
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium-catalyzed intermolecular alkene–alkyne couplings in biologically relevant media†
Alejandro
Gutiérrez-González
 a,
Daniel
Marcos-Atanes
a,
Leonard G.
Cool
a,
Fernando
López
*ab and
José L.
Mascareñas
*a
a,
Daniel
Marcos-Atanes
a,
Leonard G.
Cool
a,
Fernando
López
*ab and
José L.
Mascareñas
*a
aCentro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS), Departamento de Química Orgánica, Universidad de Santiago de Compostela, 15782, Santiago de Compostela, Spain. E-mail: joseluis.mascarenas@usc.es
bMisión Biológica de Galicia (MBG), Consejo Superior de Investigaciones Científicas (CSIC), 36080, Pontevedra, Spain. E-mail: fernando.lopez@csic.es
First published on 19th May 2023
Abstract
Cationic cyclopentadienyl Ru(II) catalysts can efficiently promote mild intermolecular alkyne–alkene couplings in aqueous media, even in the presence of different biomolecular components, and in complex media like DMEM. The method can also be used for the derivatization of amino acids and peptides, therefore proposing a new way to label biomolecules with external tags. This C–C bond-forming reaction, based on simple alkene and alkyne reactants, can now be added to the toolbox of bioorthogonal reactions promoted by transition metal catalysts.
Introduction
The development of bioorthogonal reactions has brought a paradigm shift in the potential of synthetic chemistry to impact the fields of chemical biology and biomedicine.1 Within the “toolbox” of bioorthogonal reactions, those involving transition metal catalysis are particularly appealing, as they avoid the need of strained reactants and benefit from the versatility and tuning possibilities of the transition metal reagents.2 Progress in this area has been sluggish, mostly because of the established notion that transition metal catalysts are not compatible with aqueous and biological milieu. However, recent years have witnessed a substantial growth of the field, especially in the development of uncaging reactions entailing bond-breaking processes, such as the removal of N-alloc groups.3Related bioorthogonal reactions involving bond-forming processes are much less common and, so far, mainly restricted to the construction of carbon–heteroatom bonds, especially using Click-like cycloadditions.4 Accordingly, the development of transition metal catalyzed reactions that forge carbon–carbon bonds in biorelevant media has clearly lagged behind. Most reported examples consist of Suzuki and related C–C cross-couplings promoted by palladium catalysts.5 A handful of other reactions that form C–C bonds in biological settings, including gold-promoted cyclizations or ruthenium-catalyzed metathesis, have also been sporadically described.6,7 In this context, we have recently reported a ruthenium catalyzed carbon–carbon bond-forming reaction using alkynes as reaction partners. Specifically, we demonstrated the viability of carrying out formal (2 + 2 + 2) annulation between diynes and alkynes, under biologically relevant environments, using CpRu-based catalysts (Fig. 1a).6e The excellent bioorthogonality of the reaction stems from the absence of alkyne functional groups in native biomolecules, and from the intrinsic metal chelating effect of tethered 1,n-diynes, which are well posed to generate the required ruthenacyclic intermediates I (Fig. 1a).
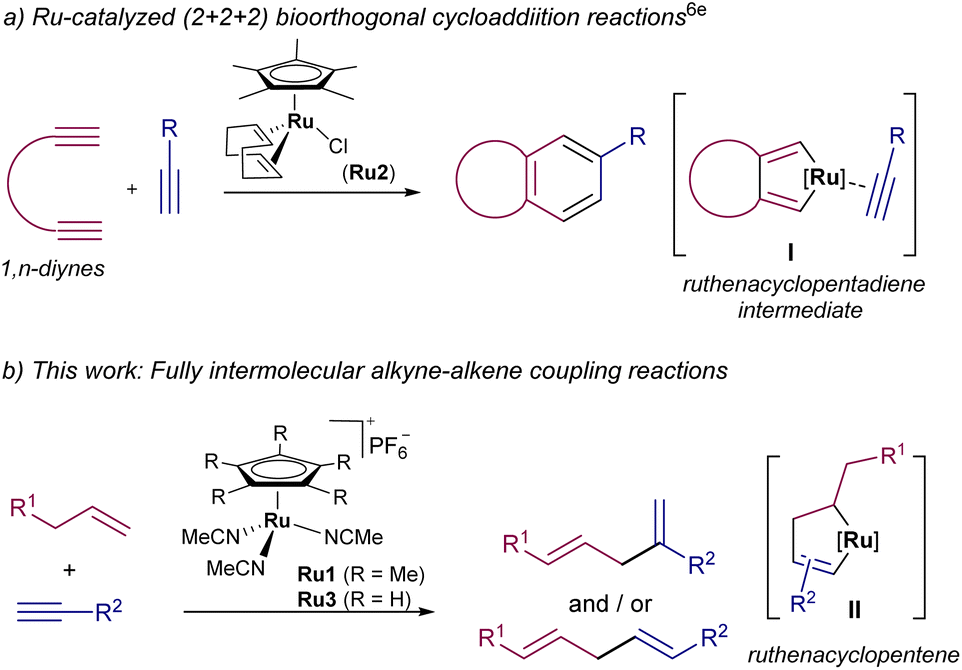 | ||
| Fig. 1 Ru-catalyzed biocompatible C–C bond forming processes involving ruthenacyclic intermediates. | ||
We next questioned whether simpler, monounsaturated alkyne or alkene precursors could also be used for fully intermolecular C–C ligation reactions in aqueous and biological buffers. Towards this aim, we paid attention to the ruthenium-promoted cross-coupling between alkenes and alkynes, an Alder-ene type of process that proceeds via ruthenacyclopentane intermediates of type II (Fig. 1b).8 Although the reaction has been widely used in synthetic chemistry, in organic solvents, it has also proven compatible with protic solvents and with small amounts of water,8c which encouraged us to explore the viability of this C–C bond forming reaction in biorelevant aqueous media.9 In addition to the intrinsic challenges of biorthogonality and aqueous compatibility, the reaction may also present chemo- and regioselectivity issues. However, in case of success, it would be a valuable new ligation tool in chemical biology, particularly considering the simplicity of the coupling partners, and the ease with which alkenes and alkynes can be directly incorporated into different types of biomolecules.10
Herein, we demonstrate the viability of the approach, by demonstrating that the intermolecular coupling between alkenes (1) and alkynes (2) to deliver 1,4-dienes (3 and or 3′) can be carried out in aqueous buffers or in complex media like DMEM, using the Ru(II) complex Ru1. Interestingly, the regioselectivity of the process [i.e. formation of branched (3) vs. linear (3′) isomers] can be controlled by appropriate selection of the catalyst and/or the type of reactants. Finally, we also show that the reaction can be used to selectively label peptides in water at low micromolar concentrations, which provides good prospects for its further use as bioconjugation tool.
