
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceResurgence and advancement of photochemical hydrogen atom transfer processes in selective alkane functionalizations
Liang
Chang
 a,
Shun
Wang
b,
Qing
An
b,
Linxuan
Liu
b,
Hexiang
Wang
b,
Yubo
Li
b,
Kaixuan
Feng
b and
Zhiwei
Zuo
*b
a,
Shun
Wang
b,
Qing
An
b,
Linxuan
Liu
b,
Hexiang
Wang
b,
Yubo
Li
b,
Kaixuan
Feng
b and
Zhiwei
Zuo
*b
aSchool of Pharmacy, Nanjing University of Chinese Medicine, Nanjing 210023, China
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China. E-mail: zuozhw@sioc.ac.cn
First published on 22nd May 2023
Abstract
The selective functionalization of alkanes has long been recognized as a prominent challenge and an arduous task in organic synthesis. Hydrogen atom transfer (HAT) processes enable the direct generation of reactive alkyl radicals from feedstock alkanes and have been successfully employed in industrial applications such as the methane chlorination process, etc. Nevertheless, challenges in the regulation of radical generation and reaction pathways have created substantial obstacles in the development of diversified alkane functionalizations. In recent years, the application of photoredox catalysis has provided exciting opportunities for alkane C–H functionalization under extremely mild conditions to trigger HAT processes and achieve radical-mediated functionalizations in a more selective manner. Considerable efforts have been devoted to building more efficient and cost-effective photocatalytic systems for sustainable transformations. In this perspective, we highlight the recent development of photocatalytic systems and provide our views on current challenges and future opportunities in this field.
Introduction
Simple alkanes constitute one of the Earth's most abundant reserves of carbon-based feedstock, providing the basic starting materials for chemical production.1–4 Equipped with chemically inert C(sp3)–H bonds and C(sp3)–C(sp3) bonds, alkanes are traditionally transformed into reactive building blocks such as alkenes, alkyl halides, and carbonyl compounds to construct polymeric materials, pharmaceuticals, and agrochemicals.5 The direct conversion of simple alkanes into high-value chemicals would make syntheses more sustainable in terms of atom and step-economy as well as ecological perspectives; this has become a long-standing goal in the synthetic community.6–8 Moreover, the recent increasing attention to reduce emissions has raised a compelling need for sustainable methodologies to utilize hydrocarbons that are otherwise wasted by gas flare.9,10 Nevertheless, the intrinsic inertness of C(sp3)–H bonds in simple alkanes and the susceptibility of functionalized products to overoxidation have presented formidable challenges in the development of selective transformations.11–13 Despite tremendous progress in the selective functionalization of C(sp3)–H bonds assisted by the direction or proximal functional groups, selective functionalization of simple and abundant alkanes remains underdeveloped and continues to provide empowering opportunities.Hydrogen atom transfer (HAT), an elementary and ubiquitous process in chemical reactions and biological systems, can provide a conceptually distinct and synthetically useful strategy to cleave C(sp3)–H bonds of feedstock alkanes.14–17 With the suitable choice of a HAT agent, such as halogen radicals and oxygen- or nitrogen-centered radicals, alkanes can be converted into highly reactive, nucleophilic carbon-centered radicals to enable functional group installation. For example, Hoechst methane chlorination, established in 1923, remains a state-of-the-art process for producing chloromethane at multiple millions of tons per year.18 Although, in principle, the resultant alkyl radical could undergo various additions, the radical chain mechanism and excess radical-generating reagents X2 and tert-butyl hydroperoxide (TBHP) have created substantial obstacles in the development of diversified functionalizations in the traditional realm of radical chemistry. The generation of heteroatom centered radicals via high energy UV irradiation or high temperature conditions also poses difficulties in functional group compatibility. Additionally, the rather limited access to structurally diversified HAT agents has imposed severe limitations on the tuning of reactivity and selectivity, despite our understanding of the critical factors that govern the reactivity and selectivity of HAT processes.16,19–21
Although appealing, the utilization of HAT for selective alkane functionalization became a focus of research only in the last decade. The recent emergence of photoredox catalysis has provided a versatile platform to exploit radical intermediates and achieve otherwise unattainable transformations under mild conditions.22–26 In striking contrast to the radical chain mechanism, photoinduced electron-transfer processes generate radical intermediates in a controllable fashion, offering unique opportunities to develop novel and selective transformations. Notably, the direct generation of heteroatom-centered radical X from H–X or X− (X = Cl, NR2, OR, etc.) could enable the catalytic employment of various operationally simple materials such as HAT agents.27–29 Although the generation of heteroatom-centered radicals such as a chlorine radical, and aminium and alkoxy radicals is among the most difficult to achieve due to the high oxidation potential of X–H or X−, highly oxidizing photocatalysts and catalytic strategies such as proton-coupled electron transfer (PCET) and ligand-to-metal charge transfer (LMCT) have provided viable solutions.30,31
Radical-mediated additions or radical trapping events during the bond-forming step of the HAT activation manifold have provided a facile and popular approach for installing functional groups. Although effective in the interception of reactive alkyl radicals, the capacity to expand reaction scope and achieve impactful selective oxidation, dehydrogenation, and selectivity remains limited. Nature provides inspiration to overcome such limitations. Methane monooxygenase employs HAT to generate a methyl radical and more importantly creates a radical rebound mechanism with iron clusters to generate methanol with high selectivity, avoiding nonselective free-radical aerobic oxidation (Scheme 1).11,32 Metallaphotoredox provides a unique strategy to enforce the interaction of alkyl radicals with transition-metal complexes for various cross-coupling reactions.33–36 The synergistic incorporation of radical-mediated HAT processes and bond-forming steps enabled by transition metals may provide new opportunities to address challenging problems such as alkane metathesis and dehydrogenation reactions. Moreover, the utilization of photonic energy provides bonus energy input to enable endothermic alkane transformations.37,38
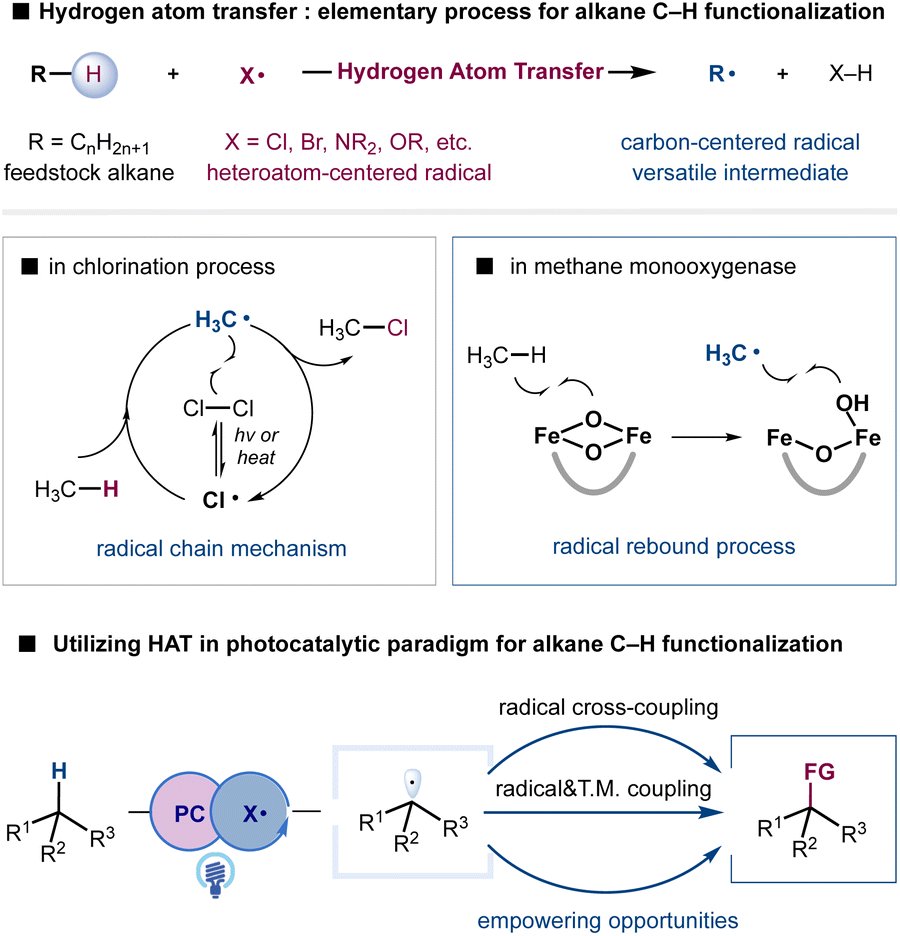 | ||
| Scheme 1 Functionalization of C(sp3)–H bonds via HAT. | ||
The last decade has witnessed a considerable amount of reports on photocatalytic transformations enabled by HAT processes, and several comprehensive reviews have been published,16,39–46 although such discussions are typically dominated by substrates with activated C(sp3)–H bonds rather than simple hydrocarbons. Considering the critical importance and recent research interest in alkane-upgrading conversions, we found it valuable to provide a brief review that exclusively focuses on advancements made with feedstock alkanes. This perspective briefly summarizes the factors that govern HAT event reactivity and selectivity. We also provide an overview of the key advances and potential impact of photocatalytic or photochemical alkane functionalization. Most reactions constitute simple hydrocarbon substrates or functionalized substrates with more activated C–H bonds; however, in this review, only the results of feedstock alkanes are discussed. We end the review by offering insights into the challenges and opportunities of this field. The nature of heteroatom-centered radicals typically plays a central role in functionalization; hence, our content is organized along the different radical or radical-like species to provide a clear comparison.
Governing factors of hydrogen atom transfer
Bond strength is a crucial regulator of radical-mediated HAT reactivities. Furthermore, the strengths of both the bond being broken and the bond being formed are closely correlated with the HAT rate, described as the Evans–Polanyi relationship.19,47 This empirically extrathermodynamic relationship could be defined as the reaction driving force, indicating that a reaction becomes faster if it is more exothermic. For C–H bonds in alkanes, a decrease in bond strength generally leads to a lower activation energy, enhancing the rate of the HAT step, even for some cases when the overall HAT event is endothermic. An example is the HAT event between a bromine radical (bond dissociation energy (BDE) of Br–H: 87 kcal mol−1) and tertiary C–H bonds (BDE of C–H: 97 kcal mol−1). The BDE values of structurally diversified X–H and C–H bonds are illustrated in Scheme 2A.20,48–51 Compared to the C–H bonds adjacent to the functional groups, the C–H bonds in alkanes bear higher BDE values. The thermodynamic stability of the alkyl radicals follows a declining trend of tertiary radicals > secondary radicals > primary radicals > methyl radicals. This trend is due to the hyperconjugation effect as the electron density of the C–C or C–H σ bond overlaps with the half-filled orbital to stabilize the alkyl radicals. The BDE values of X–H bonds in alcohols, HCl, and amide are among the highest, providing a sufficient thermodynamic driving force for almost any aliphatic C–H bond in alkanes.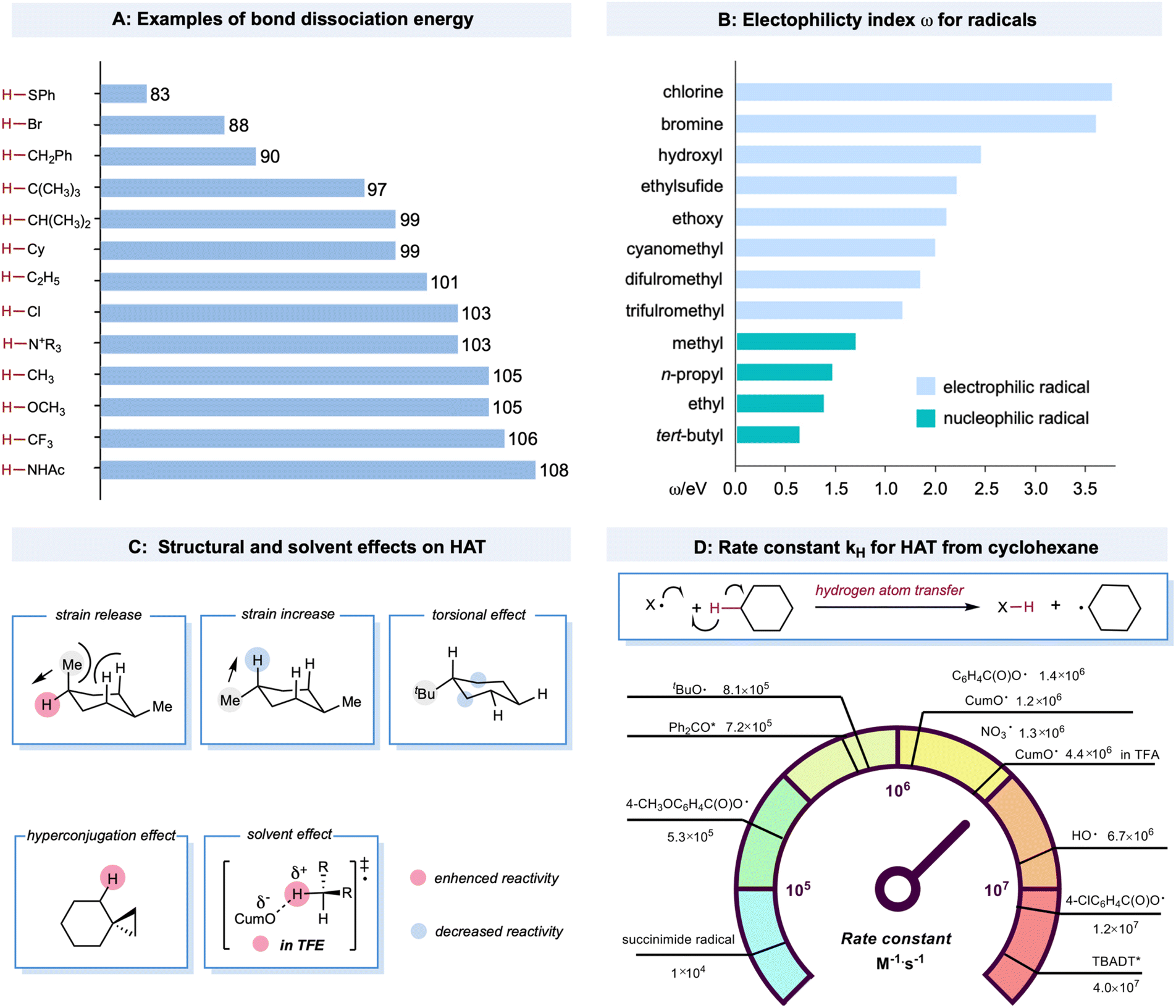 | ||
| Scheme 2 Factors that govern HAT from alkanes. | ||
HAT rates and selectivities are also dictated by polar effects that can override the preference of HAT for the weakest bonds.52 These trends can be well explained using empirically defined electrophilic or nucleophilic radicals that depend on the relatively higher or lower electron density of the open-shell species. Electron-rich C(sp3)–H bonds of alkanes are much more readily abstracted by electrophilic radicals, such as chlorine radicals, rather than nucleophilic methyl radicals, although the driving forces in bond strengths are comparable. A more theoretical mechanistic interpretation for this HAT bias was the significantly lower transition-state energy when the electron-deficient character of the radicals increased.20,53–55 Similarly, increasing electron-withdrawing substituents on the open-shell species could accelerate the HAT process with the electron-rich C(sp3)–H bond in alkanes. In contrast, the electron-rich character elicits the opposite effect. For example, in a kinetic study of the HAT event between cyclohexane and aryloxy radicals with different substituents (Scheme 2D), a significant rate enhancement was observed for 4-ClC6H4C(O)O˙ (kH = 1.2 × 107 M−1 s−1 for 4-ClC6H4C(O)O˙ and kH = 1.4 × 106 M−1 s−1 for C6H5C(O)O˙) in contrast to 4-CH3OC6H4C(O)O˙, which is 1–2 orders of magnitude slower (4-CH3OC6H4C(O)O˙, kH = 5.3 × 105 M−1 s−1).56,57 De Proft and co-workers systematically investigated the electrophilicity index for various radicals (Scheme 2B).58
HAT reactivity and selectivity on alkanes can also be tuned by structural and solvent effects (Scheme 2C).20 Bietti and co-workers observed increased HAT rates on equatorial tertiary sites of 1,4-dimethyl cyclohexane based on the release of a 1,3-diaxial strain related to the planarization of the carbon-centered radical thus formed.59,60 Based on this logic, the increased torsional strain could deactivate the tertiary axial C–H bond. For tert-butyl cyclohexane, the HAT event could selectively occur from the β and γ positions to the tertiary carbon center on the ring due to the deactivation of the proximal C–H bond and the decrease in kH by torsional effects.21,61 Hyperconjugative effects could increase the C(sp3)–H bond vicinal to the cyclopropyl ring. This could be explained by the overlap between the cyclopropyl C–C σ-bonding orbital and the proximal C–H antibonding orbital to activate the adjacent position.62,63 Solvent effects are important factors that affect HAT reactivity. Bietti and co-workers discovered that the kH values of HAT between CumO˙ and cyclohexane were enhanced in protic solvent trifluoroethanol (TFE) (kH = 4.4 × 106 M−1 s−1) compared to in cyclohexane (kH = 1.2 × 106 M−1 s−1). This effect is associated with the transition state in which TFE hydrogen bonds to the oxygen atom to provide greater stabilization.64,65
Despite challenges in the kinetic study of radical-mediated HAT with alkane substrates, several rate constants for HAT from cyclohexane, using radicals or excited species, have provided valuable kinetic data. A brief comparison of rate constants (kH) for HAT between cyclohexane and various radials or radical-like species is shown in Scheme 2D. The excited decatungstate salts66 offer the highest efficiency in the homolytic activation of C–H bonds, which are two orders of magnitude greater than that of excited ketones.67,68 The alkoxy radicals have comparable rates, mostly between 9.6 × 105 M−1 s−1 and 6.7 × 106 M−1 s−1.20,64,69–72 The kH values of a succinimide radical is relatively low (1 × 104 M−1 s−1).73
Transformations mediated by halogen radicals
The chlorine radical is one of the oldest and perhaps the most well-understood tools for photochemical alkane functionalization and is still being used for commercial-scale methane chlorination.2,74–76 The utilization of a chlorine radical in alkane functionalization has a rich history since the pioneering discovery of the photochlorination of natural gas using Cl2 by Dumas in 1840.77 A chlorine radical abstracts a hydrogen atom from electron-rich C–H bonds at fast rates; however, unpronounced selectivities are generally achieved between different C–H bonds in hydrocarbons, which could be explained by the overall similar HAT rate constants between primary, secondary, and tertiary C–H bonds.1,78 Traditionally, chlorine radicals are generated in an elementary chlorine system via thermolysis or photolysis of weak Cl–Cl bonds. Simultaneously, industrial utilization of Cl2 is cost-competitive (setting aside environmental and health concerns), and a stoichiometric excess of Cl2 is a well-known radical trapping reagent, rendering this method useful only for C–H chlorination, preventing its use for diversified alkane functionalization.Much effort has been expended to build upon photocatalytic or photochemical chlorine-radical generation under mild reaction conditions, including (a) photochemical conditions with a stoichiometric oxidant; (b) single electron oxidation of Cl− by a photoexcited catalyst or its photoexcited radical cation;79 and (c) LMCT catalysis, via metal–chlorine bond homolysis. Theoretically, chloride as a chlorine-radical precursor in chlorine-radical-mediated reactions is expected to be the most atom-efficient and usually catalytic, while oxidative inorganic chloro-compounds are generally applicable in alkane oxidation.
