
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA chemical probe unravels the reactive proteome of health-associated catechols†
Angela
Weigert Muñoz
 a,
Kevin M.
Meighen-Berger
b,
Stephan M.
Hacker
c,
Matthias J.
Feige
b and
Stephan A.
Sieber
*a
a,
Kevin M.
Meighen-Berger
b,
Stephan M.
Hacker
c,
Matthias J.
Feige
b and
Stephan A.
Sieber
*a
aCenter for Functional Protein Assemblies, Department of Bioscience, TUM School of Natural Sciences, Technical University of Munich, Ernst-Otto-Fischer-Straße 8, D-85748 Garching, Germany. E-mail: stephan.sieber@tum.de
bCenter for Functional Protein Assemblies, Department of Bioscience, TUM School of Natural Sciences, Technical University of Munich, Lichtenbergstraße 4, D-85748 Garching, Germany
cLeiden Institute of Chemistry, Leiden University, Einsteinweg 55, 2333 CC Leiden, Netherlands
First published on 22nd July 2023
Abstract
Catechol-containing natural products are common constituents of foods, drinks, and drugs. Natural products carrying this motif are often associated with beneficial biological effects such as anticancer activity and neuroprotection. However, the molecular mode of action behind these properties is poorly understood. Here, we apply a mass spectrometry-based competitive chemical proteomics approach to elucidate the target scope of catechol-containing bioactive molecules from diverse foods and drugs. Inspired by the protein reactivity of catecholamine neurotransmitters, we designed and synthesised a broadly reactive minimalist catechol chemical probe based on dopamine. Initial labelling experiments in live human cells demonstrated broad protein binding by the probe, which was largely outcompeted by its parent compound dopamine. Next, we investigated the competition profile of a selection of biologically relevant catechol-containing substances. With this approach, we characterised the protein reactivity and the target scope of dopamine and ten biologically relevant catechols. Strikingly, proteins associated with the endoplasmic reticulum (ER) were among the main targets. ER stress assays in the presence of reactive catechols revealed an activation of the unfolded protein response (UPR). The UPR is highly relevant in oncology and cellular resilience, which may provide an explanation of the health-promoting effects attributed to many catechol-containing natural products.
Introduction
The catechol motif is part of many bioactive molecules including catecholamine hormones, plant secondary metabolites, and drugs. Plant-derived catechols are often associated with health-promoting effects such as anti-inflammatory, anti-tumour, and neuroprotective activities, among others.1–11 Moreover, they have been reported to directly bind to or act on specific proteins, but the proposed target proteins and pathways are diverse.2,9,12,13 Examples include protein disulphide isomerases (PDIs),10,14 matrixmetalloproteases,7 COX-2,11 protein deglycase DJ-1 (also known as Parkinson disease protein 7, PARK7),15 a number of kinases,16 and more.6,7,11,15,17–21 Overall, the full target scopes and biological modes of action of catechols are not fully understood.Covalent binding to proteins is facilitated by the tendency of catechols to oxidise to reactive ortho-quinones in aqueous conditions at physiological pH,22 which are then subject to nucleophilic attack by amine or thiol side-chains in proteins. Specifically, this has been studied in depth for dopamine (DA), where aberrant protein modification has been implicated as a potential driver of neuron loss in the pathogenesis of Parkinson's disease (PD).19,20 However, much remains unknown about the identity of the specific protein targets, the binding sites, and the reactive molecular species.17 Chemical activity-based probes23,24 have recently been published of dopamine (DA),25 capsaicin (CP),11n-octyl caffeate,14 3,4-dihydroxyphenyl acetic acid (DPA),26 and 6-hydroxydopamine (6-OHDA),27 the latter being a neurotoxic oxidation product of DA. In general, the analysis of protein modification by DA-based catechols using mass spectrometry (MS) methods is challenged by the precipitation of DA-protein adducts,20 which interferes with their detection.17DA quinone (DAQ) is a reactive molecule that can undergo further chemical reactions or covalently modify cellular structures. For instance, it can cyclise via intramolecular nucleophilic attack by the amine side chain to form leukodopaminochrome, which may further polymerise to form the insoluble natural pigment eumelanin. DAQ is also subjected to nucleophilic attack by protein side chains such as cysteine in a Michael-type addition and results in protein post-translational modification (PTMs) which can deactivate enzyme activity and cause protein aggregation.17 Protein-bound DA can be oxidised again and add to further DA molecules via their nucleophilic amine, forming an insoluble protein–melanin conjugate termed neuromelanin (Fig. 1a).17
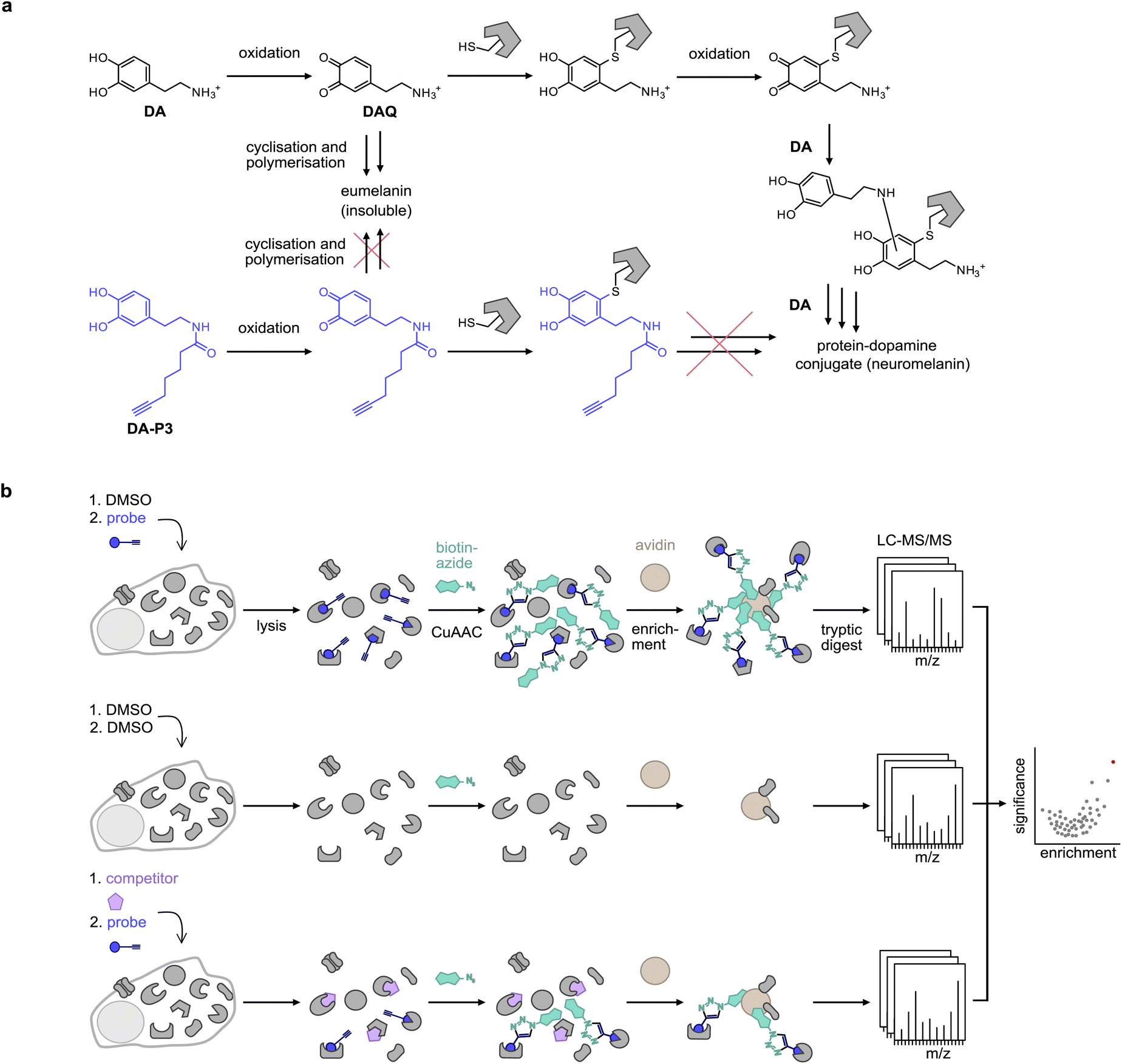 | ||