Results and discussion
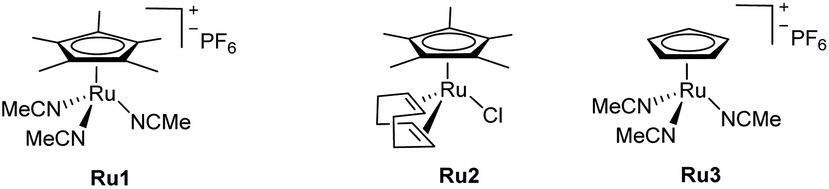
Our first experiments were carried out with the allyl ether 1a and the propargyl benzyl ether 2a as model substrates, using the cationic complex Ru1 as catalyst (10 mol%).8 In consonance with the reported precedents,8 we observed that the reaction could be performed in organic solvents, such as THF, acetone and CH2Cl2 (75 mM) although the yields of 3aa were modest, from 30 to 40% (Table 1, entries 1–3). The reaction can also be performed in water, with similar yields (entry 4), but more importantly, the incorporation of THF as co-solvent (water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF = 8:2) allowed to increase the yield up to a good 70% (entry 5). In all these cases, the regioselectivity was high, favouring the formation of the branched product 3aa, which was exclusively obtained as E-isomer.
:THF = 8:2) allowed to increase the yield up to a good 70% (entry 5). In all these cases, the regioselectivity was high, favouring the formation of the branched product 3aa, which was exclusively obtained as E-isomer.
|
|
|||||
|---|---|---|---|---|---|
| Entry | 2 | Solvent | [Ru] (x mol%) | Regio (3:3′) |
3, % yield |
a Conditions: alkene 1a (0.075 mmol), alkyne 2 (0.075 mmol), the degassed solvent (1.0 mL) and the [Ru] catalyst (x mol%) were stirred under N2 at 37 °C for 16 h, under otherwise noted. Yields and branched to linear (3:3′) ratios determined by 1H-NMR using dimethylsulfone as internal standard.
b 61% isolated yield.
c Carried out with non-degassed solvents.
d Carried out under air and non-degassed solvents.
e 33% isolated yield.
f 78% isolated yield.
|
|||||
| 1 | 2a | THF | Ru1, 10 | >9:1 |
3aa, 32 |
| 2 | 2a | Acetone | Ru1, 10 | >9:1 |
3aa, 30 |
| 3 | 2a | CH2Cl2 | Ru1, 10 | >9:1 |
3aa, 40 |
| 4 | 2a | H2O | Ru1, 10 | >9:1 |
3aa, 36 |
| 5 | 2a | H2O/THF (8:2) |
Ru1, 10 | >9:1 |
3aa, 70b |
| 6 | 2a | H2O/THF (8:2) |
Ru2, 10 | >9:1 |
3aa, 0 |
| 7 | 2a | H2O/THF (9:1) |
Ru1, 5 | >9:1 |
3aa, 56 |
| 8 | 2b | H2O/THF (8:2) |
Ru1, 5 | >9:1 |
3ab, 99 |
| 9 | 2b | H2O/THF (9:1) |
Ru1, 5 | >9:1 |
3ab, 68 |
| 10 | 2b | H2O/EtOH (8:2) |
Ru1, 5 | 5:1 |
3ab, 88 |
| 11 | 2b | H2O/tBuOH (8:2) |
Ru1, 5 | >9:1 |
3ab, 77 |
| 12 | 2b | H2O/DMSO (8:2) |
Ru1, 5 | >9:1 |
3ab, 45 |
| 13 | 2b | H2O | Ru1, 10 | >9:1 |
3ab, 53 |
| 14c | 2b | H2O/THF (8:2) |
Ru1, 10 | >9:1 |
3ab, 97 |
| 15d | 2b | H2O/THF (8:2) |
Ru1, 10 | >9:1 |
3ab, 78 |
| 16 | 2a | H2O/THF (8:2) |
Ru3, 10 | 1:6 |
3aa′, 40e |
| 17 | 2b | H2O/THF (8:2) |
Ru3, 10 | 1:7 |
3ab′, 99f |
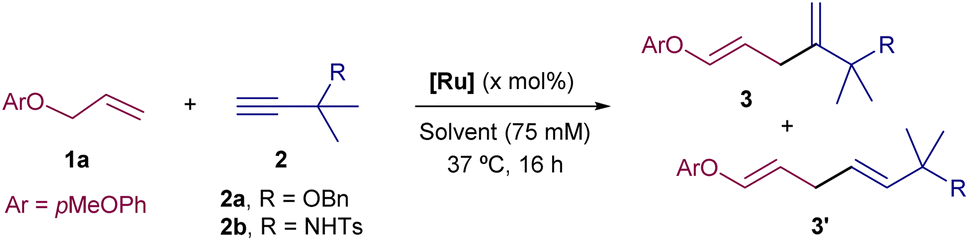