Photochemical conditions have been recently developed for the partial oxidation of gaseous alkanes into value-added liquid products (Scheme 3A). One of the major challenges in partial alkane oxidation is the overoxidation of weak bonds in the products.8,80 Thus, a product protection strategy is generally adopted in partial methane oxidation by converting the product into stable compounds, more commonly via solvation in strongly acidic systems8,81 to achieve superior selectivity. Another tactic is physically separating products from the reaction mixture once it is formed, such as in membrane-based techniques.82,83 Ohkubo and co-workers reported a photochemical method for partially oxidizing methane to formic acid and methanol under ambient conditions (298 K and 1 atm) using NaClO2 as an oxidant.84 This study exploited a perfluorohexane and water two-phase system to physically protect the highly susceptible products-formic acid and methanol-via in situ phase separation from the oxidant in the organic phase. The key step is the photolysis of the in situ-formed ClO2, generating the chlorine radical along with the oxidant O2. This physical protection enabled 99% conversion of methane to formic acid (85%) and methanol (14%) with excellent selectivity. Apart from physical protection, in situ solvation is the most common strategy for protecting susceptible products. Drawing upon their previously established thermal alkane oxidation conditions,85,86 Groves, Gunnoe, and co-workers reported a photochemical light alkane oxidation protocol (Scheme 3A).87 This method used stoichiometric ammonium iodate as the oxidant, chloride as the catalyst, trifluoroacetic acid as the solvent, and a reagent at room temperature. Under batch conditions, mercury lamp irradiation produces a high yield of methyl or ethyl trifluoracetic ester (∼50% based on methane and ∼60% based on ethane) with comparable selectivities (>95% toward alkyl ester versus alkyl chloride byproduct) but much slower reaction times when compared to thermal conditions. A kinetic isotope effect of 1.06 was obtained in a competition experiment between cyclohexane and cyclohexane d12, unlike a previous thermal protocol but is consistent with other examples of chlorine-radical HAT. Based on these results, the authors proposed a chlorine-radical-mediated HAT mechanism. Recently, Schelter, Goldberg, and co-workers demonstrated an aerobic methane iodination reaction (Scheme 3A).88 In stop-flow microtubing charged with high-pressure methane (500 psi) and an air-saturated MeCN/H2O solution of a stoichiometric amount of I2 and a catalytic loading of n-Bu4NCl, UV irradiation yielded a maximum turnover number (TON) of 10 (relative to chloride). The authors proposed that photolysis of the electron donor–acceptor (EDA) complex of I2 and Cl− initiated the chlorine radical. In addition, I2 was the radical trapping reagent and a redox mediator; however, they did not conclusively demonstrate an efficient yield enhancement role for water.
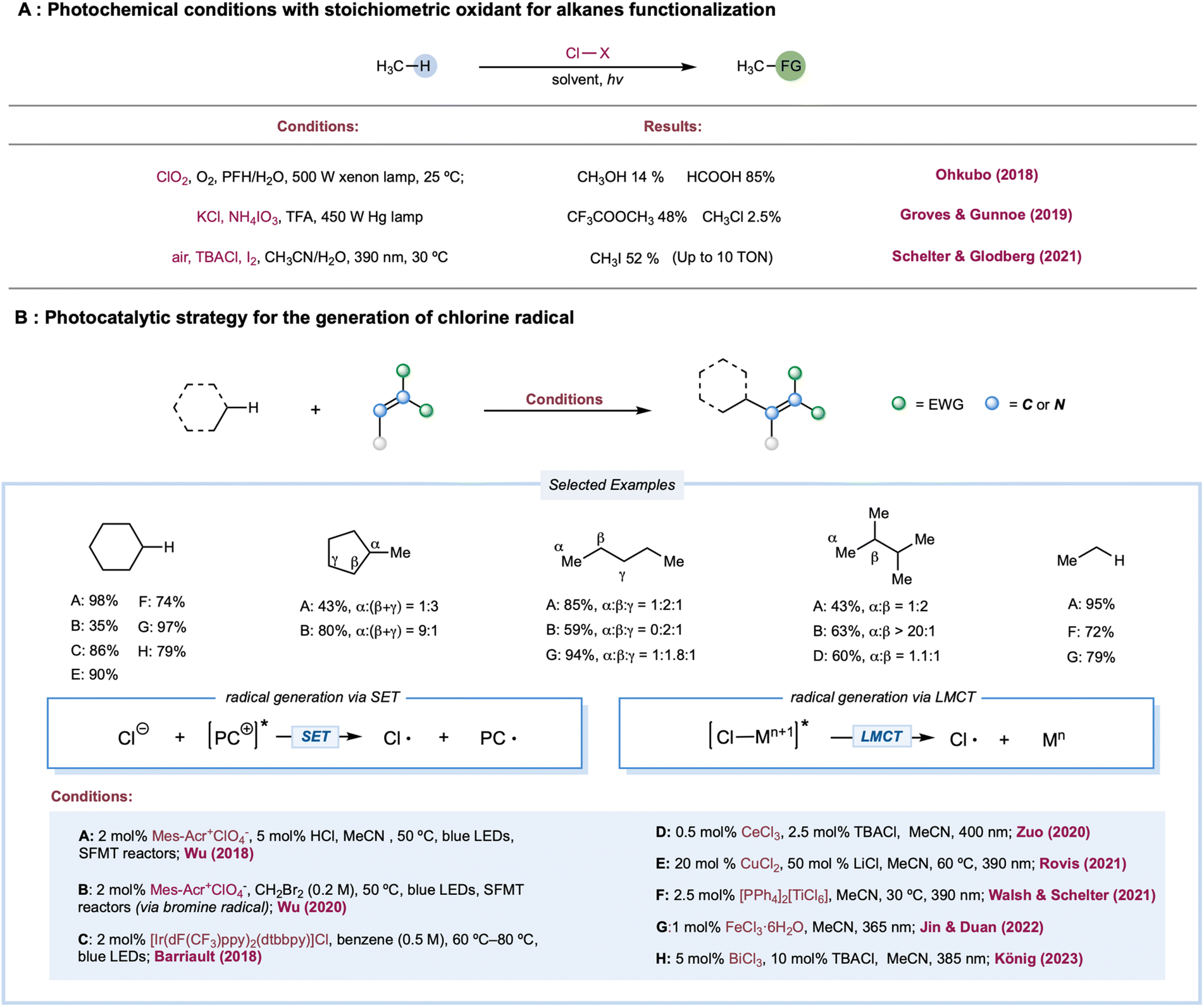 | ||
| Scheme 3 Photochemical and photocatalytic transformations mediated by halogen radicals. | ||
A cationic photocatalyst with high redox potential could oxidize Cl− to Cl˙ [E1/2(Cl˙/Cl−) = +2.03 V versus SCE]. In 2011, Fukuzumi and co-workers disclosed the first photocatalytic method for generating Cl˙ from Cl− by excited Mes–Acr–Me+* [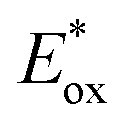 (Mes–Acr–Me+*/Mes–Acr–Me˙) = +2.06 V versus SCE].89 Using this method, they reported C–H bond oxidation of cycloalkanes in the presence of O2 and HCl. Oxygenated products cycloalkanols and cyclic ketones were formed with a TON of 7. In two simultaneous reports, Wu and Barriault reported chlorine-radical-mediated C–H alkylation (Scheme 3B).90,91 Both reports detailed the strategy of catalytic loading of chloride as the chlorine-radical precursor. Wu and co-workers employed a photoexcited Fukuzumi catalyst (Mes–Acr–Me+*) to generate chlorine radicals from HCl in a microtubing reactor.90 With methylene malononitrile radical acceptors, this approach enabled efficient Giese reactions toward secondary C–H bonds in simple cycloalkane substrates, producing good to excellent yields of alkylated malononitrile. Good product yields were obtained for linear alkanes such as pentane, although site-selectivity was unremarkable. Additionally, allyl sulfones proved to be good alkyl radical traps to afford the C–H alkylated products. The SET event between the photoexcited Ir[dF(CF3)ppy]2(dtbbpy)+ catalysts and its chloride counter anion was explored for chlorine atom generation.91 Although moderate regioselectivity was observed with adamantane, alkylated products from cycloalkanes are generally obtained in good yields. Compared to the chlorine radical, the HAT reactivities of a bromine radical decrease while selectivity increases toward tertiary C–H bonds, correlating with the stabilities of the resulting alkyl radicals. Recently, Wu and co-workers developed a photocatalytic alkane alkylation protocol using a bromine radical as a hydrogen atom abstractor.92 Dibromomethane was employed as the solvent and bromine radical precursor. Accordingly, a broad range of alkanes was converted to their corresponding alkylated products in good yields, with highly tertiary C–H bond selectivities.
(Mes–Acr–Me+*/Mes–Acr–Me˙) = +2.06 V versus SCE].89 Using this method, they reported C–H bond oxidation of cycloalkanes in the presence of O2 and HCl. Oxygenated products cycloalkanols and cyclic ketones were formed with a TON of 7. In two simultaneous reports, Wu and Barriault reported chlorine-radical-mediated C–H alkylation (Scheme 3B).90,91 Both reports detailed the strategy of catalytic loading of chloride as the chlorine-radical precursor. Wu and co-workers employed a photoexcited Fukuzumi catalyst (Mes–Acr–Me+*) to generate chlorine radicals from HCl in a microtubing reactor.90 With methylene malononitrile radical acceptors, this approach enabled efficient Giese reactions toward secondary C–H bonds in simple cycloalkane substrates, producing good to excellent yields of alkylated malononitrile. Good product yields were obtained for linear alkanes such as pentane, although site-selectivity was unremarkable. Additionally, allyl sulfones proved to be good alkyl radical traps to afford the C–H alkylated products. The SET event between the photoexcited Ir[dF(CF3)ppy]2(dtbbpy)+ catalysts and its chloride counter anion was explored for chlorine atom generation.91 Although moderate regioselectivity was observed with adamantane, alkylated products from cycloalkanes are generally obtained in good yields. Compared to the chlorine radical, the HAT reactivities of a bromine radical decrease while selectivity increases toward tertiary C–H bonds, correlating with the stabilities of the resulting alkyl radicals. Recently, Wu and co-workers developed a photocatalytic alkane alkylation protocol using a bromine radical as a hydrogen atom abstractor.92 Dibromomethane was employed as the solvent and bromine radical precursor. Accordingly, a broad range of alkanes was converted to their corresponding alkylated products in good yields, with highly tertiary C–H bond selectivities.
In 1983, a pioneering work by Condorelli and co-workers described LMCT excitation of CeCl62− to generate chlorine radicals.93 In a systematic study of photocatalytic C–H functionalization, Zuo and co-workers reported the amination of 2,3-dimethylbutane from the catalytic combination of CeCl3 and TBACl (Scheme 3B).94 However, the cerium-LMCT-generated chlorine radical was not chosen as the HAT reagent due to its low observed efficiency (with a low quantum efficiency of 0.03–0.05) and poor site-selectivity, compared to the better results obtained with a methoxy radical. Subsequently, Schelter, Walsh, and co-workers extended the alkane scope of the chlorine-radical-mediated alkane amination protocol with CeCl62− as the enabling catalyst.95 Recently, a seminal communication by Rovis and co-workers detailed a photocatalytic C–H alkylation reaction using copper salt as a photocatalyst (Scheme 3B).96 A chlorine radical was generated from an LMCT excited state of CuCl3− formed via in situ coordination of simple chloride salt and CuCl2. Linear alkanes proved to be effective substrates in this protocol, providing good yields with a mixture of regioisomers. Moreover, various electron-deficient alkenes and di-tert-butyl azodicarboxylate were demonstrated to be capable radical acceptors for this catalytic manifold. Analogous LMCT strategies employing other homoleptic metal–chloride complexes, including iron,97–100 titanium,101 and bismuth,102 have also been recently reported (Scheme 3B).
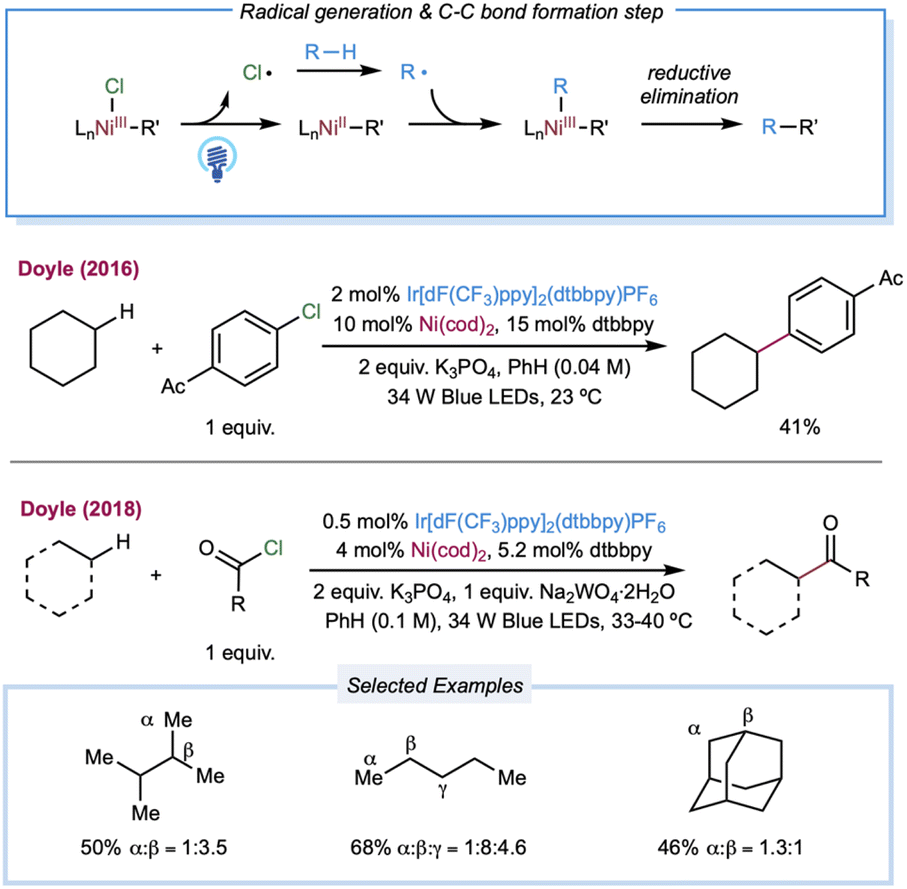
In 2016, Doyle and co-workers reported the first chlorine-radical-mediated C(sp3)–H cross-coupling reaction with aryl chloride as the coupling partner (Scheme 4).103 The proposed mechanism is the halogen photoelimination pathway,104 wherein the key step is the charge transfer of the photoexcited Ni(III)–Cl complex, in which Ni(III)–Cl bond homolysis yields a Ni(II) complex and chlorine radical. This mechanism is supported by quenching studies and stoichiometric experiments using an isolable Ni(II) aryl chloride complex. By controlling the parameters of the oxidant and light, it was concluded that photoelimination occurs on the Ni(III)–Cl complex. A large excess of cyclohexane (10 equiv.) is required to provide a synthetically valuable yield due to the chlorine-radical-mediated HAT between the unactivated alkane C–H bond (BDE = 100 kcal mol−1) and the weaker benzylic C–H bond of the product (BDE = 89 kcal mol−1). This problem could be addressed by employing electro-withdrawing coupling partners. To this end, chloroformates were explored as coupling partners in a C(sp3)–H formylation protocol (Scheme 4).105 Moderate to good yields of ester derivatives were obtained over a broader cycloalkane substrate scope, which covered ring sizes between 5 and 15. For linear alkanes, such as pentane and 2-methyl butane, the C–H formylation regioselectivity is poor, which is in accordance with the prior studies of Cl˙ mediated HAT. Hong and co-workers further expanded the scope of coupling partners with N-acylsuccinimides106 and in situ-formed N-acylpyridinium compounds107 by oxidative addition of nickel into amide C–N bonds.