| Fig. 1 Probe design and workflow for the identification of catechol protein targets. (a) DA is oxidised to DAQ, which cyclises to aminochrome by intramolecular nucleophilic attack of the amine. Aminochrome polymerises to form insoluble eumelanin. DAQ may also react with a nucleophilic amino acid residue of a protein such as a cysteine. Following protein modification, DA can undergo multiple further reactions with other DA molecules or proteins, leading to the formation of insoluble protein–dopamine conjugates (neuromelanin).4,5 An acylated probe such as DA-P3 lacks the nucleophilic amine, trapping it in the quinone state and impeding the formation of insoluble probe-protein aggregates. (b) Chemical proteomics workflow applied for target identification. Live cells are treated with the chemical probe (blue circle), DMSO, or an excess of the catechol of interest (purple pentagon) plus the chemical probe. Following protein extraction, the labelled proteome is ligated by CuAAC to biotin azide (cyan shape), enriched on avidin beads (brown circle), digested, and peptides are analysed by LC-MS/MS. | ||
Bearing in mind the propensity of DA and DA-protein adducts for precipitation, we designed novel minimalist catechol probes for global target identification by functionalisation of the DA amino group with an acyl alkyne handle. Since these probes lack a nucleophilic residue, they are readily oxidised to an ortho-quinone but, unlike DA, cannot undergo side-reactions such as cyclisation or polymerisation to insoluble DA-protein aggregates. With a preserved native protein reactivity, this probe design facilitated the direct target identification of DA by chemical proteomics in live cells by applying the probe in competition with DA and with a suite of structurally diverse catechols (Fig. 1b). Chemical proteomics revealed ER-associated proteins among the top hits of several catechol natural products which was confirmed by cellular unfolded protein response (UPR) assays. The modulation of ER stress pathways by some of these compounds provides an intriguing explanation for their anticancer activities.
Results and discussion
DA probe design and synthesis
In order to prevent undesired side reactions, we devised a novel probe design based on alkynylated DA derivatives lacking the free amine. This strategy enables quinone formation, ensures the desired protein reactivity, and prevents polymerisation, which are important prerequisites for selective target identification in competitive studies with diverse catechols. Following this approach, we synthesised three DA-probes, DA-P1, DA-P2, and DA-P3, with a varying chain length of five to seven carbons by standard amide coupling using EDC HCl and HOBt. For a negative control lacking the 3′OH group, DA-P4, we acylated tyramine with 5-hexanoic acid (Fig. 2a and S1a†). Next, we tested general protein reactivity of the probes with purified DJ-1, which has three reactive cysteines28 and has been reported to be modified by DAQ.15,17,29 DJ-1 (5 μM) was incubated with a 100-fold excess of DA-P2, DA-P3, DA-P4, DA, or the equivalent amount of DMSO as control, and analysed by high resolution MS (Fig. S1b†). Indeed, DJ-1 was modified by two to three molecules of DA-P2 and DA-P3. DA-P4, lacking an intact catechol group, showed no protein binding. Furthermore, no modification was observed on DJ-1 treated with DA, which is in line with previous studies that failed to detect DA-modifications on proteins by MS methods.17,18 As DA modifications have been proposed to be reversible18 and may lead to protein precipitation,17 labelling was performed with DA-P3 in competition with DA to reveal DA modification by probe displacement. For this, DJ-1 (1 μM) was treated with different concentrations of DA (12.5–200 μM) followed by incubation with DA-P3 (25 μM). After copper-catalysed azide–alkyne cycloaddition (CuAAC)30,31 to rhodamine azide, labelled DJ-1 was separated by SDS-PAGE and fluorescence intensity of the corresponding protein band indeed decreased with increasing DA concentration. No protein precipitation was observed after probe treatment and competition with up to 50 μM DA (Fig. S1c†). However, lower Coomassie band intensities were visible at 100–200 μM DA, indicating the formation of insoluble DA-protein aggregates25 at high concentrations.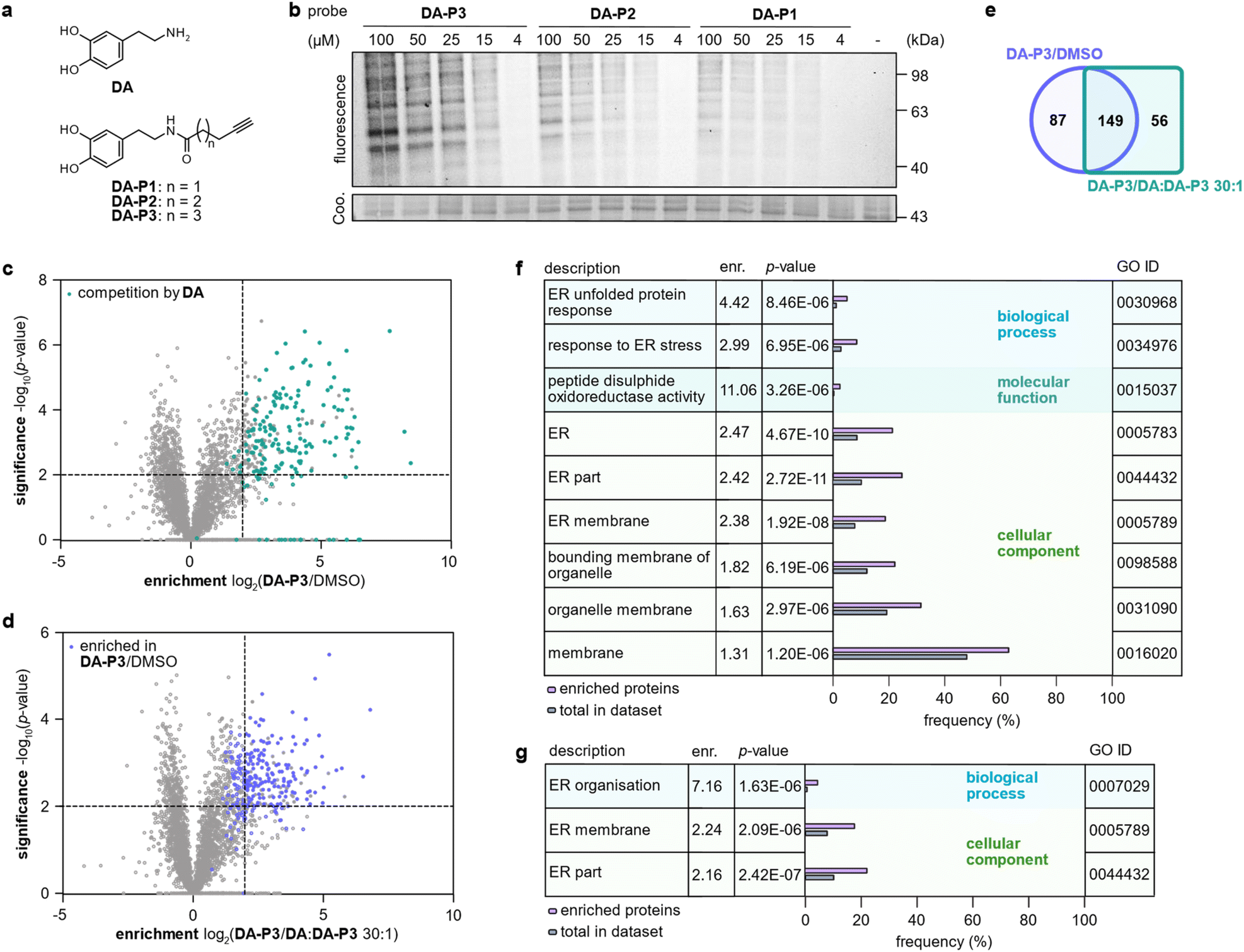 | ||