Under otherwise identical reaction conditions, the use of the neutral complex Cp*Ru(cod)Cl (Ru2) led to complete recovery of the starting materials (entry 6). With the cationic trisacetonitrile complex Ru1, we observed that the amounts of co-solvent and catalyst could be decreased down to 10% vol (THF) and 5 mol% (Ru1), without causing a severe impact on the yield and/or selectivity (entry 7). On the other hand, the use of a more polar alkyne precursor, such as the sulphonyl amide derivative 2b, led to a more efficient reaction, probably due to its higher solubility in aqueous mixtures. Thus, the coupling of 1a with 2b in a H2O:THF mixture (8:2), promoted by 5 mol% of Ru1, led to the corresponding diene product 3ab in an excellent 99% yield (entry 8). Lowering the amount of THF (entry 9), using other cosolvents such as EtOH, tBuOH or DMSO (entries 10–12), and even performing the reaction in pure water (entry 13) was also possible, leading in all cases to the desired product, 3ab, in moderate to excellent yields. On the other hand, although slightly higher yields are obtained under an atmosphere of N2, or using degassed solvents, the reaction is perfectly efficient under air and open-bottle solvents (entries 14 and 15). Interestingly, the regioselectivity of the coupling could be inverted by using the related catalyst Ru3, which features a less bulky cyclopentadienyl (Cp) ligand, instead of the pentamethyl derivate (Cp*, entries 16 and 17). This divergence can be tentatively rationalized considering the putative ruthenacycle intermediates that respectively evolve to the branched (3) and linear (3′) isomers (Int1 and Int-1′, Fig. 2). In particular, the steric clash in Int-1′, between the bulkier Cp* ligand and the alkyne propargylic substituents, would hamper the evolution towards the more stable linear isomer, 3′ (Fig. 2).11
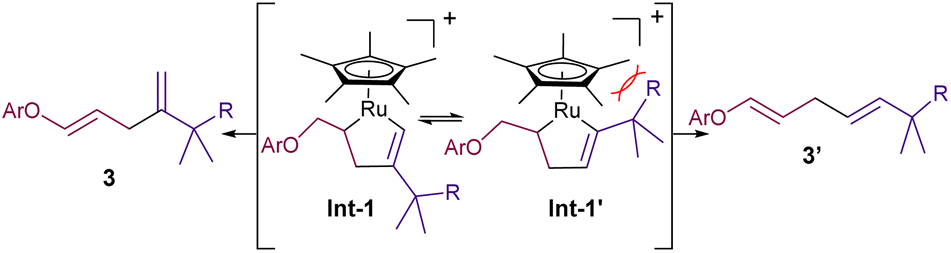 | ||
| Fig. 2 Proposed ruthenacyclic intermediates leading to 3 and 3′ regioisomers. | ||
With these optimal conditions in hand, we analysed the scope of the Ru-catalyzed coupling with different alkene and alkyne partners (Scheme 1). Regarding the alkyne, several other propargylic systems, including sulphonamides 2b–2d, other ethers like 2e and propargylic alcohols (2f–2j), efficiently participate in the coupling with the model alkene 1a, providing their respective diene products 3ab–3aj, in good to excellent yields and with high branched selectivity. Therefore, both alkyl and aryl substituents are allowed at the propargylic position.
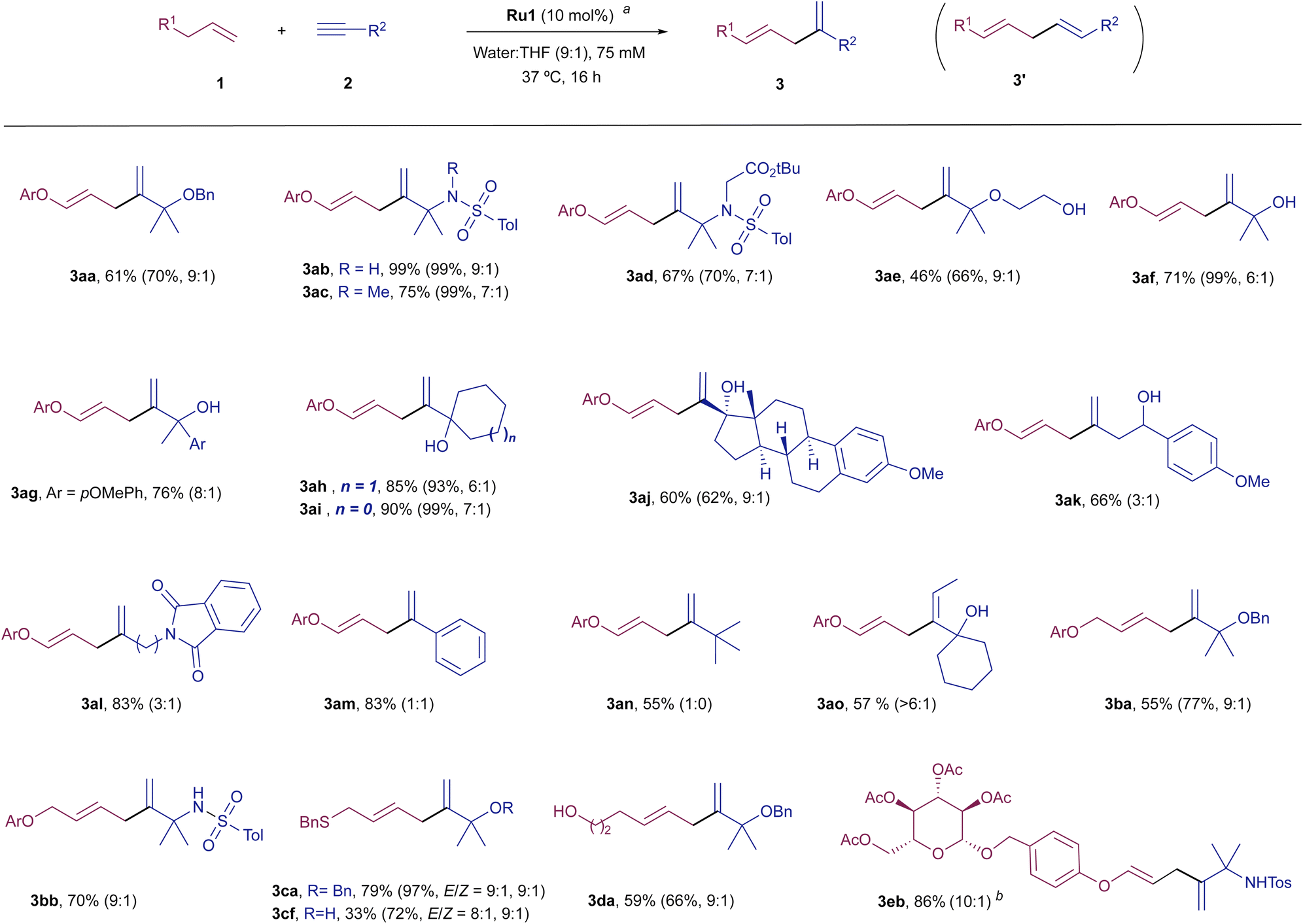 | ||
| Scheme 1
a
Conditions: alkene (1, 0.15 mmol), alkyne (2, 0.15 mmol), H2O:THF (8:2, 2.0 mL) and Ru1 (10 mol%). Yields of pure branched product 3 (3:3′ > 10:1) unless otherwise noted; only the branched structure is represented. Ar = pMeO(C6H4). NMR yield and branched: linear ratios (3:3′) using dimethyl sulfone as internal standard are given in parenthesis. bCarried out using 2.0 equiv. of alkene (0.30 mmol). | ||