 | ||
| Scheme 4 Nickel-catalyzed C(sp3)–H cross coupling enabled by catalytic generation of a chlorine radical. | ||
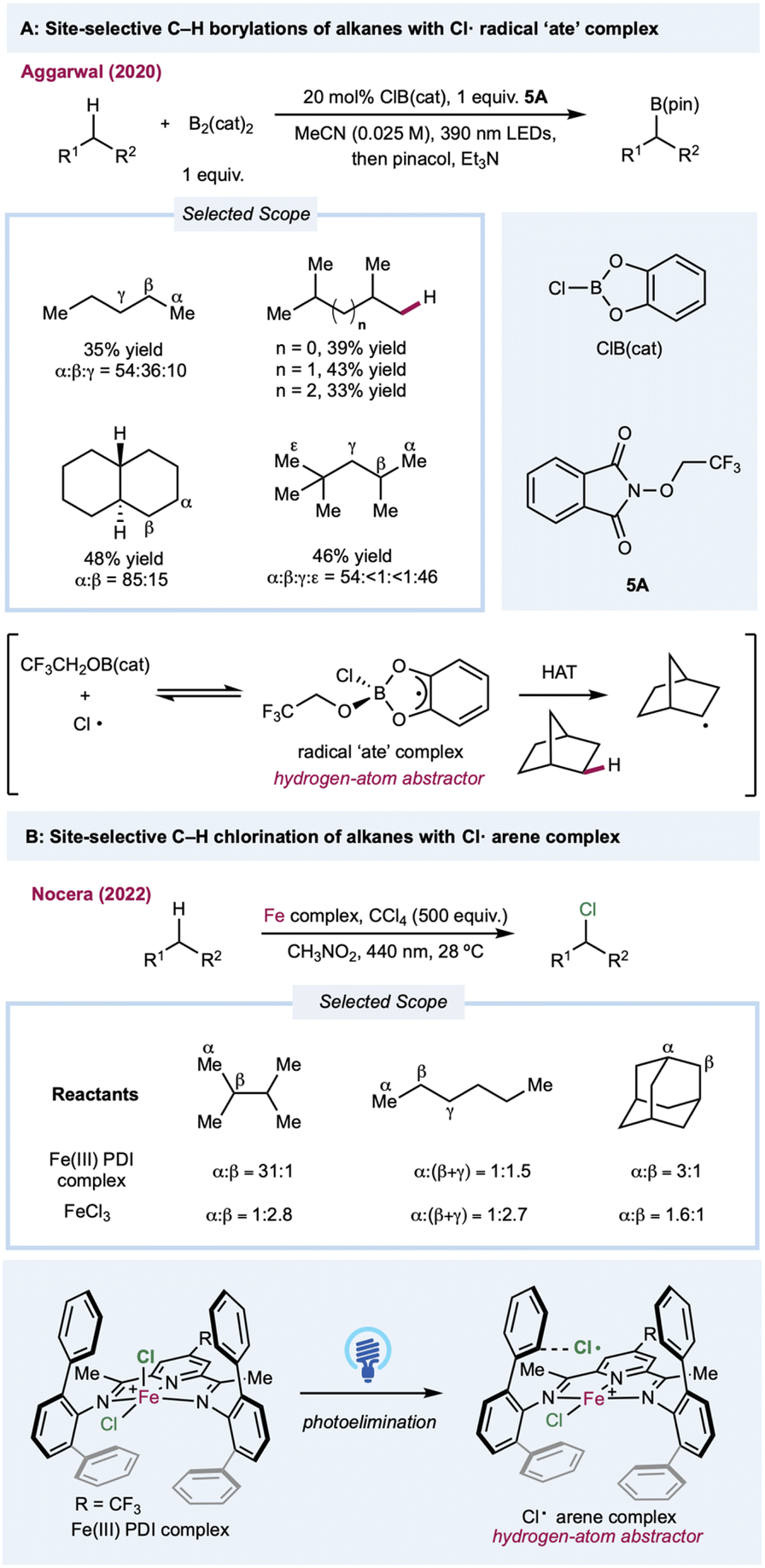
In most cases, the site selectivities of chlorine-radical-mediated protocols are generally unsatisfying. Thus, methods with inherently tuned site-selectivity would be extremely appealing. In 2020, Aggarwal and co-workers described a seminal chlorine-radical-mediated photocatalytic C(sp3)–H borylation reaction (Scheme 5A).108 Using stoichiometric amounts of B2cat2 and alkoxy phthalimide with a catalytic amount of CIB(cat), this metal-free protocol provides a unique borylation reaction preferentially skewed toward primary C–H bonds over weaker secondary or tertiary C–H bonds. Under 390 nm LED irradiation, SET between photoexcited trifluoroethoxy phthalimide and B2(cat)2 generates a trifluoroethoxy radical. The sterically demanding HAT agent radical “ate” complex forms via the addition of a trifluoroethoxy radical with CIB(cat) in a chlorine-radical equilibrium. Recently, Nocera and co-workers demonstrated a ligand-controlled C–H photochlorination and bromination protocol with a presynthesized iron(III) chloride-pyridinediimine complex for chlorine-radical generation (Scheme 5B).109,110 Significant selectivity toward the less steric-hindered methyl group was enabled by transient coordination between the chlorine radical and its vicinal aromatic groups on the pyridinediimine ligand, which provides the steric bulk needed for positional discrimination. The characterization was supported through transient absorption spectroscopy and photocrystallography. Although complexation suppresses the original HAT efficiency, this approach provides a novel method for tuning the site-selectivity of a chlorine radical.
 | ||
| Scheme 5 Site-selective alkane functionalization enabled by a chlorine radical complex. | ||
Transformations mediated by nitrogen-centered radicals
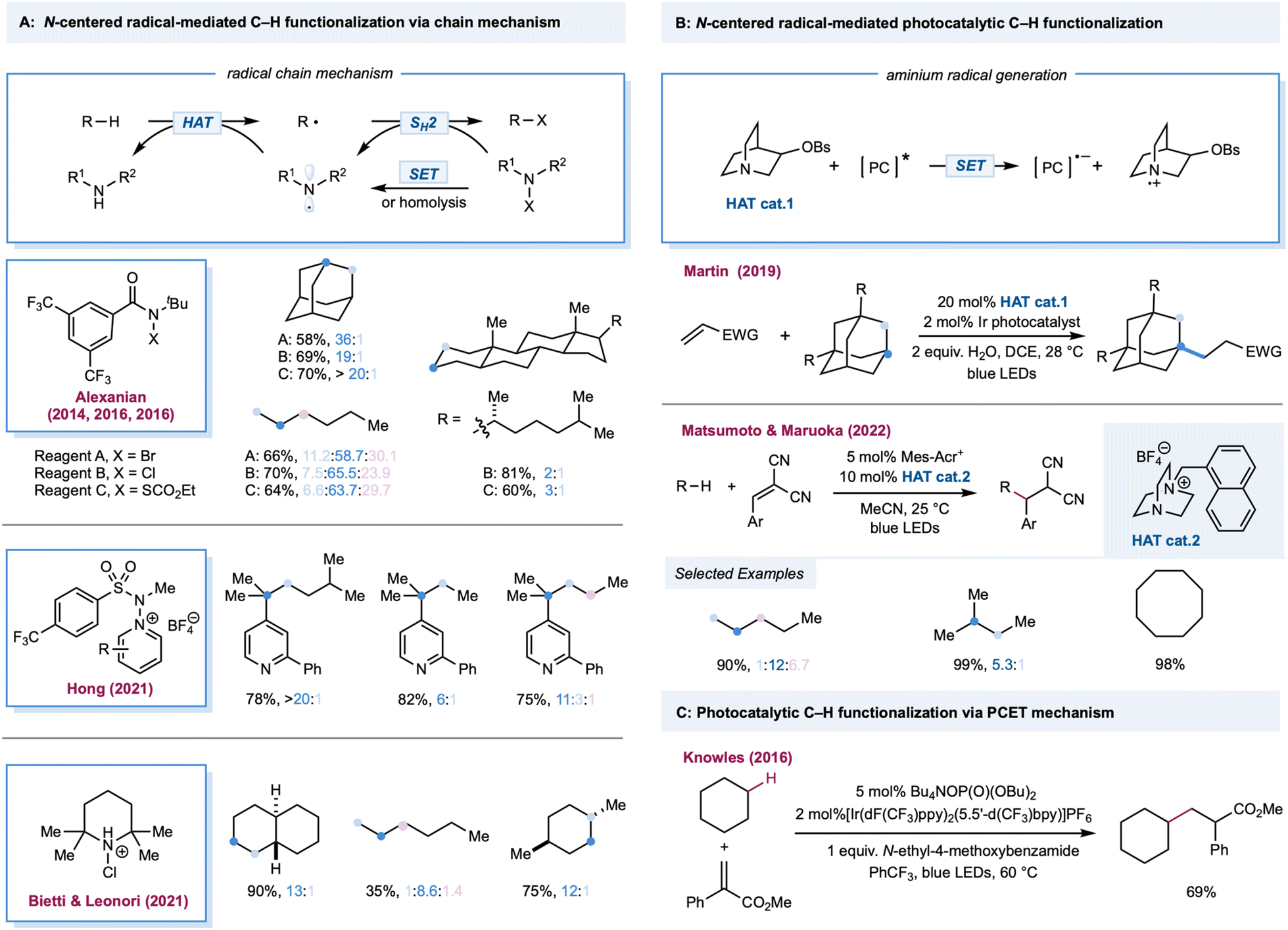
Among the most extensively explored HAT reagents in alkane functionalization, an aminium radical cation and amidyl radicals are versatile intermediates for HAT.111–113 Both can be generated from radical precursors bearing a weak N–X bond (X could be Cl, Br, O, N, or S) via single electron reduction or direct photolysis. Notably, unlike simple radicals (chlorine radical, bromine radical, etc.), the site-selectivity of different C–H bonds can be tuned by modifying the steric and electronic features of aminium radical or amidyl radical precursors, thus offering a practical toolbox for alkane functionalizations. Additionally, N–X radical precursors can be employed as radical trapping reagents in the radical chain pathway, thereby providing additional reagent-controlled site-selectivity. The amine radical cation from a quinuclidine-type cage motif (N–H BDE = 100 kcal mol−1) can be easily generated from the corresponding tertiary amines via photoinduced single electron oxidation.Amidyl-radical-mediated photochemical chlorination of alkanes was first reported in 1975 by Greene, wherein alkyl N-chloro-N-alkylacetamide was used as a radical precursor. The steric effects of chlorination were briefly discussed, wherein steric bulky N-chloroamides favor chlorination in primary C–H bonds in 2,3-dimethylbutane.114 The site-selectivity outcomes are generally poor for the classical alkane bromination protocols using Br2 or N-bromosuccinimide systems. Site-selective bromination of aliphatic C–H bonds was realized by Alexanian and co-workers using visible light and N-t-Bu, N-bromobenzamides (Scheme 6A).115 In this system, N-bromobenzamides act as amidyl radical precursors and bromination reagents, which are bench stable and can be prepared easily from the corresponding N-benzamides. By increasing the steric bulk and electronic-withdrawing substituents on N-bromobenzamide, a discriminating bromination could be achieved where bromination of secondary C–H bonds occurs preferentially over the weakest tertiary and least hindered primary C–H bonds. This site-selectivity was believed to be dictated by the sterically and electronically demanding nature of the HAT step and the C–Br bond-forming step. Moderate to good yields (45–75%) of alkyl bromides were obtained over a broad substrate range covering linear and cyclic alkanes. Photochemical methane bromination with NBS at room temperature and atmospheric pressure has been reported.116 In 2016, a site-selective C–H chlorination strategy using N-t-Bu, N-chloroamide was detailed by Alexanian and co-workers (Scheme 6A).117 Remarkably, this protocol for selective chlorination of 5α-cholestane provides an 81% yield of the chlorinated adduct with C3 selectivity (C3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C2 = 2:1). In the ongoing effort to expand the functional group beyond halogenation, they next explored the C–H xanthylation of alkanes using N-t-Bu, N-xanthylamide118 and further developed the platform for C–H xanthylation of polyolefin.119
:C2 = 2:1). In the ongoing effort to expand the functional group beyond halogenation, they next explored the C–H xanthylation of alkanes using N-t-Bu, N-xanthylamide118 and further developed the platform for C–H xanthylation of polyolefin.119
 | ||
| Scheme 6 Transformations mediated by nitrogen-centered radicals. | ||
For the Minisci-type alkane heteroarylation process, apart from the difficulties in site-selective HAT on alkanes, the positional discrimination on heteroarenes (such as C-2 versus C-4 functionalization on pyridine derivatives) is also expected to be challenging. One would expect that a mixture of isomers would be obtained if either the HAT or arylation step is not selective. Recently, Hong and co-workers reported a selective photocatalytic alkane pyridination (Scheme 6A).120 Their use of N-aminopyridinium salts provided impressive site-selective coupling between C-4 pyridines and weaker C–H bonds in alkanes. Compared to other alkane pyridination protocols, the scope of the alkane substrate was extensively explored and diverse linear and cyclic alkanes were used to form tertiary or quaternary carbon centers.
Bietti, Leonori, and co-workers successfully demonstrated site-selective C(sp3)–H bond chlorination by an aminium-radical-mediated chain mechanism (Scheme 6A).121 The alkyl radical was photochemically generated from a trace amount of chlorine, triggering chain propagation by SH2 addition to the in situ-formed N-chloroamium to form a C–Cl bond while releasing the aminium radical. Steric bulky aminium radicals offer high site-selectivity and maintain efficiency via electronic and steric effects. With 1 equiv. of alkane substrates, the mono-chlorinated product was obtained in moderate to excellent yields. With trans-decalin, employing 2,2,6,6-tetramethylpiperidine as the precursor, chlorination occurs at a more sterically accessible secondary C–H bond over a weak tertiary position furnishing the product in 90% yield. The yield was 28% with the more sterically hindered cis-decalin.
In 2019, Martin and co-workers reported a photocatalytic alkylation of adamantane using a quinuclidine-derivatized HAT catalyst (Scheme 6B).122 Compared to quinuclidine, electron-withdrawing substituted derivatives provide greater efficiency. Moderate to good yields (51–91%) of alkylated adamantane were obtained over a broad substrate scope covering a range of Michael acceptors, vinyl sulfones, and adamantyl derivatives with alkyl, ketyl, aryl, and hydroxy when using Ir(dF(CF3)ppy)2(d(CF3)bpy) with a catalytic quinuclidine derivative. This HAT catalyst selectively activates the tertiary C–H in the adamantane rather than the other weaker tertiary or activated C–H bonds. Follow-up work by the same group extended radical traps from Michael acceptors to imines and hydrazones.123 Additionally, a cationic DABCO-based tertiary amine was also developed as an efficient HAT catalyst by Matsumoto, Maruoka, and co-workers in a photoredox-catalyzed C–H alkylation (Scheme 6B).124 Mechanistic studies indicated that the aromatic group tethered on the DABCO core was crucial in generating the HAT reagent, a tertiary amine radical cation, via intramolecular electron transfer between the aromatic radical cation and tertiary amine. In addition, substituents adjacent to a nitrogen atom could improve the site-selectivity toward weaker tertiary or secondary C–H bonds. Both aliphatic and cyclic alkanes were alkylated in good to excellent yields with benzal malononitrile, dimethyl fumarate, 1,1-bis(phenylsulfonyl)ethylene, and ethyl coumarin-3-carboxylate as radical acceptors. In 2016, Knowles and Rovis independently reported a PCET-mediated strategy for the catalytic generation of amidyl radicals from strong N–H bonds in aryl amides.28,29 Notably, in Knowles' work, one example of intermolecular C–H alkylation of cyclohexane indicated that this protocol may be more broadly adopted in alkane functionalization (Scheme 6C).