| Fig. 2 Labelling in HEK293 cells in competition with DA. (a) Structures of DA and chemical probes. (b) Fluorescence visualisation of HEK293 proteome separated by SDS-PAGE after labelling in situ with different concentrations of DA-P1, DA-P2, and DA-P3, lysis, and ligation to rhodamine azide. (c) Volcano plot showing proteins labelled by DA-P3 (15 μM) in live HEK293 compared to a DMSO-treated control. Proteins that were outcompeted by a 30-fold excess of DA are highlighted in cyan. (d) Volcano plot showing proteins labelled by DA-P3 (15 μM) in live HEK293 cells compared to samples pre-treated with a 30-fold DA excess plus DA-P3 (15 μM). (c and d) MS data from three replicates were analysed by MaxLFQ and filtered for proteins identified in three replicates in at least one condition. Samples were compared using a two-sided two-sample t-test. Proteins that were enriched against DMSO are highlighted in blue, proteins that were outcompeted by DA are highlighted in cyan. Proteins were considered significant when they were enriched more than four-fold (log2(enrichment) ≥ 2) with a p-value of less than 0.01 (−log10(p-value) ≥ 2). Proteins with missing values are shown with −log10(p-value) = 0. Significant hits are shown as full circles; not enriched proteins are shown as open circles. (e) Overlap of proteins significantly enriched against DMSO (blue circle) and against a 30-fold excess of DA (cyan square). Only proteins that were identified in all samples of all three conditions were considered. (f) GO term enrichment analysis of proteins significantly enriched in HEK293 cells by 15 μM DA-P3 compared to DMSO and (g) compared to a 30-fold excess of DA. Threshold of GO term enrichment analysis: p-value ≤ 10−5. See Table S3† for details on identified proteins. | ||
To analyse overall protein binding in live cells, HEK293 cells were treated in situ with all three catechol probes (Fig. 2a). A fluorescence reporter was appended to the labelled proteins by CuAAC after lysis, and labelled proteins were visualised by fluorescent SDS-PAGE (Fig. 2b). With 1 h treatment, concentration-dependent labelling was visible for all probes starting from 15 μM. Overall, DA-P3 showed the strongest labelling and DA-P1 the weakest with a comparable labelling pattern across the probes. Importantly, no significant protein precipitation was observed in the coomassie staining even at 100 μM concentration. Next, to facilitate a MS-based comparison of probes DA-P1–3, the proteome was treated with 15 μM compound for 1 h and labelled proteins were subsequently ligated to a biotin handle after cell lysis. Following enrichment of the labelled proteome on avidin beads and tryptic digest, the resulting peptides were analysed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) with label-free quantification (Fig. 1b).34 As already observed via gel-based analysis, the MS data revealed a large overlap of identified hits across all probes and the length of the alkyl chain correlated with the number of significant hits whereas it largely did not influence the identity of enriched proteins (Fig. S2a–c, Table S1†). We next extended treatment to 3 h to account for potential differences in uptake or labelling kinetics (Fig. S3a–c, Table S2†). While the correlation between chain length and the number of identified hits remained, the overall number of labelled proteins diminished from 1 h to 3 h, indicating that the modifications introduced on the proteins are relatively short-lived. We chose treatment with DA-P3 for 1 h for all following experiments as these conditions resulted in the strongest labelling.
Competitive labelling experiments reveal DA binding proteins
HEK293 cells produce no endogenous DA (ref. 20 and 32) but have been reported to take up DA and other monoamine neurotransmitters.33 We therefore labelled intact HEK293 cells with DA-P3 in competition with DA. Treatment of HEK293 cells with 15 μM DA-P3 for 1 h resulted in an at least fourfold enrichment of 236 proteins (−log10(p-value) ≥ 2) compared to a DMSO-treated control (Fig. 2c, Table S3†). Moreover, a 30-fold excess of DA added to the cells 1 h prior to probe addition displaced the binding of 205 proteins (Fig. 2d, Table S3†). Of all proteins enriched by DA-P3 compared to the DMSO control, 63% were significantly outcompeted by DA (Fig. 2e). These data support that DA-P3 does indeed mimic the reactivity of DA/DAQ well.To link the identified targets of DA to cellular functions, we performed a gene ontology (GO) term analysis of significantly enriched proteins using the GOrilla tool.35,36 Among the proteins identified solely by DA-P3, proteins involved in ER stress and UPR (“response to ER stress”, “ER unfolded protein response”) as well as PDIs (“peptide disulphide oxidoreductase activity”) were enriched at least 3-fold (Fig. 2f). Similarly, GO terms related to the endoplasmic reticulum (ER) also stood out in the competition experiment with DA (Fig. 2g).
SH-SY5Y is a catecholaminergic neuroblastoma cell line frequently used in DA-associated neurodegeneration research.37–39 Surprisingly, whilst DA-P3 showed strong labelling in SH-SY5Y where it labelled 205 proteins compared to a DMSO control at 4 μM (Fig. S4a, Table S4†), we observed no competition by a ten-fold excess of DA (Fig. S4b, Table S4†). It is very plausible that the presence of endogenous catechols or corresponding metabolites40 may interfere with competitive experiments. Noteworthy, Hurben et al. have reported a similar experimental set up using an alkylated DA-probe (DAyne) where no competition by the parent compound DA could be observed.25 We therefore chose HEK293 for all following experiments.