The presence of two of these substituents at the propargylic center is key to achieve a good branched-to-linear ratio. Thus, the Ru-catalyzed coupling of 1a with alkynes that hold secondary carbons at the propargylic position, such as 2k or 2l, provided the corresponding dienes, 3ak and 3al, with a lower branched-to-linear ratio (3:3′ = 3:1), although in good overall yield (66 and 83%, respectively). The presence of an aromatic moiety, such as in phenylacetylene led to a non-regioselective coupling but proceeded in good overall yield (3am:3am′ = 1:1, 83% yield). Gratifyingly, we were pleased to observe that the presence of heteroatoms at the propargylic position is not mandatory to achieve good selectivity. Thus, the reaction of 1a with 3,3-dimethylbut-1-yne, 2n, provided the diene product 3an. Indeed, even an internal alkyne can engage quite efficiently in the reaction, provided that it bears an adjacent fully substituted propargylic center, such as in 3ao.12
With respect to the alkene partner, the reaction of model alkynes 2a and 2b could also be performed with an homoallyl ether like 1b, rendering their respective products (3ba and 3bb) in good yields (9:1 ratio). The use of a homoallyl thioethers is also possible, so that products 3ca and 3cf were obtained in moderate to good yields. Curiously, in these cases, small amounts of their respective Z-isomers could also be detected. Structurally simple alkenyl precursors, such as hex-5-en-1-ol, or more complex derivatives, like the pyranoside 1e are equally efficient partners, so that their corresponding products, 3da and 3eb were respectively obtained in good yields (59 and 86% yield) and high regioselectivities.13
Next, we focused on studying the bioorthogonality of the above cross-couplings, by using aqueous media containing biologically relevant additives (Scheme 2). Gratifyingly, the coupling between 1a and 2b, promoted by Ru1 (10 mol%), proceeds with similar yields in the presence of 0.1 equivalents of different amino acids like tyrosine or cysteine, and of biorelevant thiols such as glutathione. The presence of other additives such as glucose or vitamins like riboflavin neither affected the reaction. Moreover, increasing the amount of any these biomolecules up to 1 equivalent (10 times the amount of catalyst) did not compromise the alkyne–alkene coupling, so that the corresponding product, 3ab, was obtained in yields varying from 46 to 99%.
 | ||
| Scheme 2 Bioorthogonality of the Ru-catalyzed alkene–alkyne coupling. a Conditions: alkene (1, 0.075 mmol), alkyne (2, 0.075 mmol), water:THF (8:2, 1.0 mL) and Ru1 (10 mol%) unless otherwise noted. NMR yield using dimethylsulphone as internal standard; b PBS used as solvent instead of water; c DMEM used as solvent (DMEM = Dulbecco's modified eagle medium); d DMEM* used as solvent (DMEM* = DMEM + 10 fetal bovine serum + 1% antibiotics). | ||
Importantly, we found that the reaction is also feasible in PBS (phosphate buffer solution 1×. pH = 7.4), as well as in a cell culture milieu like DMEM, to give in this case 3ab in an excellent 90% yield. When the reaction was carried out in DMEM*, which includes 10% of fetal bovine serum and a few antibiotics, the product could still be obtained, albeit in a lower 30% yield, which is still satisfactory considering that this serum is a cocktail of hormones, lipids, and different type of proteins.