Transformations mediated by oxygen-centered radicals
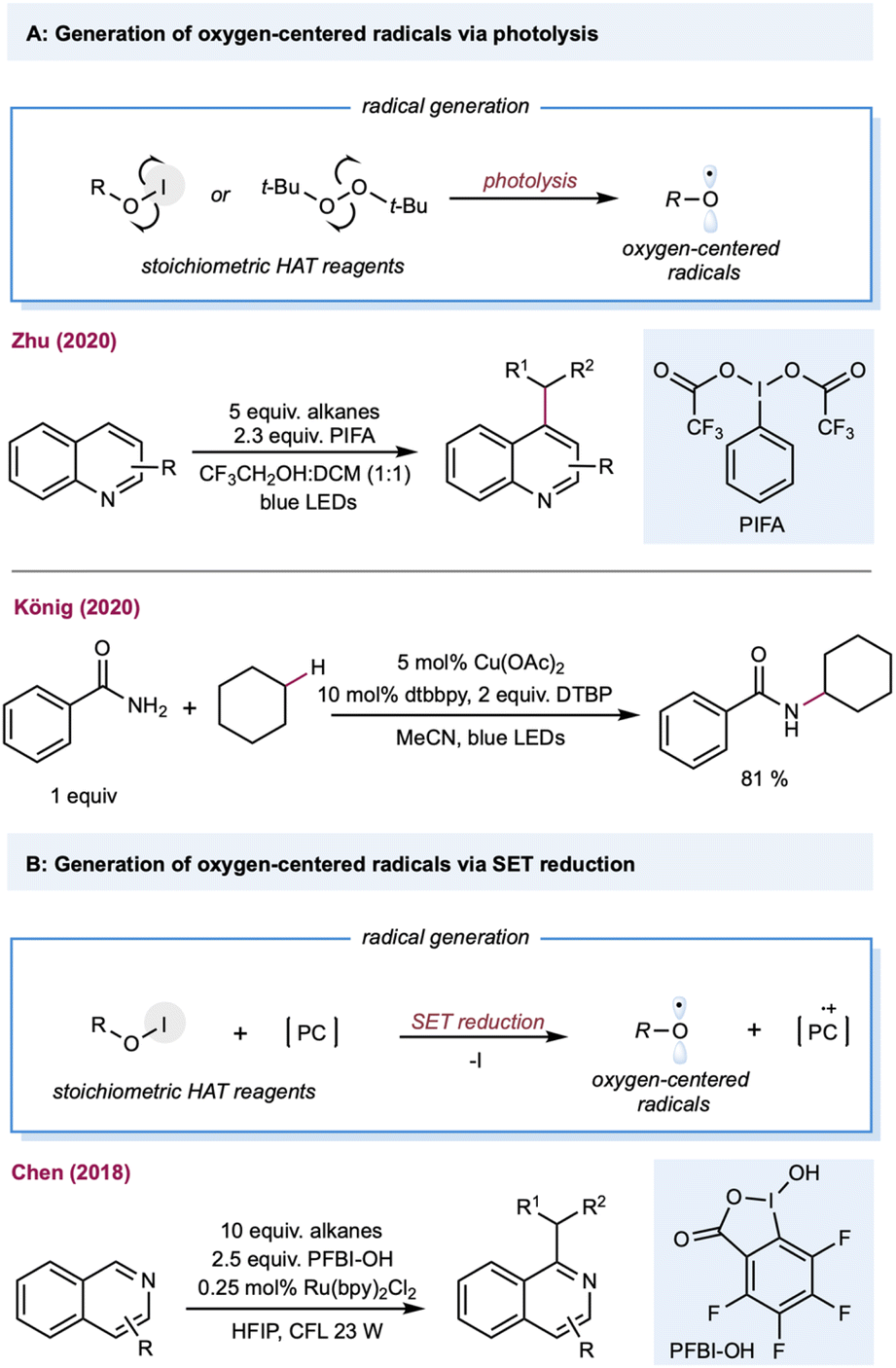
Oxygen-centered radicals have a rich history in C(sp3)–H functionalization. The classical methods require stoichiometric amounts of peroxide oxidants, such as peroxides, peroxol, peroxyacid, or molecular oxygen, which have provided unique opportunities in alkane oxidations, including Fenton-like chemistry (Fe2+/H2O2) and the later Gif-Barton system (iron catalysts with t-BuOOH or H2O2).125 Furthermore, the incorporation of photonic energy facilitates oxygen-centered radical generation, including photolysis of peroxide, and Suárez conditions. With the rapid advancement of photoredox catalysis, oxygen-centered radicals could be easily generated under mild reaction conditions using: (1) bench-stable O–X precursors via single electron reduction; (2) oxygen anions (carboxylates, phosphate, and pyridine N-oxides) via single electron oxidation; (3) free alcohols via LMCT catalysis.23Alkoxy-radical-mediated HAT has been reported by Zhu and co-workers for the heteroarylation of alkanes (Scheme 7A).126 By employing the Suárez condition for alkoxy-radical generation, several heteroarenes, including pyridine, isoquinoline, pyrazine, benzopyrazine, pyrimidine, and benzopyrimidine can serve as substrates. Peroxides can also be used under direct photolysis conditions or as a stoichiometric electron acceptor to generate oxygen-centered radicals, providing opportunities in net oxidative photocatalytic alkane functionalization. The merging of alkoxy-radical-mediated intermolecular HAT with copper catalysis was recently demonstrated by König and co-workers for the C–H aminations of alkanes (Scheme 7A).127 This protocol employed stoichiometric di-tert-butyl peroxide (DTBP) as the alkoxy-radical precursor, and Cu(OAc)2, and 1,10-Phen as the copper catalyst. Mechanistic studies showed that the t-butoxy radical forms via classical photolysis of DTBP. Various aminating reagents, including amides, sulfonamides, indole, indazole, and pyrazole, were used for copper-catalyzed C–N coupling with cyclohexane. In addition to DTBP, H2O2 is also a useful HAT reagent for synthesizing phenanthridine derivatives with alkanes.128 In a similar vein, persulfate salts were used as precursors for oxygen-centered radicals to achieve a Minisci-type alkane heteroarylation.129 The generation of a benzoyloxy radical from hypervalent iodine (PFBI-OH) by single electron reduction was reported by Chen and co-workers,130 whose method was used to produce C–H alkylation of heteroarenes (Scheme 7B).131 A range of alkanes and cycloalkanes was demonstrated in the Minisci-type alkylation of heterocycles, including quinoline and pyridine.
 | ||
| Scheme 7 Transformations enabled by the generation of oxygen-centered radicals from a stoichiometric amount of precursors. | ||
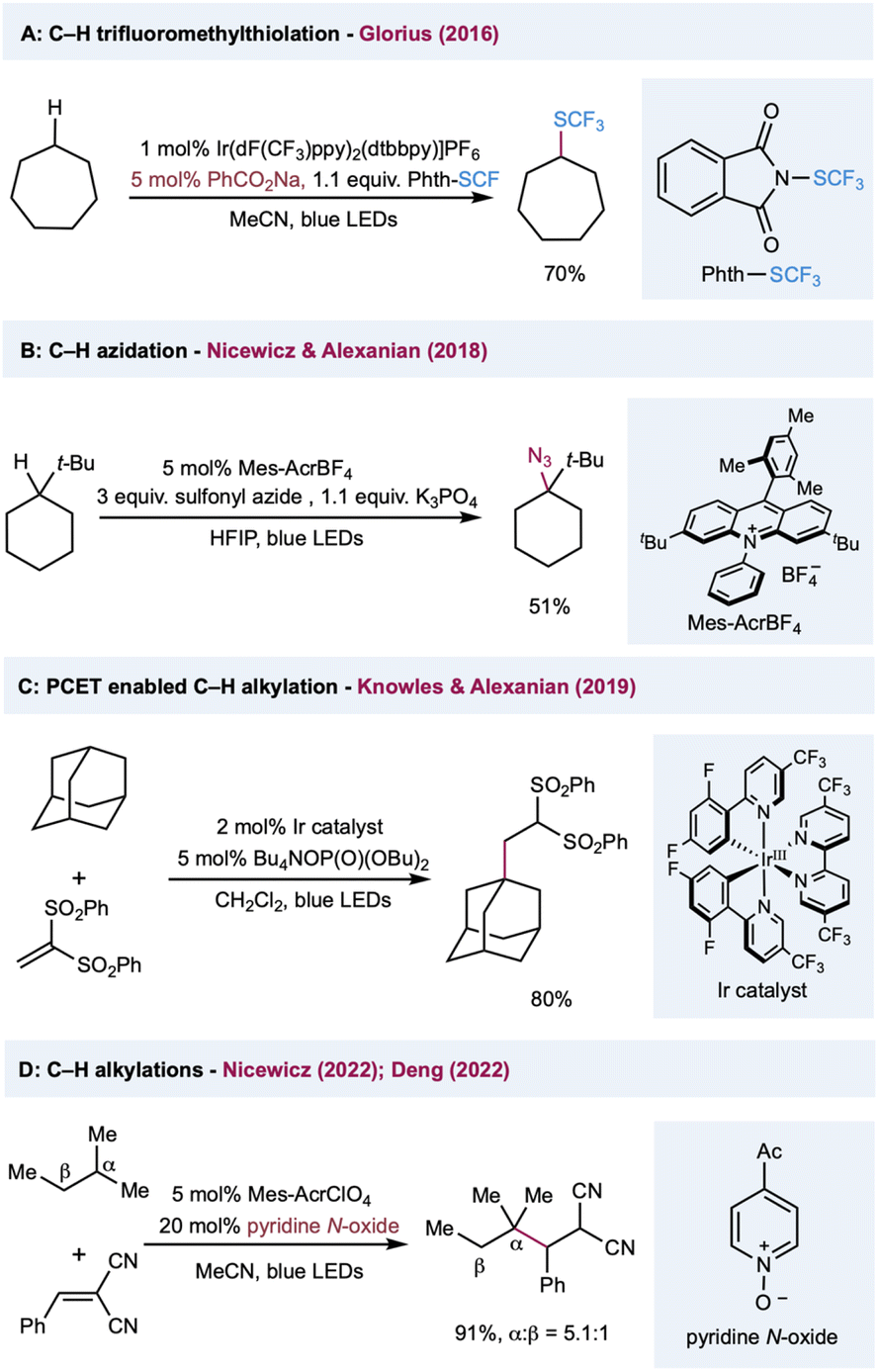
Among various oxygen-centered radicals, carboxyl radicals obtained from carboxylic acid or the corresponding precursors have remarkable synthetic utility and were extensively explored in decarboxylative functionalization. In comparison, their application in HAT catalysis in synthetic transformations has been underdeveloped. In 2016, Glorius and co-workers reported C(sp3)–H trifluoromethylthiolation by harnessing the intermolecular HAT reactivities of a benzoyloxy radical (Scheme 8A).132 The HAT catalyst sodium benzoate was converted to a benzoyloxy radical via reductive quenching of photoexcited Ir(III). The intermolecular HAT outcompeted the slow unimolecular decarboxylation of a benzoyloxy radical, preventing the decomposition of the HAT catalyst. Nicewicz, Alexanian, and co-workers described a protocol for C(sp3)–H azidation using K3PO4 as the HAT catalyst (Scheme 8B).133 The highly oxidizing acridinium photocatalyst (E1/2(cat*/cat˙) = +2.08 V versus SCE) oxidizes the phosphate salt, perhaps via a preassociated acridinium–phosphate ion pair, to form an oxygen-centered radical that serves as a hydrogen atom abstractor to activate C(sp3)–H bonds. Diverse radical trapping reagents were well-suited to this protocol, allowing the streamlined synthesis of halogenated, trifluoromethylthiolated, and alkylated adducts from simple hydrocarbons. In a mechanistically different protocol that uses phosphate salt in C(sp3)–H functionalization, Knowles, Alexanian, and co-workers developed a PCET-based C–H alkylation using an Ir(III) photocatalyst and n-Bu4N+(n-BuO)2P(O)O− cocatalyst (Scheme 8C).134 Five- to eight-membered cycloalkanes and adamantane provided moderate to good yields when using 1,1-bis-(phenylsulfonyl)ethylene as a radical acceptor. Mechanistic studies showed that the steps leading to this reactivity may involve a preassociation between the 3,3′-bipyridyl protons of the Ir photocatalyst and the phosphate oxygen. In the presence of ethyl benzene and phosphate salt, the emission is quenched with Stern–Volmer kinetics at a rate constant of 2.3 × 106 M−1 s−1 and kinetic isotope effect of kH/kD = 2.0 ± 0.2 with deuterated ethyl benzene. Based on these results, the author proposed a concerted PCET activation of the C(sp3)–H bond, from which the electron was transferred to the photoexcited catalyst and the proton was delivered to the phosphate salt in a single transient state. Very recently, Nicewicz and Deng independently reported the C(sp3)–H alkylations using pyridine N-oxides as the HAT catalyst. In both methods, a highly oxidizing photoexcited acridinium photocatalyst was used in the oxidation of pyridine N-oxides into an electrophilic N-oxy radical to activate the alkane into an alkyl radical via a HAT event (Scheme 8D). Remarkably, the HAT catalyst pyridine N-oxide can be easily regenerated from the protonated pyridine N-oxide product through proton transfer, facilitating the turnover of the HAT catalytic cycle.135,136
 | ||
| Scheme 8 Transformations enabled by the catalytic generation of oxygen-centered radicals or a PCET event from oxides. | ||
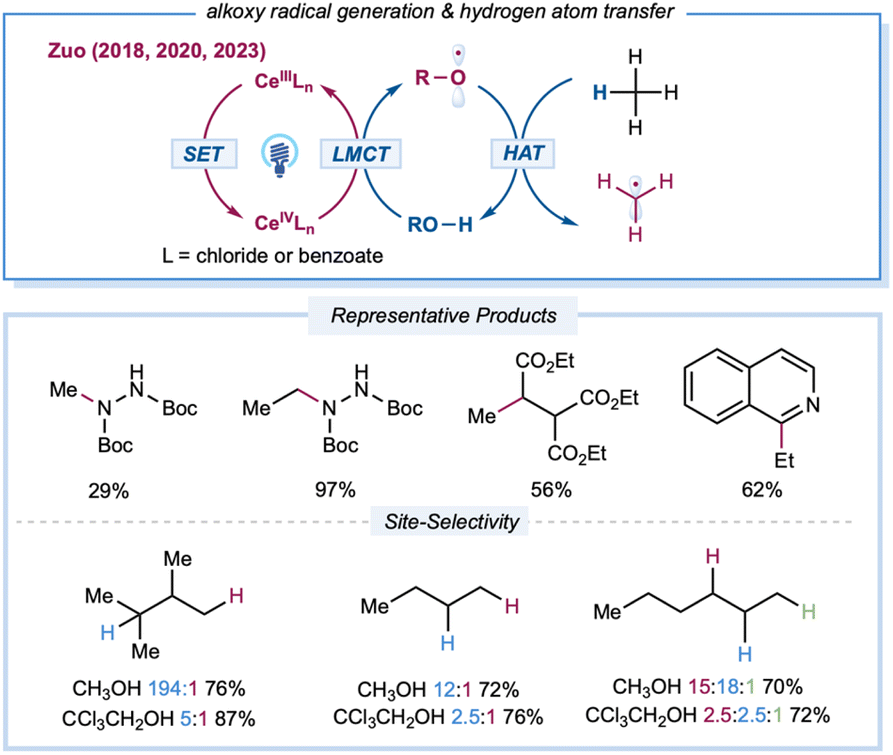
Extensive kinetic investigations of t-butyoxyl and cumyoxyl radicals by Bietti and others have revealed various key factors that govern HAT selectivity.20 Compared to other types of electrophilic open-shell species, alkoxy-radical-mediated C–H functionalizations were once hampered by a dearth of facile routes for direct initiation of these highly reactive species from abundant alcohols.23,25 Recently, the Zuo group developed a practical platform that employs LMCT excitation to access alkoxy radicals directly from abundant free alcohols using cerium catalysts and LED irradiation (Scheme 9). The structural diversity of alcohol precursors allows for fine-tuning of the steric and electronic parameters of the resultant alkoxy radicals to provide intriguing opportunities for site-selectivity studies in the elementary HAT step. In 2018, the Zuo group described selective C–H functionalization of feedstock alkanes.137 The inert C–H bonds in gaseous alkanes, such as methane and ethane, can be activated under ambient conditions or even at 0 °C. Methane amination can be achieved with 20 mol% 2,2,2-trichloro-ethanol (TCE) and 0.01 mol% Ce(OTf)4 catalysts, furnishing the N-methylhydrazine product at 29% yield with a TON of 2900. Ethane with primary C–H bonds gave rise to the aminated product at 97% yield with a TON of 9700 from 0.01 mol% CeCl3 and 20 mol% TCE. This method was also applied to C–H alkylation and heteroarylation, demonstrating the versatility of the established protocol. To tune reactivity and site-selectivity by using sterically and electronically different alcohol precursors, Zuo et al. created a photocatalytic protocol for the site-selective amination of higher alkanes.94 After extensive evaluation of the alcohol HAT catalyst, the methanol/CeCl3 catalytic system offered excellent site-selectivity toward steric tertiary C–H bonds. Various aliphatic amines could be prepared from feedstock alkanes at good yields (60–84%) when incorporated with a simple hydrogenation workup. Moreover, 9,10-diphenylanthracene was identified as a synergistic cocatalyst to accelerate the turnover of the cerium catalytic cycle to achieve efficient C–H alkylation of hydrocarbons. The Zeng group reported a merging of cerium LMCT catalysis with anodic oxidation to achieve heteroarylation of alkanes without the requirement for an additional oxidant.138 By employing CeCl3·7H2O as the photocatalyst and MeOH as a methoxy radical precursor, a range of alkylated heteroarenes could be prepared in an undivided cell with a carbon felt anode and nickel cathode pair at constant current (2 mA).
