2-R and 3-R, and demonstrate the accessibility of previously identified reaction pathways for bismuth-mediated CH activation upon CE2 elimination. The sensitivity of the CH activation 1-H → 4-H towards the polarity of the reaction medium32 motivated us to investigate the effect of the CO2 insertion on the polarity of the bismuth species by DFT methods. Indeed, the CO2-insertion-product 2-H shows a high dipole moment of 6.1 D, exceeding those of pyridine (2.3 D) or 1,2-difluorobnezene (2.6 D), which also facilitate the CH activation of 1-H when used as solvents.29 These findings indicate that an increase of the polarity of the solvent medium as a result of CO2 insertion is an important factor facilitating the CO2-catalyzed CH activation 1-H–(cat. 2-H) → 4-H.
When solutions of compounds 2-R were irradiated with a low-pressure Hg-lamp, only mixtures of products were obtained, which could not be identified to date.29 While compounds 3-R are stable in the dark, even at elevated temperature (vide supra), it quickly became evident that they are light-sensitive: at 23 °C, exposure of THF solutions of 3-R to ambient light induced a quantitative and highly selective transformation over five days with concomitant precipitation of a dark solid. Irradiation of THF solutions of 3-R with a low-pressure Hg-vapor lamp dramatically accelerated these reactions without diminishing the excellent selectivities (full conversion after 45 min).29 The THF-soluble products of these reactions were identified as benzothiazolethiones 5-R (Scheme 4, top). This corresponds to a C–S bond formation, formally resulting from the reductive elimination of BiI(OTf), which we suggest as a logical intermediate. DFT calculations confirm that a concerted mechanism only involving closed-shell species is too high in energy to be viable.29 In contrast, DFT calculations suggest that the light-induced Bi–S homolysis in 3-R is feasible and gives a short-lived intermediate (Int-5-S; ESI†), from which the elimination of Bi(thf)2(OTf) proceeds with a free activation energy of only 9.0 kcal mol−1.29 It should be noted in this context that BiI(OTf) has recently been isolated as a cyclic alkyl(amino) carbene adduct and proved to be unstable in solution.36 In general, non-stabilized bismuthinidenes BiX (X = monoanionic, monodentate ligand) are unstable in solution,16,37 with disproportionations such as 3 BiX → 2 Bi0 + BiX3 being a typical degradation pathway, which accounts for the commonly observed dark precipitate (“bismuth black”).38 In order to trap the potential intermediate BiI(OTf) in photochemical reactions of cationic bismuth species, 3,5-di-tert-butyl-o-benzoquinone (6) was added to a THF solution of 3-H. 1H NMR spectroscopy revealed that the quinone coordinated 3-H, but no reaction occurred at ambient temperature in the dark. Upon irradiation of the reaction mixture containing the quinone (Scheme 4, bottom), the precipitation of a black solid was not observed and quantitative conversion to 5-H and a new compound (later identified as 7) was detected by NMR spectroscopy. Despite similar solubilities of these two products, compound 7 could be isolated in moderate yield and was fully characterized.29
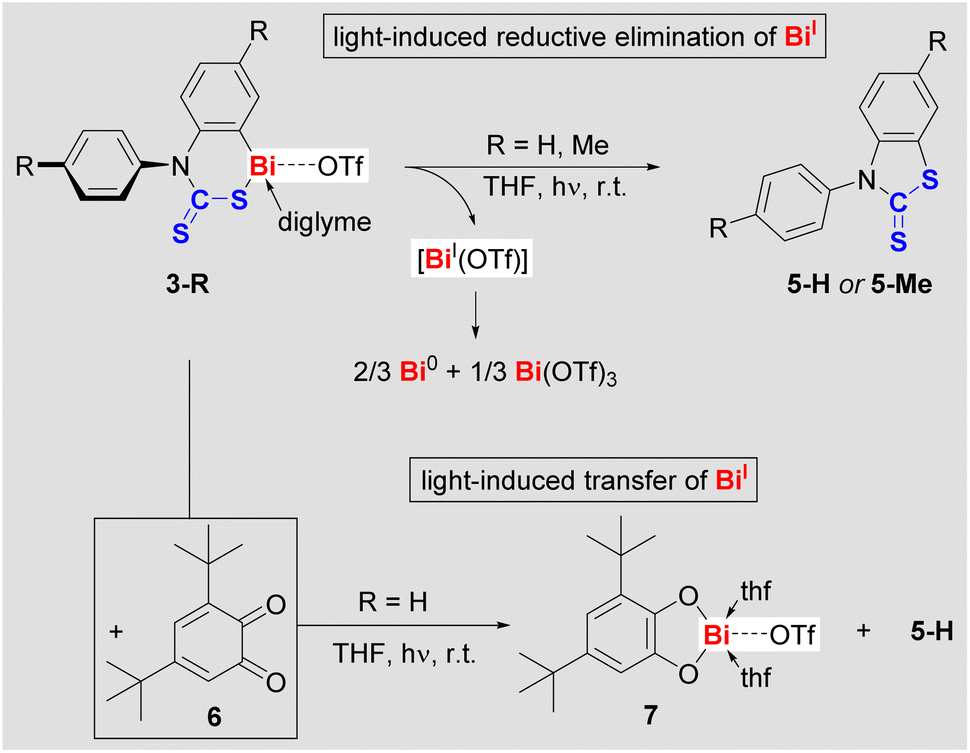 | ||
| Scheme 4 Photochemically-induced elimination of BiOTf from 3-R to give heterocycle 5-R in the absence (top) and presence (bottom) of 3,5-di-tert-butyl-o-benzoquinone (6), which is introduced as bismuthinidene trapping reagent. | ||