Overall, the observed efficiencies compare favorably with other bioorthogonal reactions catalyzed by ruthenium complexes, such as the hydrosylilation of alkynes or alkene cross metatheses.14
Considering the good orthogonality to biological components, we next checked whether the alkene–alkyne cross-coupling could also be used as bioconjugation tool, to chemoselectively modify amino acids containing either an alkene or an alkyne moiety. Gratifyingly, as can be deduced from Scheme 3, the coupling of N-Boc O-allyl tyrosine 1f with 2i proceeded efficiently to provide the diene 3fi in 62% yield (Scheme 3a). Moreover, the use of amino acids bearing an alkyne moiety is also compatible with the process. Thus, dipeptide 2p, bearing a pendant alkyne at the N-terminal position, or peptide 2q, featuring a propargyl glycine, participated efficiently in the coupling with 1a, with just 10 mol% of the Ru catalyst, leading to their respective products, 3ap and 3aq, in moderate to excellent yields (Scheme 3b and c).
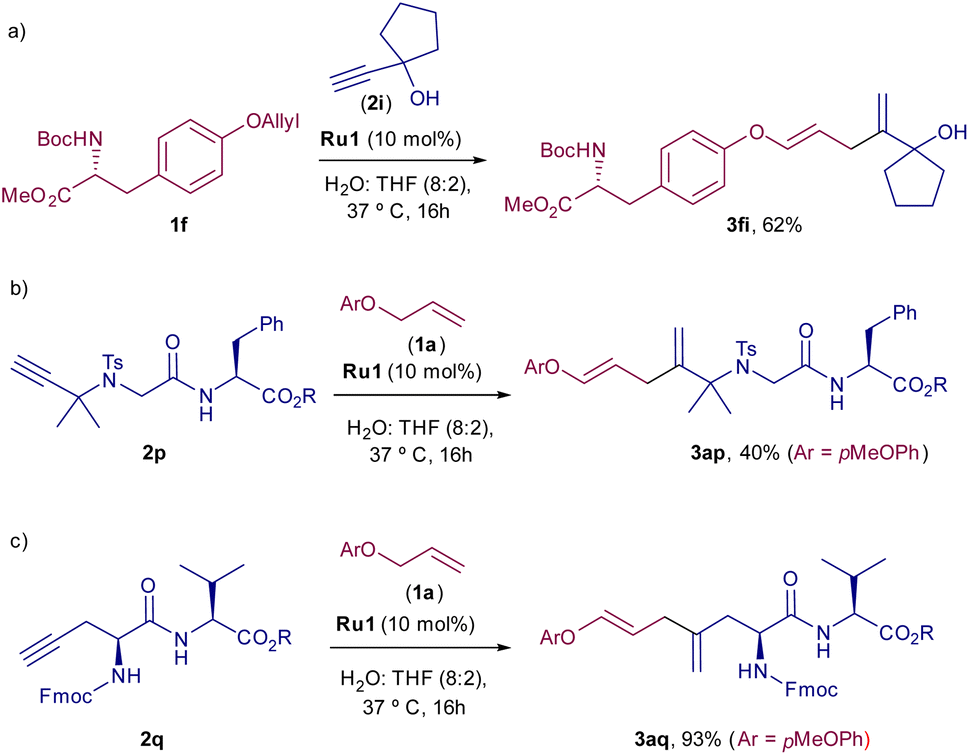 | ||
| Scheme 3 (a) Reaction using O-allyl tyrosine 1f. (b) Reaction using dipeptide 2p. (c) Reaction using dipeptide 2q. | ||
Encouraged by these results, we next checked whether the method could also be used to label larger peptides in water. Peptide 2r, which bears a N-terminal propargyl glycine was easily prepared by solid phase synthesis. Gratifyingly, after a short optimization of the coupling conditions between this peptide and alkene 1c, we were able to observe full conversion in water, using a peptide concentration as low as 200 μM, 2.5 fold excess of alkene and a Ru1 concentration of 400 μM (Scheme 4a). Almost full conversion of the peptide and the exclusive formation of the expected diene peptide product was observed by HPLC-MS. Moreover, other alkenes like 1f, 1d or the O-allyl pyranoside 1g could also react to give the corresponding peptide derivatives 3fr–3gr, as the only coupling products observed by HPLC-MS. The use of a peptide bearing the propargyl glycine residue at an internal position (i.e.2s) was also tolerated, providing efficiently the products resulting from the coupling with different types of alkene partners (e.g.3cs, 3fs, Scheme 4b). Overall, these results argue well for the application of this C–C coupling for bioconjugation and highlight the great potential of Ru catalysts to promote nontrivial C–C bond forming transformations under biorelevant conditions.