Analysis of proteome-wide DA-P3 modifications
To uncover the residues modified by DA-P3 (15 μM, in situ) as well as the mass of their modification, we clicked labelled HEK293 proteome to isotopically labelled desthiobiotin azide (isoDTB) tags.41 Proteins were subsequently digested and peptides enriched on avidin beads, eluted, and modified peptides were detected by LC-MS/MS (Fig. 3a, Table S5†).42,43 An unbiased analysis43–45 revealed the added masses of 754.4120 and 760.4206 corresponding to DA-P3 plus a heavy or a light isoDTB tag, respectively, and an additional methyl group (Fig. 3b, Table S5†). In human cells, catechol compounds such as catecholamine neurotransmitters, catechol oestrogens, or xenobiotics are methylated by the catechol-O-methyltransferase (COMT) as part of a degradation pathway explaining the mass adduct.46 The modification was highly selective for cysteines which constituted 98% of all detected modified residues (116 total, Fig. 3c, Table S5†). Recently, covalent binding of CP to certain proteins has been reported.11 So far, it remains unknown if CP protein reactivity requires demethylation and ortho-quinone formation or if a direct nucleophilic attack is also possible. In fact, our data revealed that 3-O-methylated catechols directly bind to cysteine residues. This observation is in line with an oxidation to the quinone methide, followed by a nucleophilic cysteine attack (Fig. 3d). An analogous addition of glutathione to enzymatically oxidised CP has been reported supporting this notion.47–49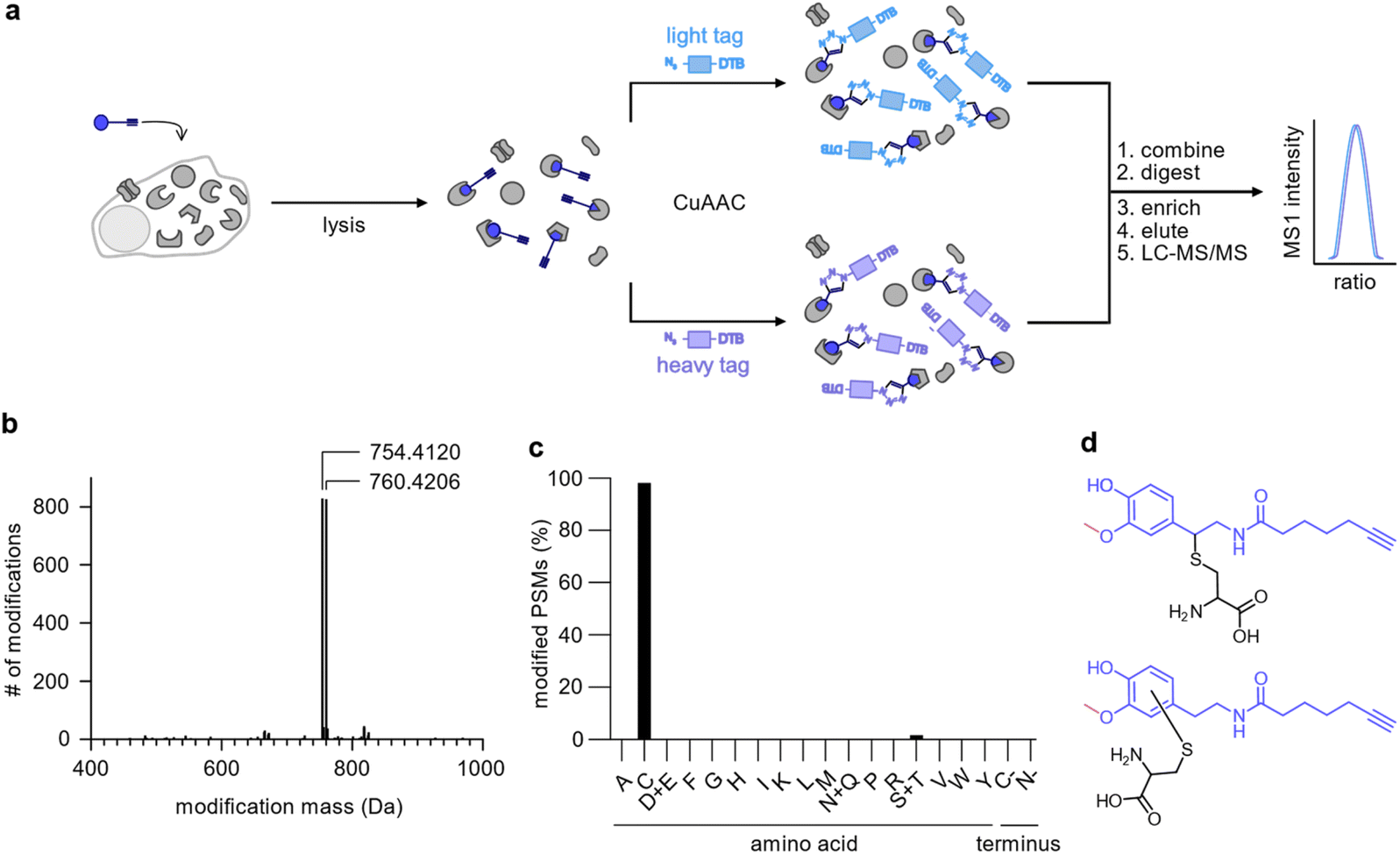 | ||
| Fig. 3 Analysis of the modifications introduced by DA-P3 on the HEK293 proteome. (a) Schematic of the workflow using isoDTB tags. HEK293 cells treated in situ with DA-P3 (15 μM) were lysed, the lysate split in two, and ligated to either light- (blue rectangle) or heavy-labelled (purple rectangle) isoDTB azide. Differentially labelled proteomes were combined, proteins were precipitated, and digested. Following peptide enrichment on avidin and elution, modified peptides were analysed by LC-MS/MS. (b) Unbiased, proteome-wide analysis of the masses of modification introduced by DA-P3 plus the light or heavy isoDTB tag, respectively. (c) Overview of amino acids modified by DA-P3. PSM, peptide spectrum matches. (d) Potential structures of the modified cysteine regioisomers corresponding to the observed modification mass. All data are based on duplicates. See Table S5† for proteomics data. | ||
Although it is possible that unmethylated catechol modifications may have escaped our MS detection (despite their identification on DJ-1 with our method), the observation of 3-O-methyl catechols as protein adducts is an intriguing and unprecedented observation.