 | ||
| Scheme 9 Transformation enabled by the catalytic LMCT strategy for the generation of alkoxy radicals from free alcohols. | ||
Zuo and co-workers conducted a thorough mechanistic investigation and conclusively defined the generation of alkoxy radicals via Ce-LMCT catalysis based on a series of experiments, including electron paramagnetic resonance and transient absorption (TA) spectroscopy, kinetic analysis, regioselectivity studies, and DFT calculations.139 Moreover, the formation of a chlorine radical or chlorine–radical–alcohol complex was ruled out based on regioselectivity studies, TA experiments, and high-level calculations. Detailed computational studies by Liu, Bietti, Houk, and co-workers elucidated the critical influence of thermodynamics, polarity, and unsaturation on the reactivity and selectivity of HAT from C(sp3)–H bonds to different alkoxy radicals.140
Transformations mediated by excited species
For alkane functionalization, the HAT event may also be accomplished via the catalytic utilization of exciting species, such as aromatic ketones, a decatungstate anion [W10O32]4−, and a uranyl cation [UO2]2+. In the ground state, a general structural motif for this type of photocatalyst includes an oxo group (X = O) that, when excited by photonic energy, generates the electrophilic radical-like oxygen center that acts as a HAT reagent for alkane substrates. Depending on the catalytic system, the HAT-generated reduced form of the photocatalyst could undergo a reverse HAT (RHAT) or stepwise SET/proton transfer to regenerate the photocatalyst (Scheme 10).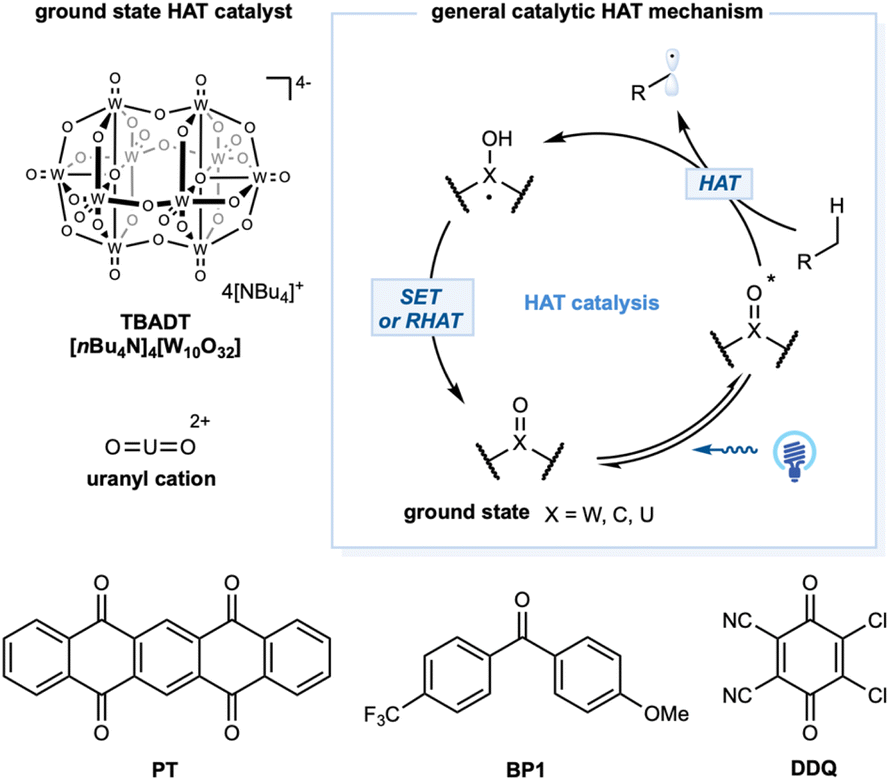 | ||
| Scheme 10 Photocatalytic HAT enabled by excited species. | ||
Tetrabutylammonium decatungstate, TBADT (nBu4N)4[W10O32], belongs to the polyoxometalate family and has unique electrochemical and photochemical properties.45 Upon photoinduced LMCT, the activated LMCT state has a long lifetime.141 Remarkably, the excited state showed a significant radical character on the oxygen center and strong oxidation potential (ranging from +2.26 to +2.61 V versus SCE), enabling the development of intriguing photocatalytic transformations. Depending on the substrates, SET or more common polarity-matched HAT events could occur. Kinetic studies demonstrated a HAT rate constant with cyclohexane of kH = 4 × 107 M−1 s−1 (Scheme 1).
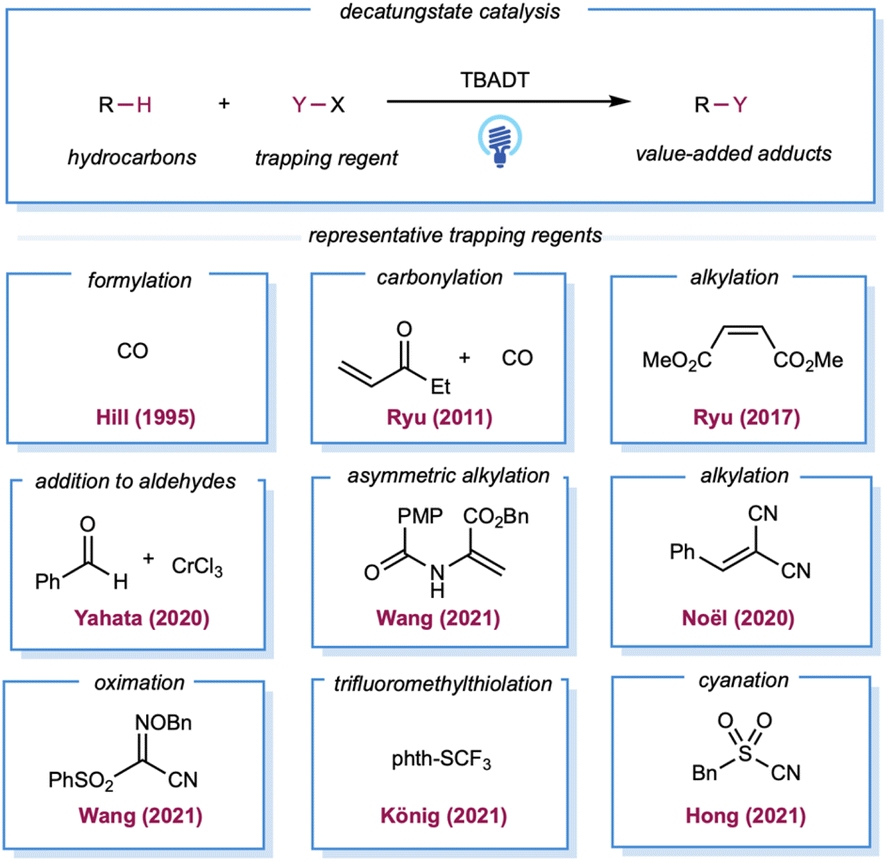
In the late 1980s, pioneering studies by Hill and co-workers demonstrated that TBADT,142,143 which can be easily prepared from Na2WO4·2H2O and TBAB,144 could be employed as a HAT photocatalyst for the generation of alkyl radicals from the corresponding alkanes. Initial reports demonstrated aerobic oxidation,142 dehydrogenation,143 and carbonylation145 of alkanes by decatungstate catalysis. However, these examples required the substrate alkanes to be used as solvents. In 2011, Ryu, Fagnoni, and co-workers reported three-component coupling reactions of alkanes, CO, and electron-deficient alkenes.146 For simple cycloalkanes, this protocol offers good yields of monoalkylated adducts. Ryu and co-workers also reported C–H alkylation with decatungstate catalysis, for which the substrate loading was reduced to 5 equiv.147 In 2020, asymmetric C–H alkylation was reported by Wang and co-workers using chiral phosphoric acid in combination with a TBADT catalyst.148 The chiral phosphoric acid was proposed as a chiral proton-transfer shuttle to an enol intermediate during the proton-transfer step. Analogous to Giese-type alkylation, radical addition to imine derivatives149 or in situ-formed iminium ion150 was also demonstrated (Scheme 11). Yahata and co-workers demonstrated direct aliphatic C–H coupling between alkanes and aldehydes by merging decatungstate HAT photocatalysis with the organochromium equivalent (Scheme 11).151 In 2020, Noël and co-workers described the alkylation of gaseous light alkanes (methane, ethane, propane, and isobutane) with Michael acceptors in a continuous flow and observed good yields.152 Several reports of alkane heteroarylation,153 cyanation,154 trifluoromethylthiolation,155 allylic alkylation,156 oxime etherification,157 and alkenylation158 have been published under similar decatungstate catalysis using various radical acceptors (Scheme 11).
 | ||
| Scheme 11 Various trapping reagents for decatungstate-catalyzed transformations. | ||
Until recently, the predominant strategy for regenerating the decatungstate catalytic cycle has relied on RHAT with classical radical trapping reagents. One would expect that a broader coupling partner could be productively engaged if decatungstate could participate in an electron-transfer event. In 2018, MacMillan and co-workers disclosed the merging of decatungstate catalysis with nickel catalysis to realize the arylation of unactivated C(sp3)–H bonds (Scheme 12).159 The proposed mechanism starts with the HAT event between a photoexcited decatungstate catalyst and alkanes. The resulting alkyl radical is captured by the Ni(0) species formed in situ to generate an alkyl Ni(I) intermediate. Subsequent oxidative addition of the aryl bromide results in a Ni(III) intermediate that undergoes reductive elimination to furnish a cross-coupling product and Ni(I). Disproportionate reduction of the decatungstate intermediate could regenerate the ground state decatungstate and the reductive hexa-anion species. The latter could then reduce Ni(I) to Ni(0), closing the nickel catalytic cycle. Linear and cyclic alkanes were compatible with this method and could be coupled with a range of phenyl, pyridyl, pyrimidyl, and thiazole derivatives. The method was applied to a pharmaceutically relevant aryl bromide, in which a 67% yield of the cyclohexyl-coupled celecoxib derivative was synthesized in one step. In a related study, Noël and co-workers recently reported a decatungstate/nickel dual-catalytic acylation and arylation of cycloalkanes (mostly cyclohexane) with acyl chloride or aryl bromides in a continuous flow, greatly reducing reaction times (5–15 min versus 12–24 h in a batch).160 This dual-catalytic strategy was further explored by Kong and co-workers in a three-component radical cross-coupling reaction to achieve alkyl-arylation of alkenes, with alkanes as alkyl precursors that were activated by TBADT photocatalysis in combination with electron-deficient alkenes and aryl bromides.161 Wang and co-workers merged decatungstate catalysis with nickel catalysis for alkane amination/amidation using nitroxide coupling partners.162 In 2015, Sorensen and co-workers demonstrated direct dehydrogenation of alkanes using dual-decatungstate cobalt catalysis.163 Along a similar line, Wu and co-workers demonstrated a dehydrogenative alkenylation of alkanes with styrenes by merging decatungstate and cobalt catalysis.164 This method enables the use of alkyl C–H partners as limiting reagents to achieve direct E-selective alkenylation of alkanes by cobalt-catalyzed, light-promoted β-H elimination.
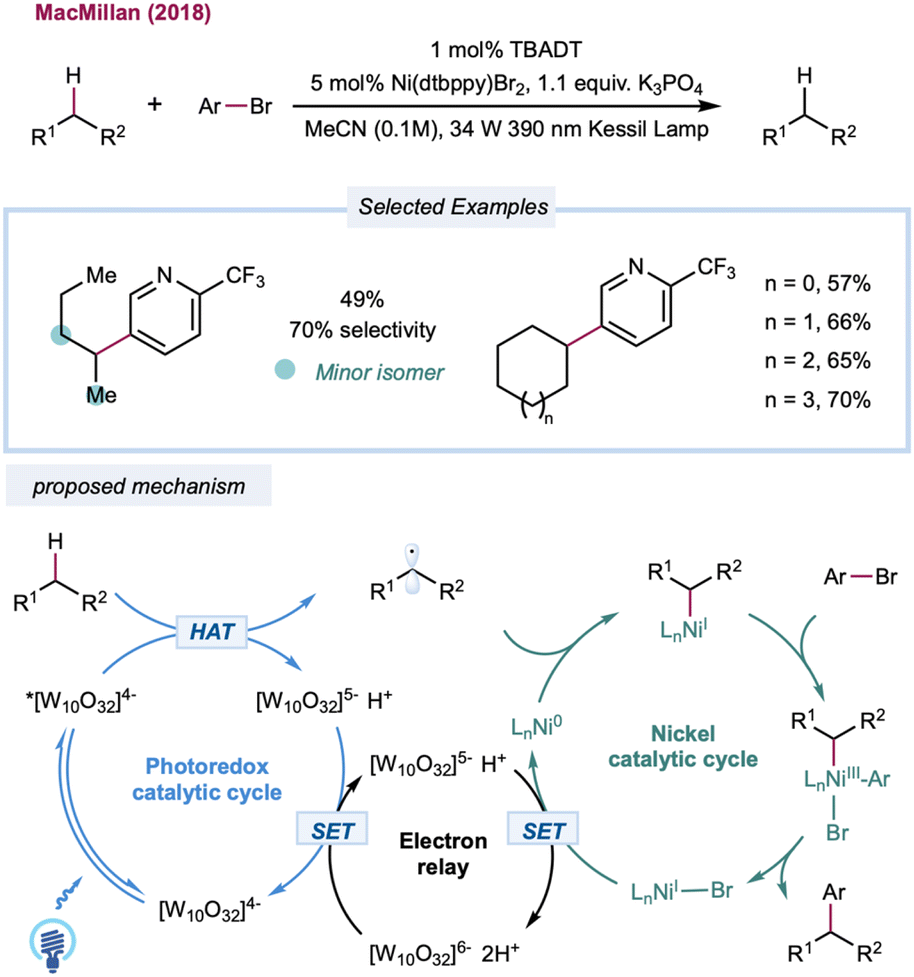 | ||
| Scheme 12 C–H arylation via nickel/decatungstate dual catalysis. | ||
Bearing a HAT mechanism similar to the decatungstate anion, UO22+ was also identified as a potent HAT catalyst. It can be photochemically activated via UV and near-UV irradiation, generating a long-lived (μs scale) excited state with oxidation potentials exceeding +2.6 V. Subsequently, the LMCT excited state from U(5f) ← O(2p) transitions generated an intermediate with a reactive oxygen-centered radical for HAT reactivity.165 The HAT ability of photoexcited UO22+ was described by Sorensen and co-workers, who demonstrated C(sp3)–H fluorination.166 With N-fluorobenzene sulfonimide (NFSI) as the fluorination reagent, moderate-to-high yields of monofluorinated alkanes were achieved. A mixture of monofluorinated regioisomers was obtained with linear alkanes. Similar to decatungstate catalysis, the alkyl radical generated via the excited-state UO22+-mediated HAT could be incorporated with classical radical acceptors, such as electron-deficient alkanes, to obtain alkylated adducts.167,168 In a recent report, benzophenone-catalyzed alkane activation was incorporated in controlled poly(ethylene glycol) synthesis to introduce an alkyl functionality at a polymer chain end.169 The versatility of the triplet ketone HAT process has also been demonstrated in alkane functionalization, including oxygenation,170 sulfonylation,171 and dehydrogenation.172
Excited-state benzophenone derivatives rapidly abstract hydrogen atoms from aliphatic C–H bonds. Photoexcitation of the n → π* transition of benzophenone derivatives generates the reactive triplet-excited-state diradical with a lifetime in the microsecond range. These triplet states generate a stable ketyl radical after the HAT event. The HAT catalytic cycle could turnover via RHAT from a weak O–H bond in the ketyl radical (O–H ca. BDE = 16 kcal mol−1) or stepwise electron transfer and proton-transfer mechanisms to regenerate the benzophenone derivatives. Photophysical properties and the theoretical aspects of excited-state benzophenone derivatives were established in the 1960s.173 Early examples of alkane dehydrogenation were reported in the 1970s;174 however, this topic has remained underdeveloped until recently. In 2016, Kamijo and co-workers reported photocatalytic C–H allylation using 5,7,12,14-pentacenetetrone.175 The C–H alkylation mechanism depends on a benzophenone photocatalyst with a catalytic amount of Cu(OAc)2. In this study, Cu(II) functions as a redox shuttle additive and prevents the polymerization of Michael acceptors.176 The HAT reactivity of triplet-excited eosin-Y was also demonstrated by Wu and co-workers to enable cyclohexane alkylation.177 In 2019, Li and co-workers detailed aliphatic C–H heteroarylation utilizing triplet diacetyl as a stoichiometric HAT reagent in the presence of DTBP.178 In 2018, Martin and co-workers demonstrated a dual-catalytic strategy based on triplet ketone and nickel cross-coupling to achieve arylation and alkylation of aliphatic C–H bonds (Scheme 13a).179 In this method, C(sp3)–C(sp2) and C(sp3)–C(sp3) linkages are easily forged between the solvent quantity of cycloalkanes and aryl or alkyl bromide coupling partners.179 The synergistic merging of this activation mode with Cu-based chiral Lewis acid catalysis was achieved by Gong and co-workers to allow both site- and enantioselective functionalization of unactivated hydrocarbons with N-sulfonylimines (Scheme 13b).46 In this dual-catalytic strategy, 5,7,12,14-pentacenetetrone was employed as the HAT photocatalyst to activate the weakest C–H bond in the alkanes. The high enantioselective C(sp3)–C(sp3) bond formation step was achieved with a chiral Cu–based complex containing a bisoxazoline (BOX) ligand. In 2020, Lei and co-workers detailed a merging of triplet-excited-state HAT catalysis with manganese catalysis to enable aliphatic C–H azidation in an electrophotocatalysis protocol (Scheme 13c).180 Using sodium azide as the azide building block, ketones (9-fluorenone, DDQ, or 4,4′-dimethoxybenzophenone) as the HAT photocatalyst, MnF2, and 1,10-phenantroline as the catalyst, a range of azide products were obtained in moderate to excellent yields with a carbon felt anode and platinum cathode pair at a constant current of 8 mA.