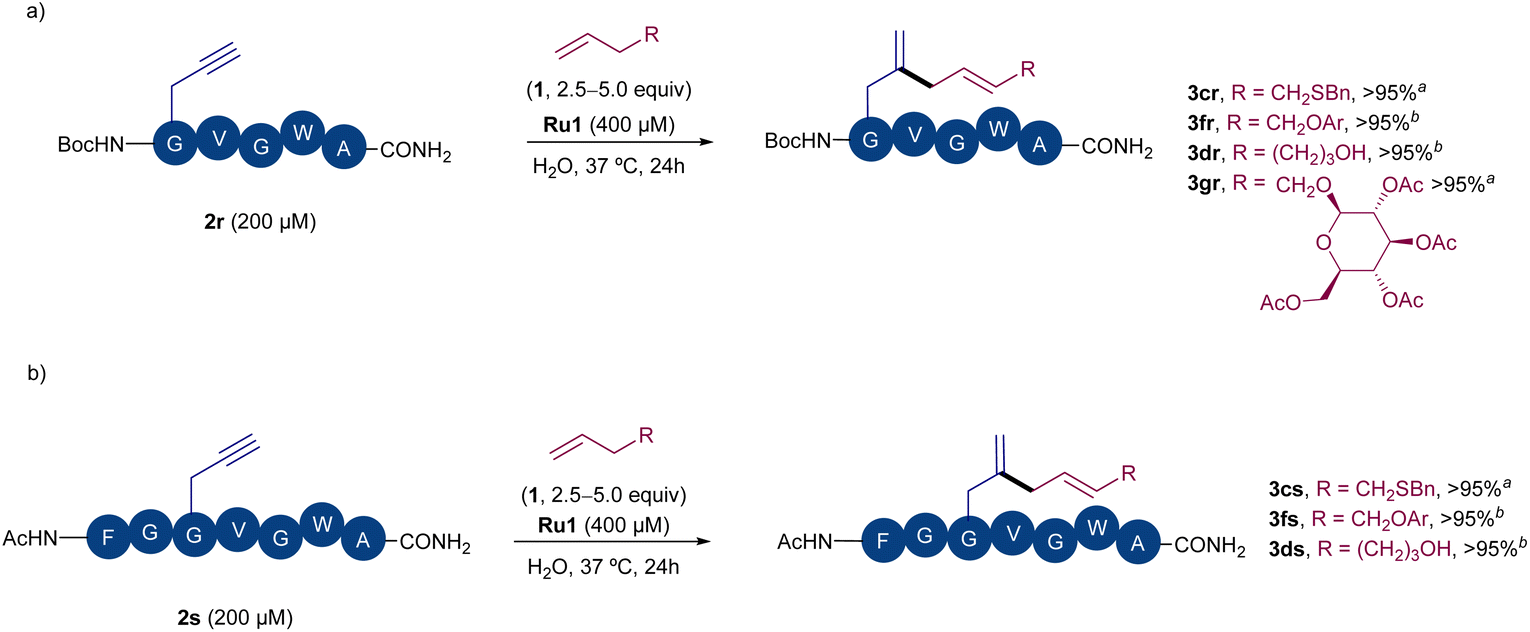 | ||
| Scheme 4 Ru-promoted derivatization of peptides in water: (a) reaction with peptide 2r. (b) Reaction with peptide 2s, bearing the alkyne in an internal position. Conversions of the precursor peptide (2) to the corresponding diene product determined by HPLC-MS (see ESI† for details). Ar = pMeOPh. a 5 equiv. of alkene 1 were used. b 2.5 equiv. of alkene 1 were used. | ||
Conclusions
In conclusion, we have demonstrated that the ruthenium catalysed Alder-ene coupling between alkenes and alkynes, originally developed in organic solvents, can be efficiently promoted in aqueous and biologically relevant environments, in high yields and with good to excellent regioselectivities. The reaction proved to be tolerant to the presence of a variety of functional groups at the alkyne and alkene partners, and has also been shown to proceed efficiently in the presence of different types of biomolecules as well as in cell cultured complex media. Despite its fully intermolecular nature, the reaction does not generally need excess of any of the two partners and proceeds with low catalyst loadings. Finally, we showed that by adjusting the reaction conditions, it can be applied to the modification of alkyne containing peptides. The structural simplicity of alkene and alkynes, and the ease with which these groups can be incorporated into a wide variety of biomolecules, argue well for the applicability of the method as a new bioconjugation tool in chemical biology.Data availability
General and synthetic procedures, orthogonality assays, HPLC data and NMR spectra are available in the ESI.†Author contributions
A. G.-G., F. L. and J. L. M. conceived the study, analyzed the results, and wrote the paper. A. G.-G., D. M.-A., L. G. C. performed the experimental work. All authors discussed the results and edited the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received financial support from Spanish grants (Grants PID2020-118579GB-I00 and PID2019-108624RB-I00 funded by MCIN/AEI/10.13039/501100011033 and by “ERDF A way of making Europe”, and ORFEO-CINQA network RED2018-102387-T), the Consellería de Cultura, Educación e Ordenación Universitaria (Grant ED431C 2021/25 and Grant ED431G 2019/03: Centro Singular de Investigación de Galicia accreditation 2019–2022) and the European Union (European Regional Development Fund-ERDF corresponding to the multiannual financial framework 2014–2020). We thank the Ministerio de Universidades for the FPU fellowship to A. G. and D. M.-A. (FPU17/00711 and FPU18/04495).Notes and references
- For reviews on bioorthogonal reactions, see: (a) M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef PubMed; (b) H. W. Shih, D. N. Kamber and J. A. Prescher, Curr. Opin. Chem. Biol., 2014, 21, 103–111 CrossRef CAS PubMed; (c) R. D. Row and J. A. Prescher, Acc. Chem. Res., 2018, 51, 1073–1081 CrossRef CAS PubMed; (d) X. Ji, Z. Pan, B. Yu, L. K. de la Cruz, Y. Zheng, B. Ke and B. Wang, Chem. Soc. Rev., 2019, 48, 1077–1094 RSC; (e) N. K. Devaraj, ACS Cent. Sci., 2018, 4, 952–959 CrossRef CAS PubMed; (f) S. L. Scinto, D. A. Bilodeau, R. Hincapie, W. Lee, S. S. Nguyen, M. Xu, C. W. am Ende, M. G. Finn, K. Lang, Q. Lin, J. P. Pezacki, J. A. Prescher, M. S. Robillard and J. M. Fox, Nat. Rev. Methods Primers, 2021, 1, 30 CrossRef CAS PubMed; (g) O. Vázquez, M. I. Sánchez, J. Martinez-Costas, M. E. Vázquez and J. L Mascareñas, Org. Lett., 2010, 12, 216–219 CrossRef PubMed; (h) H. Wu and N. K. Devaraj, in Cycloadditions in Bioorthogonal Chemistry, ed. M. Vrabel and T. Carell, Springer International Publishing, Cham, 2016, pp. 109–130 Search PubMed; (i) B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- For recent reviews, see: (a) P. Destito, C. Vidal, F. López and J. L. Mascareñas, Chem.–Eur. J., 2021, 27, 4789–4816 CrossRef CAS PubMed; (b) M. O. N. van de L'Isle, M. C. Ortega-Liebana and A. Unciti-Broceta, Curr. Opin. Chem. Biol., 2021, 61, 32–42 CrossRef PubMed; (c) Y. Liu, K. L. Lai and K. Vong, Eur. J. Inorg. Chem., 2022, 2022, e202200215 CAS; (d) S. Gutiérrez, M. Tomás-Gamasa and J. L. Mascareñas, Chem. Sci., 2022, 13, 6478–6495 RSC; (e) Y. Liu and Y. Bai, ACS Appl. Bio Mater., 2020, 3, 4717–4746 CrossRef CAS PubMed.
