
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReplacing the BO in BODIPY: unlocking the path to SBDIPY and BIDIPY chromophores†
André
Korzun
a,
Stefano
Crespi
 b,
Christopher
Golz
a and
Alessandro
Bismuto
*ac
b,
Christopher
Golz
a and
Alessandro
Bismuto
*ac
aInstitut für Organische und Biomolekulare Chemie, Georg-August-Universität Göttingen, Tammannstr. 2, 37077, Göttingen, Germany. E-mail: alessandro.bismuto@chemie.uni-goettingen.de
bDepartment of Chemistry – Ångström Laboratory Uppsala University, Box 523, 751 20, Uppsala, Sweden
cInstitut für Anorganische Chemie, Gerhard-Domagk-Straße 1, 53121 Bonn, Germany. E-mail: bismuto@uni-bonn.de
First published on 7th June 2023
Abstract
Boron-based dipyrrin chromophores (BODIPY) have found widespread application over the last twenty years in fields as diverse as medicine and materials. Thus, several efforts have been placed to exchange boron with other elements, with the aim of developing materials with complementary luminescent properties. However, despite these attempts, the incorporation of other main-group elements in dipyrrin scaffolds remains still rare. We have successfully synthesized and characterized novel chromophores based on antimony and bismuth, SBDIPY and BIDIPY. Solution stabilities have been investigated by VT-UV/vis spectroscopy and the fluorescence emission studied and supported by computational analysis. We were also able to isolate the first direct analogue of BODIPY containing fluoride handles, disclosing preliminary luminescent features.
The luminescent properties of light main-group elements have been long investigated with great advances in the development of materials with optoelectronic properties.1 In particular, the strong rigidity imposed by the boron atom provides crucial features for the design of potent luminescent materials, limiting the decay thorough non-radiative pathways.2 Among those, BODIPY chromophores have become ubiquitous in several fields besides chemistry.3 The reason for their success can be attributed to the stability and the simple synthetic routes to access them, which allow for a fine tuning of the luminescent properties, reaching a range of absorbance wavelengths exceeding 100 nanometers (Fig. 1A).4 BODIPY chromophores have thus found a large number of applications as labelling reagents,5 photocatalysts,6 chemosensors,7 materials and many others.8
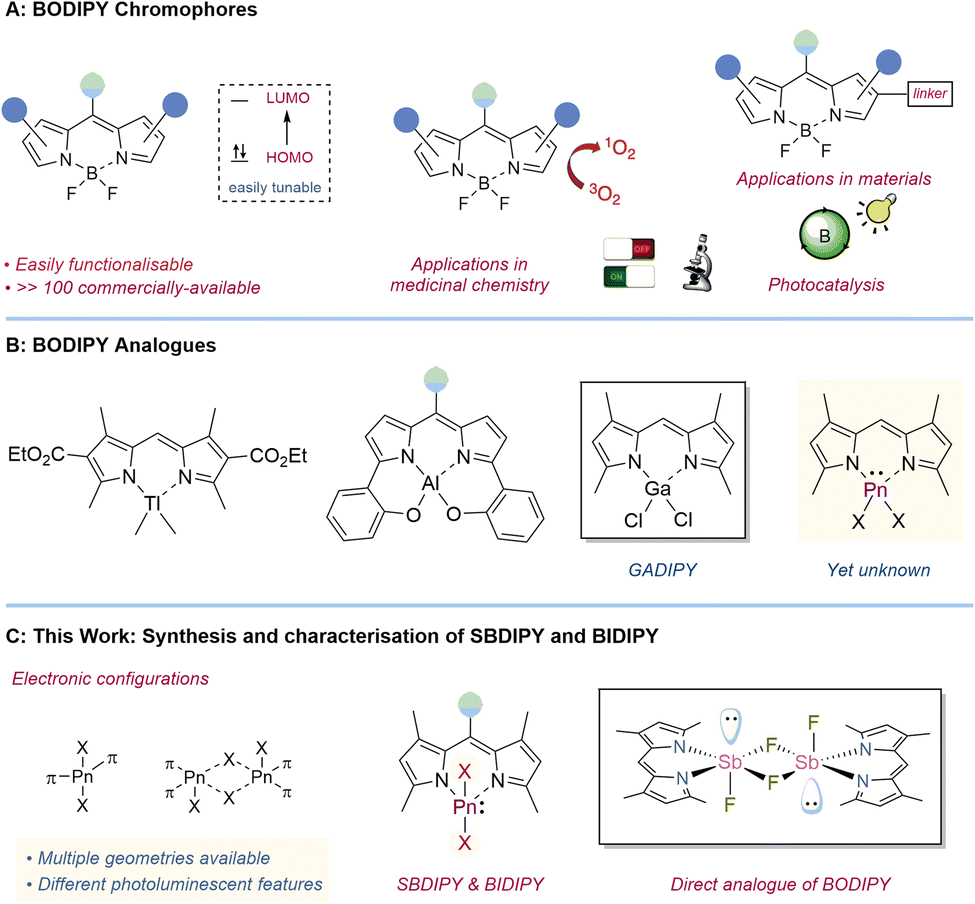 | ||
| Fig. 1 Context of the work. (A) BODIPY chromophores (B) previous heavier BODIPY analogues. (C) Synthesis and characterisation of SBDIPY and BIDIPY. | ||
While the combination of dipyrrins and boron was key to the broad success of BODIPY, the corresponding analogues containing other main-group elements are comparably underexplored.9 A rare example of a monomeric, heteroleptic dipyrromethene complex was obtained using dimethyl thallium chloride (Fig. 1B).10 Attempts with other elements in the group led to poly-substituted dipyrromethene adducts showcasing the challenges in isolating direct BODIPY analogues.11 A modification in the α position gave access to the N2O2-dipyrrin scaffolds which allowed for the isolation of several single-ligated main-group chromophores, such as aluminium and silicon ones.12 Despite continuous efforts, the first examples of a heavy main-group element analogue of BODIPY, containing chloride functionalities, were only reported three years ago by Smith and co-workers.13 They synthesized and characterized GADIPY (based on Ga) and provided preliminary data on the cationic PHODIPY (based on P(III)). Although the synthesis of the group XIII analogue proceeded smoothly, accessing