
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A highly enantioselective intramolecular 1,3-dipolar cycloaddition yields novel pseudo-natural product inhibitors of the Hedgehog signalling pathway†
Jie
Liu‡
 a,
Ruirui
Zhang‡
a,
Shubhadip
Mallick
a,
Sohan
Patil
a,
Chantal
Wientjens
b,
Jana
Flegel
a,
Anna
Krupp
c,
Carsten
Strohmann
c,
Corentin
Grassin
d,
Christian
Merten
d,
Axel
Pahl
ae,
Michael
Grigalunas
a and
Herbert
Waldmann
*ab
a,
Ruirui
Zhang‡
a,
Shubhadip
Mallick
a,
Sohan
Patil
a,
Chantal
Wientjens
b,
Jana
Flegel
a,
Anna
Krupp
c,
Carsten
Strohmann
c,
Corentin
Grassin
d,
Christian
Merten
d,
Axel
Pahl
ae,
Michael
Grigalunas
a and
Herbert
Waldmann
*ab
aDepartment of Chemical Biology, Max Planck Institute of Molecular Physiology, Otto-Hahn-Street 11, 44227 Dortmund, Germany. E-mail: Herbert.waldmann@mpi-dortmund.mpg.de
bFaculty of Chemistry, Chemical Biology, Technical University Dortmund, Otto-Hahn-Street 6, 44221 Dortmund, Germany
cFaculty of Chemistry, Inorganic Chemistry, Technical University Dortmund, Otto-Hahn-Street 6, 44221 Dortmund, Germany
dFaculty of Chemistry and Biochemistry, Organic Chemistry II, Ruhr University Bochum, University-Street 150, 44801 Bochum, Germany
eCompound Management and Screening Center, Otto-Hahn-Street 11, 44227 Dortmund, Germany
First published on 21st June 2023
Abstract
De novo combination of natural product (NP) fragments by means of efficient, complexity- and stereogenic character-generating transformations to yield pseudo-natural products (PNPs) may explore novel biologically relevant chemical space. Pyrrolidine- and tetrahydroquinoline fragments rarely occur in combination in nature, such that PNPs that embody both fragments might represent novel NP-inspired chemical matter endowed with bioactivity. We describe the synthesis of pyrrolo[3,2-c]quinolines by means of a highly enantioselective intramolecular exo-1,3-dipolar cycloaddition catalysed by the AgOAc/(S)-DMBiphep complex. The cycloadditions proceeded in excellent yields (up to 98%) and with very high enantioselectivity (up to 99% ee). Investigation of the resulting PNP collection in cell-based assays monitoring different biological programmes led to the discovery of a structurally novel and potent inhibitor of the Hedgehog signalling pathway that targets the Smoothened protein.
Introduction
Compound classes endowed with pronounced bioactivity are often inspired by natural product (NP) structure and have high stereogenic content, which positively correlates with performance in drug discovery.1 To identify such novel NP-inspired bioactive matter, principles including pseudo-natural product (PNP) design,2 biology-oriented synthesis (BIOS)3 and the complexity-to-diversity (CtD) approach4 have been developed as guiding strategies. In PNP design and synthesis, NP fragments are combined in novel arrangements not found in nature by means of efficient, complexity- and stereogenic character-generating transformations, and for such syntheses enantioselective 1,3-dipolar cycloadditions have proven to be of high value.2d,5The pyrrolidine-derived and tetrahydroquinoline alkaloid-derived fragments frequently occur individually in NPs, but rarely in combination,6 such that PNPs that embody both fragments might represent novel NP-inspired chemical matter that is endowed with bioactivity. We have recently developed asymmetric syntheses of pyrrolo[2,3-c]quinolines 1 (ref. 7) and pyrrolo[3,4-c]quinolines 2 (ref. 8) and shown