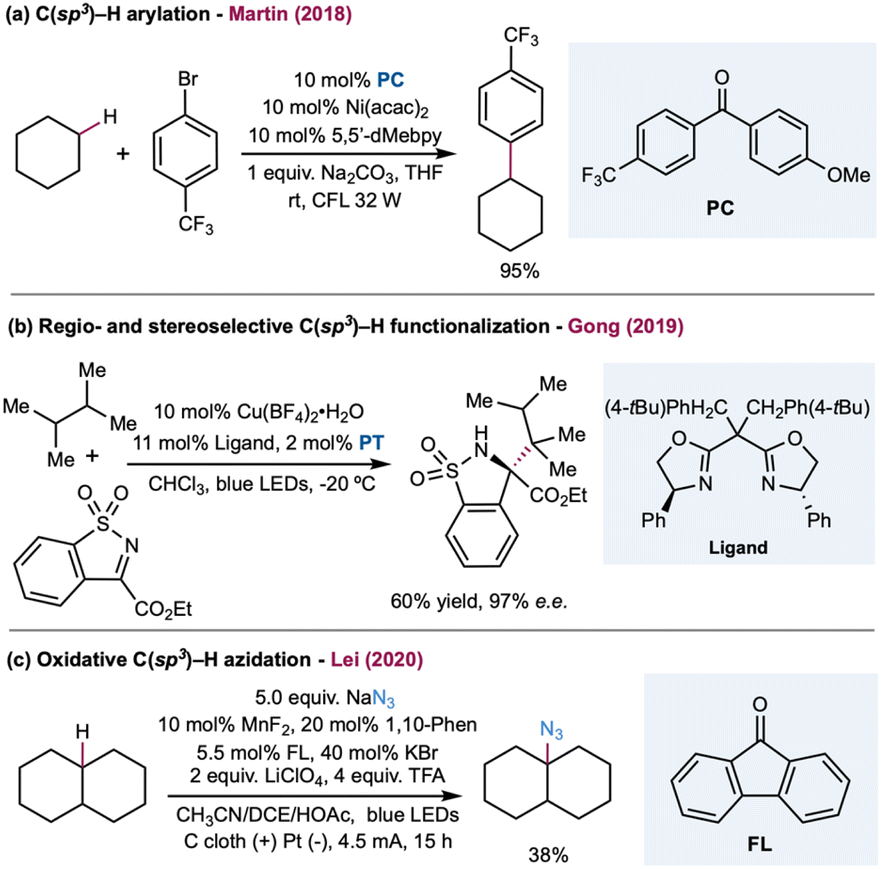 | ||
| Scheme 13 Dual catalysis for alkane functionalization enabled by triplet ketone. | ||
Applications in a continuous-flow setup
Due to the low solubility of gaseous alkanes in an organic solvent, gaseous methane, ethane, and other light alkanes are extremely challenging substrates for activation and application in flow chemistry devices. In a demonstration of Ce-catalyzed C–H amination, Zuo's group employed a gaseous continuous-flow device with 10 parallel perfluoro alkoxy alkane (PFA) tubing microreactors for a total volume of 4.5 mL (inner diameter: 3.17 mm) to provide scalable C–H amination of methane, ethane, and other light alkanes (Scheme 14A).137 With 1 mol% loading of the cerium catalyst, methane (1800 kPa) was efficiently converted to amination products with 15% yields and productivities up to 0.6 mmol d−1. Under similar conditions, ethane and higher alkanes were functionalized at good to excellent yields (56–90%) and good productivities (up to 2 mmol h−1 for ethane and 1.2 mmol h−1 for propane). Enhanced catalytic efficiency could be obtained with a DPA cocatalyst. Zuo and co-workers then developed a method for rapid access to 3-amino-1-adamantanol for synthesizing the antidiabetic agent vildagliptin.94 Here, the scale-up synthesis of 3-amino-1-adamantanol from hydroxy adamantane was implemented in two glass microreactors (Corning low flow reactor: 2 × 8.2 mL internal volume) placed in series. The tertiary C–H bonds in the hydroxy adamantanes were site-selectively aminated at a productivity rate of 0.79 mmol h−1 with a flow rate of 0.25 mL min−1 and residence time of 66 min.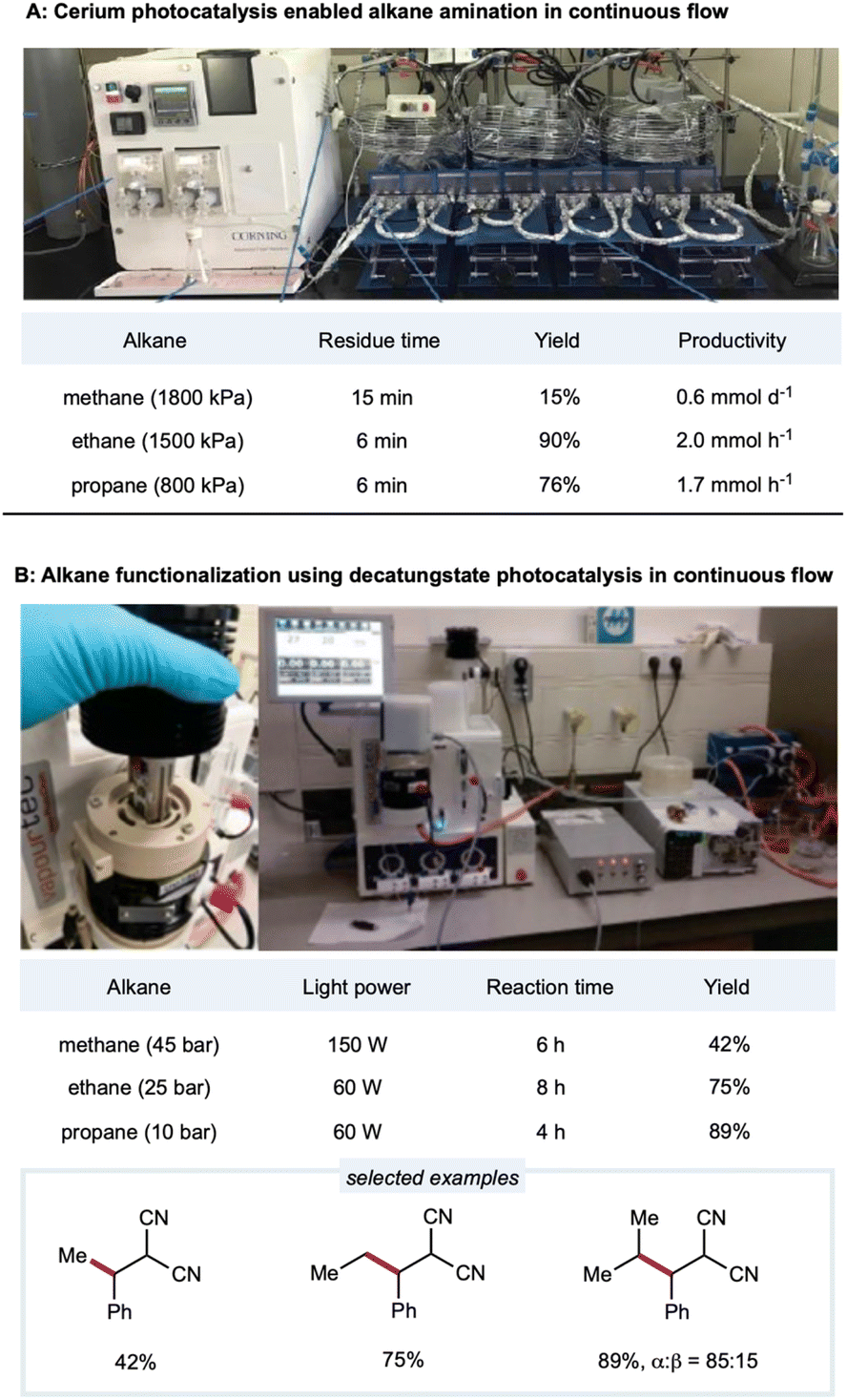 | ||
| Scheme 14 HAT enabled alkane functionalizations under continuous-flow conditions. (A) Picture of the set-up. Adapted with permission from ref. 137 copyright 2018, The American Association for the Advancement of Science; (B) picture of the reactor (left) and set-up (right). Reprinted with permission from ref. 152 Copyright 2020, The American Association for the Advancement of Science. | ||
Noël and co-workers developed the decatungstate-catalyzed C(sp3)–H functionalization of light alkanes in a flow setting (Scheme 14B).152 Employing a Vapourtec UV-150 flow reactor consisting of PFA tubing (inner diameter: 0.75 mm; total volume: 3–5 mL) with a back-pressure regulator, ethane, propane, and isobutane could be alkylated in high yields with a 4 h residence time. For the less reactive methane, higher catalyst loading with a more powerful light source was used in the deuterated acetonitrile/water solvent to provide a 42% yield of the alkylated product. To further improve the efficiency of this protocol, Noël and co-workers employed a commercially available oscillatory flow HANU reactor (volume 10 mL) for the alkylation of cyclohexane and achieved remarkably higher productivity (36.7 mmol h−1) in 7.5 min.181 Using azodicarboxylates as amination reagents, they extended the decatungstate-catalyzed protocol to C(sp3)–H amination to synthesize pyrazoles, phthalazinones, and free amines. In a lab-engineered flow setup (inner diameter: 750 μm; productivity: 314 mmol h−1) equipped with high-intensity LEDs, C(sp3)–H amination could be scaled up to 2.15 kg day−1 in a single microreactor of only 11 mL volume.182 Noël and co-workers reported a dual-decatungstate and nickel catalytic process for alkane acylation and arylation (Vapourtec system: 0.76 mm inner diameter; volume: 5 mL). Notably, compared to a batch system, much shorter reaction times with comparable yields were observed under flow (5–15 min) conditions than under batch (12–48 h) conditions.160 The Vapourtec UV-150 photochemical reactor (PFA: inner diameter: 0.75 mm; volume: 3) allows optimized reproducibility and scalability of decatungstate-catalyzed allylation of C(sp3)–H bonds.183
Outlook
The selective functionalization of alkanes has long been recognized as a predominant challenge, and it is still an arduous task in organic synthesis. Despite the tremendous progress made in transition metal catalysis, general methods for the selective functionalization of the C(sp3)–H bond of simple alkanes remain largely elusive. With the ever-increasing need for more sustainable and economic synthetic processes, the development of new strategies has drawn significant research attention. The past decade has witnessed significant advances in photochemical and photocatalytic alkane C–H functionalizations using hydrogen atom transfer as the enabling activation strategy.From a historical perspective, the use of the HAT process for alkane C–H functionalization is not new and finds research practices in the early work of enzymatic catalysis, the alkane halogenation process, etc. In recent years, the application of photoredox catalysis provides exciting opportunities for alkane C–H functionalization under extremely mild conditions to trigger HAT processes and achieve radical-mediated functionalization processes in a more selective manner. Considerable efforts have been devoted to building more efficient and cost-effective photocatalytic systems for sustainable transformations. Along this way, the use of economic photocatalysts including non-noble metal complexes or organocatalysts could offer exciting opportunities to conduct the functionalization at a low cost while maintaining the desired performance. It is with these motivations that radical-mediated C–H functionalization has evolved as an efficient catalytic process by applying a range of HAT reagents under the paradigm of photocatalysis. The versatility of this approach is evidenced by the direct installation of functionalities that would otherwise be difficult to realize. Moreover, the merging of photocatalysis with transition metal catalysis has enabled the development of versatile cross-coupling methods for alkane C–H transformation.
Although recent development has demonstrated photo-induced HAT as an appealing strategy to activate inert alkanes into reactive radical intermediates, the exploitation of this catalytic manifold is still in the early stage. Given the ever-increasing research interests in this field, several critical issues remain to be addressed.
(1) Radical trapping reagents
First, the radical trapping reagents employed in photochemical alkane C–H transformation reactions often function as terminal oxidants. While applying these reagents can conveniently install functional groups to facilitate fine chemical production and late-stage functionalization of complex molecules, it is of paramount importance to identify systems that allow for the use of economic and sustainable oxidants such as O2, N2, H2O, or electricity to access value-added products from simple alkanes. These oxidants are not without drawbacks, as reagent activation, catalyst deactivation, over-oxidation, and selectivity issues can all pose potential issues. For example, the catalytic oxidation of alkanes using O2 as the oxidant to afford value-added alcohols or carboxylic acids would be highly attractive. Although O2 is a well-established trapping agent for alkyl radicals (kinetic rate of the reaction between a methyl radical and O2, k = 4.7 ×109 M−1 s−1),184 the selectivity of this radical-mediated addition pathway is severely compromised by peroxide decomposition and product overoxidation, rendering the selective aerobic oxidation of feedstock alkanes an arduous task. The challenge ahead now lies in the development of selective and long-lived catalytic systems that are compatible with the employed low-cost and environmentally benign oxidants. The development of such a highly efficient methodology could pave the way to provide bulk chemicals from naturally abundant alkanes at a low cost.An intriguing catalytic strategy to avoid the use of radial trapping reagents or oxidants is dehydrogenation to produce alkenes and H2. Both products could find broad applications in chemical synthesis and the hydrogen evolution strategy could enable the alkane functionalizations under oxidant-free conditions. One main obstacle arises from the weakened allylic C(sp3)–H bond of the alkene products which might lead to thorough dehydrogenation or allylic radical-mediated side reactions. In this regard, while current studies have provided proof of concept for the acceptor-less dehydrogenation of alkanes,185–187 new catalytic systems are required to enhance the catalytic performance and achieve selective functionalization of alkanes over more reactive alkene products.