the corresponding group XV derivative was synthetically more demanding. Presumably this can be partially attributed to the cationic nature of this species promoting deleterious side reactions, and the more contorted geometry caused by the extra lone pair. Preliminary photophysical investigations showed an unusual hypsochromic shift in the PHODIPY absorption (λabs = 468 nm) compared to that of previously reported P(V) dipyrromethene systems14 and BODIPY fluorophores (λabs = 500 nm), where the first π → π* transition is ligand centred. We thus wondered whether this effect would be even more prominent with heavier pnictogens. It is indeed well-known that the incorporation of a heavy atom in a chromophore scaffold can have a dramatic effect on the luminescent and optoelectronic properties.15,16 Unfortunately, this study is hampered by the lack of synthetic protocols to isolate Sb and Bi analogues.17
Here, we report the synthesis and characterisation of the first examples of SBDIPY and BIDIPY and a thorough study on the stability and on the luminescent properties. Tuning of the halogen handles has provided access to the first direct analogue of BODIPY containing fluoro substituents (Fig. 1C).
We began our investigation using the archetypal dipyrrin lithium complex for our synthesis.18 Upon addition of it to a pre-cooled solution of SbCl3 in THF, the formation of a crystalline solid was observed, giving SBDIPY-1-Cl in excellent yield (Fig. 2). The identity of this compound was confirmed by single-crystal X-ray analysis (Fig. 3) and 1H, 13C NMR spectroscopy. A modification of this protocol led to the isolation of further two meso-substituted analogues SBDIPY-2-Cl and SBDIPY-3-Cl in 80% and 67% yields, respectively.
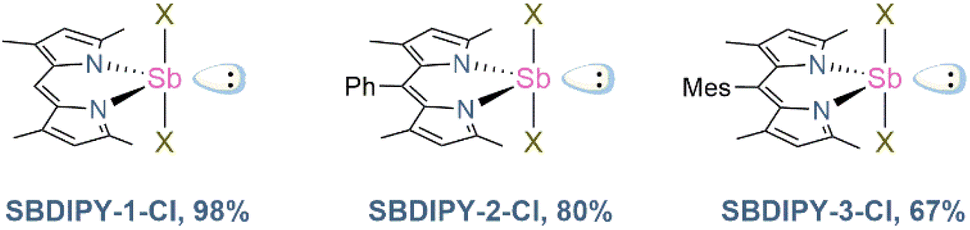 | ||
| Fig. 2 Isolated SBDIPY complexes. | ||
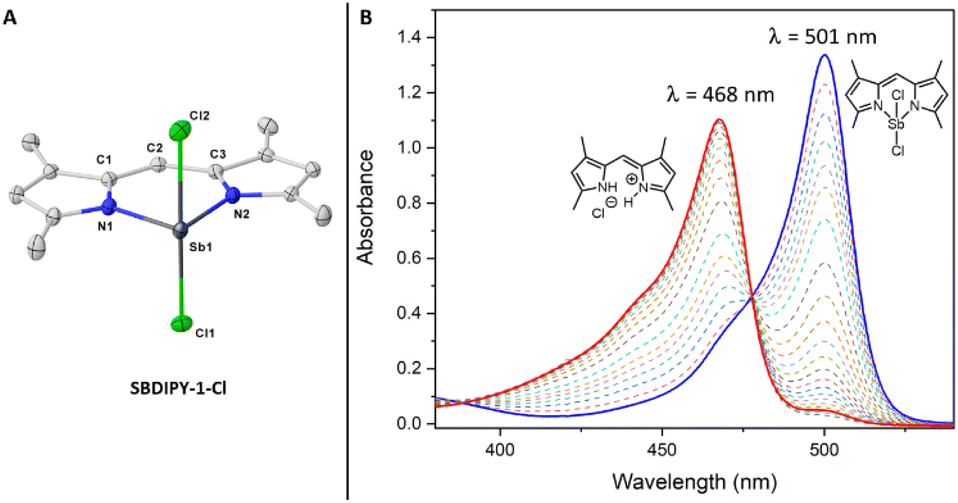 | ||
| Fig. 3 (A) Single crystal X-ray structure of SBDIPY-1-Cl, displacement ellipsoids are drawn at 50% probability level; hydrogen atoms are omitted for clarity. (B) UV/vis absorbance spectra over time at 40 °C (marked in blue the starting measurement and in red the final one). | ||
In contrast to BODIPY, the presence of the lone pair on the pnictogen atom imposes a completely different coordination environment with the Sb atom adopting a distorted pseudo-trigonal bipyramidal geometry. The immediate consequence of the longer N–Sb bonds is the formation of an irregular six-membered ring when compared to that of BODIPY (for SBDIPY N(1)–Sb–N(2) = 88.2°(1), for BODIPY N(1)–B–N(2) = 107.1°).19 Both boron and antimony lie in a near planar conformation with the ligand; however, in the latter case the halogen atoms are placed in an apical position with only a small distortion away from 180° (Fig. 3A). The geometry and structural parameters, including the Sb–Cl and Sb–N bond lengths and angles, across all the SBDIPY complexes differ only slightly, indicating very little effect upon modification of the meso position in the solid-state.20
We next focused on investigating the luminescent properties of these complexes in solution using absorption and emission spectroscopies. The absorption spectrum of SBDIPY-1-Cl displayed a band (λabs = 501 nm, CH2Cl2) with shape and values comparable to the one of BODIPY, with a small hypsochromically shifted shoulder (ca. 470 nm), usually identified as a vibronic transition.21 However, upon evaluating other solvents, a new blue-shifted band became more pronounced, suggesting a potentially different phenomenon. VT-UV/vis was used to elucidate the origin of this effect (see ESI†). Interestingly, measuring the kinetics at T = 40 °C, we observed a more rapid decrease over time of the band at λabs = 501 nm with the concomitant formation of a new band at λabs = 468 nm (Fig. 3B), near the vibronic shoulder of SBDIPY-1-Cl, creating ambiguity in the assignment of the principal transition.13