that the combination of pyrrolidine and tetrahydroquinoline fragments in different arrangements and with different connectivities that are not found in nature yields chemically and biologically diverse PNP classes.9 However, the regioisomeric pyrrolo[3,2-c]quinolines 3 (Fig. 1a) were only accessible in racemic9,10 and/or cis form.11 In principle, this scaffold would be available by means of an enantioselective intramolecular 1,3-dipolar cycloaddition, but such transformations have rarely been described. Recently, del Pozo and Adrio et al. reported a catalytic enantioselective intramolecular 1,3-dipolar cycloaddition to afford trans-configured fluorinated pyrroloquinolines (Fig. 1b), but with limited exploration of substrate scope.12
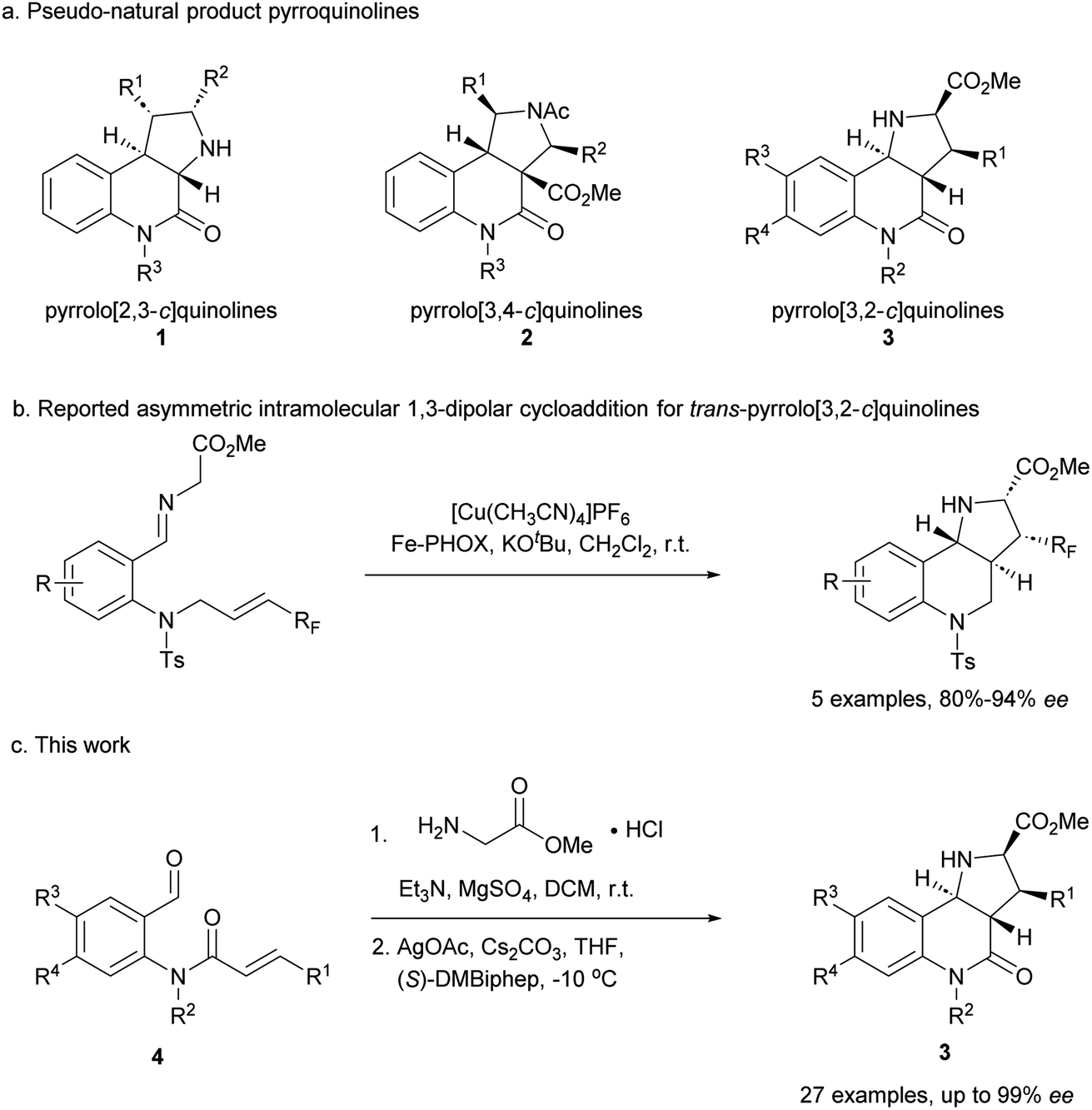 | ||
| Fig. 1 Asymmetric synthesis of pyrrolo[3,2-c]quinolines. (a) Reported pyrroquinolines with different connectivities. (b) Reported asymmetric synthesis of trans-pyrrolo[3,2-c]quinolines. (c) This work. | ||
We have now developed a new enantioselective intramolecular 1,3-dipolar cycloaddition catalysed by a chiral AgOAc/DMBiphep complex that yields trans-pyrrolo[3,2-c]quinolines in high yields and with excellent enantioselectivity (Fig. 1c). Investigation of these pseudo-NPs in different biological assays identified a novel inhibitor of Hedgehog signalling targeting the protein Smoothened (SMO).
Results and discussion
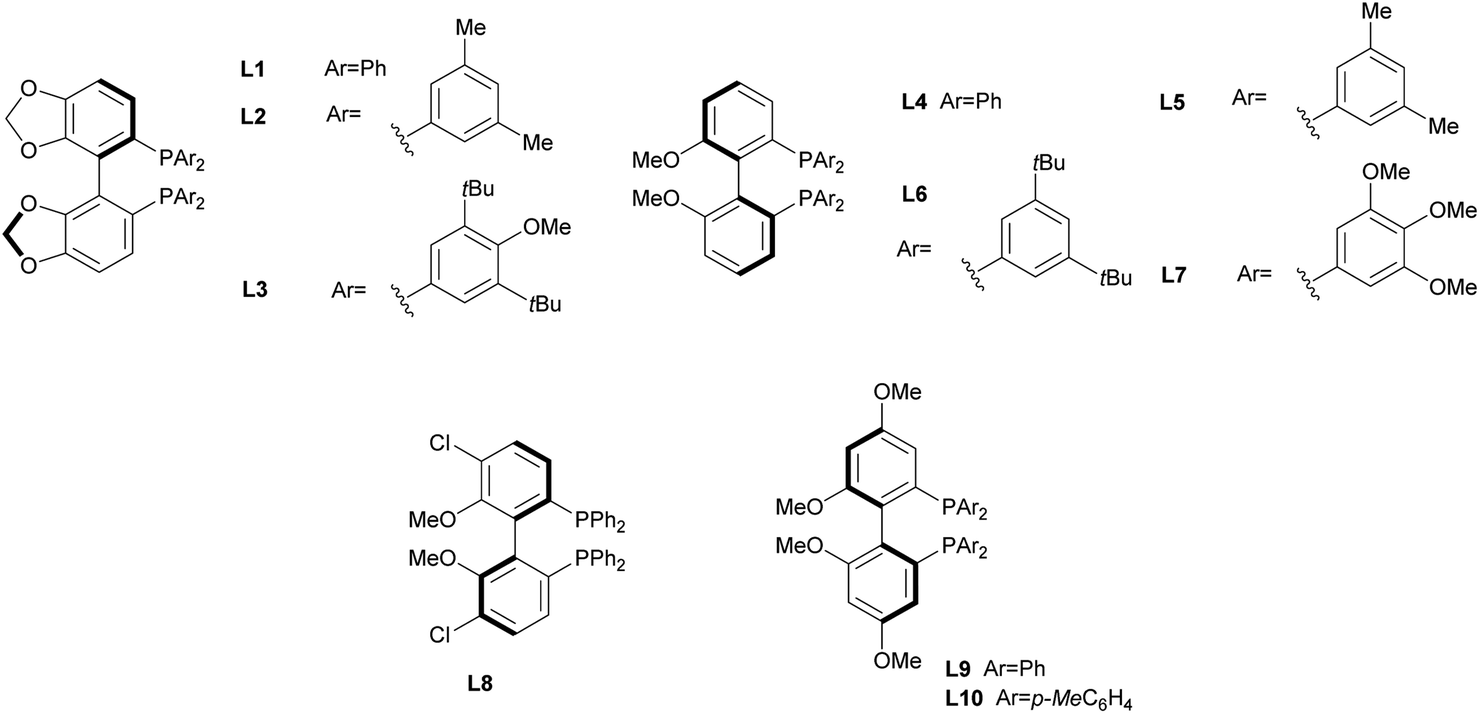
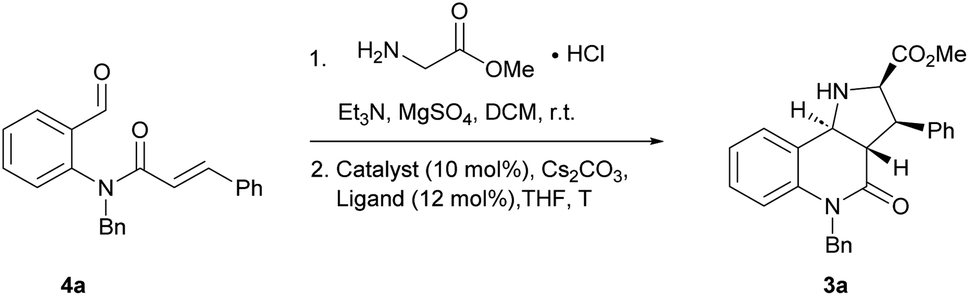
In order to identify favourable reaction conditions, aldehyde 4a was condensed with glycine methyl ester and the resulting iminoester was subjected to intramolecular cycloaddition without further purification in the presence of different chiral metal complexes (Table 1). Initial solvent screening revealed that the transformations proceeded best in THF (Table S1†). When Segphos was used as ligand (L1) and AgOAc as metal salt, the cycloadduct was isolated with 79% ee (Table 1, entry 1). Increasing steric hindrance of the substituents on Segphos did not improve the enantioselectivity (L2 and L3, Table 1, entries 2 and 3). When the biaryl substituent was changed, only DMBiphep L5 with moderate steric demand gave a slight increase of enantioselectivity to 80% ee (Table 1, entry 5). Further increasing or decreasing the steric demand of the substituents led to lower enantioselectivity (Table 1, entries 4–10). Changing the metal catalyst from AgOAc to either a Cu(I) or a Cu(II) salt led to greatly reduced enantioselectivity (Table 1, entries 11 and 12). Finally, lowering the temperature to −10 °C yielded the desired cycloadduct 3a in 90% ee and in viable yield (Table 1, entry 13).|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Ligand | T [°C] | Yield [%] | ee [%] |
| a The reaction was performed on 0.1 mmol scale. | |||||
| 1 | AgOAc | L1 | r.t. | 90 | 79 |
| 2 | AgOAc | L2 | r.t. | 65 | 74 |
| 3 | AgOAc | L3 | r.t. | 64 | 5 |
| 4 | AgOAc | L4 | r.t. | 80 | 73 |
| 5 | AgOAc | L5 | r.t. | 83 | 80 |
| 6 | AgOAc | L6 | r.t. | 66 | 17 |
| 7 | AgOAc | L7 | r.t. | 85 | 6 |
| 8 | AgOAc | L8 | r.t. | 85 | 26 |
| 9 | AgOAc | L9 | r.t. | 77 | 71 |
| 10 | AgOAc | L10 | r.t. | 85 | 67 |
| 11 | [Cu(CH3CN)4]BF4 | L5 | r.t. | 70 | 51 |
| 12 | Cu(OAc)2 | L5 | r.t. | 50 | 56 |
| 13a | AgOAc | L5 | −10 | 67 | 90 |
|
|||||