(2) Inherent reactivity of unstabilized alkyl radicals
Dimerization of alkyl radicals formed by HAT can provide an atom-economical means for the homologation of simple gaseous alkanes into long-chain liquid alkanes. Despite the well-explored activities of alkyl radicals, such radical dimerizations in catalytic processes have been scarce with the exception of stabilized radicals (e.g., benzyl radical). Indeed, during efforts in our group towards the catalytic utilization of an LMCT–HAT sequence for alkane functionalizations, alkane dimerization products have never been detected in any appreciable quantities, as non-persistent alkyl radicals are generated in an imperceptibly low concentration in photocatalytic HAT processes. These radicals are prone to react with stoichiometric oxidants, rendering radical dimerization obsolete and ineffective. In order to achieve photocatalytic alkane homologation in the HAT activation manifold, innovative strategies to override the inherent preferences of reactive alkyl radicals are needed.To realize these challenging transformations, the merging of photocatalytic HAT with the rich chemistry endowed by transition metal catalysis could provide revolutionary solutions. Taking the photoredox/nickel dual catalysis as an example, alkyl radicals generated from a HAT process engage in nickel catalyzed C–C coupling reactions. This enabling metallaphotoredox paradigm would offer exciting opportunities to exploit metal–alkyl complexes for divergent C–H functionalizations otherwise unattainable.17,33,188 For example, mechanistic investigations have revealed that certain high-valent metal dialkyl complexes are prone to undergo bond-forming reductive eliminations that are challenging at lower oxidation states, indicating that the synergistic incorporation of single electron chemistry with transition metal catalysis can provide intriguing opportunities for challenging homologation processes such as alkane metathesis.189–194
(3) Regioselectivity of HAT
The regioselectivity of the HAT process toward different C–H bonds of linear alkanes is another restriction that must be addressed. Currently, methods to allow flexible tuning of site-selectivity in the HAT pathway remain largely elusive. Regioselectivity could be governed by the properties of hydrogen abstractors, BDEs of C–H bonds, and steric effects, which present difficulties in achieving selective functionalization. Currently established HAT processes mediated by heteroatom centred radicals showed preferential selectivity of tertiary and secondary C–H bonds over primary C–H bonds. In the case of linear alkanes, methods to realize site-selective C–H functionalizations remain elusive owing to the trivial difference in either steric or electronic properties. In particular, the selective functionalization of primary C–H bonds of simple alkanes is highly desirable for the large-scale synthesis of commodity chemicals such as plasticizer and detergent alcohols.4 Accordingly, much effort has been devoted to tuning the site selectivity of C–H functionalization by employing newly developed HAT agents.16 For example, sterically hindered radical abstractors have has shown a certain degree of preference toward primary C–H bonds.23,195(4) Scalability of photochemical transformations
Moreover, in addition to the above-mentioned problems, the successful application of any photocatalytic transformation necessitates the development of efficient and practical upscaling techniques. As the efficiency of photon penetration and absorption is a critical factor, photochemical transformations pose inherent challenges to scale up relative to thermally-promoted reactions. Nevertheless, significant progress has been made in the industrial applications of radical chain reactions promoted by mercury vapor lamps, such as photochemical chlorination, sulfochlorination, nitrosation, etc.196 Rapid advancement in LED lighting technology and flow chemistry has greatly simplified the scaling up of photochemical reactions.197 Recently, photocatalytic processes have been successfully scaled up from the milligram scale in a batch to the multikilogram scale in the pharmaceutical industry using continuous-flow technology.198 Furthermore, large scale prototype photocatalytic water splitting systems have been demonstrated, providing inspirational concepts of sunlight-driven reactors and photochemical plants for alkane functionalizations.199 Although the use of a continuous-flow setup imposes a strict requirement on catalytic efficiency as well as quantum efficiency, it is safe to state that the upscaling of photocatalytic alkane functionalizations should no longer be a daunting task in the future.200The area of C–H functionalization will continue to be of high interest within the chemical community due to the fundamental and potentially ground-breaking innovations that are yet to be discovered. Breakthroughs leading to an abundance of novel transformations for alkane functionalization would arise from combining metal catalysis or electrocatalysis and photocatalysis. Additional detailed mechanistic studies, including intermediate characterization, TA spectroscopy, and high-level calculations, are needed to understand activation and functionalization as well as to provide opportunities for finely tuned catalytic reactivity. Finally, due to ubiquitous aliphatic C–H bonds in organic molecules, the successful development of the photocatalytic processes using alkane substrates will represent a paradigm shift in synthetic chemistry, allowing the development of novel C–H functionalization processes with pharmaceutical agents, natural products, and other abundant organic molecules.
Author contributions
Z. Z. conceived the topic and structure of the manuscript. All authors contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Key R&D Program of China (no. 2021YFA1500100), the National Natural Science Foundation of China (no. 22125111 and 21971163), the Shanghai Pilot Program for Basic Research – Chinese Academy of Science, Shanghai Branch, and SIOC for financial support.References
- A. A. Fokin and P. R. Schreiner, Chem. Rev., 2002, 102, 1551–1594 CrossRef CAS PubMed.
- R. H. Crabtree, Chem. Rev., 1995, 95, 987–1007 CrossRef CAS.
- J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2–24 CrossRef CAS PubMed.
- X. Tang, X. Jia and Z. Huang, Chem. Sci., 2018, 9, 288–299 RSC.
- C. Li and G. Wang, Chem. Soc. Rev., 2021, 50, 4359–4381 RSC.
- M. Ravi, M. Ranocchiari and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2017, 56, 16464–16483 CrossRef CAS PubMed.
- J. F. Hartwig and M. A. Larsen, ACS Cent. Sci., 2016, 2, 281–292 CrossRef CAS PubMed.
- N. J. Gunsalus, A. Koppaka, S. H. Park, S. M. Bischof, B. G. Hashiguchi and R. A. Periana, Chem. Rev., 2017, 117, 8521–8573 CrossRef CAS PubMed.
- N. Yassaa, B. Youcef Meklati, A. Cecinato and F. Marino, Atmos. Environ., 2001, 35, 1843–1851 CrossRef CAS.
- M. Misz-Kennan and M. J. Fabiańska, Int. J. Coal Geol., 2011, 88, 1–23 CrossRef CAS.
- T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362–3374 CrossRef CAS PubMed.
- V. C. C. Wang, S. Maji, P. P. Y. Chen, H. K. Lee, S. S. F. Yu and S. I. Chan, Chem. Rev., 2017, 117, 8574–8621 CrossRef CAS PubMed.
- R. H. Crabtree, J. Organomet. Chem., 2004, 689, 4083–4091 CrossRef CAS.
- L. Capaldo and D. Ravelli, Eur. J. Org. Chem., 2017, 2056–2071 CrossRef CAS PubMed.
- L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed.
- M. Galeotti, M. Salamone and M. Bietti, Chem. Soc. Rev., 2022, 51, 2171–2223 RSC.
- J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62–101 CrossRef CAS PubMed.
- V. Paunović and J. Pérez-Ramírez, Catal. Sci. Technol., 2019, 9, 4515–4530 RSC.
- J. M. Tedder, Angew. Chem., Int. Ed., 1982, 21, 401–410 CrossRef.
- M. Salamone and M. Bietti, Acc. Chem. Res., 2015, 48, 2895–2903 CrossRef CAS PubMed.
- M. Milan, M. Salamone, M. Costas and M. Bietti, Acc. Chem. Res., 2018, 51, 1984–1995 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- L. Chang, Q. An, L. Duan, K. Feng and Z. Zuo, Chem. Rev., 2022, 122, 2429–2486 CrossRef CAS PubMed.
- K. Kwon, R. T. Simons, M. Nandakumar and J. L. Roizen, Chem. Rev., 2022, 122, 2353–2428 CrossRef CAS PubMed.
- E. Tsui, H. Wang and R. R. Knowles, Chem. Sci., 2020, 11, 11124–11141 RSC.
- P. Bellotti, H.-M. Huang, T. Faber and F. Glorius, Chem. Rev., 2023, 123, 4237–4252 CrossRef CAS PubMed.
- A. Hu, J.-J. Guo, H. Pan, H. Tang, Z. Gao and Z. Zuo, J. Am. Chem. Soc., 2018, 140, 1612–1616 CrossRef CAS PubMed.
- J. C. K. Chu and T. Rovis, Nature, 2016, 539, 272–275 CrossRef PubMed.
- G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268–271 CrossRef CAS PubMed.
- P. R. D. Murray, J. H. Cox, N. D. Chiappini, C. B. Roos, E. A. McLoughlin, B. G. Hejna, S. T. Nguyen, H. H. Ripberger, J. M. Ganley, E. Tsui, N. Y. Shin, B. Koronkiewicz, G. Qiu and R. R. Knowles, Chem. Rev., 2022, 122, 2017–2291 CrossRef CAS PubMed.
- F. Juliá, ChemCatChem, 2022, 14, e202200916 CrossRef.
- C. E. Tinberg and S. J. Lippard, Acc. Chem. Res., 2011, 44, 280–288 CrossRef CAS PubMed.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- A. Lipp, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2021, 60, 1714–1726 CrossRef CAS PubMed.
- F.-D. Lu, G.-F. He, L.-Q. Lu and W.-J. Xiao, Green Chem., 2021, 23, 5379–5393 RSC.
- S. B. Beil, T. Q. Chen, N. E. Intermaggio and D. W. C. MacMillan, Acc. Chem. Res., 2022, 55, 3481–3494 CrossRef CAS PubMed.
- N. E. S. Tay, D. Lehnherr and T. Rovis, Chem. Rev., 2022, 122, 2487–2649 CrossRef CAS PubMed.
- A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022–4047 CrossRef CAS PubMed.
- N. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2022, 122, 1925–2016 CrossRef CAS PubMed.
- L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed.
- Z. Ye, Y. M. Lin and L. Gong, Eur. J. Org. Chem., 2021, 2021, 5545–5556 CrossRef CAS.
- H. Cao, X. Tang, H. Tang, Y. Yuan and J. Wu, Chem Catal., 2021, 1, 523–598 CrossRef CAS.
- S. Sarkar, K. P. S. Cheung and V. Gevorgyan, Chem. Sci., 2020, 11, 12974–12993 RSC.
- L. Capaldo, L. L. Quadri and D. Ravelli, Green Chem., 2020, 22, 3376–3396 RSC.
- D. Ravelli, M. Fagnoni, T. Fukuyama, T. Nishikawa and I. Ryu, ACS Catal., 2018, 8, 701–713 CrossRef CAS.
- S. Cheng, Q. Li, X. Cheng, Y.-M. Lin and L. Gong, Chin. J. Chem., 2022, 40, 2825–2837 CrossRef CAS.
- M. Salamone, M. Galeotti, E. Romero-Montalvo, J. A. van Santen, B. D. Groff, J. M. Mayer, G. A. DiLabio and M. Bietti, J. Am. Chem. Soc., 2021, 143, 11759–11776 CrossRef CAS PubMed.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- B. Ruscic, J. V. Michael, P. C. Redfern, L. A. Curtiss and K. Raghavachari, J. Phys. Chem. A, 1998, 102, 10889–10899 CrossRef CAS.
- R. M. Borges dos Santos, V. S. F. Muralha, C. F. Correia, R. C. Guedes, B. J. Costa Cabral and J. A. Martinho Simões, J. Phys. Chem. A, 2002, 106, 9883–9889 CrossRef CAS.
- F. G. Bordwell and G. Z. Ji, J. Am. Chem. Soc., 1991, 113, 8398–8401 CrossRef CAS.
- B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25–35 RSC.
- A. A. Zavitsas and C. Chatgilialoglu, J. Am. Chem. Soc., 1995, 117, 10645–10654 CrossRef CAS.
- A. Pross and S. S. Shaik, Acc. Chem. Res., 1983, 16, 363–370 CrossRef CAS.
- A. Ruffoni, R. C. Mykura, M. Bietti and D. Leonori, Nat. Synth., 2022, 1, 682–695 CrossRef.
- J. Chateauneuf, J. Lusztyk and K. U. Ingold, J. Am. Chem. Soc., 1988, 110, 2877–2885 CrossRef CAS.
- J. Chateauneuf, J. Lusztyk and K. U. Ingold, J. Am. Chem. Soc., 1988, 110, 2886–2893 CrossRef CAS.
- F. De Vleeschouwer, V. Van Speybroeck, M. Waroquier, P. Geerlings and F. De Proft, Org. Lett., 2007, 9, 2721–2724 CrossRef CAS PubMed.
- M. Salamone, V. B. Ortega and M. Bietti, J. Org. Chem., 2015, 80, 4710–4715 CrossRef CAS PubMed.
- M. Salamone, T. Martin, M. Milan, M. Costas and M. Bietti, J. Org. Chem., 2017, 82, 13542–13549 CrossRef CAS PubMed.
- M. Milan, M. Bietti and M. Costas, ACS Cent. Sci., 2017, 3, 196–204 CrossRef CAS PubMed.
- M. S. Chen and M. C. White, Science, 2010, 327, 566–571 CrossRef CAS PubMed.
- E. Proksch and A. de Meijere, Angew. Chem., Int. Ed., 1976, 15, 761–762 CrossRef.
- M. Bietti, R. Martella and M. Salamone, Org. Lett., 2011, 13, 6110–6113 CrossRef CAS PubMed.
- M. Salamone, I. Giammarioli and M. Bietti, J. Org. Chem., 2011, 76, 4645–4651 CrossRef CAS PubMed.
- D. Dondi, M. Fagnoni and A. Albini, Chem. - Eur. J., 2006, 12, 4153–4163 CrossRef CAS PubMed.
- C. Coenjarts and J. C. Scaiano, J. Am. Chem. Soc., 2000, 122, 3635–3641 CrossRef CAS.
- L. Giering, M. Berger and C. Steel, J. Am. Chem. Soc., 1974, 96, 953–958 CrossRef CAS.
- D. V. Avila, C. E. Brown, K. U. Ingold and J. Lusztyk, J. Am. Chem. Soc., 1993, 115, 466–470 CrossRef CAS.
- S. M. Saunders, D. L. Baulch, K. M. Cooke, M. J. Pilling and P. I. Smurthwaite, Int. J. Chem. Kinet., 1994, 26, 113–130 CrossRef CAS.
- M. Weber and H. Fischer, J. Am. Chem. Soc., 1999, 121, 7381–7388 CrossRef CAS.
- S. Mitroka, S. Zimmeck, D. Troya and J. M. Tanko, J. Am. Chem. Soc., 2010, 132, 2907–2913 CrossRef CAS PubMed.
- C. Walling, G. M. El-Taliawi and C. Zhao, J. Am. Chem. Soc., 1983, 105, 5119–5124 CrossRef CAS.
- A. T. Droege and F. P. Tully, J. Phys. Chem. A, 1987, 91, 1222–1225 CAS.
- P. Xu, P.-Y. Chen and H.-C. Xu, Angew. Chem., Int. Ed., 2020, 59, 14275–14280 CrossRef CAS PubMed.
- C.-Y. Huang, J. Li and C.-J. Li, Nat. Commun., 2021, 12, 4010 CrossRef CAS PubMed.
- J. Dumas, Justus Liebigs Ann. Chem., 1840, 33, 259–300 CrossRef.
- S. M. Aschmann and R. Atkinson, Int. J. Chem. Kinet., 1995, 27, 613–622 CrossRef CAS.
- P. Li, A. M. Deetz, J. Hu, G. J. Meyer and K. Hu, J. Am. Chem. Soc., 2022, 144, 17604–17610 CrossRef CAS PubMed.
- M. Ravi, M. Ranocchiari and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2017, 56, 16464–16483 CrossRef CAS PubMed.
- R. A. Periana, D. J. Taube, S. Gamble, H. Taube, T. Satoh and H. Fujii, Science, 1998, 280, 560–564 CrossRef CAS PubMed.
- C. Duan, M. Luo and X. Xing, Bioresour. Technol., 2011, 102, 7349–7353 CrossRef CAS PubMed.
- S. G. Lee, J. H. Goo, H. G. Kim, J.-I. Oh, Y. M. Kim and S. W. Kim, Biotechnol. Lett., 2004, 26, 947–950 CrossRef CAS PubMed.
- K. Ohkubo and K. Hirose, Angew. Chem., Int. Ed., 2018, 57, 2126–2129 CrossRef CAS PubMed.
- G. C. Fortman, N. C. Boaz, D. Munz, M. M. Konnick, R. A. Periana, J. T. Groves and T. B. Gunnoe, J. Am. Chem. Soc., 2014, 136, 8393–8401 CrossRef CAS PubMed.
- N. A. Schwartz, N. C. Boaz, S. E. Kalman, T. Zhuang, J. M. Goldberg, R. Fu, R. J. Nielsen, W. A. Goddard, III, J. T. Groves and T. B. Gunnoe, ACS Catal., 2018, 8, 3138–3149 CrossRef CAS.
- N. S. Liebov, J. M. Goldberg, N. C. Boaz, N. Coutard, S. E. Kalman, T. Zhuang, J. T. Groves and T. B. Gunnoe, ChemcatChem, 2019, 11, 5045–5054 CrossRef CAS.
- N. A. Hirscher, N. Ohri, Q. Yang, J. Zhou, J. M. Anna, E. J. Schelter and K. I. Goldberg, J. Am. Chem. Soc., 2021, 143, 19262–19267 CrossRef CAS PubMed.
- K. Ohkubo, A. Fujimoto and S. Fukuzumi, Chem. Commun., 2011, 47, 8515–8517 RSC.
- H.-P. Deng, Q. Zhou and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 12661–12665 CrossRef CAS PubMed.
- S. Rohe, A. O. Morris, T. McCallum and L. Barriault, Angew. Chem., Int. Ed., 2018, 57, 15664–15669 CrossRef CAS PubMed.
- P. Jia, Q. Li, W. C. Poh, H. Jiang, H. Liu, H. Deng and J. Wu, Chem, 2020, 6, 1766–1776 CAS.
- L. L. Costanzo, S. Pistarà and G. Condorelli, J. Photochem., 1983, 21, 45–51 CrossRef CAS.
- Q. An, Z. Wang, Y. Chen, X. Wang, K. Zhang, H. Pan, W. Liu and Z. Zuo, J. Am. Chem. Soc., 2020, 142, 6216–6226 CrossRef CAS PubMed.
- Q. Yang, Y.-H. Wang, Y. Qiao, M. Gau, P. J. Carroll, P. J. Walsh and E. J. Schelter, Science, 2021, 372, 847–852 CrossRef CAS PubMed.
- S. M. Treacy and T. Rovis, J. Am. Chem. Soc., 2021, 143, 2729–2735 CrossRef CAS PubMed.
- Q. Zhang, S. Liu, J. Lei, Y. Zhang, C. Meng, C. Duan and Y. Jin, Org. Lett., 2022, 24, 1901–1906 CrossRef CAS PubMed.
- Y. Jin, L. Wang, Q. Zhang, Y. Zhang, Q. Liao and C. Duan, Green Chem., 2021, 23, 9406–9411 RSC.
- Y. Jin, Q. Zhang, L. Wang, X. Wang, C. Meng and C. Duan, Green Chem., 2021, 23, 6984–6989 RSC.
- Z.-Y. Dai, S.-Q. Zhang, X. Hong, P.-S. Wang and L.-Z. Gong, Chem Catal., 2022, 2, 1211–1222 CrossRef CAS.