Our results suggest that the raising band could be associated with the formation of a decomposition product, with spectral properties similar to the dipyrrin ligand. We were able to crystallize the species responsible for the band and identified it as the hydrochloride salt of the dipyrrin scaffold 1 which features no appreciable fluorescence emission.22 The low temperature UV/vis analysis revealed SBDIPY-1-Cl to be more stable at 5 °C (see ESI,† spectroscopic studies section), thus we collected fluorescence emission spectra at this temperature which displayed a similar shape to the one of BODIPY and a maximum of emission at λem = 516 nm with a Stokes shift of 580 cm−1. At this temperature we were also able to calculate the molar absorption coefficient (ε = 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 460 M−1 cm−1) and the photoluminescence quantum yield (ϕ = 0.03), which resulted to be in line with previously synthesised BODIPY analogues.12
460 M−1 cm−1) and the photoluminescence quantum yield (ϕ = 0.03), which resulted to be in line with previously synthesised BODIPY analogues.12
We then wondered whether the addition of the substituent in meso position would influence the stability of the complex. However, as observed for the solid-state, UV/vis analysis confirmed very little influence of the meso substituent on the stability of these compounds. The initial absorbance at λabs = 501 nm for SBDIPY-2-Cl and SBDIPY-3-Cl decreased with the concurrent formation of a second band over time at λabs = 486 nm and λabs = 480 nm, hinting to a similar decomposition event into the corresponding dipyrrin byproducts. The intensity of the fluorescence emission of SBDIPY-2-Cl is considerably lower than that of SBDIPY-1-Cl, due to the nonradiative decay of the excited state attributed to the freely rotating phenyl ring, a phenomenon already observed with BODIPY.23 In contrast, the addition of methyl groups in the ortho positions of the phenyl group led to a comparable emission intensity to that of SBDIPY-1-Cl but with a slightly larger Stokes shift (840 cm−1). This contrasts with what was previously observed for PHODIPY, where the absorbance band was found at much lower wavelengths than of BODIPY and featuring a comparably larger Stokes shift.13
Intrigued by this discrepancy, we computed the first electronic transition of the different substituted dipyrrin derivatives at the SCS-CIS(D)/def2-TZVPP//r2SCAN-3c level (Fig. 4).24 As expected from our experimental evidence, the predicted main transition is qualitatively and quantitatively similar to the π → π* of the fluorinated BODIPY, both in nature and energy (see computational analysis section, ESI†). Despite the introduction of the heavy atom, we found the central atom and the halogens to have only a minimal influence on the character of the transition, as can be seen by modifying the BF2 group with SbF2 (SBDIPY-1-F), SbCl2 (SBDIPY-1-Cl) or BiCl2 (BIDIPY-1-Cl). Our calculations, which take into consideration also solvent effects, are in agreement with the one previously reported,13 where no significant differences were found between BODIPY, GADIPY and PHODIPY in their computed transition. Based on this evidence, we believed that the replacement of boron with a heavier analogue does not lead to a significant change in the Stokes shift but can potentially result in unexpected decomposition events.