- (a) E. Latocheski, G. M. Dal Forno, T. M. Ferreira, B. L. Oliveira, G. J. L. Bernardes and J. B. Domingos, Chem. Soc. Rev., 2020, 49, 7710–7729 RSC; (b) J. Wang, X. Wang, X. Fan and P. R. Chen, ACS Cent. Sci., 2021, 7, 929–943 CrossRef CAS PubMed; (c) J. Tu, M. Xu and R. M. Franzini, ChemBioChem, 2019, 20, 1615–1627 CrossRef CAS PubMed; (d) Y. Li and H. Fu, ChemistryOpen, 2020, 9, 835–853 CrossRef CAS PubMed; (e) V. Sabatino, V. B. Unnikrishnan and G. J. L. Bernardes, Chem Catal., 2022, 2, 39–51 CrossRef CAS; (f) B. Lozhkin and T. R. Ward, Bioorg. Med. Chem., 2021, 45, 116310 CrossRef CAS PubMed.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS; (b) L. Li and Z. Zhang, Molecules, 2016, 21, 1393 CrossRef PubMed; (c) A. Gutiérrez-González, P. Destito, J. R. Couceiro, C. Pérez-González, F. López and J. L. Mascareñas, Angew. Chem., Int. Ed., 2021, 60, 16059–16066 CrossRef PubMed; (d) M. Li, N. Zheng, J. Li, Y. Zheng and W. Song, Green Chem., 2020, 22, 2394–2398 RSC; (e) R. Chen, L. Zeng, Z. Lai and S. Cui, Adv. Synth. Catal., 2019, 361, 989–994 CrossRef CAS; (f) S. Gutiérrez, M. Tomás-Gamasa and J. L. Mascareñas, Angew. Chem., Int. Ed., 2021, 60, 22017–22025 CrossRef PubMed; (g) P. Destito, J. R. Couceiro, H. Faustino, F. Lopez and J. L. Mascareñas, Angew. Chem., Int. Ed., 2017, 56, 10766–10770 CrossRef CAS PubMed; (h) C. Vidal, M. Tomas-Gamasa, A. Gutierrez-Gonzalez and J. L. Mascareñas, J. Am. Chem. Soc., 2019, 141, 5125–5129 CrossRef CAS PubMed.
- For relevant examples, see: (a) J. M. Chalker, C. S. C. Wood and B. G. Davis, J. Am. Chem. Soc., 2009, 131, 16346–16347 CrossRef CAS PubMed; (b) R. M. Yusop, A. Unciti-Broceta, E. M. V. Johansson, R. M. Sánchez-Martín and M. Bradley, Nat. Chem., 2011, 3, 239 CrossRef CAS PubMed; (c) P. Destito, A. Sousa-Castillo, J. R. Couceiro, F. López, M. A. Correa-Duarte and J. L. Mascareñas, Chem. Sci., 2019, 10, 2598–2603 RSC; (d) J. Li, S. Lin, J. Wang, S. Jia, M. Yang, Z. Hao, X. Zhang and P. R. Chen, J. Am. Chem. Soc., 2013, 135, 7330–7338 CrossRef CAS PubMed; (e) F. Wang, Y. Zhang, Z. Du, J. Ren and X. Qu, Nat. Commun., 2018, 9, 1209 CrossRef PubMed.
- (a) C. Vidal, M. Tomas-Gamasa, P. Destito, F. López and J. L. Mascarenas, Nat. Commun., 2018, 9, 1913 CrossRef PubMed; (b) Y. Long, B. Cao, X. Xiong, A. S. C. Chan, R. W.-Y. Sun and T. Zou, Angew. Chem., Int. Ed., 2021, 60, 4133–4141 CrossRef CAS PubMed; (c) Y. A. Lin, O. Boutureira, L. Lercher, B. Bhushan, R. S. Paton and B. G. Davis, J. Am. Chem. Soc., 2013, 135, 12156–12159 CrossRef CAS PubMed; (d) L. Adriaenssens, L. Severa, J. Vávra, T. Šálová, J. Hývl, M. Čížková, R. Pohl, D. Šaman and F. Teplý, Collect. Czech. Chem. Commun., 2009, 74, 1023–1034 CrossRef CAS; (e) J. Miguel-Ávila, M. Tomás-Gamasa and J. L. Mascareñas, Angew. Chem., Int. Ed., 2020, 59, 17628–17633 CrossRef PubMed.