While the chemistry of mononuclear bismuth compounds used to be centered around BiIII and BiV species, recent advances have begun to uncover an unexpectedly rich redox chemistry for this class of compounds.7,11,14,39 This includes examples of reductive elimination reactions for selective bond formation in stoichiometric or catalytic regimes, which operate under thermal conditions.15,40 While the light-sensitivity of molecular bismuth compounds has been phenomenologically described,41 the exploitation of their photochemistry for selective bond formations is still in its infancy.23,24 Similarly, the field of cationic bismuth(I) compounds is virtually unexplored, with a single example of an isolable species36 and the reactivity of these species being entirely unknown.17,42 Along these lines, reactions of compounds 3-R demonstrate for the first time that cationic bismuth compounds may be exploited both, for the light-driven construction of organic heterocycles and for photochemically-induced transfer of low-valent Bi(I) building blocks.
Conclusions
In summary we have shown, that ring-strained bismuth amide cations selectively insert CO2 and CS2. The insertion products show striking differences in their reactivity: the CO2 activation products undergo thermochemical CO2 elimination, which goes along with a CH activation to furnish previously reported cationic bisma-cycles. These reactions were transferred to the catalytic regime, which formally corresponds to an unprecedented CO2-catalyzed CH activation reaction. In the case of the CS2 activation product, the highly selective, photochemical elimination of low-valent BiOTf (a so-called bismuthinidene) is observed, while benzothiazolethiones are generated through selective C–S bond formation. Chemical trapping of the photochemically released BiOTf was realized for the first time, opening up the field of light-driven bismuthinidene transfer reactions, which are subject of future investigations in our laboratories.Data availability
Experimental data is provided in the ESI.†Author contributions
Synthesis: K. O. (exclusively); X-ray diffraction analyses: K. O., A. H. (equal); DFT calculations: J. P. (lead); F. M. B., C. L. (supporting); DOSY studies: X. X. (lead); K. O., C. L. (supporting) composition of the manuscript: K. O., C. L. (lead); J. P. (supporting); discussion of results and correction of manuscript: all authors; project administration: C. L. (lead); acquisition of funding: K. O., J. P., F. M. B., C. L.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Benedikt Ritschel for his contributions. Financial support by the FCI, the DFG (LI2860/3-1, LI2860/5-1), the LOEWE Program, the Cusanuswerk, the Netherlands Organisation of Scientific Research (NWO), and the Spanish Government (PID-2019-106830GB-I00, CEX2021-001202-M), and the Catalan Government (2021SGR442) is gratefully acknowledged. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement No. 946184).Notes and references
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- M.-A. Légaré, C. Pranckevicius and H. Braunschweig, Chem. Rev., 2019, 119, 8231–8261 CrossRef PubMed.
- C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS.
- P. P. Power, Chem. Rev., 2003, 103, 789–810 CrossRef CAS PubMed.
- D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 RSC.
- D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed.