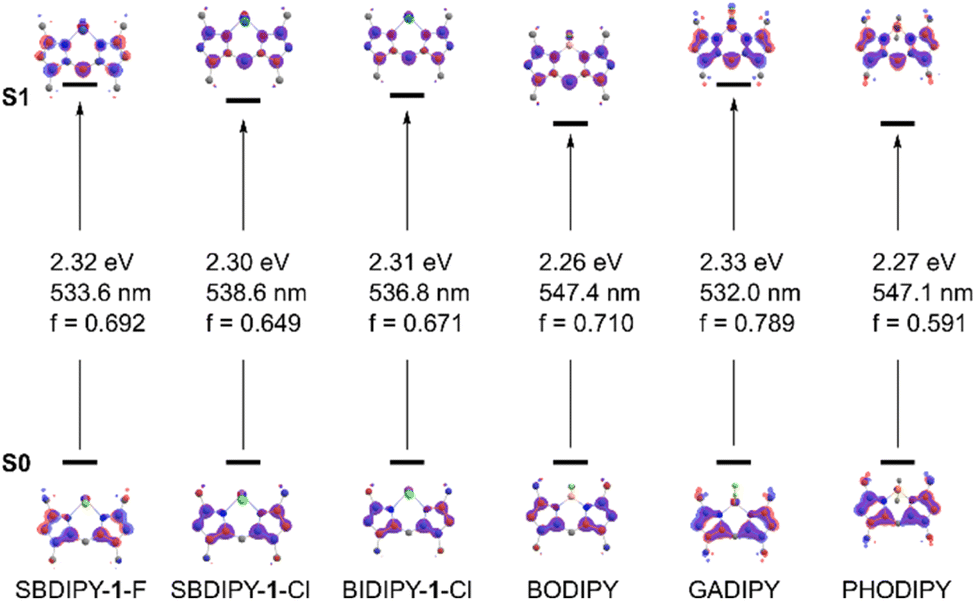 | ||
| Fig. 4 Computed first electronic transition of different dipyrrin derivatives at the CPCM-SCS-CIS(D)/def2-TZVPP//r2SCAN-3c level. The Natural Transition Orbitals involved in the main π → π* excitations are displayed together with the predicted energies and oscillator strengths. | ||
Due to the potential formation of dimers or higher order oligomers via halogen bridging, we evaluated experimentally the effect of tuning the halide handles. Using SbBr3 as the metal precursor, we were able to isolate the corresponding SBDIPY-1-Br in 91% yield. Single-crystal X-ray analysis showed a geometry comparable to SBDIPY-1-Cl, however, the Sb was observed to bend considerably out of plane due to the elongated Sb–halogen bonds (Fig. 5A, see ESI† for further details). The VT-UV/vis analysis showed enhanced stability with a longer half-life (140 min vs. 600 min, see ESI†) compared to that of SBDIPY-1-Cl.
 | ||
| Fig. 5 (A) Single crystal X-ray of complex SBDIPY-1-Br; (B) SBDIPY-1-F. Displacement ellipsoids are drawn at 50% probability level; hydrogen atoms are omitted for clarity. | ||
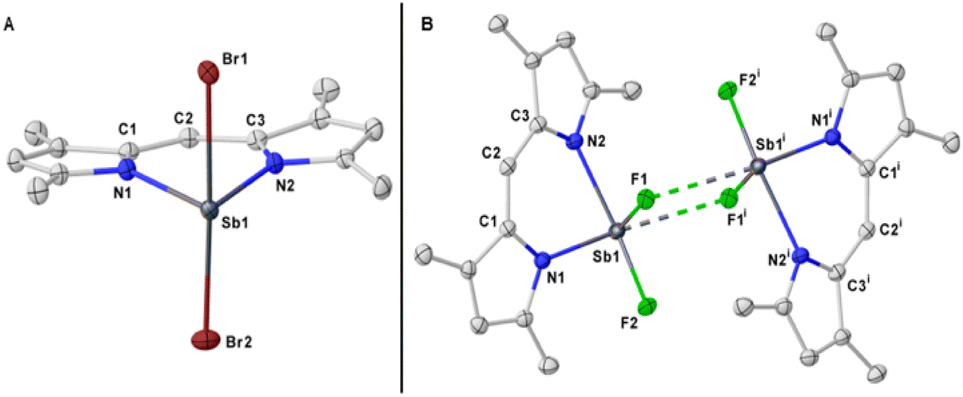
To determine whether the opposite effect would occur with a smaller halogen, we set to synthesize the yet unknown fluorinated analogue of BODIPY. To the best of our knowledge, no fluorinated version of BODIPY with other main-group elements is known in the literature. Upon mixing a solution of the lithium salt of the simple dipyrrin at −35 °C with a solution of SbF3 in THF led to formation of a crystalline material in 94% yield. The solid-state structure revealed the SBDIPY-1-F to adopt a completely different geometry to that of SBDIPY-1-Cl with the antimony arranged in a distorted pseudo-octahedral fashion (Fig. 5B). Bridging fluorides formed a dimeric structure and accounted for the octahedral geometry. We observed strikingly different Sb–N bonds (Sb(1)–N(1) = 2.098 (1) Å, Sb(1)–N(2) = 2.249 (1) Å) potentially due to a stronger trans-influence of the fluoride. Furthermore, the Sb atom is more bent out of plane than that of SBDIPY-1-Cl (see ESI,† for further details).
To further elucidate whether the distorted geometry observed in the solid-state structure was only a consequence of crystal packing, we computed SBDIPY-1-Cl and SBDIPY-1-F at the r2SCAN-3c level both in the gas and solvent model. In SBDIPY-1-Cl the difference in Gibbs free energy between the linear and the distorted geometry is 10 kcal mol−1 favouring the linear. Instead for SBDIPY-1-F it becomes only 3 kcal mol−1, indicating that the difference in the X-ray structure could not only arise from crystal packing but also from an increased trans-influence (the DOI to the figshare repository of the computed structures can be found in the ESI†).
As predicted, the stability of SBDIPY-1-F in solution was very limited with only one band present at λabs = 468 nm (17 μM), which is most likely the decomposition product. Upon increasing the concentration (84 μM) and measuring at −10 °C, we were able to observe an absorbance band at λabs = 496 nm which resulted in a fluorescence emission at λem = 522 nm. At the current stage, we cannot confidently discern whether the fluorescence band is caused by an aggregation-induced phenomenon and, thus, by the dimer/oligomer or due to the increased stability of the complex at higher concentrations. In order to further investigate the aggregation state in solution, we have performed a VT 19F-NMR analysis where we observed a splitting of the fluorine resonance which supports a dimer or a higher aggregate state (Fig. 36, ESI†). However, the computed structure of dimeric SBDIPY-1-F in toluene (CPCM) 9.1 kcal mol−1 less stable than two separated optimised monomers, indicating a preferred monomeric form. Thus, we hypothesise that the aggregation of these compounds is highly dependent on the concertation and on the temperature.