With favourable conditions identified, the substrate scope of this asymmetric reaction was explored (Table 2). When chloro-substituents were introduced into the phenyl ring of the cinnamic acid unit (R1), enantioselectivity was lower regardless of the substitution site (Table 2, entries 2–4). Also, differently functionalised N-benzyl substituents or an N-methyl group (R2) led to a decrease of ee to 79–88% (Table 2, entries 5–8). Conversely, introduction of a substituent meta to the aldehyde in starting compound 4 (i.e. R3), which corresponds to the C6 position in the final quinoline scaffold, increased the enantioselectivity. Diverse substituents with different electronic properties or steric demand were well tolerated and afforded the desired cycloadducts 3 in good yields and with excellent enantioselectivity (Table 2, entries 9–14). A decreased enantioselectivity was observed when the substituents were introduced para to the aldehyde in 4a, i.e. R4 at the C7 position of the quinoline (Table 2, entries 15 and 16). The relative and absolute configuration of the cycloadducts were determined by X-ray crystallography for rac-3j and computed vibrational circular dichroism (VCD) spectra13 for 3l, respectively, and were assigned by analogy to the other cycloadducts (see Fig. S1† for details).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Product | R1 | R2 | R3 | R4 | Yield [%] | ee [%] |
| 1 | 3a | Ph | Bn | H | H | 67 | 90 |
| 2 | 3b | p-ClC6H4 | Bn | H | H | 92 | 85 |
| 3 | 3c | m-ClC6H4 | Bn | H | H | 81 | 76 |
| 4 | 3d | o-ClC6H4 | Bn | H | H | 73 | 75 |
| 5 | 3e | Ph | o-Me benzyl | H | H | 62 | 88 |
| 6 | 3f | Ph | m-Br benzyl | H | H | 47 | 85 |
| 7 | 3g | Ph | p-Cl benzyl | H | H | 59 | 85 |
| 8 | 3h | Ph | Me | H | H | 56 | 79 |
| 9 | 3i | Ph | Bn | F | H | 75 | 94 |
| 10 | 3j | Ph | Bn | Cl | H | 76 | 97 |
| 11 | 3k | Ph | Bn | Br | H | 84 | 96 |
| 12 | 3l | Ph | Bn | Me | H | 98 | 97 |
| 13 | 3m | Ph | Bn | MeO | H | 63 | 98 |
| 14 | 3n | Ph | Bn | CF3O | H | 69 | 96 |
| 15 | 3o | Ph | Bn | H | F | 68 | 83 |
| 16 | 3p | Ph | Bn | H | Br | 82 | 80 |
| 17 | 3q | p-ClC6H4 | Bn | Me | H | 59 | 96 |
| 18 | 3r | m-ClC6H4 | Bn | Me | H | 67 | 97 |
| 19 | 3s | o-ClC6H4 | Bn | Me | H | 92 | 94 |
| 20 | 3t | Ph | o-Me benzyl | Br | H | 59 | 97 |
| 21 | 3u | Ph | o-Me benzyl | Me | H | 52 | 98 |
| 22 | 3v | Ph | m-Br benzyl | F | H | 61 | 98 |
| 23 | 3w | Ph | m-Br benzyl | Me | H | 65 | 98 |
| 24 | 3x | Ph | p-Cl benzyl | MeO | H | 58 | 99 |
| 25 | 3y | Ph | Me | F | H | 66 | 90 |
| 26 | 3z | Ph | Bn | F | F | 70 | 96 |
| 27 | 3aa | Ph | Bn | –C4H4– | 59 | 93 | |
| 28 | 3bb | p-CNC6H4 | Bn | H | H | 91 | 4 |
| 29 | 3cc | Ph | p-CN benzyl | H | H | 94 | 88 |
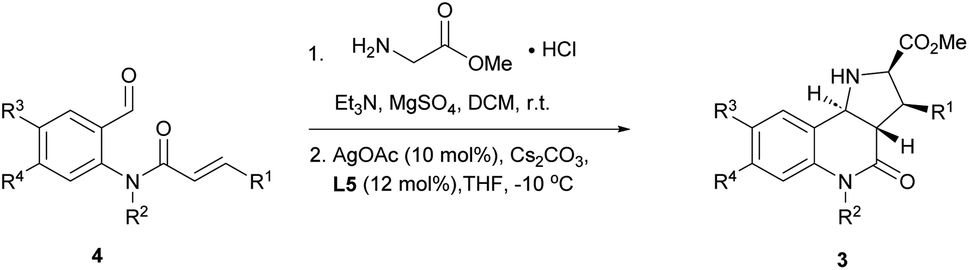
To further explore the positive impact of the quinoline C6 substituent (R3) on enantioselectivity, diverse substituent combinations were investigated. In the presence of a methyl group at R3, enantioselectivity improved from 75%–85% to 94%–97% for regioisomeric chlorophenyl substituents at R1 (Table 2, entries 2–4 and 17–19). By analogy, excellent ee values were recorded for different substitutions of the benzyl group at R2 as long as there are substituents at the final quinoline C6 position (Table 2, entries 20–25). Even when R2 is a methyl group, a sterically less demanding substitution with a fluorine atom at the C6 position improved the enantioselectivity from 79% to 90% (Table 2, entries 8 and 25). When both R3 and R4 were fluorine or represented a fusion with a benzene ring, enantioselectivity was also very high (Table 2, entries 26 and 27).