Competitive labelling reveals targets of health-associated catechols
Catechol groups not only play crucial roles in neurotransmitters but are also widely found in plant-derived foods attributed with health-promoting properties. Previous findings have indicated that different catechols may share a common protein target space as labelling of β-actin with a DPA-probe could be outcompeted by certain flavonoids.26 Nevertheless, insights into the molecular targets are sparse. We thus took advantage of the broad DA-P3 reactivity and utilised it as a minimalist catechol probe in competition with a selection of health-associated catechol compounds that are often found in plant-derived foods/beverages (quercetin, QC; taxifolin, TF; epicatechin, EC; luteolin, LU; epigallocatechin gallate, EG; caffeic acid, KS) or drugs (dobutamine, DB; carbidopa, CD). CP with a methylated catechol group was included for comparison (Fig. 4a). Labelling was performed with DA-P3 (15 μM) and a 10-fold excess of the respective catechol in live HEK293 cells followed by enrichment and quantitative LC-MS/MS analysis with label-free quantification.34 Of the ten compounds tested, LU, CP, OL, QC, DB, and EG outcompeted probe binding at 180–286 proteins whereas CD, TF, EC, and KS showed no competition at all (Fig. 4b–e and S5, Table S6†). Interestingly, CP, carrying a 3-O-methylcatechol group, was among the reactive compounds, corroborating our results with methylated catechols (Fig. 4d–e, Table S6†). Metabolic activity assays following treatment with competitive catechols only revealed a moderate decrease in cell viability in the presence of EG and CP, substantiating that the observed protein enrichment is not the result of toxicity (Fig. S6†).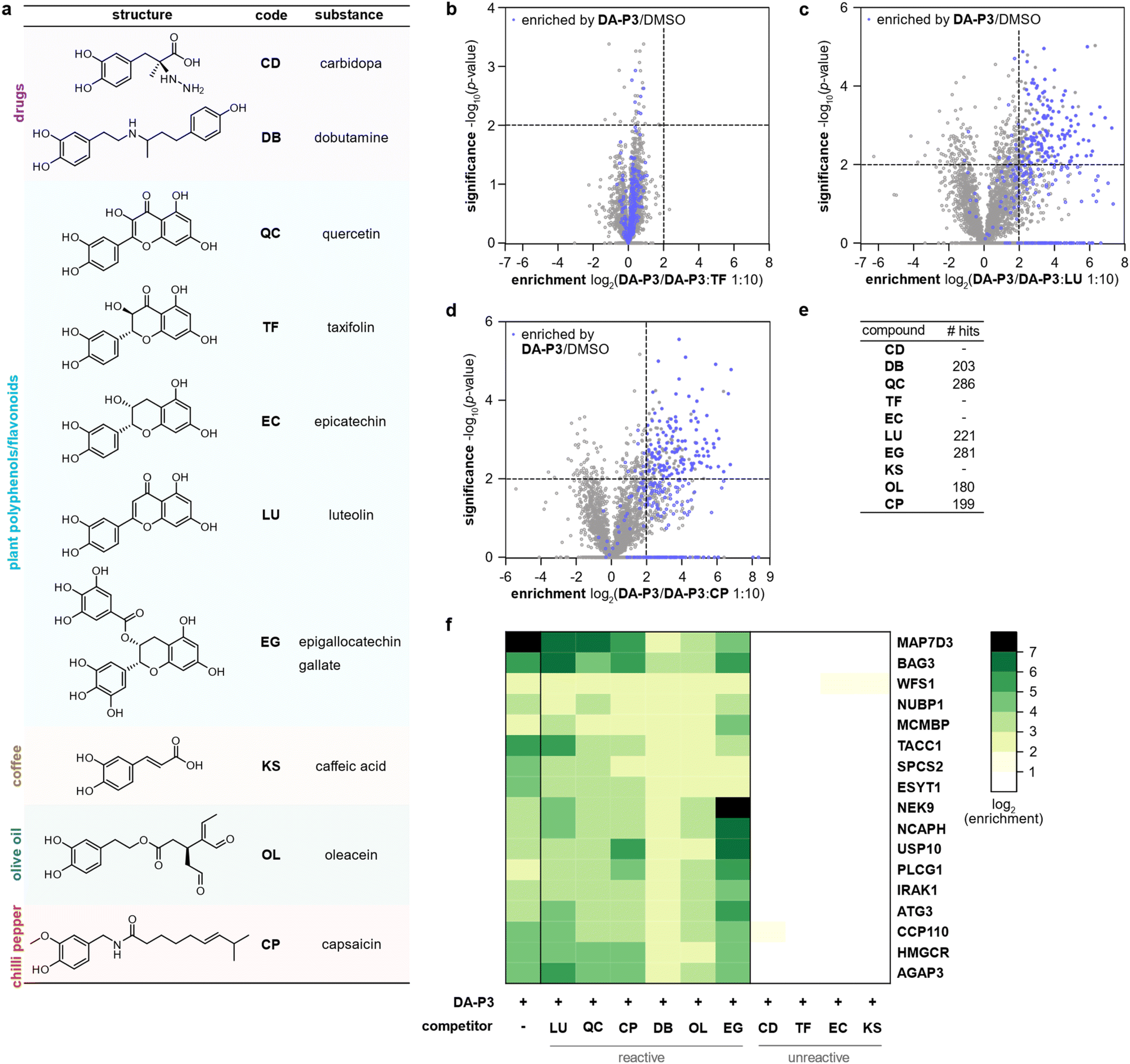 | ||
| Fig. 4 Labelling in HEK293 cells with DA-P3 in competition with a 10-fold excess of different catechol compounds. (a) Overview of the catechol compounds applied as competitors. (b–d) Example volcano plots of catechols showing differential protein reactivity, and CP. MS data from three replicates were analysed by MaxLFQ and filtered for proteins identified in three replicates in at least one condition. Samples were compared using a two-sided two-sample t-test. Proteins were considered significant when they were enriched more than four-fold (log2(enrichment) ≥ 2) with a p-value of less than 0.01 (−log10(p-value) ≥ 2); proteins that were above the cut-off in the enrichment experiment (against DMSO) are highlighted in blue. Proteins with missing values are shown with −log10(p-value) = 0. Significant hits are shown as full circles; not enriched proteins are shown as open circles. (e) Table shows number of protein hits obtained with different catechol compounds. (f) Heat map shows the enrichment of protein hits that were consistently significant in reactive catechol samples. See Table S6† for details on identified proteins and Fig. S5† for volcano plots of other compounds. | ||
We next tested competition by two exemplary unreactive catechols at higher concentrations. Indeed, we observed competition of DA-P3 labelling by a 100-fold excess of CD and KS with 113 and 259 proteins, respectively (Fig. S7a–b, Table S7 and S8†). This indicates that these compounds are less reactive or cell permeable and facilitate competition solely at high concentrations. Given that these higher concentrations could lead to unspecific effects, we decided to focus on competition in a 10-fold excess in the following experiments.