The preliminary structural and spectroscopic data gathered, set the ground for discovering novel pnictogen-based direct analogues of BODIPY, a completely unknown field. As last functionalisation of the halogen handles, we wondered whether we could exchange them with a weakly-coordinating counterion such as NaBArF24. Upon mixing a solution of SBDIPY-1-Cl and the salt in CH2Cl2, we observed a net colour change from orange to bright purple (Fig. 6A). The reaction was completed in 1 hour, leading to SBDIPY-1+-Cl in 86% yield, as confirmed by single-crystal X-ray analysis (Fig. 6B). Crystals were grown from a saturated CH2Cl2/benzene mixture, with a strong coordination of the aromatic solvent to stabilize the cationic charge. The distance from the centroid is 3.212(1) Å with the shortest Sb–C distance of 3.250(4) and the longest of 3.702(5) Å, hinting a potential asymmetric coordination best described as η3. The cationic complex features a bathochromic shift in solution compared to the SBDIPYs and a smaller Stokes shift (λabs = 513 nm, 188 cm−1).
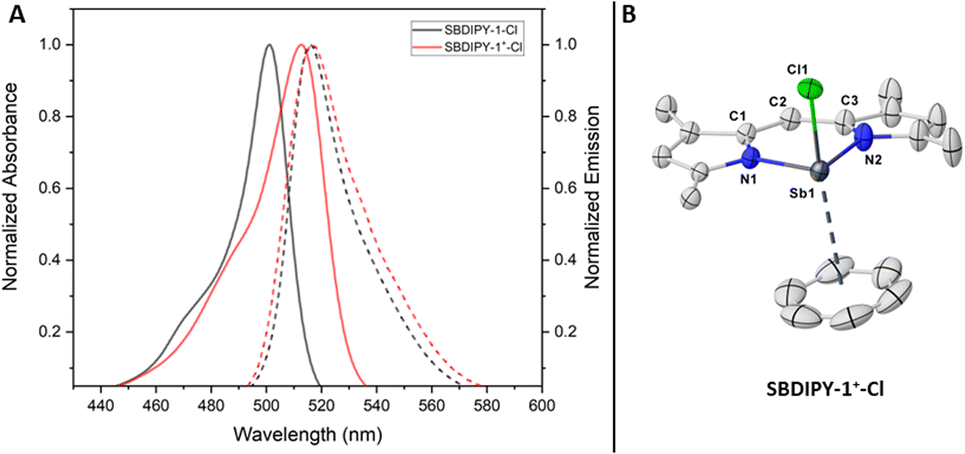 | ||
| Fig. 6 Characterisation of the differently substituted halo SBDIPY. (A) Comparison of absorption and emission profile of SBDIPY-1-Cl (black) and SBDIPY-1+-Cl (red); (B) single crystal X-ray of complex SBDIPY-1+-Cl; displacement ellipsoids are drawn at 50% probability level; hydrogens atoms are omitted for clarity. | ||
Once we had successfully synthesised and characterised the SBDIPY complexes, we were curious to see the effect of changing the Sb atom with the heaviest stable pnictogen, bismuth. We envisaged that the strong electrophilicity, and the relativistic effect,25 may lead to materials with completely different properties both in solution- and in solid-state. Unfortunately, the previously developed synthetic procedure did not lead to any Bi incorporation. However, using sodium as a counterion led to a smoother transmetallation reaction. Addition of BiCl3 to a solution of the sodium dipyrrinato salt gave immediate formation of a red crystalline material in 94% yield (Fig. 7). This protocol was then successfully applied to the Ph and Mes-substituted scaffolds, giving complexes BIDIPY-2-Cl and -3-Cl in 70% and 80% yield, respectively.
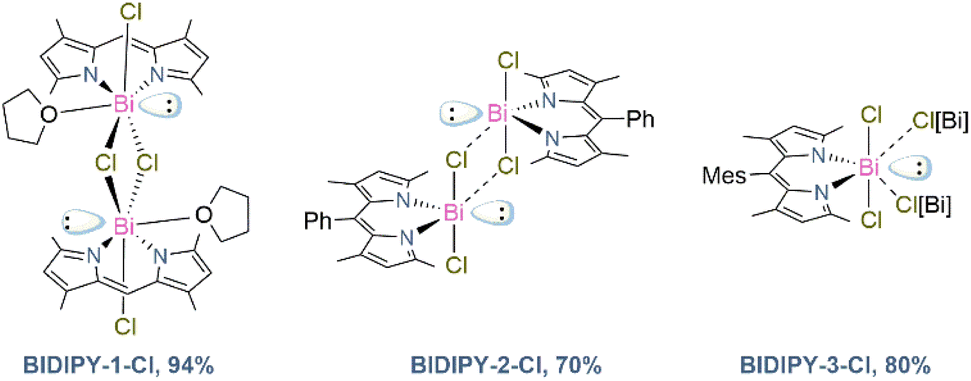 | ||
| Fig. 7 Isolated BIDIPY chromophores (dotted lines show halogen interactions in the solid-state). | ||
Single-crystal X-ray analysis confirmed the product to be the first example of BIDIPY (Fig. 8A). The structure featured an even more irregular six-membered ring when compared to that of BODIPY (for BIDIPY N(1)–Bi–N(2) = 81.5(1)°, for BODIPY N(1)–B–N(2) = 107.1°) with the Bi bent substantially out of the plane due to the bigger size of the pnictogen. In contrast to what was observed with SBDIPY-1-Cl, BIDIPY-1-Cl is a dimer with two bridging chlorides connecting two bismuth centers.