- For alternative strategies based on metalloenzymes instead of discrete metal catalysts, see: (a) M. Jeschek, R. Reuter, T. Heinisch, C. Trindler, J. Klehr, S. Panke and T. R. Ward, Nature, 2016, 537, 661–665 CrossRef CAS PubMed; (b) S. Eda, I. Nasibullin, K. Vong, N. Kudo, M. Yoshida, A. Kurbangalieva and K. Tanaka, Nat. Catal., 2019, 2, 780–792 CrossRef CAS; (c) R. K. Zhang, K. Chen, X. Huang, L. Wohlschlager, H. Renata and F. H. Arnold, Nature, 2019, 565, 67–72 CrossRef CAS PubMed; (d) A. M. Knight, S. B. J. Kan, R. D. Lewis, O. F. Brandenberg, K. Chen and F. H. Arnold, ACS Cent. Sci., 2018, 4, 372–377 CrossRef CAS PubMed; (e) K. Chen and F. H. Arnold, J. Am. Chem. Soc., 2020, 142, 6891–6895 CrossRef CAS PubMed; (f) Z. Liu, J. Huang, Y. Gu, D. S. Clark, A. Mukhopadhyay, J. D. Keasling and J. F. Hartwig, J. Am. Chem. Soc., 2022, 144, 883–890 CrossRef CAS PubMed; (g) J. Huang, Z. Liu, B. J. Bloomer, D. S. Clark, A. Mukhopadhyay, J. D. Keasling and J. F. Hartwig, Nat. Chem., 2021, 13, 1186–1191 CrossRef CAS PubMed.
- (a) B. M. Trost and F. D. Toste, Tetrahedron Lett., 1999, 40, 7739–7743 CrossRef CAS; (b) B. M. Trost, H. C. Shen and A. B. Pinkerton, Chem.–Eur. J., 2002, 8, 2341–2349 CrossRef CAS PubMed; (c) B. M. Trost and A. Indolese, J. Am. Chem. Soc., 1993, 115, 4361–4362 CrossRef CAS; (d) B. M. Trost, A. F. Indolese, T. J. J. Mueller and B. Treptow, J. Am. Chem. Soc., 1995, 117, 615–623 CrossRef CAS; For reviews, see: (e) B. M. Trost, M. U. Frederiksen and M. T. Rudd, Angew. Chem., Int. Ed., 2005, 44, 6630–6666 CrossRef CAS PubMed; (f) B. M. Trost, F. D. Toste and A. B. Pinkerton, Chem. Rev., 2001, 101, 2067–2096 CrossRef CAS PubMed.
- For an isolated example with a metalloprotein, which proceeds in an aqueous/CH3CN mixtures at 60 °C, see: A. Thiel, D. F. Sauer, U. Markel, M. A. S. Mertens, T. Polen, U. Schwaneberg and J. Okuda, Org. Biomol. Chem., 2021, 19, 2912–2916 RSC.
- (a) J. A. Marchand, M. E. Neugebauer, M. C. Ing, C. I. Lin, J. G. Pelton and M. C. Y. Chang, Nature, 2019, 567, 420–424 CrossRef CAS PubMed; (b) M. A. Shandell, Z. Tan and V. W. Cornish, Biochemistry, 2021, 60, 3455–3469 CrossRef CAS PubMed; (c) B. Bhushan, Y. A. Lin, M. Bak, A. Phanumartwiwath, N. Yang, M. K. Bilyard, T. Tanaka, K. L. Hudson, L. Lercher, M. Stegmann, S. Mohammed and B. G. Davis, J. Am. Chem. Soc., 2018, 140, 14599–14603 CrossRef CAS PubMed; (d) W. Song, Y. Wang, Z. Yu, C. I. R. Vera, J. Qu and Q. Lin, ACS Chem. Biol., 2010, 5, 875–885 CrossRef CAS PubMed; (e) Z. Zhang, L. Wang, A. Brock and P. G. Schultz, Angew. Chem., Int. Ed., 2002, 41, 2840–2842 CrossRef CAS.
- The formation of intermediate Int-1 with catalyst Ru3 has been proposed to be hampered when using alkynes bearing propargylic groups, due to the steric constrains associated to the ruthenacyclic C–C bond formation. See ref. 8.
- B. M. Trost and J. J. Cregg, J. Am. Chem. Soc., 2015, 137, 620–623 CrossRef CAS PubMed.
- Preliminary results suggest that the presence of additional substituents in the allylic carbon of the alkene partner (1) hamper the process. See the ESI† for details.
- (a) T. T. L. Kwan, O. Boutureira, E. C. Frye, S. J. Walsh, M. K. Gupta, S. Wallace, Y. Wu, F. Zhang, H. F. Sore, W. R. J. D. Galloway, J. W. Chin, M. Welch, G. J. L. Bernardes and D. R. Spring, Chem. Sci., 2017, 8, 3871–3878 RSC; (b) Y. A. Lin, J. M. Chalker, N. Floyd, G. J. L. Bernardes and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 9642–9643 CrossRef CAS PubMed; (c) A. Gutierrez-González, F. López and J. L. Mascareñas, Helv. Chim. Acta, 2023, e202300001 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01254a |
| This journal is © The Royal Society of Chemistry 2023 |