The reaction was incompatible with a strong electron-withdrawing group at the R1 position. Thus, in the case of a para-cyanide substituent hardly any enantioselectivity was observed under standard conditions (Table 2, entry 28). However, when the cyanide substituent was at the R2 position on the benzyl group, only a minor effect was observed and the reaction proceeded in excellent yield and with good enantioselectivity (Table 2, entry 29).
The direction of the stereoselection and the pronounced influence of the substituent meta to the aldehyde in starting material 4 (R3) may be rationalised by the transition state proposed in Fig. 2. The silver ion most likely would be chelated by the bidentate phosphine ligand (S)-DMBiphep and the iminoester intermediate, which would be deprotonated to the azomethine ylide by Cs2CO3. In this transition state, the dipolarophile would preferably approach the 1,3-dipole from the back side to avoid steric repulsion between the substrate backbone and the ligand. In particular, in the presence of a substituent R3, the repulsion would be pronounced, thus leading to higher enantioselectivity. In comparison, no improved enantioselectivity was recorded if a substituent R4 was introduced because this position does not point towards the ligand. We also note that the intramolecular cycloaddition proceeds with an exo approach due to the steric demands and conformational rigidity in the transition state.
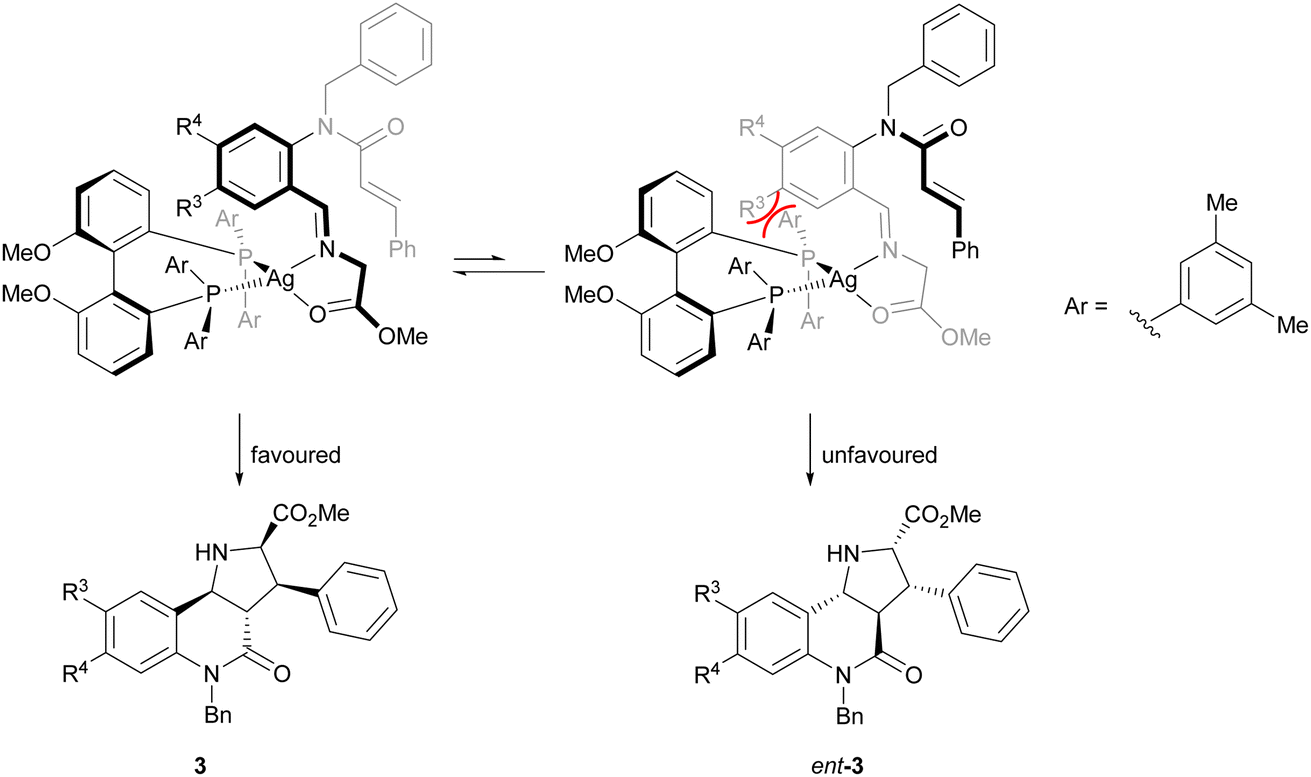 | ||
| Fig. 2 Proposed transition state for the 1,3-dipolar cycloaddition. | ||
In order to investigate whether the cycloadducts 3 are endowed with bioactivity, they were subjected to cell-based assays monitoring different biological programmes, such as oncogenic signalling, autophagy and immunometabolism. Gratifyingly, the compound collection defines a new inhibitor chemotype of signal transduction through the Hedgehog pathway. The Hedgehog (Hh) signalling pathway plays a critical role in the regulation of embryonic development, post-embryonic tissue homeostasis and regeneration in vertebrates.14 Excessive activity of the pathway is associated with malignancy, such as medulloblastoma,15 basal cell carcinoma16 and rhabdomyosarcoma.17 Therefore, inhibitors of the Hh signalling pathway have emerged as attractive therapeutic options in oncology.18
Initially, racemic pseudo-NPs were subjected to a phenotypic Hh-dependent osteoblast differentiation assay to monitor their possible impact on the Hh signalling pathway upon activation by purmorphamine, a pathway agonist binding to the Smoothened protein.19 Gratifyingly, most of the compounds showed potent inhibition of Hh signalling (see Table S2†), with the most potent compound rac-3a displaying a half-maximal inhibitory concentration (IC50) of 0.29 ± 0.05 μM (Fig. 3a). Interestingly, 3a (90% ee) showed an IC50 value of 4.83 ± 1.81 μM, while its enantiomer ent-3a (90% ee) displayed an approximately 30-fold lower IC50 value of 0.15 ± 0.03 μM. This tendency was also observed in the orthogonal Gli-dependent reporter gene assay in Shh-LIGHT2 cells20 with an IC50 of 0.17 ± 0.02 μM for ent-3a while 3a was inactive (IC50 > 30 μM, Fig. 3b). Hh target gene expression was explored to validate the inhibition of the Hh pathway. Compound ent-3a reduced the expression of Hh target genes Ptch1 and Gli1 dose dependently (Fig. 3c and d); at 1 μM, Ptch1 and Gli1 gene expression was suppressed by 83% and 96%, respectively. The activity of ent-3a was comparable to the Hh pathway inhibitor vismodegib at the same concentration, which is clinically approved for the treatment of basal cell carcinoma.21