- C. Lichtenberg, Angew. Chem., Int. Ed., 2016, 55, 484–486 CrossRef CAS PubMed.
- L. Greb, F. Ebner, Y. Ginzburg and L. M. Sigmund, Eur. J. Inorg. Chem., 2020, 2020, 3030–3047 CrossRef CAS.
- X. Yang, E. J. Reijerse, K. Bhattacharyya, M. Leutzsch, M. Kochius, N. Nöthling, J. Busch, A. Schnegg, A. A. Auer and J. Cornella, J. Am. Chem. Soc., 2022, 144, 16535–16544 CrossRef CAS PubMed.
- J. Ramler, J. Poater, F. Hirsch, B. Ritschel, I. Fischer, F. M. Bickelhaupt and C. Lichtenberg, Chem. Sci., 2019, 10, 4169–4176 RSC.
- H. W. Moon and J. Cornella, ACS Catal., 2022, 12, 1382–1393 CrossRef CAS PubMed.
- M. Kořenková, M. Hejda, M. Erben, R. Jirásko, R. Jambor, A. Růžička, E. Rychagova, S. Ketkov and L. Dostál, Chem.–Eur. J., 2019, 25, 12884–12888 CrossRef PubMed.
- (a) R. J. F. Berger, D. Rettenwander, S. Spirk, C. Wolf, M. Patzschke, M. Ertl, U. Monkowius and N. W. Mitzel, Phys. Chem. Chem. Phys., 2012, 14, 15520–15524 RSC; (b) S. Ishida, F. Hirakawa, K. Furukawa, K. Yoza and T. Iwamoto, Angew. Chem., Int. Ed., 2014, 53, 11172–11176 CrossRef CAS PubMed; (c) T. Y. Lai, L. Tao, R. D. Britt and P. P. Power, J. Am. Chem. Soc., 2019, 141, 12527–12530 CrossRef CAS PubMed; (d) J. Ramler and C. Lichtenberg, Chem.–Eur. J., 2020, 26, 10250–10258 CrossRef CAS PubMed; (e) D. Dakternieks, D. J. Henry and C. H. Schiesser, Organometallics, 1998, 17, 1079–1084 CrossRef CAS; (f) J. Ramler, F. Fantuzzi, F. Geist, A. Hanft, H. Braunschweig, B. Engels and C. Lichtenberg, Angew. Chem., Int. Ed., 2021, 60, 24388–24394 CrossRef CAS PubMed; (g) L. D. Freedman and G. O. Doak, Chem. Rev., 1982, 82, 15–57 CrossRef CAS; (h) G. P. Smith and R. Patrick, Int. J. Chem. Kinet., 1983, 15, 167–185 CrossRef CAS.
- C. Lichtenberg, Chem. Commun., 2021, 57, 4483–4495 RSC.
- K. Oberdorf, A. Hanft, J. Ramler, I. Krummenacher, F. M. Bickelhaupt, J. Poater and C. Lichtenberg, Angew. Chem., Int. Ed., 2021, 60, 6441–6445 CrossRef CAS PubMed.
- D. P. Mukhopadhyay, D. Schleier, S. Wirsing, J. Ramler, D. Kaiser, E. Reusch, P. Hemberger, T. Preitschopf, I. Krummenacher, B. Engels, I. Fischer and C. Lichtenberg, Chem. Sci., 2020, 11, 7562–7568 RSC.
- J. Heine, B. Peerless, S. Dehnen and C. Lichtenberg, Angew. Chem., Int. Ed., 2023, e202218771 Search PubMed.
- (a) Y. Peng, B. D. Ellis, X. Wang, J. C. Fettinger and P. P. Power, Science, 2009, 325, 1668–1670 CrossRef CAS PubMed; (b) T. Y. Lai, J.-D. Guo, J. C. Fettinger, S. Nagase and P. P. Power, Chem. Commun., 2019, 55, 405–407 RSC; (c) Y. Peng, M. Brynda, B. D. Ellis, J. C. Fettinger, E. Rivard and P. P. Power, Chem. Commun., 2008, 6042–6044 RSC; (d) Y. Peng, B. D. Ellis, X. Wang and P. P. Power, J. Am. Chem. Soc., 2008, 130, 12268–12269 CrossRef CAS PubMed; (e) S. Wang, T. J. Sherbow, L. A. Berben and P. P. Power, J. Am. Chem. Soc., 2018, 140, 590–593 CrossRef CAS PubMed.