Across all reactive compounds, 17 proteins were consistently significantly targeted by DB, OL, QC, CP, LU, and EG, revealing an unanticipated broad overlap of target proteins susceptible to catechol modification despite the diversity of the chemical structures (Fig. 4f). Interestingly, four proteins were associated with the ER, namely ESYT1 (tethers the ER to the plasma membrane),50,51 SPCS2 (contributes to cotranslational translocation of nascent proteins into the ER),52 WFS1 (ER membrane glycoprotein involved in the regulation of cellular Ca2+ homeostasis),53 and HMGCR. HMGCR is localised in the ER membrane, where it catalyses the rate-determining step in the biosynthesis of cholesterol and other isoprenoids.54
A comparison of the frequency of GO terms35,36 associated with significantly enriched proteins revealed 57 biological process, 21 molecular function, and 27 cellular component terms that were overrepresented in at least one competition condition (Fig. S8†). Across all categories, terms related to structural proteins (e.g., microtubule, cytoskeleton, cell adhesion) were consistently represented. Furthermore, as already observed in the previous analyses for DA-P3 and DA, ER-associated terms were enriched for selected catechols.
As the catechols appeared to target ER proteins, we exemplarily chose this crucial organelle for validation and hypothesised that these compounds could induce ER stress. To test this hypothesis, HEK293T cells were treated with DA-P3, DA, compounds classified as “reactive” (QC, DB, OL, CP) or “unreactive” (TF and KS), and tested for UPR induction at 100 μM. DA was additionally tested at 450 μM as this was the concentration applied in the MS experiments. The analysis was performed in three biological replicates which were in qualitative agreement (Fig. 5 and S9a–b†). ER stress is characterised by an accumulation of unfolded proteins which triggers the activation of three signalling pathways mediated by the receptors ATF6, PERK, and IRE1α in mammalian cells.55 The UPR is moreover typically accompanied by an increase in the expression of the immunoglobulin heavy chain-binding protein (BiP), a chaperone of the 70 kilodalton heat shock protein (Hsp70) family.56,57 Immunoblotting of ATF6, which is cleaved upon activation, revealed no formation of the 50 kDa N-terminal cytosolic fragment in the presence of any of the tested catechols, indicating that this pathway is not activated at our experimental conditions. We next investigated the phosphorylation of eIF2α by immunoblotting which monitors the activation of the PERK sensor. Treatment with the positive control tunicamycin, DA-P3, TF, QC, and OL visibly increased eIF2α phosphorylation. Moreover, the expression of BiP was upregulated in the presence of TF, OL, QC, DA, and DA-P3. Finally, we tested for the splicing of XBP1 mRNA, a process triggered by the UPR sensor IRE1α. RT-PCR of the XBP1 mRNA revealed the formation of a spliced 416 bp fragment58 in the presence of tunicamycin, DA-P3, QC, OL, and, to a lesser extent, DB and TF. Overall, these data revealed a very strong activation of the UPR by DA-P3, and a weaker one by other compounds including QC, OL, DA, DB, and TF. No UPR activation was observed for CP and KS. The lack of UPR activation by CP despite its broad protein competition may be due to its structural differences, i.e. the 3-O-methylation, compared to the catechols. UPR activation by DA-P3, DB, QC, OL, and DA and the inactivity of KS are well in line with the results of the MS experiments.
 | ||
| Fig. 5 UPR assessment of HEK293T cells treated with the compounds of interest, the positive control tunicamycin (Tun.), or DMSO (as a vehicle control). Immunoblotting was performed against ATF6, eIF2α, phosphorylated eIF2α, BiP or Hsc70. XBP1 splicing was analysed using agarose gel electrophoresis. Cells were treated with 100 μM compound for 16 h unless otherwise indicated. | ||
Conclusions
Here, we have applied a broadly reactive minimalist catechol probe to elucidate the protein reactivity of biologically relevant catechol compounds in live cells. The direct comparison of structurally diverse catechols revealed large variations in their protein reactivities. The origin of these differences remains to be resolved. Among the catechols with high protein reactivity were compounds, for which, to our knowledge, broad protein reactivity has not been shown previously, for example the inotrope and clinically applied drug DB and the olive oil constituent OL. The tendency to covalently modify proteins is one of the reasons that catechols are known as “Pan Assay Interference Compounds” (PAINS),60–62 a term describing compound classes that frequently recur as screening hits as a consequence of unspecific interference with biological assays.60,61 Catechol-containing natural products have widely been reported as bioactive compounds in a variety of disease contexts63 but the reports often fail to take into account the promiscuity in terms of protein binding.60 Our results illustrate the scope of proteins modified by catechols as well as the low degree of selectivity.A potential limitation of our and other reactivity-based labelling tools is that protein modifications could be substoichiometric,25,59 therefore, certain proteins may have escaped our competitive labelling method. Moreover, certain proteins addressed by our panel of catechols and DA-P3 may differ and thus escape probe detection in the competition experiments. Yet, our data reveals that a significant number of proteins is susceptible to catechol modification regardless of their structure and we have revealed an unexpectedly broad protein reactivity of certain catechols.
Another important finding was the direct modification of cysteines by methylated catechols. This protein modification has not been reported previously, but corroborates the recently reported cysteine-reactivity of the 3-O-methyl catechol CP.11
To date, only selected catechol targets have been reported whereas our work highlights the broad reactivity of catechols in live cells. Specifically, we were able to show that certain catechol compounds target ER proteins, including PDIs critical for ER protein folding and UPR regulation,64–66 which results in increased ER stress. The UPR is a cellular response during ER stress caused by an accumulation of unfolded proteins. As a consequence, cells adjust protein synthesis, folding, and degradation to reduce the burden of unfolded proteins or else undergo apoptosis if unsuccessful. Due to increased protein turnover, many cancer cells experience constant ER stress and are more sensitive to UPR and PDI inhibition compared to healthy cells.67,68 Furthermore, recent studies report the inhibition of PDIs by DA, the flavonoid isoquercetin, and n-octyl caffeate.10,14,25 Altogether, the modulation of ER-associated cellular processes highlights one intriguing facet on how catechols could promote health-beneficial effects.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository69 with the dataset identifier PXD043348.Author contributions
A. W. M. planned and performed probe synthesis, mass spectrometry experiments, cytotoxicity assays, and data analysis; K. M. B. performed ER stress assays; S. M. H. analysed isoDTB data; M. J. F. supervised ER stress assays; S. A. S. supervised the project and acquired funding. A. W. M. wrote the manuscript with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. W. M. was supported by the German Academic Scholarship Foundation (Studienstiftung des Deutschen Volkes). S. M. H. acknowledges funding by the Fonds der Chemischen Industrie through a Liebig Fellowship. S. A. S and M. J. F. received funding through Sonderforschungsbereich 1035, project number 201302640, projects A09 and B11. We thank Elena Kratz for help with labelling experiments, Dr Jonas Drechsel for the donation of DJ-1, and Mona Wolff and Katja Bäuml for technical assistance.References
- Y. Cao and R. Cao, Nature, 1999, 398, 381 CrossRef CAS PubMed.
- C. Ramassamy, Eur. J. Pharmacol., 2006, 545, 51–64 CrossRef CAS.
- F. L. Palhano, J. Lee, N. P. Grimster and J. W. Kelly, J. Am. Chem. Soc., 2013, 135, 7503–7510 CrossRef CAS.