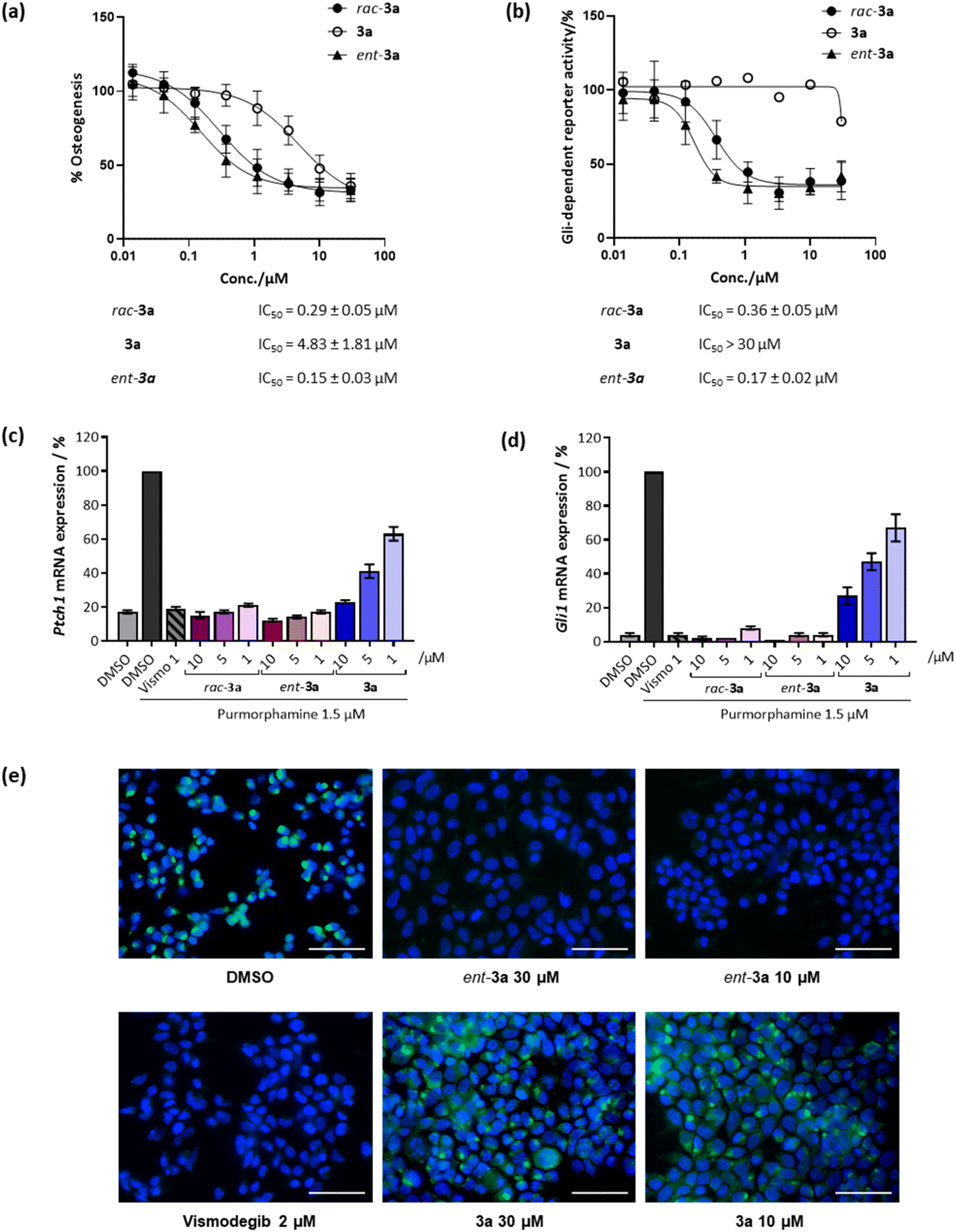 | ||
| Fig. 3 Biological characterisation of selected compounds for Hh pathway inhibition. (a) Osteoblast differentiation assay. C3H10T1/2 cells were treated with 1.5 μM purmorphamine together with DMSO as a control or compounds for 96 h. The activity of alkaline phosphatase was quantified as a measure of Hh pathway activity. Values for cells treated with purmorphamine and DMSO were set to 100%. Data are mean values ± SD (n = 3). (b) Gli-responsive reporter gene assay. Shh-LIGHT2 cells were treated with 2 μM purmorphamine together with DMSO as a control or compounds for 48 h. The Gli-responsive firefly luciferase signals were normalised to the signal for Renilla luciferase. The value for the purmorphamine/DMSO control was set to 100%. Data are mean values ± SD and are representative of three biological replicates (n = 3). (c) and (d) Expression of the Hh target genes Ptch1 (c) and (d) Gli1. C3H10T1/2 cells were incubated with 1.5 μM purmorphamine and DMSO, 1 μM vismodegib (Vismo) or compounds for 96 h prior to RT-qPCR. Data are mean values ± SD (n = 3). (e) Smoothened binding assay. HEK293T cells were transfected with a SMO-expressing plasmid and 48 h later cells were fixed and incubated with 5 nM BODIPY-cyclopamine (green) and treated with either DMSO (negative control), vismodegib (2 μM, positive control) or compounds at 30 μM or 10 μM for 4 h. The cells were stained with 4′,6-diamidino-2-phenylindole (DAPI, blue) to visualise the nuclei. Images are overlayed images of DAPI and BODIPY signals, representative of three biological replicates (n = 3). Scale bar: 50 μm. | ||
The Hh pathway is activated by the binding of Hh ligands to the trans-membrane receptor Patched1 (Ptch1). Upon binding, cellular internalisation of Ptch1 relieves the Ptch1-mediated inhibition of the Smoothened (SMO) protein, allowing for SMO translocation to the membrane, which triggers a signalling cascade that ultimately activates transcription of Hh target genes such as Ptch1 and glioma-associated oncogene homolog 1 (Gli1).14 The protein SMO is a clinically validated therapeutic target and many relevant Hh pathway inhibitors act by SMO antagonism, including vismodegib and cyclopamine, which specifically bind to the heptahelical bundle of SMO leading to pathway inhibition.22 To detect whether 3a also binds to SMO, a competitive SMO-binding assay was performed with a BODIPY-labelled derivative of the SMO binder cyclopamine.19 HEK293T cells transfected with a SMO-expressing plasmid were incubated with the compounds and BODIPY-cyclopamine at 5 nM. Less potent compound 3a did not decrease BODIPY-related fluorescence (Fig. 3e). In contrast, vismodegib and the potent enantiomer ent-3a decreased BODIPY fluorescence, indicating competition with cyclopamine for SMO binding and the displacement of BODIPY-cyclopamine from SMO (Fig. 3e). Thus, strong inhibitory activity exhibited by ent-3a on the Hedgehog pathway most likely is the result of direct binding to SMO.