- S.-F. Yin, J. Maruyama, T. Yamashita and S. Shimada, Angew. Chem., Int. Ed., 2008, 47, 6590–6593 CrossRef CAS PubMed.
- (a) L. Dostál, R. Jambor, A. Růžička, R. Jirásko, E. Černošková, L. Beneš and F. de Proft, Organometallics, 2010, 29, 4486–4490 CrossRef; (b) L. Dostál, R. Jambor, A. Růžička, M. Erben, R. Jirásko, E. Černošková and J. Holeček, Organometallics, 2009, 28, 2633–2636 CrossRef; (c) G. Strîmb, A. Pöllnitz, C. I. Raţ and C. Silvestru, Dalton Trans., 2015, 44, 9927–9942 RSC.
- R. J. Schwamm, M. Lein, M. P. Coles and C. M. Fitchett, Angew. Chem., Int. Ed., 2016, 55, 14798–14801 CrossRef CAS PubMed.
- (a) T. A. Hanna, A. L. Rieger, P. H. Rieger and X. Wang, Inorg. Chem., 2002, 41, 3590–3592 CrossRef CAS PubMed; (b) S. Roggan, C. Limberg, B. Ziemer and M. Brandt, Angew. Chem., Int. Ed., 2004, 43, 2846–2849 CrossRef CAS PubMed; (c) C. Limberg, Angew. Chem., Int. Ed., 2003, 42, 5932–5954 CrossRef CAS PubMed.
- J. Ramler, I. Krummenacher and C. Lichtenberg, Chem.–Eur. J., 2020, 26, 14551–14555 CrossRef CAS PubMed.
- J. Ramler, J. Schwarzmann, A. Stoy and C. Lichtenberg, Eur. J. Inorg. Chem., 2022, 2022, e202100934 CAS.
- B. Ritschel, J. Poater, H. Dengel, F. M. Bickelhaupt and C. Lichtenberg, Angew. Chem., Int. Ed., 2018, 57, 3825–3829 CrossRef CAS PubMed.
- B. Ritschel and C. Lichtenberg, Synlett, 2018, 29, 2213–2217 CrossRef CAS.
- K. Oberdorf, P. Grenzer, N. Wieprecht, J. Ramler, A. Hanft, A. Rempel, A. Stoy, K. Radacki and C. Lichtenberg, Inorg. Chem., 2021, 60, 19086–19097 CrossRef CAS PubMed.
- F. Ando, T. Hayashi, K. Ohashi and J. Koketsu, J. Inorg. Nucl. Chem., 1975, 37, 2011–2013 CrossRef CAS.
- For details see the ESI.†.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- (a) S. D. Cosham, M. S. Hill, G. A. Horley, A. L. Johnson, L. Jordan, K. C. Molloy and D. C. Stanton, Inorg. Chem., 2014, 53, 503–511 CrossRef CAS PubMed; (b) D. R. Kindra, I. J. Casely, M. E. Fieser, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2013, 135, 7777–7787 CrossRef CAS PubMed; (c) S.-F. Yin and S. Shimada, Chem. Commun., 2009, 1136–1138 RSC; (d) Y. Chen, R. Qiu, X. Xu, C.-T. Au and S.-F. Yin, RSC Adv., 2014, 4, 11907–11918 RSC; (e) K. Marczenko and S. Chitnis, ChemRxiv, 2021, preprint, DOI:10.26434/chemrxiv.13619180.v2. This work has not been subjected to peer review.
- The polarity of the reaction medium appears to be crucial for the transformation of 1-H to 4-H. 1-H is stable in dichloromethane (dipole moment = 1.6 D) at 60 °C (bath temperature) for at least 3 d and in THF (dipole moment = 1.8 D) at 60 °C for at least 4 d. However, 1-H is transformed into 4-H in quantitative spectroscopic yield, when heated to 80 °C for 16 h in pyridine (dipole moment = 2.3 D) or 1,2-difluorobenzene (dipole moment = 2.6 D).
- (a) Y. Sugawara, W. Yamada, S. Yoshida, T. Ikeno and T. Yamada, J. Am. Chem. Soc., 2007, 129, 12902–12903 CrossRef CAS PubMed; (b) D. Riemer, B. Mandaviya, W. Schilling, A. C. Götz, T. Kühl, M. Finger and S. Das, ACS Catal., 2018, 8, 3030–3034 CrossRef CAS.