- J. Bieschke, J. Russ, R. P. Friedrich, D. E. Ehrnhoefer, H. Wobst, K. Neugebauer and E. E. Wanker, Proc. Natl. Acad. Sci. U.S. A., 2010, 107, 7710–7715 CrossRef CAS PubMed.
- J. Li, M. Zhu, A. B. Manning-Bog, D. A. Di Monte and A. L. Fink, FASEB J., 2004, 18, 962–964 CrossRef CAS.
- H. Tachibana, K. Koga, Y. Fujimura and K. Yamada, Nat. Struct. Mol. Biol., 2004, 11, 380–381 CrossRef CAS PubMed.
- S. Garbisa, S. Biggin, N. Cavallarin, L. Sartor, R. Benelli and A. Albini, Nat. Med., 1999, 5, 1216 CrossRef CAS PubMed.
- Y. C. Liang, S. Y. Lin-Shiau, C. F. Chen and J. K. Lin, J. Cell. Biochem., 1999, 75, 1–12 CrossRef CAS.
- V. Fattori, M. S. N. Hohmann, A. C. Rossaneis, F. A. Pinho-Ribeiro and W. A. Verri, Molecules, 2016, 21, 844 CrossRef PubMed.
- J. I. Zwicker, B. L. Schlechter, J. D. Stopa, H. A. Liebman, A. Aggarwal, M. Puligandla, T. Caughey, K. A. Bauer, N. Kuemmerle, E. Wong, T. Wun, M. McLaughlin, M. Hidalgo, D. Neuberg, B. Furie and R. Flaumenhaft, JCI Insight, 2019, 4, e125851 CrossRef PubMed.
- Q. Zhang, P. Luo, F. Xia, H. Tang, J. Chen, J. Zhang, D. Liu, Y. Zhu, Y. Liu, L. Gu, L. Zheng, Z. Li, F. Yang, L. Dai, F. Liao, C. Xu and J. Wang, Cell Chem. Biol., 2022, 29, 1248–1259.e1246 CrossRef CAS.
- A. M. Chapa-Oliver and L. Mejía-Teniente, Molecules, 2016, 21, 931 CrossRef.
- N. Khan, F. Afaq, M. Saleem, N. Ahmad and H. Mukhtar, Cancer Res., 2006, 66, 2500–2505 CrossRef CAS PubMed.
- R. M. Robinson, L. Reyes, R. M. Duncan, H. Bian, A. B. Reitz, Y. Manevich, J. J. McClure, M. M. Champion, C. J. Chou, M. E. Sharik, M. Chesi, P. L. Bergsagel and N. G. Dolloff, Leukemia, 2019, 33, 1011–1022 CrossRef CAS PubMed.
- V. S. Van Laar, A. J. Mishizen, M. Cascio and T. G. Hastings, Neurobiol. Dis., 2009, 34, 487–500 CrossRef CAS PubMed.
- R. Boly, T. Gras, T. Lamkami, P. Guissou, D. Serteyn, R. Kiss and J. Dubois, Int. J. Oncol., 2011, 38, 833–842 CAS.
- E. Monzani, S. Nicolis, S. Dell'Acqua, A. Capucciati, C. Bacchella, F. A. Zucca, E. V. Mosharov, D. Sulzer, L. Zecca and L. Casella, Angew. Chem., Int. Ed. Engl., 2019, 58, 6512–6527 CrossRef CAS PubMed.
- J. M. Bruning, Y. Wang, F. Oltrabella, B. Tian, S. A. Kholodar, H. Liu, P. Bhattacharya, S. Guo, J. M. Holton, R. J. Fletterick, M. P. Jacobson and P. M. England, Cell Chem. Biol., 2019, 26, 674–685.e676 CrossRef CAS.
- L. F. Burbulla, P. Song, J. R. Mazzulli, E. Zampese, Y. C. Wong, S. Jeon, D. P. Santos, J. Blanz, C. D. Obermaier, C. Strojny, J. N. Savas, E. Kiskinis, X. Zhuang, R. Krüger, D. J. Surmeier and D. Krainc, Science, 2017, 357, 1255–1261 CrossRef CAS.
- M. J. LaVoie, B. L. Ostaszewski, A. Weihofen, M. G. Schlossmacher and D. J. Selkoe, Nat. Med., 2005, 11, 1214–1221 CrossRef CAS PubMed.
- J. Jankun, S. H. Selman, R. Swiercz and E. Skrzypczak-Jankun, Nature, 1997, 387, 561 CrossRef CAS PubMed.
- N. Umek, B. Geršak, N. Vintar, M. Šoštarič and J. Mavri, Front. Mol. Neurosci., 2018, 11, 467 CrossRef CAS PubMed.
- M. Fonović and M. Bogyo, Expert Rev. Proteomics, 2008, 5, 721–730 CrossRef PubMed.
- B. F. Cravatt, A. T. Wright and J. W. Kozarich, Annu. Rev. Biochem., 2008, 77, 383–414 CrossRef CAS PubMed.
- A. K. Hurben, L. N. Erber, N. Y. Tretyakova and T. M. Doran, ACS Chem. Biol., 2021, 16, 2581–2594 CrossRef CAS PubMed.
- T. Ishii, M. Ishikawa, N. Miyoshi, M. Yasunaga, M. Akagawa, K. Uchida and Y. Nakamura, Chem. Res. Toxicol., 2009, 22, 1689–1698 Search PubMed.
- A. Farzam, K. Chohan, M. Strmiskova, S. J. Hewitt, D. S. Park, J. P. Pezacki and D. Özcelik, Redox Biol, 2020, 28, 101377 Search PubMed.
- J. Drechsel, F. A. Mandl and S. A. Sieber, ACS Chem. Biol., 2018, 13, 2016–2019 CrossRef CAS PubMed.
- S. Zhang, R. Wang and G. Wang, ACS Chem. Neurosci., 2019, 10, 945–953 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed. Engl., 2002, 41, 2596–2599 CrossRef CAS.
- V. Nawaratne, S. P. McLaughlin, F. P. Mayer, Z. Gichi, A. Mastriano and L. Carvelli, Front. Cell. Neurosci., 2021, 15, 681539 CrossRef CAS PubMed.
- K. H. Boxberger, B. Hagenbuch and J. N. Lampe, Drug Metab. Dispos., 2014, 42, 990–995 CrossRef PubMed.
- J. Cox, M. Y. Hein, C. A. Luber, I. Paron, N. Nagaraj and M. Mann, Mol. Cell. Proteomics, 2014, 13, 2513–2526 CrossRef CAS PubMed.
- E. Eden, R. Navon, I. Steinfeld, D. Lipson and Z. Yakhini, BMC Bioinform, 2009, 10, 48 CrossRef PubMed.
- E. Eden, D. Lipson, S. Yogev and Z. Yakhini, PLoS Comput. Biol., 2007, 3, e39 CrossRef PubMed.
- T. Alberio, M. Colapinto, M. Natale, R. Ravizza, M. B. Gariboldi, E. M. Bucci, L. Lopiano and M. Fasano, IUBMB Life, 2010, 62, 688–692 CrossRef CAS PubMed.