- G. B. Panetti, Q. Yang, M. R. Gau, P. J. Carroll, P. J. Walsh and E. J. Schelter, Chem Catal., 2022, 2, 853–866 CrossRef CAS.
- D. Birnthaler, R. Narobe, E. Lopez-Berguno, C. Haag and B. König, ACS Catal., 2023, 13, 1125–1132 CrossRef CAS.
- B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719–12722 CrossRef CAS PubMed.
- S. J. Hwang, D. C. Powers, A. G. Maher, B. L. Anderson, R. G. Hadt, S.-L. Zheng, Y.-S. Chen and D. G. Nocera, J. Am. Chem. Soc., 2015, 137, 6472–6475 CrossRef CAS PubMed.
- L. K. G. Ackerman, J. I. Martinez Alvarado and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 14059–14063 CrossRef CAS PubMed.
- G. S. Lee, J. Won, S. Choi, M.-H. Baik and S. H. Hong, Angew. Chem., Int. Ed., 2020, 59, 16933–16942 CrossRef CAS PubMed.
- G. S. Lee, B. Park and S. H. Hong, Nat. Commun., 2022, 13, 5200 CrossRef CAS PubMed.
- C. Shu, A. Noble and V. K. Aggarwal, Nature, 2020, 586, 714–719 CrossRef CAS PubMed.
- M. I. Gonzalez, D. Gygi, Y. Qin, Q. Zhu, E. J. Johnson, Y.-S. Chen and D. G. Nocera, J. Am. Chem. Soc., 2022, 144, 1464–1472 CrossRef CAS PubMed.
- D. Gygi, M. I. Gonzalez, S. J. Hwang, K. T. Xia, Y. Qin, E. J. Johnson, F. Gygi, Y.-S. Chen and D. G. Nocera, J. Am. Chem. Soc., 2021, 143, 6060–6064 CrossRef CAS PubMed.
- S. Z. Zard, Synlett, 1996, 1148–1154 CrossRef CAS.
- L. Stella, Angew. Chem., Int. Ed., 1983, 22, 337–350 CrossRef.
- S. Z. Zard, Chem. Soc. Rev., 2008, 37, 1603–1618 RSC.
- R. A. Johnson and F. D. Greene, J. Org. Chem., 1975, 40, 2192–2196 CrossRef CAS.
- V. A. Schmidt, R. K. Quinn, A. T. Brusoe and E. J. Alexanian, J. Am. Chem. Soc., 2014, 136, 14389–14392 CrossRef CAS PubMed.
- S. Huo, R. Wu, M. Li, H. Chen and W. Zuo, Ind. Eng. Chem. Res., 2020, 59, 22690–22695 CrossRef CAS.
- R. K. Quinn, Z. A. Könst, S. E. Michalak, Y. Schmidt, A. R. Szklarski, A. R. Flores, S. Nam, D. A. Horne, C. D. Vanderwal and E. J. Alexanian, J. Am. Chem. Soc., 2016, 138, 696–702 CrossRef CAS PubMed.
- W. L. Czaplyski, C. G. Na and E. J. Alexanian, J. Am. Chem. Soc., 2016, 138, 13854–13857 CrossRef CAS PubMed.
- J. B. Williamson, W. L. Czaplyski, E. J. Alexanian and F. A. Leibfarth, Angew. Chem., Int. Ed., 2018, 57, 6261–6265 CrossRef CAS PubMed.
- W. Lee, S. Jung, M. Kim and S. Hong, J. Am. Chem. Soc., 2021, 143, 3003–3012 CrossRef CAS PubMed.
- A. J. McMillan, M. Sieńkowska, P. Di Lorenzo, G. K. Gransbury, N. F. Chilton, M. Salamone, A. Ruffoni, M. Bietti and D. Leonori, Angew. Chem., Int. Ed., 2021, 60, 7132–7139 CrossRef CAS PubMed.
- H.-B. Yang, A. Feceu and D. B. C. Martin, ACS Catal., 2019, 9, 5708–5715 CrossRef CAS.
- W. K. Weigel, H. T. Dang, H.-B. Yang and D. B. C. Martin, Chem. Commun., 2020, 56, 9699–9702 RSC.
- A. Matsumoto, M. Yamamoto and K. Maruoka, ACS Catal., 2022, 12, 2045–2051 CrossRef CAS.
- D. H. R. Barton and D. Doller, Acc. Chem. Res., 1992, 25, 504–512 CrossRef CAS.
- X. Shao, X. Wu, S. Wu and C. Zhu, Org. Lett., 2020, 22, 7450–7454 CrossRef CAS PubMed.
- Y.-W. Zheng, R. Narobe, K. Donabauer, S. Yakubov and B. König, ACS Catal., 2020, 10, 8582–8589 CrossRef CAS.
- Y. Zhang, Y. Jin, L. Wang, Q. Zhang, C. Meng and C. Duan, Green Chem., 2021, 23, 6926–6930 RSC.
- C. Huang, J.-H. Wang, J. Qiao, X.-W. Fan, B. Chen, C.-H. Tung and L.-Z. Wu, J. Org. Chem., 2019, 84, 12904–12912 CrossRef CAS PubMed.
- G.-X. Li, C. A. Morales-Rivera, F. Gao, Y. Wang, G. He, P. Liu and G. Chen, Chem. Sci., 2017, 8, 7180–7185 RSC.
- G.-X. Li, X. Hu, G. He and G. Chen, ACS Catal., 2018, 8, 11847–11853 CrossRef CAS.
- S. Mukherjee, B. Maji, A. Tlahuext-Aca and F. Glorius, J. Am. Chem. Soc., 2016, 138, 16200–16203 CrossRef CAS PubMed.
- K. A. Margrey, W. L. Czaplyski, D. A. Nicewicz and E. J. Alexanian, J. Am. Chem. Soc., 2018, 140, 4213–4217 CrossRef CAS PubMed.
- C. M. Morton, Q. Zhu, H. Ripberger, L. Troian-Gautier, Z. S. D. Toa, R. R. Knowles and E. J. Alexanian, J. Am. Chem. Soc., 2019, 141, 13253–13260 CrossRef CAS PubMed.
- M. Schlegel, S. Qian and D. A. Nicewicz, ACS Catal., 2022, 12, 10499–10505 CrossRef CAS.
- B. Wang, C. Ascenzi Pettenuzzo, J. Singh, G. E. McCabe, L. Clark, R. Young, J. Pu and Y. Deng, ACS Catal., 2022, 12, 10441–10448 CrossRef CAS.
- A. Hu, J.-J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668–672 CrossRef CAS PubMed.
- Z. Tan, X. He, K. Xu and C. Zeng, ChemSusChem, 2022, 15, e202102360 CAS.
- Q. An, Y.-Y. Xing, R. Pu, M. Jia, Y. Chen, A. Hu, S.-Q. Zhang, N. Yu, J. Du, Y. Zhang, J. Chen, W. Liu, X. Hong and Z. Zuo, J. Am. Chem. Soc., 2022, 145, 359–376 CrossRef PubMed.
- F. Liu, S. Ma, Z. Lu, A. Nangia, M. Duan, Y. Yu, G. Xu, Y. Mei, M. Bietti and K. N. Houk, J. Am. Chem. Soc., 2022, 144, 6802–6812 CrossRef CAS PubMed.
- D. C. Duncan, T. L. Netzel and C. L. Hill, Inorg. Chem., 1995, 34, 4640–4646 CrossRef CAS.
- C. L. Hill, R. F. Renneke and L. Combs, Tetrahedron, 1988, 44, 7499–7507 CrossRef CAS.
- R. F. Renneke and C. L. Hill, Angew. Chem., Int. Ed., 1988, 27, 1526–1527 CrossRef.
- A. Chemseddine, C. Sanchez, J. Livage, J. P. Launay and M. Fournier, Inorg. Chem., 1984, 23, 2609–2613 CrossRef CAS.
- B. S. Jaynes and C. L. Hill, J. Am. Chem. Soc., 1995, 117, 4704–4705 CrossRef CAS.
- I. Ryu, A. Tani, T. Fukuyama, D. Ravelli, M. Fagnoni and A. Albini, Angew. Chem., Int. Ed., 2011, 50, 1869–1872 CrossRef CAS PubMed.
- T. Fukuyama, K. Yamada, T. Nishikawa, D. Ravelli, M. Fagnoni and I. Ryu, Chem. Lett., 2017, 47, 207–209 CrossRef.
- Z.-Y. Dai, Z.-S. Nong and P.-S. Wang, ACS Catal., 2020, 10, 4786–4790 CrossRef CAS.
- V. I. Supranovich, V. V. Levin and A. D. Dilman, Org. Lett., 2019, 21, 4271–4274 CrossRef CAS PubMed.
- X.-K. Qi, L. Guo, L.-J. Yao, H. Gao, C. Yang and W. Xia, Org. Lett., 2021, 23, 4473–4477 CrossRef CAS PubMed.
- K. Yahata, S. Sakurai, S. Hori, S. Yoshioka, Y. Kaneko, K. Hasegawa and S. Akai, Org. Lett., 2020, 22, 1199–1203 CrossRef CAS PubMed.
- G. Laudadio, Y. Deng, K. van der Wal, D. Ravelli, M. Nuño, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 369, 92–96 CrossRef CAS PubMed.
- L. Capaldo, L. L. Quadri, D. Merli and D. Ravelli, Chem. Commun., 2021, 57, 4424–4427 RSC.
- K. Kim, S. Lee and S. H. Hong, Org. Lett., 2021, 23, 5501–5505 CrossRef CAS PubMed.
- T. E. Schirmer, A. B. Rolka, T. A. Karl, F. Holzhausen and B. Konig, Org. Lett., 2021, 23, 5729–5733 CrossRef CAS PubMed.
- Y. Shen, Z.-Y. Dai, C. Zhang and P.-S. Wang, ACS Catal., 2021, 11, 6757–6762 CrossRef CAS.
- X. Wang, M. Yu, H. Song, Y. Liu and Q. Wang, Org. Lett., 2021, 23, 8353–8358 CrossRef CAS PubMed.
- Y.-T. Wang, Y.-L. Shih, Y.-K. Wu and I. Ryu, Adv. Synth. Catal., 2022, 364, 1–6 CrossRef.
- I. B. Perry, T. F. Brewer, P. J. Sarver, D. M. Schultz, D. A. DiRocco and D. W. C. MacMillan, Nature, 2018, 560, 70–75 CrossRef CAS PubMed.
- D. Mazzarella, A. Pulcinella, L. Bovy, R. Broersma and T. Noël, Angew. Chem., Int. Ed., 2021, 60, 21277–21282 CrossRef CAS PubMed.
- S. Xu, H. Chen, Z. Zhou and W. Kong, Angew. Chem., Int. Ed., 2021, 60, 7405–7411 CrossRef CAS PubMed.
- Q. Wang, S. Ni, L. Yu, Y. Pan and Y. Wang, ACS Catal., 2022, 12, 11071–11077 CrossRef CAS.
- J. G. West, D. Huang and E. J. Sorensen, Nat. Commun., 2015, 6, 10093 CrossRef PubMed.
- H. Cao, Y. Kuang, X. Shi, K. L. Wong, B. B. Tan, J. M. C. Kwan, X. Liu and J. Wu, Nat. Commun., 2020, 11, 1956 CrossRef CAS PubMed.
- B. E. Cowie, J. M. Purkis, J. Austin, J. B. Love and P. L. Arnold, Chem. Rev., 2019, 119, 10595–10637 CrossRef CAS PubMed.
- J. G. West, T. A. Bedell and E. J. Sorensen, Angew. Chem., Int. Ed., 2016, 55, 8923–8927 CrossRef CAS PubMed.
- L. Capaldo, D. Merli, M. Fagnoni and D. Ravelli, ACS Catal., 2019, 9, 3054–3058 CrossRef CAS.
- J. Yu, C. Zhao, R. Zhou, W. Gao, S. Wang, K. Liu, S. Chen, K. Hu, L. Mei, L. Yuan, Z. Chai, H. Hu and W. Shi, Chem. - Eur. J., 2020, 26, 16521–16529 CrossRef CAS PubMed.
- E. E. Stache, V. Kottisch and B. P. Fors, J. Am. Chem. Soc., 2020, 142, 4581–4585 CrossRef CAS PubMed.
- K. Ohkubo, K. Hirose and S. Fukuzumi, Photochem. Photobiol. Sci., 2016, 15, 731–734 CrossRef CAS PubMed.
- S. Zhang, S. Cao, Y.-M. Lin, L. Sha, C. Lu and L. Gong, Chin. J. Catal., 2022, 43, 564–570 CrossRef CAS.
- M.-J. Zhou, L. Zhang, G. Liu, C. Xu and Z. Huang, J. Am. Chem. Soc., 2021, 143, 16470–16485 CrossRef CAS PubMed.
- C. Walling and M. J. Gibian, J. Am. Chem. Soc., 1965, 87, 3361–3364 CrossRef CAS.
- G. H. Jones, D. W. Edwards and D. Parr, J. Chem. Soc., Chem. Commun., 1976, 969–970 RSC.
- S. Kamijo, K. Kamijo, K. Maruoka and T. Murafuji, Org. Lett., 2016, 18, 6516–6519 CrossRef CAS PubMed.
- B. Abadie, D. Jardel, G. Pozzi, P. Toullec and J.-M. Vincent, Chem. - Eur. J., 2019, 25, 16120–16127 CrossRef CAS PubMed.
- X.-Z. Fan, J.-W. Rong, H.-L. Wu, Q. Zhou, H.-P. Deng, J. D. Tan, C.-W. Xue, L.-Z. Wu, H.-R. Tao and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 8514–8518 CrossRef CAS PubMed.
- C.-Y. Huang, J. Li, W. Liu and C.-J. Li, Chem. Sci., 2019, 10, 5018–5024 RSC.
- Y. Shen, Y. Gu and R. Martin, J. Am. Chem. Soc., 2018, 140, 12200–12209 CrossRef CAS PubMed.
- L. Niu, C. Jiang, Y. Liang, D. Liu, F. Bu, R. Shi, H. Chen, A. D. Chowdhury and A. Lei, J. Am. Chem. Soc., 2020, 142, 17693–17702 CrossRef CAS PubMed.
- Z. Wen, A. Maheshwari, C. Sambiagio, Y. Deng, G. Laudadio, K. Van Aken, Y. Sun, H. P. L. Gemoets and T. Noël, Org. Process Res. Dev., 2020, 24, 2356–2361 CrossRef CAS PubMed.
- T. Wan, Z. Wen, G. Laudadio, L. Capaldo, R. Lammers, J. A. Rincón, P. García-Losada, C. Mateos, M. O. Frederick, R. Broersma and T. Noël, ACS Cent. Sci., 2022, 8, 51–56 CrossRef CAS PubMed.
- L. Capaldo, S. Bonciolini, A. Pulcinella, M. Nuño and T. Noël, Chem. Sci., 2022, 13, 7325–7331 RSC.
- J. K. Thomas, J. Phys. Chem., 1967, 71, 1919–1925 CrossRef CAS.
- Y. Wang, L. Qian, Z. Huang, G. Liu and Z. Huang, Chin. J. Chem., 2020, 38, 837–841 CrossRef CAS.
- Y. Su, F. Yu, G. Liu and Z. Huang, Chin. J. Chem., 2022, 40, 2263–2268 CrossRef CAS.
- X. Sun, J. Xue, Y. Ren, X. Li, L. Zhou, B. Li and Z. Zhao, Chin. J. Chem., 2021, 39, 661–665 CrossRef CAS.
- D. Wu, L. Wu, P. Chen and G. Liu, Chin. J. Chem., 2022, 40, 1699–1704 CrossRef CAS.
- M. W. Milbauer, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2022, 144, 21030–21034 CrossRef CAS PubMed.
- M. P. Lanci, M. S. Remy, D. B. Lao, M. S. Sanford and J. M. Mayer, Organometallics, 2011, 30, 3704–3707 CrossRef CAS.
- M. P. Lanci, M. S. Remy, W. Kaminsky, J. M. Mayer and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 15618–15620 CrossRef CAS PubMed.
- X. Wang, N. Luo and F. Wang, Chin. J. Chem., 2022, 40, 1492–1505 CrossRef CAS.
- D. Chen, J. Jiang and J.-P. Wan, Chin. J. Chem., 2022, 40, 2582–2594 CrossRef CAS.
- L. Zhu, Y. Fang and C. Li, Chin. J. Chem., 2020, 38, 787–789 CrossRef CAS.
- Z.-L. Wang and F. Wang, Chin. J. Chem., 2022, 40, 1751–1753 CrossRef CAS.
- M. Fischer, Angew. Chem., Int. Ed., 1978, 17, 16–26 CrossRef.
- L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Chem. Rev., 2022, 122, 2752–2906 CrossRef CAS PubMed.
- E. B. Corcoran, J. P. McMullen, F. Lévesque, M. K. Wismer and J. R. Naber, Angew. Chem., Int. Ed., 2020, 59, 11964–11968 CrossRef CAS PubMed.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- G. Fang, D.-Z. Lin and K. Liao, Chin. J. Chem., 2023, 41, 1075–1079 CrossRef.
| This journal is © The Royal Society of Chemistry 2023 |