Molecular docking was performed to determine the potential binding mode of ent-3a to SMO (Fig. S2†). Seven different crystal structures of human SMO (PDB ID: 4JKV, 4N4W, 4O9R, 4QIM, 4QIN, 5L7I and 5V56) were processed for the modelling. Only structure 5L7I could differentiate well between 3a and ent-3a according to the docking scores, which suggests that ent-3a may adopt a similar binding mode, as observed for the ligand vismodegib in 5L7I.23 The binding pose suggests that ent-3a occupies a space closer to the entrance of the pocket, while vismodegib resides deeper inside. For this model, the key interactions are hydrogen bonding between the ester carbonyl oxygen of ent-3a to Arg400 and of an amino proton to Gln477 (Fig. S2b†). Additionally, this binding pose suggests a stabilising π-stacking interaction with Phe484. This binding mode supports the substituent preference observed in the structure–activity relationship investigation (Table S2†). Any additional substituents at R3 would impair the π–π interaction, and only a small fluorine substitution at R3 afforded potency comparable to rac-3a (Table S2,† entry 9). Hydrogen bonding between ent-3a and SMO Gln477 proves to be vital for the binding, as acetylation of 3a totally abolishes activity (Table S2,† entry 16). The incompatibility of different substituents at R1 or R2 may be attributed to the rigidity of the scaffold, where a distant substitution leads to conformational change of the entire molecule.
SMO mutations have been associated with Hh pathway-driven oncogenicity and acquired resistance to treatment with drugs, limiting the clinical success of SMO antagonists.24 The first and only characterised mechanism of resistance to date is a mutation of Asp473 to a histidine (D473H), observed in a relapsed patient with metastatic medulloblastoma after vismodegib treatment.24b,25 It is believed that D473H mutation impairs the tight binding of vismodegib through an indirect pathway.26 The cation–π interaction between Arg400 and the pyridine ring of vismodegib is a key source of high affinity, where Asp473 forms a tight hydrogen bond with Arg400 (Fig. S2a†). Therefore, D473H mutation can disrupt the orientation of Arg400 for interaction and lead to loss of binding. In the model of SMO binding to ent-3a, Arg400 also forms a hydrogen bond with the ester motif, and thus for this compound it is most likely that a similar resistance might evolve. Combination therapies with downstream antagonists may be an option to circumvent such drug resistance.
The pharmacokinetic properties of vismodegib and ent-3a were compared by calculating their physicochemical properties using the webtool SwissADME.27 Notably, ent-3a resides in the physiochemical space predicted for good oral bioavailability (Fig. S3a†), while vismodegib has one violation in the dimension of INSATU (fraction of carbons in the sp3 hybridisation) due to its relatively high degree of saturation (Fig. S3b†). Considering the application of SMO inhibitors for medulloblastoma treatment, penetrating the blood–brain barrier (BBB) is another major challenge. While the calculation suggests that vismodegib may not permeate the BBB, for pseudo-NP ent-3a the prediction is different (Fig. S3a†), indicating that ent-3a may have more favourable properties.
The unsaturated pyrrolo[3,2-c]quinoline scaffold was previously reported as a lead chemotype for SMO binding.28 Therefore, the aromatised planar pyrrolo[3,2-c]quinoline 5a derived from 3a was also tested in the purmorphamine-induced osteogenesis assay. However, a more than five-fold decrease of inhibition was observed (Table S2,† entry 15), indicating that in the case under investigation, an increase in saturation is beneficial among these structurally related chemotypes. This observation may be regarded as an example that an “escape from flatland”1a,d may lead to more advantageous interactions between related small molecules and their cellular targets.
For comparison of the structural and chemical properties of ent-3a with previously described SMO antagonists, ent-3a and a reference set of 578 reported SMO antagonists (see ESI† for curation details)29 were subjected to cheminformatic analyses computed using the open-source software RDKit.30 Molecular shape was evaluated by a principal moments of inertia analysis.31 Most of the reported SMO antagonists have a rod-like shape, while ent-3a occupies a unique position with a more disc-like topology that is very sparsely populated by the reference set (Fig. S4a†). An NP-likeness score32 and a quantitative estimate of drug likeness (QED)33 were calculated and visualised in a two-dimensional plot to evaluate fragment compositions and drug-like properties, respectively. Ent-3a occupies an unpopulated area that has a high NP-likeness score and high QED score relative to the previously developed SMO antagonists (Fig. S4b†). This indicates that the pseudo-NP may be simultaneously more NP-like and drug-like than other SMO antagonists. Overall, these analyses suggest that the pseudo-NP ent-3a is characterised by a unique combination of molecular shape, atom connectivity and chemical properties relative to known SMO antagonists and represents a new chemotype for SMO inhibition.