- M. Fasano, T. Alberio, M. Colapinto, S. Mila and L. Lopiano, Parkinsonism Relat Disord, 2008, 14(Suppl 2), S135–S138 CrossRef PubMed.
- C. Gómez-Santos, I. Ferrer, A. F. Santidrián, M. Barrachina, J. Gil and S. Ambrosio, J. Neurosci. Res., 2003, 73, 341–350 CrossRef.
- M. A. Mena, J. Garcia de Yebenes, A. Dwork, S. Fahn, N. Latov, J. Herbert, E. Flaster and D. Slonim, Brain Res., 1989, 486, 286–296 CrossRef CAS PubMed.
- P. R. A. Zanon, L. Lewald and S. M. Hacker, Angew. Chem., Int. Ed. Engl., 2020, 59, 2829–2836 CrossRef CAS PubMed.
- P. W. A. Allihn, M. W. Hackl, C. Ludwig, S. M. Hacker and S. A. Sieber, Chem. Sci., 2021, 12, 4763–4770 RSC.
- A. Weigert Muñoz, E. Hoyer, K. Schumacher, M. Grognot, K. M. Taute, S. M. Hacker, S. A. Sieber and K. Jung, Proc. Natl. Acad. Sci. U.S. A., 2022, 119, e2118227119 CrossRef.
- P. R. A. Zanon, F. Yu, P. Z. Musacchio, L. Lewald, M. Zollo, K. Krauskopf, D. Mrdović, P. Raunft, T. E. Maher, M. Cigler, C. J. Chang, K. Lang, F. D. Toste, A. I. Nesvizhskii and S. M. Hacker, ChemRxiv, 2021, preprint, DOI:10.26434/chemrxiv.14186561.v1.
- A. T. Kong, F. V. Leprevost, D. M. Avtonomov, D. Mellacheruvu and A. I. Nesvizhskii, Nat. Methods, 2017, 14, 513–520 CrossRef CAS PubMed.
- B. T. Zhu, Curr. Drug Metab., 2002, 3, 321–349 CrossRef CAS PubMed.
- K. Bley, G. Boorman, B. Mohammad, D. McKenzie and S. Babbar, Toxicol. Pathol., 2012, 40, 847–873 CrossRef CAS.
- C. A. Reilly and G. S. Yost, Drug Metab. Rev., 2006, 38, 685–706 CrossRef CAS PubMed.
- C. A. Reilly, F. Henion, T. S. Bugni, M. Ethirajan, C. Stockmann, K. C. Pramanik, S. K. Srivastava and G. S. Yost, Chem. Res. Toxicol., 2013, 26, 55–66 Search PubMed.
- F. Giordano, Y. Saheki, O. Idevall-Hagren, S. F. Colombo, M. Pirruccello, I. Milosevic, E. O. Gracheva, S. N. Bagriantsev, N. Borgese and P. De Camilli, Cell, 2013, 153, 1494–1509 CrossRef CAS PubMed.
- C. L. Chang, T. S. Hsieh, T. T. Yang, K. G. Rothberg, D. B. Azizoglu, E. Volk, J. C. Liao and J. Liou, Cell. Rep., 2013, 5, 813–825 CrossRef CAS.
- A. M. Liaci, B. Steigenberger, P. C. Telles de Souza, S. Tamara, M. Gröllers-Mulderij, P. Ogrissek, S. J. Marrink, R. A. Scheltema and F. Förster, Mol. Cell, 2021, 81, 3934–3948.e3911 CrossRef CAS PubMed.
- D. Takei, H. Ishihara, S. Yamaguchi, T. Yamada, A. Tamura, H. Katagiri, Y. Maruyama and Y. Oka, FEBS Lett., 2006, 580, 5635–5640 CrossRef CAS PubMed.
- K. L. Luskey and B. Stevens, J. Biol. Chem., 1985, 260, 10271–10277 CrossRef CAS.
- C. Hetz, Nat. Rev. Mol. Cell Biol., 2012, 13, 89–102 CrossRef CAS PubMed.
- A. S. Lee, Methods, 2005, 35, 373–381 CrossRef CAS PubMed.
- N. Sato, F. Urano, J. Yoon Leem, S. H. Kim, M. Li, D. Donoviel, A. Bernstein, A. S. Lee, D. Ron, M. L. Veselits, S. S. Sisodia and G. Thinakaran, Nat. Cell Biol., 2000, 2, 863–870 CrossRef CAS.
- A. M. Fribley, J. R. Miller, T. E. Reist, M. U. Callaghan and R. J. Kaufman, in Methods in Enzymology, ed. P. M. Conn, Academic Press, 2011, vol. 491, pp. 57–71 Search PubMed.
- E. N. Pierce, S. C. Piyankarage, T. Dunlap, V. Litosh, M. I. Siklos, Y. T. Wang and G. R. Thatcher, Chem. Res. Toxicol., 2016, 29, 1151–1159 Search PubMed.
- J. B. Baell, J. Nat. Prod., 2016, 79, 616–628 CrossRef CAS PubMed.
- J. B. Baell and G. A. Holloway, J. Med. Chem., 2010, 53, 2719–2740 CrossRef CAS PubMed.
- J. Baell and M. A. Walters, Nature, 2014, 513, 481–483 CrossRef CAS PubMed.
- J. Bisson, J. B. McAlpine, J. B. Friesen, S.-N. Chen, J. Graham and G. F. Pauli, J. Med. Chem., 2016, 59, 1671–1690 CrossRef CAS PubMed.
- M. J. Feige and L. M. Hendershot, Curr. Opin. Cell Biol., 2011, 23, 167–175 CrossRef CAS.
- J. Yu, T. Li, Y. Liu, X. Wang, J. Zhang, X. e. Wang, G. Shi, J. Lou, L. Wang, C. c. Wang and L. Wang, EMBO J., 2020, 39, e103841 CrossRef CAS PubMed.
- O. B. Oka, M. van Lith, J. Rudolf, W. Tungkum, M. A. Pringle and N. J. Bulleid, EMBO J., 2019, 38, e100990 CrossRef PubMed.
- M. Wang and R. J. Kaufman, Nature, 2016, 529, 326–335 CrossRef CAS PubMed.
- S. Xu, S. Sankar and N. Neamati, Drug Discov, 2014, 19, 222–240 CAS.
- Y. Perez-Riverol, A. Csordas, J. Bai, M. Bernal-Llinares, S. Hewapathirana, D. J. Kundu, A. Inuganti, J. Griss, G. Mayer, M. Eisenacher, E. Pérez, J. Uszkoreit, J. Pfeuffer, T. Sachsenberg, S. Yilmaz, S. Tiwary, J. Cox, E. Audain, M. Walzer, A. F. Jarnuczak, T. Ternent, A. Brazma and J. A. Vizcaíno, Nucleic Acids Res., 2019, 47, 442–450 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, supporting figures, supporting tables, and compound characterisation. See DOI: https://doi.org/10.1039/d3sc00888f |
| This journal is © The Royal Society of Chemistry 2023 |