- K. Oberdorf, P. Pfister, P. Grenzer, J. Ramler, A. Hanft, A. Stoy, X. Xie and C. Lichtenberg, Organometallics, 2023 Search PubMed , accepted.
- K. B. Wiberg and Y. Wang, Arkivoc, 2011, 2011, 45–56 Search PubMed.
- M. M. Siddiqui, S. K. Sarkar, M. Nazish, M. Morganti, C. Köhler, J. Cai, L. Zhao, R. Herbst-Irmer, D. Stalke, G. Frenking and H. W. Roesky, J. Am. Chem. Soc., 2021, 143, 1301–1306 CrossRef CAS PubMed.
- (a) E. H. Fink, K. D. Setzer, D. A. Ramsay and M. Vervloet, Chem. Phys. Lett., 1991, 179, 95–102 CrossRef CAS; (b) E. Fink, K. Setzer, D. Ramsay, M. Vervloet and J. Brown, J. Mol. Spectrosc., 1990, 142, 108–116 CrossRef CAS.
- J. Bresien, A. Schulz, M. Thomas and A. Villinger, Eur. J. Inorg. Chem., 2019, 2019, 1279–1287 CrossRef CAS.
- (a) C. Lichtenberg, Chem.–Eur. J., 2020, 26, 9674–9687 CrossRef CAS PubMed; (b) C. Helling and S. Schulz, Eur. J. Inorg. Chem., 2020, 2020, 3209–3221 CrossRef CAS; (c) C. Lichtenberg, Radical Compounds of Antimony and Bismuth in EIBC, ed. R. A. Scott, Wiley VCH, 2020, pp. 1–12 Search PubMed.
- (a) F. Wang, O. Planas and J. Cornella, J. Am. Chem. Soc., 2019, 141, 4235–4240 CrossRef CAS PubMed; (b) Y. Pang, M. Leutzsch, N. Nöthling and J. Cornella, J. Am. Chem. Soc., 2020, 142, 19473–19479 CrossRef CAS PubMed; (c) Y. Pang, M. Leutzsch, N. Nöthling, F. Katzenburg and J. Cornella, J. Am. Chem. Soc., 2021, 143, 12487–12493 CrossRef CAS PubMed; (d) W.-C. Xiao, Y.-W. Tao and G.-G. Luo, Int. J. Hydrogen Energy, 2020, 45, 8177–8185 CrossRef CAS; (e) O. Planas, F. Wang, M. Leutzsch and J. Cornella, Science, 2020, 367, 313–317 CrossRef CAS PubMed; (f) D. H. R. Barton, W. B. Motherwell and A. Stobie, J. Chem. Soc., Chem. Commun., 1981, 1232–1233 RSC; (g) S. M. Nabavizadeh, F. Niroomand Hosseini, N. Nejabat and Z. Parsa, Inorg. Chem., 2013, 52, 13480–13489 CrossRef CAS PubMed.
- (a) C. Lichtenberg, F. Pan, T. P. Spaniol, U. Englert and J. Okuda, Angew. Chem., Int. Ed., 2012, 51, 13011–13015 CrossRef CAS PubMed; (b) A. J. Ashe, E. G. Ludwig and J. Oleksyszyn, Organometallics, 1983, 2, 1859–1866 CrossRef CAS; (c) J. Lorberth, W. Massa, S. Wocadlo, I. Sarraje, S.-H. Shin and X.-W. Li, J. Organomet. Chem., 1995, 485, 149–152 CrossRef CAS; (d) W. Clegg, N. A. Compton, R. J. Errington, G. A. Fisher, M. E. Green, D. C. R. Hockless and N. C. Norman, Inorg. Chem., 1991, 30, 4680–4682 CrossRef CAS; (e) J. M. Wallis, G. Müller and H. Schmidbaur, J. Organomet. Chem., 1987, 325, 159–168 CrossRef CAS.
- R. Deb, P. Balakrishna and M. Majumdar, Chem.–Asian J., 2022, 17, e202101133 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, crystallographic information; computational details; cartesian coordinates of calculated compounds. CCDC 2223276–2223278. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01635h |
| This journal is © The Royal Society of Chemistry 2023 |