Conclusions
In conclusion we have developed a new enantioselective intramolecular 1,3-dipolar cycloaddition for the combination of the pyrrolidine- and tetrahydroquinoline-NP fragments. The highly challenging asymmetric cycloaddition was enabled by the use of a AgOAc/(S)-DMBiphep catalyst, resulting in excellent yields and enantioselectivity. Exploration of the substrate scope revealed that enantioselectivity in the cycloaddition is decisively influenced by the site-specific introduction of a substituent. Biological evaluation of the resulting pseudo-NP collection revealed a novel Hh signalling inhibitor, whose activity was highly dependent on absolute configuration. Compound ent-3a binds to the SMO protein and, thereby, potently suppresses Hh target gene expression to an extent similar to the clinically approved SMO antagonist vismodegib. Cheminformatic analysis of ent-3a and reported SMO antagonists revealed that the pseudo-NP has unique properties and constitutes a new chemotype for SMO inhibition.Data availability
All experimental and characterisation data, as well as NMR spectra are available in the ESI.† Crystallographic data for compound rac-3j have been deposited in the Cambridge Crystallographic Data Centre under accession number CCDC 2243515.Author contributions
J. L. and R. R. Z. contributed equally. J. L. and H. W. conceived and directed the project. J. L. and S. M. performed the organic synthesis. J. L., R. R. Z., S. P., C. W. and J. F. performed the biological evaluation of the compound. A. K. and C. S. conducted the X-ray crystallography. C. G. and C. M. performed the computation of VCD spectra. J. L. performed molecular docking and the prediction of drug-like properties. A. P. and M. G. carried out the cheminformatic analysis. J. L., R. R. Z. and H. W. wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was supported by the Max-Planck-Gesellschaft. The authors are grateful to Dr Slava Ziegler for helpful discussions on biological characterisation. The authors acknowledge Compound Management and Screening Center (COMAS) for screening the compound collections. The figure for the graphical abstract was generated by BioRender.References
- (a) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed; (b) P. A. Clemons, N. E. Bodycombe, H. A. Carrinski, J. A. Wilson, A. F. Shamji, B. K. Wagner, A. N. Koehler and S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18787–18792 CrossRef CAS PubMed; (c) B. Over, S. Wetzel, C. Grütter, Y. Nakai, S. Renner, D. Rauh and H. Waldmann, Nat. Chem., 2013, 5, 21–28 CrossRef CAS PubMed; (d) F. Lovering, MedChemComm, 2013, 4, 515–519 RSC; (e) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- (a) G. Karageorgis, D. J. Foley, L. Laraia and H. Waldmann, Nat. Chem., 2020, 12, 227–235 CrossRef CAS PubMed; (b) G. Karageorgis, D. J. Foley, L. Laraia, S. Brakmann and H. Waldmann, Angew. Chem., Int. Ed., 2021, 60, 15705–15723 CrossRef CAS PubMed; (c) M. Grigalunas, S. Brakmann and H. Waldmann, J. Am. Chem. Soc., 2022, 144, 3314–3329 CrossRef CAS PubMed; (d) G. S. Cremosnik, J. Liu and H. Waldmann, Nat. Prod. Rep., 2020, 37, 1497–1510 RSC.
- (a) S. Wetzel, R. S. Bon, K. Kumar and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 10800–10826 CrossRef CAS PubMed; (b) H. van Hattum and H. Waldmann, J. Am. Chem. Soc., 2014, 136, 11853–11859 CrossRef CAS PubMed.
- (a) K. C. Morrison and P. J. Hergenrother, Nat. Prod. Rep., 2014, 31, 6–14 RSC; (b) S. E. Motika and P. J. Hergenrother, Nat. Prod. Rep., 2020, 37, 1395–1403 RSC; (c) Y. Li, S. Cheng, Y. Tian, Y. Zhang and Y. Zhao, Nat. Prod. Rep., 2022, 39, 1970–1992 RSC.
- (a) J. Adrio and J. C. Carretero, Chem. Commun., 2019, 55, 11979–11991 RSC; (b) T. Hashimoto and K. Maruoka, Chem. Rev., 2015, 115, 5366–5412 CrossRef CAS PubMed; (c) R. Narayan, M. Potowski, Z.-J. Jia, A. P. Antonchick and H. Waldmann, Acc. Chem. Res., 2014, 47, 1296–1310 CrossRef CAS PubMed.
- (a) D. Gao, T. Zhou, L.-T. Da, T. Bruhn, L.-L. Guo, Y.-H. Chen, J. Xu and M.-J. Xu, Org. Lett., 2019, 21, 8577–8581 CrossRef CAS PubMed; (b) A. Numata, C. Takahashi, Y. Ito, T. Takada, K. Kawai, Y. Usami, E. Matsumura, M. Imachi, T. Ito and T. Hasegawa, Tetrahedron Lett., 1993, 34, 2355–2358 CrossRef CAS.
- S. R. Vidadala, C. Golz, C. Strohmann, C.-G. Daniliuc and H. Waldmann, Angew. Chem., Int. Ed., 2015, 54, 651–655 CAS.
- J. Liu, F. Otte, C. Strohmann and H. Waldmann, Tetrahedron Lett., 2021, 76, 153228 CrossRef CAS.
- J. Liu, G. S. Cremosnik, F. Otte, A. Pahl, S. Sievers, C. Strohmann and H. Waldmann, Angew. Chem., Int. Ed., 2021, 60, 4648–4656 CrossRef CAS PubMed.
- C. E. Masse, P. Y. Ng, Y. Fukase, M. Sánchez-Roselló and J. T. Shaw, J. Comb. Chem., 2006, 8, 293–296 CrossRef CAS PubMed.
- (a) M. Nyerges, I. Fejes and L. Tõke, Synthesis, 2002, 2002, 1823–1828 CrossRef; (b) P. Mahto, K. Shukla, A. Das and V. K. Singh, Tetrahedron, 2021, 87, 132115 CrossRef CAS; (c) H. Xu, S. J. Zuend, M. G. Woll, Y. Tao and E. N. Jacobsen, Science, 2010, 327, 986–990 CrossRef CAS PubMed; (d) Y.-J. Yang, H.-R. Zhang, S.-Y. Zhu, P. Zhu and X.-P. Hui, Org. Lett., 2014, 16, 5048–5051 CrossRef CAS PubMed; (e) J. Wang, Y. Li, J. Sun, H. Wang, Z. Jin and Y. R. Chi, ACS Catal., 2018, 8, 9859–9864 CrossRef CAS; (f) S. G. Davies, A. M. Fletcher, J. A. Lee, T. J. A. Lorkin, P. M. Roberts and J. E. Thomson, Org. Lett., 2013, 15, 2050–2053 CrossRef CAS PubMed.