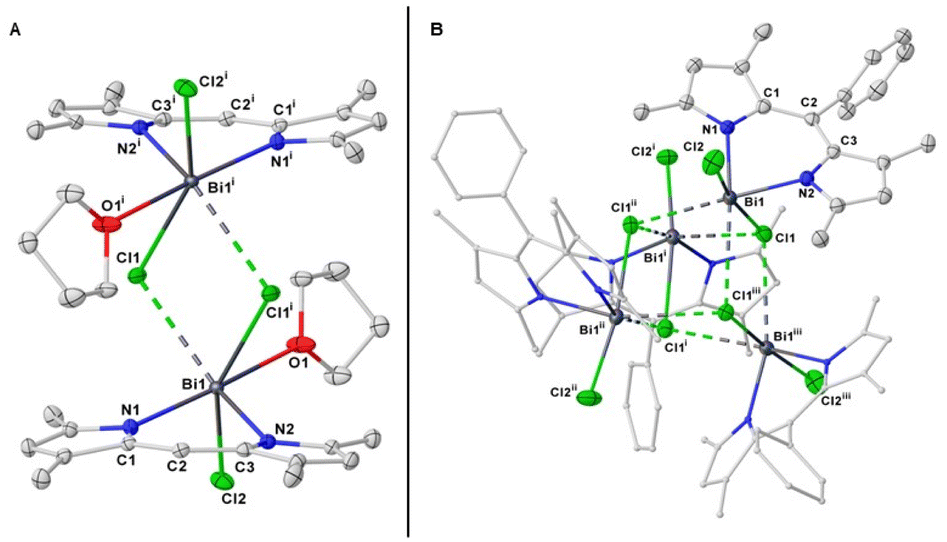 | ||
| Fig. 8 Single crystal X-ray of complex BIDIPY-1-Cl (A), and 2-Cl (B). Displacement ellipsoids are drawn at 50% probability level; hydrogen atoms are omitted for clarity. | ||
The distance of the non-bridging chloride is considerably shorter (Bi(1)–Cl(2) = 2.580(1) Å) compared to that of the two bridging and non-equivalent (2.826(1) and 3.046(1) Å). The bismuth atom is arranged in a distorted pseudo-prismatic geometry with one molecule of THF occupying one of the apical positions. Compared to the SBDIPYs, the introduction of an aryl-substituent in meso position had an important effect on the agglomeration of the solid-state structure.
BIDIPY-2-Cl featured a T-shaped Bi atom similar to SBDIPY-1-Cl, however when considering the short contact with the neighbouring chloride atoms, this can be best described as a tetramer ascribed to the Td point group (Fig. 8B). BIDIPY-3-Cl presented a similar arrangement as BIDIPY-1-Cl, but with all the chlorides bridging to the next bismuth in a polymeric arrangement (see ESI†). Due to a packing effect, all the isolated bismuth complexes feature low solubility even in tetrahydrofuran. The increased electrophilic character of the pnictogen is reflected in the higher coordination numbers found in the solid-state structures.
Overall, all the presented X-ray structures show differences in geometry when compared to the analogues recently reported by Liu, Su and co-workers, most likely due to the additional donation provided by the coordinated solvent (see ESI† for further details).17
Akin to the antimony complexes, we performed UV/vis and fluorescence analysis of the Bi complexes in solution. The increased size and the higher electrophilicity led to an even higher instability in dilute solutions. Even at lower temperatures, we observed significant amounts of decomposition for all the BIDIPY complexes by UV/vis spectroscopy. All the bismuth complexes featured a slight bathochromic shift (λabs = 504 nm, λabs = 507 nm, λabs = 506 nm). The fluorescence emission is significantly less intense than that of SBDIPY with an extreme case in BIDIPY-1-Cl where it seems completely switched-off. However, at this stage we cannot rule out whether that is related to the heavy atom effect or to any quenching effect of the more pronounced decomposition product. All the absorbance and emission bands for SBDIPY-1 and BIDIPY-1 complexes are summarized in Table 1. The absorbance and the emission values present a hypsochromic shift compared to those recently reported by Liu, Su and co-workers most likely due to the change in the ligand scaffold.17
| Complex | Absorption maximum λabsmax (nm) | Emission maximum λemmax (nm) | Stokes shift (cm−1) | Quantum yield |
|---|---|---|---|---|
| SBDIPY-1-Cl | 501 | 516 | 580 | 0.025 |
| SBDIPY-1-Br | 504 | 518 | 536 | 0.004 |
| SBDIPY-1-F | 496 | 522 | 1004 | n.a. |
| SBDIPY-1-Cl | 501 | 520 | 729 | n.a. |
| SBDIPY-3-Cl | 501 | 523 | 840 | 0.011 |
| SBDIPY-1+-Cl | 513 | 518 | 188 | n.a. |
| BIDIPY-1-Cl | 505 | n.a. | n.a. | n.a. |
| BIDIPY-2-Cl | 506 | n.a. | n.a. | n.a. |
| BIDIPY-3-Cl | 506 | 535 | 1071 | n.a. |
To conclude, we have disclosed the synthesis of yet unknown heavier group XV analogues of BODIPY, unlocking the path to SBDIPY and BIDIPY. The compounds displayed similar luminescence properties, however with notably lower stability, especially for bismuth, potentially due to the increased size and weaker Pn–N bonds.
Preliminary modifications showed a possible increase of stability by tuning the halogen handles and allowed for the isolation of the first direct analogue of pnictogen-based BODIPY. Further investigations will be carried out to fully explore the luminescence features of SBDIPY-1-F and the discovered compounds to guide the design of more stable chromophores.
Data availability
All the experimental procedures, the spectroscopic data, the computational studies, and detailed crystallographic information can be found in the ESI.† Deposition numbers 2241590 (for SBDIPY-1-Cl), 2241588 (SBDIPY-2-Cl), 2241584 (for SBDIPY-3-Cl), 2241583 (for SBDIPY-1-Br), 2241582 (for SBDIPY-1-F), 2241589 (for SBDIPY-1-Cl+), 2241585 (BIDIPY-1-Cl), 2241586 (for BIDIPY-2-Cl), 2241587 (BIDIPY-3-Cl) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.Author contributions
A. K. and A. B. conceived the idea and performed the experimental work, S. C. performed the computational analysis, C. G. performed the X-ray measurements and structure refinements. A. B. drafted the manuscript. All the author contributed with comments, revising the manuscript and all have approved the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NMR and X-ray facilities of the University of Göttingen for technical assistance. All the authors thank Prof. Dr M. Alcarazo for the continuous support and thoughtful discussion. A. B. thanks the University of Bonn and A. B. and A. K. thank the Fonds der Chemischen Industrie (FCI) for financial support (Liebig scholarship to A. B. 208/06 and PhD stipend for A. K.). S. C. thanks the Swedish Vetenskapsrådet for a Starting Grant (2021-05414) and the Swedish National Infrastructure for Computations (SNIC) for a Small Compute Grant (SNIC 2022/22-525).Notes and references
- (a) M. P. Duffy, P.-A. Bouit, and M. Hissler, in Main Gr. Strateg. Towar. Funct. Hybrid Mater., John Wiley & Sons, Ltd, Chichester, UK, 2018, pp. 295–327 Search PubMed; (b) K. Strakova, L. Assies, A. Goujon, F. Piazzolla, H. V. Humeniuk and S. Matile, Chem. Rev., 2019, 119, 10977–11005 CrossRef CAS PubMed; (c) S. M. Parke, M. P. Boone and E. Rivard, Chem. Commun., 2016, 52, 9485–9505 RSC.
- (a) C. Suksai and T. Tuntulani, Chem. Soc. Rev., 2003, 32, 192 RSC; (b) L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846–863 RSC; (c) S. K. Mellerup and S. Wang, Trends Chem., 2019, 1, 77–89 CrossRef CAS.
- Y. S. Marfin, A. V. Solomonov, A. S. Timin and E. V. Rumyantsev, Curr. Med. Chem., 2017, 24, 2745–2772 CrossRef CAS PubMed.
- (a) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed; (b) T. Rohand, M. Baruah, W. Qin, N. Boens and W. Dehaen, Chem. Commun., 2006, 266–268 RSC; (c) R. G. Clarke, and M. J. Hall, in Adv. Heterocycl. Chem., Elsevier Inc., 2019, pp. 181–261 Search PubMed; (d) T. Rohand, M. Baruah, W. Qin, N. Boens and W. Dehaen, Chem. Commun., 2006, 266–268 RSC.
- (a) J. Xu, L. Zhu, Q. Wang, L. Zeng, X. Hu, B. Fu and Z. Sun, Tetrahedron, 2014, 70, 5800–5805 CrossRef CAS; (b) M. R. Momeni and A. Brown, J. Phys. Chem. A, 2016, 120, 2550–2560 CrossRef CAS PubMed.
- P. De Bonfils, L. Péault, P. Nun and V. Coeffard, Eur. J. Org. Chem., 2021, 2021, 1809–1824 CrossRef CAS.
- B. Bertrand, K. Passador, C. Goze, F. Denat, E. Bodio and M. Salmain, Coord. Chem. Rev., 2018, 358, 108–124 CrossRef CAS.
- (a) J. Zhao, K. Xu, W. Yang, Z. Wang and F. Zhong, Chem. Soc. Rev., 2015, 44, 8904–8939 RSC; (b) E. V. Antina, E. V. Rumyantsev, N. A. Dudina, Y. S. Marfin and L. A. Antina, Russ. J. Gen. Chem., 2016, 86, 2209–2225 CrossRef CAS; (c) A. Atilgan, Y. Beldjoudi, J. Yu, K. O. Kirlikovali, J. A. Weber, J. Liu, D. Jung, P. Deria, T. Islamoglu, J. F. Stoddart, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2022, 14, 12596–12605 CrossRef CAS PubMed.
- (a) S. A. Baudron, Dalton Trans., 2013, 42, 7498–7509 RSC; (b) S. A. Baudron, Dalton Trans., 2020, 49, 6161–6175 RSC.
- A. Hsieh, C. Rogers and B. West, Aust. J. Chem., 1976, 29, 49 CrossRef CAS.
- R. Sharma, M. R. Rao and M. Ravikanth, Coord. Chem. Rev., 2017, 348, 92–120 CrossRef CAS.
- (a) M. Saikawa, M. Daicho, T. Nakamura, J. Uchida, M. Yamamura and T. Nabeshima, Chem. Commun., 2016, 52, 4014–4017 RSC; (b) N. Sakamoto, C. Ikeda, M. Yamamura and T. Nabeshima, J. Am. Chem. Soc., 2011, 133, 4726–4729 CrossRef CAS PubMed; (c) C. G. Gianopoulos, K. Kirschbaum and M. R. Mason, Organometallics, 2014, 33, 4503–4511 CrossRef CAS; (d) N. Sakamoto, C. Ikeda, M. Yamamura and T. Nabeshima, J. Am. Chem. Soc., 2011, 133, 4726–4729 CrossRef CAS PubMed; (e) C. Ikeda, S. Ueda and T. Nabeshima, Chem. Commun., 2009, 2544–2546 RSC; (f) M. Yamamura, H. Takizawa and T. Nabeshima, Org. Lett., 2015, 17, 3114–3117 CrossRef CAS PubMed; (g) V. S. Thoi, J. R. Stork, D. Magde and S. M. Cohen, Inorg. Chem., 2006, 45, 10688–10697 CrossRef CAS PubMed.
- W. Wan, M. S. Silva, C. D. McMillen, S. E. Creager and R. C. Smith, J. Am. Chem. Soc., 2019, 141, 8703–8707 CrossRef CAS PubMed.
- (a) X. D. Jiang, J. Zhao, D. Xi, H. Yu, J. Guan, S. Li, C. L. Sun and L. J. Xiao, Chem. – Eur. J., 2015, 21, 6079–6082 CrossRef CAS PubMed; (b) Y. S. Marfin, O. S. Vodyanova, S. D. Usoltsev, A. V. Kazak and E. V. Rumyantsev, Crystallogr. Rep., 2019, 64, 644–648 CrossRef CAS.
- (a) Y. P. Rey, D. G. Abradelo, N. Santschi, C. A. Strassert and R. Gilmour, Eur. J. Org. Chem., 2017, 15, 2170–2178 CrossRef; (b) M. A. Filatov, Org. Biomol. Chem., 2020, 18, 10–27 RSC.
- (a) I.-S. Ke, M. Myahkostupov, F. N. Castellano and F. P. Gabbaï, J. Am. Chem. Soc., 2012, 134, 15309–15311 CrossRef CAS PubMed; (b) J. S. Jones and F. P. Gabbaï, Acc. Chem. Res., 2016, 49, 857–867 CrossRef CAS PubMed; (c) H. Yang and F. P. Gabbaï, J. Am. Chem. Soc., 2015, 137, 13425–13432 CrossRef CAS PubMed; (d) G. R. Kumar, M. Yang, B. Zhou and F. P. Gabbaï, Mendeleev Commun., 2022, 32, 66–67 CrossRef CAS; (e) G. Park, D. J. Brock, J.-P. Pellois and F. P. Gabbaï, Chem, 2019, 5, 2215–2227 CrossRef CAS PubMed.
- During the final stage of the manuscript, a complementary approach for the synthesis of heavy pnictogen analogues of BODIPY with a bulky ligand was reported by C. Liu, Y. Dai, Q. Han, C. Liu and Y. Su, Chem. Commun., 2023, 59, 2161–2164 RSC.
- (a) J. Cipot-Wechsler, A. A.-S. Ali, E. E. Chapman, T. S. Cameron and A. Thompson, Inorg. Chem., 2007, 46, 10947–10949 CrossRef CAS PubMed; (b) T. E. Wood and A. Thompson, Chem. Rev., 2007, 107, 1831–1861 CrossRef CAS PubMed.
- R. Bandichhor, C. Thivierge, N. S. P. Bhuvanesh and K. Burgess, Acta Crystallogr., 2006, E62, o4310–o4311 Search PubMed.
- Deposition numbers 2241590 (for SBDIPY-1-Cl), 2241588 (SBDIPY-2-Cl), 2241584 (for SBDIPY-3-Cl), 2241583 (for SBDIPY-1-Br), 2241582 (for SBDIPY-1-F), 2241589 (for SBDIPY-1-Cl+), 2241585 (BIDIPY-1-Cl), 2241586 (for BIDIPY-2-Cl), 2241587 (BIDIPY-3-Cl) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- S. Chibani, B. Le Guennic, A. D. Laurent and D. Jacquemin, Chem. Sci., 2013, 4, 1950–1963 RSC.
- J. L. Sessler, L. R. Eller, W.-S. Cho, S. Nicolaou, A. Aguilar, J. T. Lee, V. M. Lynch and D. J. Magda, Angew. Chem., Int. Ed., 2005, 44, 5989–5992 CrossRef CAS PubMed.
- A. Orte, E. Debroye, M. J. Ruedas-Rama, E. Garcia-Fernandez, D. Robinson, L. Crovetto, E. M. Talavera, J. M. Alvarez-Pez, V. Leen, B. Verbelen, L. Cunha Dias de Rezende, W. Dehaen, J. Hofkens, M. Van der Auweraer and N. Boens, RSC Adv., 2016, 6, 102899–102913 RSC.
- S. Chibani, A. D. Laurent, B. Le Guennic and D. Jacquemin, J. Chem. Theory Comput., 2014, 10, 4574–4582 CrossRef CAS PubMed.
- (a) S. C. Gangadharappa, I. Maisuls, D. A. Schwab, J. Kösters, N. L. Doltsinis and C. A. Strassert, J. Am. Chem. Soc., 2020, 142, 21353–21367 CrossRef CAS PubMed; (b) L. de Azevedo Santos, T. A. Hamlin, T. C. Ramalho and F. M. Bickelhaupt, Phys. Chem. Chem. Phys., 2021, 23, 13842–13852 RSC; (c) J. M. Lipshultz, G. Li and A. T. Radosevich, J. Am. Chem. Soc., 2021, 143, 1699–1721 CrossRef CAS PubMed; (d) H. W. Moon and J. Cornella, ACS Catal., 2022, 12, 1382–1393 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, spectroscopic studies, computational analysis and crystal structures. CCDC 2241582–2241590. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01493b |
| This journal is © The Royal Society of Chemistry 2023 |