- C. Cristóbal, D. Gaviña, I. Alonso, M. Ribagorda, J. C. Carretero, C. del Pozo and J. Adrio, Chem. Commun., 2022, 58, 7805–7808 RSC.
- C. Merten, T. P. Golub and N. M. Kreienborg, J. Org. Chem., 2019, 84, 8797–8814 CrossRef CAS PubMed.
- J. Briscoe and P. P. Thérond, Nat. Rev. Mol. Cell Biol., 2013, 14, 416–429 CrossRef PubMed.
- C. Raffel, R. B. Jenkins, L. Frederick, D. Hebrink, B. Alderete, D. W. Fults and C. D. James, Cancer Res., 1997, 57, 842–845 CAS.
- J. Xie, M. Murone, S. M. Luoh, A. Ryan, Q. Gu, C. Zhang, J. M. Bonifas, C. W. Lam, M. Hynes, A. Goddard, A. Rosenthal, E. H. Epstein Jr and F. J. de Sauvage, Nature, 1998, 391, 90–92 CrossRef CAS PubMed.
- U. Tostar, C. J. Malm, J. M. Meis-Kindblom, L.-G. Kindblom, R. Toftgård and A. B. Undén, J. Pathol., 2006, 208, 17–25 CrossRef CAS PubMed.
- (a) S. Peukert and K. Miller-Moslin, ChemMedChem, 2010, 5, 500–512 CrossRef CAS PubMed; (b) D. Quaglio, P. Infante, L. Di Marcotullio, B. Botta and M. Mori, Expert Opin. Ther. Pat., 2020, 30, 235–250 CrossRef CAS PubMed.
- S. Sinha and J. K. Chen, Nat. Chem. Biol., 2006, 2, 29–30 CrossRef CAS PubMed.
- J. Taipale, J. K. Chen, M. K. Cooper, B. Wang, R. K. Mann, L. Milenkovic, M. P. Scott and P. A. Beachy, Nature, 2000, 406, 1005–1009 CrossRef CAS PubMed.
- K. D. Robarge, S. A. Brunton, G. M. Castanedo, Y. Cui, M. S. Dina, R. Goldsmith, S. E. Gould, O. Guichert, J. L. Gunzner, J. Halladay, W. Jia, C. Khojasteh, M. F. T. Koehler, K. Kotkow, H. La, R. L. LaLonde, K. Lau, L. Lee, D. Marshall, J. C. Marsters, L. J. Murray, C. Qian, L. L. Rubin, L. Salphati, M. S. Stanley, J. H. A. Stibbard, D. P. Sutherlin, S. Ubhayaker, S. Wang, S. Wong and M. Xie, Bioorg. Med. Chem. Lett., 2009, 19, 5576–5581 CrossRef CAS PubMed.
- J. K. Chen, J. Taipale, M. K. Cooper and P. A. Beachy, Genes Dev., 2002, 16, 2743–2748 CrossRef CAS PubMed.
- E. F. X. Byrne, R. Sircar, P. S. Miller, G. Hedger, G. Luchetti, S. Nachtergaele, M. D. Tully, L. Mydock-McGrane, D. F. Covey, R. P. Rambo, M. S. P. Sansom, S. Newstead, R. Rohatgi and C. Siebold, Nature, 2016, 535, 517–522 CrossRef CAS PubMed.
- (a) H. J. Sharpe, W. Wang, R. N. Hannoush and F. J. de Sauvage, Nat. Chem. Biol., 2015, 11, 246–255 CrossRef CAS PubMed; (b) R. L. Yauch, G. J. P. Dijkgraaf, B. Alicke, T. Januario, C. P. Ahn, T. Holcomb, K. Pujara, J. Stinson, C. A. Callahan, T. Tang, J. F. Bazan, Z. Kan, S. Seshagiri, C. L. Hann, S. E. Gould, J. A. Low, C. M. Rudin and F. J. de Sauvage, Science, 2009, 326, 572–574 CrossRef CAS PubMed.
- (a) C. M. Rudin, C. L. Hann, J. Laterra, R. L. Yauch, C. A. Callahan, L. Fu, T. Holcomb, J. Stinson, S. E. Gould, B. Coleman, P. M. LoRusso, D. D. Von Hoff, F. J. de Sauvage and J. A. Low, N. Engl. J. Med., 2009, 361, 1173–1178 CrossRef CAS PubMed; (b) R. L. Yauch, G. J. P. Dijkgraaf, B. Alicke, T. Januario, C. P. Ahn, T. Holcomb, K. Pujara, J. Stinson, C. A. Callahan, T. Tang, J. F. Bazan, Z. Kan, S. Seshagiri, C. L. Hann, S. E. Gould, J. A. Low, C. M. Rudin and F. J. de Sauvage, Science, 2009, 326, 572–574 CrossRef CAS PubMed.
- H. J. Sharpe, G. Pau, G. J. Dijkgraaf, N. Basset-Seguin, Z. Modrusan, T. Januario, V. Tsui, A. B. Durham, A. A. Dlugosz, P. M. Haverty, R. Bourgon, J. Y. Tang, K. Y. Sarin, L. Dirix, D. C. Fisher, C. M. Rudin, H. Sofen, M. R. Migden, R. L. Yauch and F. J. de Sauvage, Cancer Cell, 2015, 27, 327–341 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- T. Ohashi, Y. Oguro, T. Tanaka, Z. Shiokawa, S. Shibata, Y. Sato, H. Yamakawa, H. Hattori, Y. Yamamoto, S. Kondo, M. Miyamoto, H. Tojo, A. Baba and S. Sasaki, Bioorg. Med. Chem., 2012, 20, 5496–5506 CrossRef CAS PubMed.
- M. Grigalunas, S. Patil, A. Krzyzanowski, A. Pahl, J. Flegel, B. Schölermann, J. Xie, S. Sievers, S. Ziegler and H. Waldmann, Chem.–Eur. J., 2022, 28, e202202164 CrossRef CAS PubMed.
- RDKit: open-source cheminformatics, http://www.rdkit.org.
- W. H. B. Sauer and M. K. Schwarz, J. Chem. Inf. Comput., 2003, 43, 987–1003 CrossRef CAS PubMed.
- P. Ertl, S. Roggo and A. Schuffenhauer, J. Chem. Inf. Model., 2008, 48, 68–74 CrossRef CAS PubMed.
- G. R. Bickerton, G. V. Paolini, J. Besnard, S. Muresan and A. L. Hopkins, Nat. Chem., 2012, 4, 90–98 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01240a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |