
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrigin of intersystem crossing in highly distorted organic molecules: a case study with red light-absorbing N,N,O,O-boron-chelated Bodipys†
Xue
Zhang‡
 a,
Andrey A.
Sukhanov‡
b,
Xi
Liu‡
c,
Maria
Taddei
d,
Jianzhang
Zhao
*a,
Anthony
Harriman
*e,
Violeta K.
Voronkova
*b,
Yan
Wan
*c,
Bernhard
Dick
*f and
Mariangela
Di Donato
*dg
a,
Andrey A.
Sukhanov‡
b,
Xi
Liu‡
c,
Maria
Taddei
d,
Jianzhang
Zhao
*a,
Anthony
Harriman
*e,
Violeta K.
Voronkova
*b,
Yan
Wan
*c,
Bernhard
Dick
*f and
Mariangela
Di Donato
*dg
aState Key Laboratory of Fine Chemicals, Frontiers Science Center for Smart Materials, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, P. R. China. E-mail: zhaojzh@dlut.edu.cn
bZavoisky Physical-Technical Institute, FRC Kazan Scientific Center of Russian Academy of Sciences, Kazan 420029, Russia. E-mail: vio@kfti.knc.ru
cCollege of Chemistry, Beijing Normal University, Beijing 100875, P. R. China. E-mail: wanyan@bnu.edu.cn
dLENS (European Laboratory for Non-Linear Spectroscopy), Via N. Carrara 1, 50019 Sesto Fiorentino (FI), Italy. E-mail: didonato@lens.unifi.it
eMolecular Photonics Laboratory, School of Natural and Environmental Sciences, Newcastle University, Newcastle Upon Tyne, NE1 7RU, UK. E-mail: anthony.harriman@newcastle.ac.uk
fLehrstuhl für Physikalische Chemie, Institut für Physikalische und Theoretische Chemie, Universität Regensburg, D-93053 Regensburg, Germany. E-mail: Bernhard.Dick@chemie.uni-regensburg.de
gICCOM, Istituto di Chimica dei Complessi OrganoMetallici, Via Madonna del Piano 10, 50019 Sesto Fiorentino (FI), Italy
First published on 15th April 2023
Abstract
To explore the relationship between the twisted π-conjugation framework of aromatic chromophores and the efficacy of intersystem crossing (ISC), we have studied a N,N,O,O-boron-chelated Bodipy derivative possessing a severely distorted molecular structure. Surprisingly, this chromophore is highly fluorescent, showing inefficient ISC (singlet oxygen quantum yield, ΦΔ = 12%). These features differ from those of helical aromatic hydrocarbons, where the twisted framework promotes ISC. We attribute the inefficient ISC to a large singlet-triplet energy gap (ΔES1/T1 = 0.61 eV). This postulate is tested by critical examination of a distorted Bodipy having an anthryl unit at the meso-position, for which ΦΔ is increased to 40%. The improved ISC yield is rationalized by the presence of a T2 state, localized on the anthryl unit, with energy close to that of the S1 state. The electron spin polarization phase pattern of the triplet state is (e, e, e, a, a, a), with the Tz sublevel of the T1 state overpopulated. The small zero-field splitting D parameter (−1470 MHz) indicates that the electron spin density is delocalized over the twisted framework. It is concluded that twisting of π-conjugation framework does not necessarily induce ISC, but S1/Tn energy matching may be a generic feature for increasing ISC for a new-generation of heavy atom-free triplet photosensitizers.
Introduction
Triplet photosensitizers (PSs) should possess significant absorption in the visible or near-UV region, efficient intersystem crossing (ISC), and reversible electrochemistry with well-defined redox potentials.1–3 Such compounds are crucial for the development of photocatalytic cycles,4–7 including solar fuel formation via water splitting8–12 or fixation of CO2,13–15 photodynamic therapy,1,16–21 and triplet-triplet-annihilation upconversion.22–25 However, ISC is a spin forbidden process, being largely ineffective for organic compounds with extended planar π-conjugation frameworks.26 Spin orbit coupling (SOC) usually vanishes for these compounds due to the planar molecular structure,27 while a large energy gap between S1 and T1 states is also a common feature. These two factors combine to minimize ISC rates. An additional factor that inhibits ISC in aromatic compounds is the large electronic exchange energy for the frontier molecular orbitals, which increases the S1/T1 energy gap. In addition, the planarity and high symmetry of the π-conjugation systems serve to reduce the SOC matrix element (SOCME).26,28 These various parameters influence the rate constant for ISC in accordance with the Fermi Golden rule (eqn (1)): | (1) |
Within this context, we note that ISC induced by a helical π-conjugation structure might offer new perspectives.27,28,52–58 It is known that for helicenes, ISC is due to the non-vanishing SOCMEs at each molecular orbital component.27 However, helicenes are not ideal triplet PSs because of their poor visible light absorption, and their challenging derivatization.59,60 Recently, twisted π-conjugation framework-induced ISC was observed for certain chromophores, including perylenebisimide (PBI).28,52–54 We and other groups also found that twisted Bodipy derivatives show efficient ISC, as for instance, the naphtha[b]-fused Bodipy (triplet state quantum yield, ΦT = 52%)58 and the dihydronaphto[a]-fused Bodipy (singlet oxygen quantum yield, ΦΔ = 55%).53,55,61 Moreover, the electron spin selectivity of the ISC process, manifested by the electron spin polarization (ESP) phase pattern of the triplet state, is highly dependent on molecular structure.55,58,61,62 Intriguingly, twisted Bodipy structures do not always give efficient ISC, in marked contrast to the helicenes,57 and recent work by Vennapusa reports the same situation with PBI.63
In seeking to expand the range of structurally distorted chromophores absorbing in the red region, our attention has returned to the helical N,N,O,O boron-chelated dipyrromethenes (BO).64,65 The synthesis of such compounds is relatively straightforward and there remains the strong possibility that the twisted geometry will promote triplet formation.27,66 Although the helical structure was introduced as a mean to enhance fluorescence of the 3,5-arylated Bodipy derivatives,64 the fluorescence quantum yields are lower than those of the native Bodipy. Coordination of two 3,5-ortho-phenolic substituents to the central boron atom reduces the molecular symmetry and enforces helicity, thereby leading to circularly polarized luminescence.65 The same synthetic approach can be applied to develop near-IR absorbing compounds,67–71 and to construct supramolecular assemblies.72 To the best of our knowledge, however, the ISC characteristics of these compounds have not been investigated. Now, using a variety of time-resolved spectroscopic tools, together with quantum chemical calculations, we compare the photophysical properties of a N,N,O,O-boron-chelated Bodipy (Ph-BO) possessing a severely twisted π-conjugation framework with those of the corresponding compound bearing an anthryl moiety at the meso-position of the dipyrrin core (An-BO). This latter functionalization adds an additional triplet level (T2) to the excited state manifold which turns out to be crucial in terms of the propensity for ISC.
Results and discussion
Molecular structure design and synthesis
Initial studies focus on determination of the photophysical properties of a known compound, Ph-BO (Scheme 1),65 for which we find the rather modest triplet yield of ca. 12%. Two strategies can be invoked when seeking to increase triplet quantum yield, SOCT-ISC51,76–78 and minimizing the singlet-triplet energy gap by introducing an intermediate triplet state localized on a meso-substituent. Both concepts can be examined using a meso-anthryl derivative, An-BO.79,80 Synthesis of the latter is based on the known derivatization of this kind of twisted Bodipy derivative. The yield was judged to be satisfactory and the molecular structure has been verified by 1H NMR, 13C NMR and HR MS spectroscopic methods (for details, see the ESI†). | ||
| Scheme 1 Synthetic scheme for preparation of An-BO. The molecular formulae of the reference compounds are also presented, BO is a reference compound used in the computational work. (a) (i) NaH, THF, (ii) ZnCl2, (iii) Pd(OAc)2, JohnPhos, 2-chloroanisole, reflux, 24 h, yield: 49%.73,74 (b) 9-Anthraldehyde, trifluoroacetic acid, rt, 2 h, reflux, overnight; then DDQ, reflux, overnight; then triethylamine, 30 min, BF3·Et2O, reflux, 4 h, THF, overall yield: 21%.75 (c) BBr3, DCM, yield: 49%.65 | ||
Single crystals of Ph-BO were obtained by slow diffusion of n-hexane (HEX) into a dichloromethane (DCM) solution. Subsequent X-ray diffraction studies show that the angle between the planes defined by the two pyrrolic rings is 48.8°.
The two phenyl rings deviate from the mean plane of the dipyrrin by ca. 22.1° and 26.7°, confirming that the molecular structure is highly twisted (Fig. 1b). The O–B–O, N–B–N and O–B–N bond angles are 107.6°, 105.6°, 106.8°, respectively (Fig. 1c). These data show a distorted tetrahedral boron center, while angles are similar to those of previously reported analogues.64,81
 | ||
Fig. 1 ORTEP views of the molecular structure determined by single-crystal X-ray diffraction of Ph-BO. (a) Bottom view, (b) side view and (c) front view. The dihedral angle between the meso-phenyl and dipyrrin moieties and the twist angle are highlighted. Thermal ellipsoids are at 50% probability. CCDC number: 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 210911 contains detailed information for Ph-BO. 210911 contains detailed information for Ph-BO. | ||
Steady-state optical spectra
The UV/vis absorption spectra recorded for An-BO and Ph-BO are shown in Fig. 2. Both compounds exhibit a strong absorption band at ca. 620 nm, the molar absorption coefficient for An-BO, ε = 59000 M−1 cm−1, being somewhat more pronounced than the previously reported N,N,O,O-boron-chelated Bodipy analogue (ε = 46000 M−1 cm−1).64 The red-shifted absorption of the helical derivatives compared to the corresponding precursor indicates an expanded π-conjugation system.64,67 For An-BO, an additional structured absorption band was observed in the range of 330–400 nm, attributed to the anthryl moiety, indicating only minor electronic interaction between the anthracene substituent and the dipyrrin core at the ground state.83
 | ||
| Fig. 2 UV/vis absorption spectra recorded for An-BO and Ph-BO in acetonitrile. c = 1.0 × 10−5 M, 25 °C. | ||
The corresponding fluorescence spectra are presented in Fig. 3. Ph-BO has a strong emission band (fluorescence quantum yield, ΦF = 73%, Table 1), whose position and intensity are independent of solvent polarity (Fig. 3c). The strong fluorescence is noteworthy because ISC is usually efficient for helicenes. Recent results from our laboratory,55–58 and Hasobe's laboratory53 show that twisted Bodipys can also display efficient ISC. A similar fluorescence spectrum is observed for An-BO (Fig. 3b), although the fluorescence quantum yield is lower (ΦF = 48%, Table 1). Furthermore, no charge-transfer emission band could be observed for An-BO, the fluorescence emission intensity and wavelength do not change with solvent polarity, these properties are uncommon compared to previously reported anthryl-Bodipy dyads.48,62,84 The singlet state energy (E00) of the compounds, determined as the crossover point of normalized absorption and fluorescence spectra, is 1.96 eV and 1.99 eV for An-BO and Ph-BO, respectively.
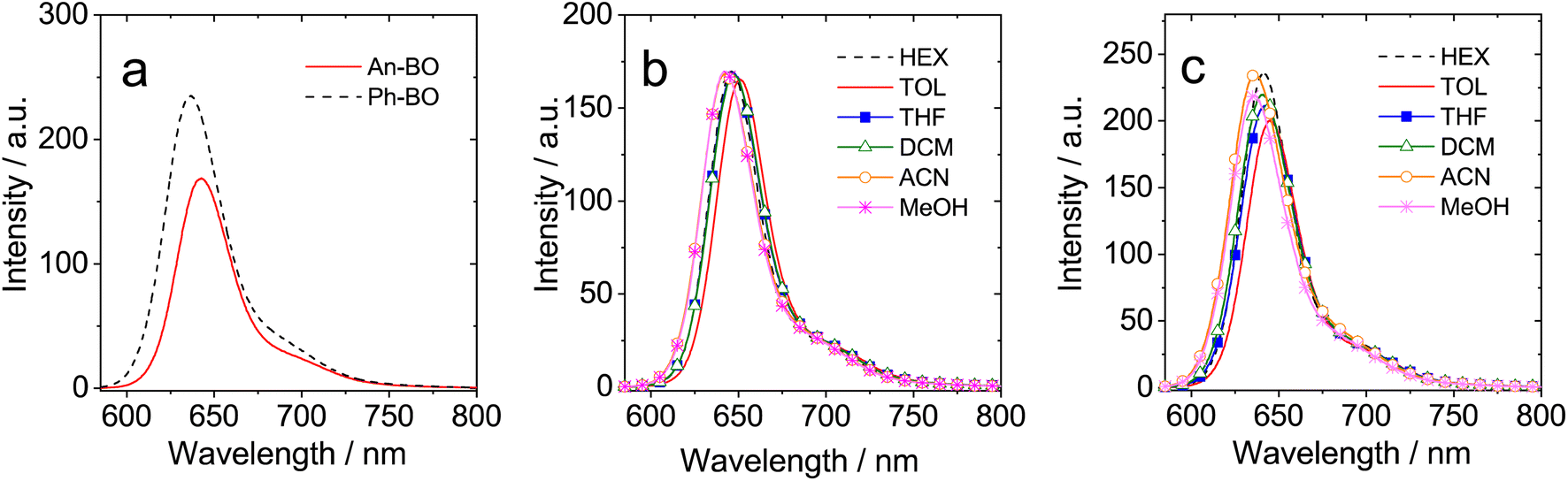 | ||
| Fig. 3 Fluorescence spectra of (a) An-BO and Ph-BO in acetonitrile, (b) An-BO and (c) Ph-BO in different solvents. Optically matched solutions were used in each panel (each of the solutions gives the same absorbance at the excitation wavelength, A = 0.100), λex = 580 nm, 25 °C. | ||
| Compounds | λ abs [nm] | ε | λ em [nm] | τ F [ns] | τ T [μs] | Φ Δ | Φ F |
|---|---|---|---|---|---|---|---|
| a In acetonitrile (1.0 × 10−5 M). b UV/vis absorption maximum. c Molar absorption coefficient, ε: 104 M−1 cm−1. d Fluorescence emission maximum. e Fluorescence lifetime. f Intrinsic triplet lifetime. g Singlet oxygen quantum yield (λex = 630 nm, methylene blue used as standard compound, ΦΔ = 57% in dichloromethane). h Absolute fluorescence quantum yield, determined with optical integrating sphere. i Parameters for BDP are taken from the literature.82 j Parameters of BDP-Me are from the literature.31 k Not observed. | |||||||
| An-BO | 623 | 5.9 | 636 | 8.3 | 226 | 40 | 48 |
| Ph-BO | 613 | 5.4 | 643 | 11.3 | 215 | 12 | 73 |
| BDP | 499 | 6.1 | 521 | 0.2 | −k | −k | 5 |
| BDP-Me | 503 | 8.2 | 515 | 3.9 | 0.02 | −k | 71 |
The fluorescence lifetimes of An-BO and Ph-BO were measured by time-correlated, single photon counting (TCSPC), using picosecond pulsed laser excitation (Fig. S13†). Both compounds gave mono-exponential decay curves, with lifetimes of 8.3 ns and 11.3 ns in ACN, respectively, for An-BO and Ph-BO. These lifetimes were found to be independent of solvent polarity. It is interesting to note that the measured lifetimes are considerably longer than those associated with conventional Bodipy emitters (τF ≈ 4.0 ns),31,85,86 and of a recently reported Bodipy with twisted structure (τF ≈ 3 ns),53,55,58 The radiative rate constants for An-BO and Ph-BO are lowered to ca. 6 × 107 s−1, which is roughly half of that reported for conventional Bodipy-based chromophores. The photophysical properties of the target compounds are summarized in Table 1.
As a preliminary evaluation of the ISC efficiency of these compounds, the quantum yields for formation of singlet molecular oxygen (ΦΔ) were measured (Table 2). For Ph-BO, ΦΔ ≤ 12% and dependent on solvent polarity; the highest yield was observed in ACN, with a much lower yield in HEX. The poor sensitization behaviour contrasts with that of the helicenes.27,60,66 and with that of the twisted Bodipy, which shows ΦΔ up to 55%.53,55,57,58 Interestingly, an improved photosensitizing capability was observed for An-BO, where ΦΔ = 40% in ACN and ΦΔ = 12% in HEX (Table 2). Based on the absorption and fluorescence data, we propose that the weak dependence of the ΦΔ values on the solvent polarity does not support the SOCT-ISC mechanism, as also demonstrated by femtosecond transient absorption spectroscopy (fs-TA, see later). However, the polarity of the solvent might affect the S1/Tn energy matching, thereby influencing the yield of ISC, as found previously for anthracene (An).87
| Compounds | HEXb | TOLc | THFd | DCMe | ACNf | MeOHg |
|---|---|---|---|---|---|---|
| a In percentage, λex = 630 nm, methylene blue was used as standard, ΦΔ = 57% in dichloromethane. b n-Hexane, ET(30) = 30.9 kcal mol−1. c Toluene, ET(30) = 33.9 kcal mol−1. d Tetrahydrofuran, ET(30) = 37.4 kcal mol−1. e Dichloromethane, ET(30) = 41.1 kcal mol−1. f Acetonitrile, ET(30) = 46.0 kcal mol−1. g Methanol, ET(30) = 55.5 kcal mol−1. | ||||||
| An-BO | 12 | 17 | 15 | 28 | 40 | 12 |
| Ph-BO | 5 | 7 | 6 | 10 | 12 | 5 |
Electrochemical properties of the compounds
The redox potentials of the compounds were studied by cyclic voltammetry (Fig. 4a). For Ph-BO, an irreversible oxidation wave was observed at +0.67 V (vs. Fc/Fc+) together with a reversible reduction wave at −1.35 V (vs. Fc/Fc+) and an irreversible reduction wave at −2.17 V (vs. Fc/Fc+). This behaviour is closely comparable with the previously reported results for the iodinated analogue of Ph-BO.64 | ||
| Fig. 4 Cyclic voltammograms determined for BO derivatives in deaerated ACN containing 0.10 M Bu4N[PF6] as supporting electrolyte and with Ag/AgNO3 as reference electrode. Scan rate: 100 mV s−1. Ferrocene (Fc) was used as internal reference (set as 0 V in the cyclic voltammograms), c = 1.0 × 10−3 M. The absorption changes of An-BO upon (b) reduction with a potential of −1.40 V applied and (c) oxidation with a potential of +0.62 V applied on the working electrode. The spectra were recorded in situ with a spectroelectrochemical cuvette (1 mm optical path). c = 1.0 × 10−4 M in deaerated DCM. 20 °C. | ||
Redox potentials were also determined for An-BO. At +1.12 eV there is a reversible oxidation wave, attributed to one-electron oxidation of the anthryl moiety,88 compared to the known oxidation potential of anthracene (+0.9 V vs. Fc/Fc+).88,89 Making use of the Weller equation (eqn (S1)–(S3)†),90–92 the Gibbs free energy change (ΔGCS) accompanying intramolecular electron transfer for An-BO (with E00 = 1.96 eV) was calculated (for details please refer to the ESI†). The main finding is a positive ΔGCS in all solvents, a result in full agreement with the solvent-independent fluorescence properties found for An-BO (Fig. 3 and Table 3). Finally, spectroelectrochemistry was used to record absorption spectra for the BO radical anion (BO−˙), with peaks at 540 nm and 700 nm, and the anthracene radical cation (An+˙) with peaks at 540 nm and 580 nm (Fig. 4b and c). This latter information, used in conjunction with ultrafast transient absorption spectral measurements, helps to rule out the occurrence of light-induced charge separation.
| E RED/V | E OX/V | ΔGCS (eV)/ECT (eV) | ||||
|---|---|---|---|---|---|---|
| HEX | TOL | DCM | ACN | |||
| a Cyclic voltammetry in N2-saturated ACN containing 0.1 M Bu4N[PF6] as supporting electrolyte. Redox potentials of the compounds were determined with ferrocene (Fc) as internal standard (0 V). Counter electrode is Pt electrode and working electrode is glassy carbon electrode, with Ag/AgNO3 couple as the reference electrode. b E 00 = 1.96 eV. E00 is the energy level of singlet excited state localized on BO moiety (1BO*) approximated as the crossing point of normalized absorption and fluorescence spectra. c Not observed. d Not applicable. | ||||||
| An | − c | +0.90 | —d | —d | —d | —d |
| Ph-BO | −1.35/−2.17 | +0.67 | —d | —d | —d | —d |
| An-BO | −1.39/−2.17 | +0.67/+1.12 | 1.22/3.17 | 1.06/3.01 | 0.61/2.56 | 0.49/2.44 |
Femtosecond transient absorption spectra: photoexcited state dynamics
The excited state dynamics of the target compounds were measured using femtosecond transient absorption (fs-TA) spectroscopy with a small series of solvents (HEX, DCM and ACN). For An-BO, excitation was made at both 360 nm, where the anthryl unit is an important absorber, and at 610 nm, where BO is the sole absorber. The fs-TA spectra of anthracene (An) in HEX were also recorded (Fig. 5a). The corresponding spectra are characterized by a strong excited state absorption (ESA) band centred at 420 nm and a weaker band centred at about 600 nm. Evolution associated difference spectra (EADS) were obtained by global analysis of the fs-TA data with a sequential model (Fig. 5b). The signal evolution on the picosecond timescale shows small changes in the spectral shape due to vibrational relaxation of the singlet excited state of An, indicating that both transient species (A and B) belong to singlet excited states and that vibrational relaxation occurs within 7 ps. The longer-living transient (species C) shows an ESA band centred at 420 nm and can be attributed to the T1 → Tn transition. ISC occurs with a time constant of 2.5 ns, in good agreement with previous reports (τISC = 2.3 ns).88,89,93–95 | ||
| Fig. 5 Femtosecond transient absorption spectra of (a) An excited at 360 nm, (c) Ph-BO excited at 610 nm in ACN, and (b) EADS of An, (d) SADS of Ph-BO obtained with global analysis of the spectra reported in panels (a) and (c), respectively. c = 1 × 10−5 M. | ||
Fig. 5c presents the fs-TA spectra of Ph-BO recorded in ACN following excitation at 610 nm. The transient spectra present an intense negative band centred at 630 nm, interpreted as the convolution between ground state bleaching (GSB) of the BO chromophore and stimulated emission (SE), with a weaker transient absorption band in the 380–600 nm range. The SE contributes mainly to the low-energy tail of the negative signal, extending beyond 740 nm. The transient bleaching gradually recovers over the timescale of the measurement, while the positive absorption band does not change significantly over time. Global analysis of the kinetic traces was applied to extract relevant time constants associated with evolution of the system (species associated difference spectra, SADS), using a target-model decay scheme comprising four species (Fig. 5d); the S1 state of Ph-BO produces the triplet state with a low probability and relaxes to the ground state by radiative and non-radiative transitions.
The first component (black dashed curve) is assigned to a singlet excited state localized on the N,N,O,O-boron-chelated Bodipy. It is expected that excitation at 610 nm will populate the S1 state with excess vibrational energy. The first ultrafast process, close to the detection limit of the instrument (155 fs) is assigned to a fast electronic relaxation bringing the system out of the initially populated Franck Condon state. Within 50 ps, the system evolves towards the third component, which retains similar spectral characteristics and is therefore ascribed to the S1 state. Over this time interval, there is a small increase in the bleaching signal, that might be related to a red shift of the emission band, together with a slight increase of ESA between 550 and 600 nm. These spectral perturbations can be assigned to relaxation of the excited state. Evolution to the final species occurs within ca. 9 ns, but the residual transient species is much longer lived than the maximum time window of the instrument. The profile of this residual signal is consistent with that of the triplet state observed in nanosecond transient absorption spectra (see below). This result indicates that the time constant for the decay of S1 state is 8.8 ns, which is roughly in agreement with the long fluorescence lifetime of Ph-BO (11.3 ns, Table 1). Ph-BO has been studied also in HEX (Fig. S14†). The recorded transient spectra and the excited state evolution are similar to those observed in ACN, shown by the comparison between the kinetic traces recorded at the maximum of the GSB signal in ACN and HEX (Fig. S16†).
The fs-TA spectra recorded for An-BO in different solvents are comparable to those recorded for Ph-BO, thereby suggesting that formation of an intramolecular charge-transfer state is unimportant for this dyad (Fig. 6 and S15†). The spectral changes, however, do not rule out fast electronic energy transfer from anthracene to BO following excitation at 360 nm. Indeed, the component A with a lifetime close to the instrument detection limit, presents no SE signal, indicating that the higher singlet excited state is located on the anthryl unit (Fig. 6b). Subsequent spectra show a negative signal, with a minimum at 630 nm, attributable to a combination of GSB and SE of the BO unit. In addition, there is a weak absorption band extending from 380 to 600 nm. Target analysis performed in both HEX and ACN shows that spectral evolution occurs on different timescales compared to Ph-BO. The two components B and C have similar spectral character and are also similar to the singlet excited state signal of Ph-BO (Fig. 5d), so that, they can be assigned to the S1 state and vibrationally relaxed S1 state, respectively. The relaxation times, possibly including a contribution from intramolecular electronic energy transfer, are 24 ps and 31 ps, respectively in HEX and ACN. The residual species D shows ESA peaks at ca. 460 nm and 660 nm, and GSB peaks at 550 nm–650 nm (olive line in Fig. 6b); this spectral pattern can be attributed to the triplet-excited state of An-BO (see below). From the kinetics study, the time constants for the decay of S1 of An-BO in HEX and ACN are 7.7 ns and 8.0 ns, respectively. When An-BO is excited at longer wavelength (610 nm, Fig. 6c and d), the first three components observed have similar shape. The GSB of the first component (black dotted line, component A, Fig. 6d) is slightly narrower than that of other two components, and has a somewhat stronger absorption at 590 nm; we attribute A to the unrelaxed singlet state. Solvation and vibrational relaxation occur within 250 fs and 50 ps, then the triplet state is produced (olive line). The lifetime of S1 state is 6.7 ns. Transient absorption spectra for An-BO have been recorded also in polar solvents (Fig. S17 and S18†). The evolution of the transient spectra and the associated kinetics show only a minimal dependence on the solvent polarity.
 | ||
| Fig. 6 Femtosecond transient absorption spectra for An-BO recorded in ACN, excited at (a) 360 nm and (c) 610 nm, the relative SADS obtained with global analysis are presented in panels (b) and (d), respectively, c = 1.0 × 10−5 M. | ||
Assuming internal conversion to the ground state is insignificant, the time constant for ISC can be approximated by τISC = τs1/(1 − ΦF). An estimate for τs1 is available from the fs-TA studies, leading to the time constant for ISC for Ph-BO in acetonitrile as being ca. 33 ns, and that for An-BO as ca. 13 ns. This indicates that the rate of ISC for An-BO is roughly twice that for Ph-BO, which helps to account for the higher ΦΔ for the former. Importantly, the ISC time constant is significantly shorter than that for conventional Bodipy derivatives.
Nanosecond transient absorption (ns-TA) spectroscopy
The triplet excited states of the target compounds were studied by ns-TA spectroscopy in deoxygenated solution. The spectra recorded for Ph-BO show an ESA in the 350–500 nm range, overlapping with the ground state band at 410 nm (Fig. S19†). A significant GSB signal centred at 613 nm was also observed. The triplet state lifetime (τT) was determined as 192 μs. We developed a kinetic model to take into account triplet–triplet annihilation (TTA) effects, allowing the determination of the intrinsic triplet state lifetime49,96,97 as being 215 μs. To the best of our knowledge, the ISC and triplet state of N,N,O,O-boron-chelated Bodipy were not studied previously. It might be noted that the red absorbing Ph-BO has a much longer triplet lifetime than the 2,6-diiodobisstyrylBodipy (τT = 1.8 μs) commonly portrayed as the prototypic red absorbing Bodipy species.98For An-BO, a strong bleaching signal is observed at 622 nm, together with weaker absorption centred at 457 nm (Fig. 7). The intrinsic triplet lifetime was determined as 226 μs. This was shortened to 0.32 μs in aerated solution (Fig. S20†), confirming that the observed transient species is a triplet excited state localized on the Bodipy core. The ESA band of the N,N,O,O-boron coordinated Bodipy is drastically different from that observed for a bis-styrylBodipy, although the latter shows a similar absorption maximum to the N,N,O,O-boron coordinated Bodipy.40,99,100 The intrinsic triplet lifetime of An-BO is much longer than that of Bodipy accessed with the heavy atom effect (153 μs),101 or of the diiodobisstyrylBodipy (1.8 μs).98 This is an important advantage of facilitating triplet population through the heavy atom-free protocal, e.g. by distorted framework approach, rather than the more conventional heavy-atom perturbation methodology.17,102
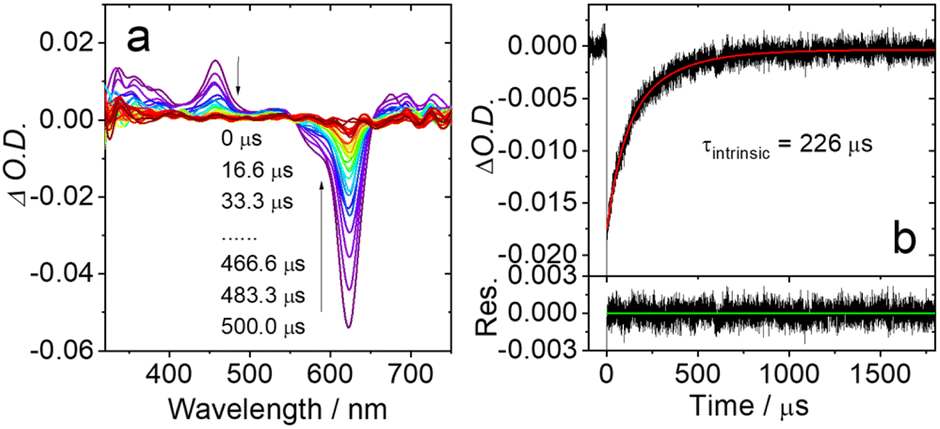 | ||
| Fig. 7 (a) Nanosecond time-resolved transient absorption spectra recorded for An-BO upon pulsed laser excitation (λex = 610 nm), (b) decay trace of An-BO at 605 nm in c = 1.0 × 10−6 M in deaerated acetonitrile, the intrinsic triplet state lifetime (τintrinsic) was obtained by fitting the decay curves to the kinetic model taking into account triplet–triplet annihilation (TTA) (see the ESI† for details), the lifetime data in (b) were measured at 5 × 10−6 M, apparent lifetime is 209 μs. 25 °C. | ||
Neither of the target compounds is phosphorescent at room temperature or at 77 K. Estimates of the triplet state energies of An-BO and Ph-BO as being 1.35 eV and 1.38 eV, respectively, were obtained by TD-DFT computations (see ESI† for details). Also, an experimental determination of the triplet energy was made by intermolecular sensitization,103 using meso-tetraphenylporphyrin (TPP, ET = 1.44 eV) as triplet donor or perylenediimide (PBI, ET = 1.20 eV) as triplet acceptor (Fig. S21 and S22†).104 With this method, the triplet state energy of An-BO was determined to be ca. 1.24 eV. The T1 state energies of these compounds are lower than that of the pristine Bodipy (ca. 1.6 eV),105 but much higher than the bis-styrylBodipy (ca. 1.0 eV).40,100 As such, promotion of the triplet-excited state through a twisted molecular structure55,58 offers a promising route towards increasing absorption in the red region whilst maintaining a viable triplet state energy and long lifetime.106
Time-resolved electron paramagnetic resonance (TR-EPR) spectroscopy
Optical spectroscopy aids the identification of intermediate species and provides critical kinetic information but cannot give useful insight about the ISC electron spin selectivity or the zero field splitting (ZFS) of the triplet state. Moreover, knowledge of the extent of localization of the excitation energy in molecules with a large π-conjugated framework is a fundamental aspect of molecular photophysics and an important criterion for the successful design of new opto-electro materials.107,108 The ZFS parameters D and E are important parameters of a chromophore, which may offer information about the spatial confinement of the triplet state wave function.78,110–112 Specifically, the zero-field splitting parameter D is a measure of the delocalization of the unpaired electron of the triplet over the molecule, while E describes the nonaxial asymmetry (rhombicity) of the delocalization.110,111The TR-EPR spectra of Ph-BO and An-BO recorded at 600 ns after the laser flash are presented in Fig. 8. The dependence of the TR-EPR and magnetophotoselection TR-EPR spectra of Ph-BO and An-BO on the delay time after photoexcitation are shown in the Fig. S25–S27†. The spectra are fully consistent with the involvement of a solitary triplet state. Furthermore, both the spectra and the consequent fitting parameters are comparable for the two compounds, clearly indicative of the localization of the triplet state on the BO fragment. The electron spin polarization (ESP) pattern for both compounds conforms to (e, e, e, a, a, a), this situation is reminiscent of that found for 2,6-diiodoBodipy,49,97,113 and 2,6-diiodobisstyrylBodipy.102 We previously observed the TR-EPR spectrum of the Bodipy triplet state, generated with the SOCT-ISC in compact anthryl-Bodipy orthogonal dyads, showing ESP phase pattern of (e, e, e, a, a, a), the SOCT-ISC mechanism can be excluded for An-BO based on the steady state fluorescence spectral study (Fig. 3b) and the fs-TA spectra (Fig. 6). Recently we found that the ESP of the triplet state TR-EPR spectrum of a twisted naphthalene-fused Bodipy is (a, e, a, e, a, e),58 this indicates that the ESP of the triplet state of twisted Bodipy derivatives is highly dependent on the molecular structures.56 This result shows the rich electron spin selectivity of the ISC of the twisted Bodipy derivatives.62,114
 | ||
| Fig. 8 Experimental TR-EPR spectra for Ph-BO, recorded at a delay time of 600 ns after laser excitation, the frozen samples (c = 3.0 × 10−4 M) were excited at 585 nm with a pulse energy of 1 mJ. For An-BO, the frozen samples (c = 2.0 × 10−4 M) were excited at 613 nm. The model TR-EPR spectrum of An calculated for D = 2150 MHz, E = 244 MHz, Pz = 0.00, Py = 0.56, Px = 0.44 (ref. 49 and 109) is also presented for comparison. Samples were dissolved in TOL/2-MeTHF (1/1, v/v), and the spectra were recorded at 80 K. Simulation parameters are presented in Table 4. | ||
Simulation of the TR-EPR spectra gives ZFS D and E parameters for Ph-BO as −1470 MHz and 477 MHz, and almost the same for An-BO (Table 4). Similar D and E parameters indicate that the T1 state of the molecules is confined on the BO chromophore. These parameters are different from those for 2,6-diiodoBodipy, for which the D and E values are −2940 MHz and 655 MHz (Table 4), and the rhombicity (non-axiality) of the spatial distribution of the triplet state wave function |E/D| is increased compared to the native Bodipy.49,113 These results show that the triplet state wave functions are delocalized over the twisted molecular framework. Interestingly, the ZFS D and E parameters of Ph-BO and An-BO are similar to the recently reported naphthalene-fused Bodipy derivative, which also has a twisted molecular structure.58 The population ratios of the sublevels of the T1 state of An-BO remain similar to those of Ph-BO (Table 4), Tz sublevel of the T1 state being overpopulated for both, which is supported by theoretical computations (see ESI† for more details).
| Compound | D (MHz) | E (MHz) |
P
x
:Py:Pz |
|---|---|---|---|
| a Obtained from simulation of the triplet-state TR-EPR spectra of the indicated molecules in a toluene/2-MeTHF glass (1/1, v/v) at 80 K. b The sign of the D parameter is negative for Bodipy. | |||
| An-BO | −1470 | 467 | 0.00:0.00:1.00 |
| Ph-BO | −1470 | 477 | 0.00:0.00:1.00 |
| 2,6-diiodoBodipy | −2940 | 655 | 0.00:0.15:1.00 |
DFT computation and the ISC mechanism
Density functional theory (DFT) calculations are often helpful in explaining photophysical behaviour. The molecular geometry of the ground state was optimized with DFT theory (Fig. 9). The dihedral angle between the dipyrrin ring and the anthryl moiety is 77° for An-BO, whereas that for Ph-BO is reduced to 47°. The twist angles between the planes defined by the two pyrrolic rings are 49° in both molecules, this being larger than the previously reported analogue (lacking the meso-substituent group, where the twist angle is ca. 10°).65 The twist angle optimized by DFT is slightly different from that obtained by X-ray diffraction from a single crystal (Fig. 1). This is probably due to intermolecular interactions in the crystal. The similar torsion angles of the core chromophore in Ph-BO and An-BO unambiguously demonstrate that the disparate ISC efficiency of An-BO and Ph-BO is not caused by any different twisting of the chromophore, rather, it is due to the tuning of the excited states, by introducing the anthryl group in An-BO. | ||
| Fig. 9 Dihedral angles of selected atoms of (a) An-BO and (b) Ph-BO at the optimized ground state geometry, at CAM-B3LYP/6-31G (d) level with Gaussian 09; spin density surfaces of (c) An-BO and (d) Ph-BO at the optimized triplet state geometry; calculated at the CAM-B3LYP/6-31G(d) level with Gaussian 09W. Iso value of the spin density surfaces is 0.0015. | ||
The electron spin density of the T1 state was computed (Fig. 9c and d). For An-BO, the spin density is almost exclusively localized on the twisted Bodipy moiety, with negligible distribution on the anthryl unit. This seems consistent with the large dihedral angle between the anthryl moiety and the twisted Bodipy core (77°), which effectively decouples the two components. For Ph-BO, part of the spin density is distributed over the meso-phenyl ring, presumably because of the smaller dihedral angle. However, this disparate distribution of the triplet state electron spin density surface between the two triplets does not affect either the transient absorption or the TR-EPR spectra. For both compounds, the difference in electron density of the triplet excitation (corresponding to a HOMO → LUMO transition, Fig. S27†) is mainly localized on the twisted dipyrrin core.68
In order to further understand the difference in triplet state quantum yield of the compounds, we also studied the native oxo-Bodipy (BO), which has a hydrogen atom at the meso position. All electronic structure calculations were performed with the ORCA programs,115,116 using DFT with the CAM-B3LYP functional and the def2-SVP atomic orbital basis set. The electronic ground state S0 was optimized with restricted DFT, the T1 state with unrestricted DFT, and all other excited states with TD-DFT. For each excited state the Hessian matrix was calculated and checked for positive definiteness. These optimized geometries and their Hessians are subsequently used in an excited state dynamic (ESD) calculation that yields the rate constant for ISC. Both Franck–Condon and Herzberg–Teller contributions were calculated for all three sublevels of each triplet state. We used a purpose-written program applying the formulae of Baiardi et al.117 or of de Souza et al.,118 which yield identical results.
With the CAM-B3LYP functional, the T2 state is found below the S1 state, hence the ISC rates for S1 → T2 ISC were also calculated. The excitation energies calculated with the CAM-B3LYP functional for S1 are substantially higher than the experimental values (see below), TD-DFT usually highly overestimates the low-lying excited states of Bodipy derivatives, hence it is possible that the true S1 → T2 energy gap is small or even negative. An exception is the T2 state of An-BO which is a locally excited state on the anthryl unit and has no counterpart in the two other compounds. At each optimized geometry, the vertical excitation energies were calculated, resulting in the level diagrams shown in Scheme 2 (refer to Table S2† for specific values). For Ph-BO, both HOMO and LUMO are localized on the dipyrrin unit. The S1 state is predominantly of HOMO → LUMO excitation (i.e., a locally excited singlet state). Optimization of the S1 state lowers its energy by ca. 1000 cm−1 with respect to the vertical excitation and shifts the S0 state by about the same amount to higher energies (Table S2†), thus predicting a rather small Stokes shift. The geometrical change is small, and the dipole moment almost unchanged on excitation. For all three compounds, the T1 state is found at about half of the energy of the S1 state, but at almost the same geometry. A rather large exchange integral is indicated by the observation that T1 also has a large contribution from the HOMO → LUMO excitation.
 | ||
| Scheme 2 Energy level diagrams for (a) An-BO, (b) Ph-BO and (c) BO at each of the optimized geometries. In each column, the optimized state is indicated by the full dot. Black lines indicate singlet states, red lines represent the triplet states, and blue arrows indicate the ISC transitions. | ||
For An-BO, the T2 state corresponds to excitation from the HOMO to the LUMO of the anthryl unit, i.e., it is a locally excited state (3An*). Optimization of the geometry of this T2 state leads to a strong stabilization accompanied by significant structural changes at the anthryl unit. At its minimum, the T2 state is almost degenerate with the Bodipy-localized T1 state. Since both the lower and the upper singly occupied orbitals in S1 and T2 are different, we expect small SOCMEs. Hence vibrationally induced SOC should be considered.28,119 Indeed, we find that Herzberg–Teller coupling accounts for 100% of the ISC rates to the M = +1/−1 substates of the triplets, and for a substantial fraction of the M = 0 substate.
The program allows calculation of the ISC rate constant as a continuous function of the energy gap. Input data were the geometries and Hessians from the ORCA optimizations, as well as the SOCMEs and their derivatives with respect to all normal coordinates. Fig. 10a shows the resulting ISC rate constants (kISC) for the S1 → T1 transition for each of the three compounds, the calculated energy gaps are indicated by a dot for each curve. We note that the rates of the S1 → T1 transition are comparable for the three compounds, indicating that the anthryl substituent is not involved in this particular process. For energy gaps of ca. 10000 cm−1, these rates are smaller than 105 s−1 and hence cannot explain the observed triplet yields. This is most probably a consequence of the fact that S1 and T1 correspond to the same HOMO → LUMO excitation and therefore have rather small SOCME. However, for An-BO, the kISC of S1 → T2 is greatly increased, which is consistent with the higher triplet yield. The S1 → T2 transition in An-BO leads directly to the triplet state localized on the anthryl moiety.
 | ||
| Fig. 10 Energy gap dependence of the ISC rate constants (Franck–Condon plus Herzberg–Teller terms). (a) Rate constants for the S1 → T1 transitions of An-BO (red line), Ph-BO (blue line) and BO (black line), the red dotted line refers the S1 → T2 transition of An-BO. The calculated energy gap is indicated by a dot in each curve. (b) Energy gap dependence of the ISC rate constant for S1 → Tn transitions. The full lines are for the second local triplet of the BO unit, which is T2 in BO and Ph-BO, but T3 in An-BO. The dotted line is for T2 of An-BO which is a local triplet on the anthryl unit. | ||
Fig. 10b shows the corresponding data for ISC from the S1 state to the second locally excited triplet on the BO unit, the relevant transitions are S1 → T2 for BO and Ph-BO, and S1 → T3 for An-BO. As in the case of the S1 → T2 transitions, these curves are comparable, indicating that these triplet states are associated with the BO unit and have similar orbital excitation character. The substituents appear not to be involved. Since orbitals other than those associated with S1 are involved, the SOCMEs are increased, thereby enhancing the rate constants by two orders of magnitude compared to S1 → T1. The rate constant for the S1 → T2 transition for An-BO shows significantly different behaviour to that of the other compounds (dotted line in Fig. 10). The final state T2 is a locally excited triplet of the anthryl unit which has no low-energy counterpart in the parent compound or the phenyl-derivative (i.e., ISC corresponds to intramolecular energy transfer). The transition is accompanied by a strong geometrical relaxation within the anthryl unit, giving a broad profile to the energy gap dependence.
The second local triplet state on the BO unit is probably at higher energy than the S1 state, i.e. the energy gap is negative. A gap in the range (1500–1000) cm−1 would still allow ISC to occur with a rate constant of ca. 108 s−1. There is little experimental information on the true energy gap between S1 and T2 in these compounds. It is hence possible that the T2 states of BO and Ph-BO in reality are higher than S1. On the other hand, the strong relaxation of T2 observed for An-BO suggests that the state is near T1 and below S1. Unsubstituted anthracene has a T1 state energy of 1.8 eV.104 This is below the experimental energy for the S1 state of Ph-BO (ca. 2.0 eV). For an energy gap in the range 4000–6000 cm−1 the calculations anticipate ISC rate constants near 5 × 108 s−1. These are in good agreement with a triplet yield in the range of 50%. It must then be concluded that the actual S1 → T2 energy gap in Ph-BO is negative, i.e., T2 is above S1. Thus, the increased rate of ISC for An-BO can be ascribed to transient population of the T2 state localized on the anthryl unit.
This is an interesting example whereby the initial and the final states of ISC are confined on different chromophores, yet the matching of the S1 and T2 state energies (at the geometry of S1) still induces efficient ISC. This result may present new understanding of singlet-to-triplet energy transfer, which is much less common than singlet–singlet or triplet–triplet energy transfer.120,121 Our theoretical studies are able to correctly predict that the Tz sublevel of the T1 states of the compounds are overpopulated, which is in agreement with the TR-EPR spectral observations (see ESI for more details, Fig. S30 and S31†). We found that the SOCME favours the transition to the Tx-substate, while the much larger vibrational contributions favour the transition to the Tz-state, which is in agreement with the TR-EPR spectral experimental results (Fig. 8 and Table 4). This principal is applicable to other twisted molecules, i.e. the ESP phase pattern of the T1 state TREPR spectrum of a compound, should be directly related to the ISC rate constants of S1 → Tx, S1 → Ty and S1 → Tz.
Because of the stated uncertainties about the S1 state energy obtained by the TD-DFT calculation, we prefer to rely on the values obtained experimentally. The absence of detectable levels of phosphorescence, however, precludes any experimental T1 state energy, and we must rely on the computed results. This leads to the simplified energy diagrams for An-BO and Ph-BO shown in Scheme 3. For Ph-BO, the T2 state energy is higher than that of S1, and there is a large energy gap between S1 and T1 states, making ISC inefficient. For An-BO, the T2 state (i.e., 1.81 eV), localized on the anthryl moiety, is lower in energy than the S1 state (i.e., 1.96 eV), thereby facilitating faster ISC through intermediary population of the anthryl triplet state. Notably, triplet population via this route involves two separate electronic energy transfer events rather than the conventional SOC mechanism.
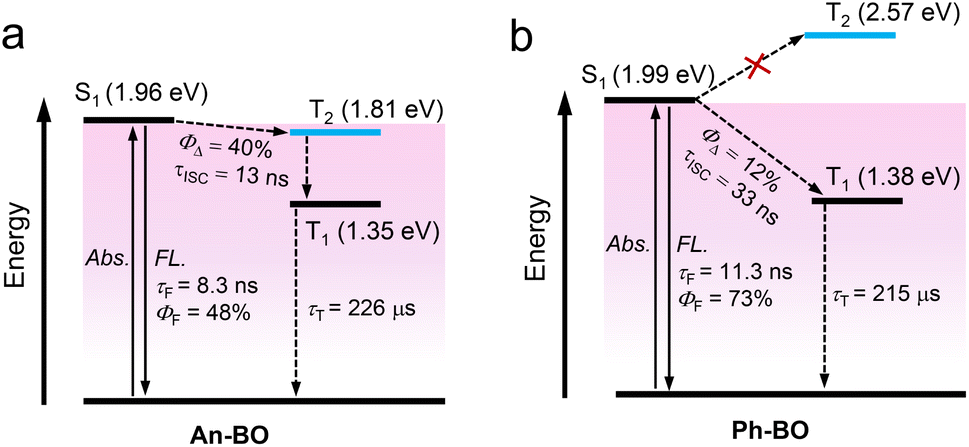 | ||
| Scheme 3 Simplified Jablonski diagram illustrating the photophysical processes involved in (a) An-BO and (b) Ph-BO upon photoexcitation. Energy level of S1 is taken as the intersection of absorption and luminescence spectra, and the triplet energy is from the TD-DFT calculation. | ||
Conclusion
This study represents an attempt to identify ways to enhance intersystem crossing (ISC), leading to triplet formation, for heavy atom-free organic chromophores, which usually have slow ISC kinetics. The common rationale for promoting ISC in such materials relies on incorporating a heavy atom into the π-system, but this has several disadvantages. The main concept under investigation here relates to the notion that a severely twisted π-conjugation framework, such as that inherent to the family of N,N,O,O-boron-chelated Bodipy chromophores, serves to promote ISC without the undesired consequences of heavy atom substituents. Our results indicate that ISC is indeed enhanced through this treatment but this is not a major effect. Thus, Ph-BO shows only a modest singlet oxygen quantum yield (ΦΔ = 12%) and a time constant for ISC of ca. 33 ns. However, this strategy pushes the absorption bands into the red region, in marked contrast with the effects of halogenation, and this is highly beneficial. The limited performance with Ph-BO, which contrasts with the ISC behaviour of the helicenes, can be traced to a large S1 → T1 energy gap, combined with weak S1 → T2 coupling. Time-resolved electron paramagnetic resonance spectra of the triplet show an electron spin polarization phase pattern of (e, e, e, a, a, a). Zero field splitting D and E parameters of the triplet state of the twisted Bodipy chromophore indicate the triplet wave function is spread over the distorted π-conjugation framework. The main finding from these various studies is that the twisted π-conjugation does not necessarily promote ISC.To overcome the large S1 → T1 energy gap, the meso-phenyl ring has been replaced with an anthryl moiety which has a locally-excited triplet state of comparable energy to the S1 state associated with the twisted Bodipy chromophore. For this dyad, An-BO, charge recombination-induced ISC is insignificant, as demonstrated by steady-state optical spectra and fs-TA measurements. The triplet quantum yield is increased substantially and An-BO exhibits a much improved quantum yield for formation of singlet molecular oxygen (ΦΔ = 40%). The greater propensity towards triplet formation must arise from increased coupling between the S1 and T2 states. It is important to stress that these two excited states are localized on different chromophores and held in a near orthogonal geometry but still manage to promote ISC. Transient population of the anthryl-based triplet state is followed by triplet–triplet energy transfer to form the T1 state localized on the Bodipy fragment. The latter process is spin-allowed and likely to be fast but the former step is relatively slow because of the molecular orientation and limited spectral overlap. Nonetheless, the process competes with fluorescence and internal conversion from S1. The overall ISC rate for An-BO is faster (ca. 13 ns) than for Ph-BO. In this respect, it is important to note that these Bodipy derivatives based on twisted π-conjugation pathways show unusually long-lived S1 states and this is a considerable benefit in terms of applications. Our results show that the twisted π-conjugation system cannot be relied upon to induce efficient ISC. An improved strategy is based on energy matching of S1/Tn (n > 1) states. Moreover, since the anthryl triplet is far from optimal as an acceptor, there is considerable scope to design a new range of heavy atom-free triplet sensitizers along these lines.
Experimental section
General method
All chemicals used for synthesis were analytically pure and used as received. Solvents were dried and distilled prior to use. Samples of Ph-BO were prepared as before and we thank Dr J. G. Knight (Newcastle University) for providing a sample for preliminary spectroscopic investigations.64,65 The UV/vis absorption spectra were recorded with a UV2550 spectrophotometer (Shimadzu Ltd, Japan). The fluorescence spectra were recorded with an FS5 spectrofluorometer (with photon counting detection method. Edinburgh Instruments, UK). The fluorescence lifetimes of compounds were measured with an OB920 luminescence lifetime spectrometer (Edinburgh Instruments, UK), an EPL picosecond pulsed laser was used for excitation, and time-correlated, single photon counting (TCSPC) was used for detection.Nanosecond transient absorption spectroscopy
The nanosecond transient absorption spectra were acquired on a LP980 laser flash photolysis spectrometer (Edinburgh Instruments, UK). The signal was digitized with a Tektronix TDS 3012B oscilloscope. Samples were excited with a nanosecond pulsed laser (Surelite I-10, USA; the wavelength being tunable in the range of 210–2400 nm). Typical laser energy was 5 mJ per pulse. The samples were deaerated with N2 for 15 min.Femtosecond transient absorption spectroscopy
Femtosecond transient absorption spectra were measured by an ultrafast transient absorption spectrometer (Harpia-TA, Light Conversion). A Ti:sapphire laser amplifier (Astrella Coherent) with 40 fs pulse duration at 800 nm and 1 kHz repetition rate was used for these experiments. The white-light-continuum probe pulses were generated by a 2 mm CaF2 nonlinear crystal, and spectrally tunable pump pulses were generated by an optical parametric amplifier (TOPAS-C, light conversion). A 1 mm pathlength quartz cuvette housed the solution. Changes in absorbance were monitored by focusing the transmitted probe light through the solution onto a broadband UV/vis detector. Data analysis was performed applying Singular Value Decomposition (SVD) and global analysis with target model, using the software GLOTARAN.122TR-EPR spectroscopy
TR-EPR spectral measurements were carried out at 80 K. Samples were excited at 532–613 nm, 1 mJ pulse,10 ns, 100 Hz using the output of an optical parametric oscillator (LP603 SOLAR Laser Systems), pumped with the output of a Nd:YAG laser (LQ629 SOLAR Laser Systems). Samples were dissolved in toluene/2-methyltetrahydrofuran (1/1, v/v), with subsequent removal of dissolved air by freeze–pump–thaw cycles. The time-resolved continuous-wave EPR spectral measurements were performed on an EPR Elexsys E-580 spectrometer (Bruker) with a flexlinel resonator (ER 4118X-MD5-W1) at X-band at 80 K. Low temperature measurements were carried out in a CF935 cryostat (Oxford Instruments) cooled by liquid nitrogen. The TR-CW EPR spectra were obtained by the summation of the data in different time windows after the laser pulse. The EPR spectra were simulated using the Easy Spin package implemented in the MATLAB programming language.123Theoretical computation
Geometries for the electronic ground state and for the lowest-excited triplet state were optimized with density functional theory (DFT) at the CAM-B3LYP/6-31G(d) level, using the Gaussian 09W program.124 All structures were minima on the potential energy surface, since all eigenvalues of the Hessians were positive. The spin orbit coupling matrix elements (SOCME), and their derivatives along the normal coordinates, were calculated between S1 and all triplet states with the ORCA program at the CAM-B3LYP/def2-SVP level. These data were subsequently used by a purpose-built program which calculates the ISC rates (both Franck–Condon and Herzberg Teller contributions) as a function of the diabatic energy gaps between S1 and each final triplet state. This program applies the formulae of Baiardi et al.117 or of de Souza et al.118 originally developed for calculation of emission spectra, but which can be modified for ISC. Specifically, we replaced the three components of the transition dipole matrix elements by the SOCME for the three sublevels, also replacing their derivatives with respect to the normal coordinates. In these modified formulae, the energy gap can be regarded as a variable parameter, such that instead of a spectrum one gets the energy-gap dependence of the ISC rate constant. The SOCME and their derivatives are calculated with the ESD module of the ORCA program. These use the RI-SOMF(1X) option of ORCA, i.e., the mean-field operator (effective potentials), including one-electron terms. Coulomb terms are computed with the resolution of identity approximation, exchange terms use one-center exact integrals including the spin-other orbit interaction, and DFT local correlation terms are not included.Data availability
All relevant data are presented in the main text and/or ESI.†Author contributions
X. Z., A. A. S. and X. L. contributed equally to this work. J. Z. conceived the research, directed the analysis of the results; X. Z. synthesized the compounds, measured and analysed steady-state optical spectra, electrochemistry, transient absorption spectra and other photophysical processes; A. A. S. and V. K. V. measured and simulated TR-EPR spectra; X. L., Y. W, M. T. and M. D. D. measured and analysed femtosecond transient absorption spectroscopy; A. H. provided guidance on the analysis of parts of the photophysical studies and revised parts of the manuscript; B. D. performed the theoretical computational analysis. All authors provided comments on preparation and modification of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. Z. thanks the NSFC (U2001222), the Research and Innovation Team Project of Dalian University of Technology (DUT2022TB10), the Fundamental Research Funds for the Central Universities (DUT22LAB610) and the State Key Laboratory of Fine Chemicals for financial support. M. D. D acknowledges support from the European Union's Horizon 2020 research and innovation program under grant agreement 871124 Laserlab-Europe. A. A. S. and V. K. V. acknowledge financial support from the government assignment for the Federal Research Center (Kazan Scientific Center of Russian Academy of Sciences). B. D. thanks the Dalian University of Technology for support through a HaiTian Professorship. X. Z. thanks the support of “the Fundamental Research Funds for the Central Universities”.Notes and references
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77–88 RSC.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- J. Zhao, K. Xu, W. Yang, Z. Wang and F. Zhong, Chem. Soc. Rev., 2015, 44, 8904–8939 RSC.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687–7697 RSC.
- D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97–113 RSC.
- S. Fukuzumi and K. Ohkubo, Chem. Sci., 2013, 4, 561–574 RSC.
- J. I. Goldsmith, W. R. Hudson, M. S. Lowry, T. H. Anderson and S. Bernhard, J. Am. Chem. Soc., 2005, 127, 7502–7510 CrossRef CAS PubMed.
- G.-G. Luo, H. Lu, X.-L. Zhang, J.-C. Dai, J.-H. Wu and J.-J. Wu, Phys. Chem. Chem. Phys., 2015, 17, 9716–9729 RSC.
- G.-G. Luo, K. Fang, J.-H. Wu and J. Mo, Chem. Commun., 2015, 51, 12361–12364 RSC.
- P. Wang, S. Guo, H.-J. Wang, K.-K. Chen, N. Zhang, Z.-M. Zhang and T.-B. Lu, Nat. Commun., 2019, 10, 3155 CrossRef.
- S. Guo, K.-K. Chen, R. Dong, Z.-M. Zhang, J. Zhao and T.-B. Lu, ACS Catal., 2018, 8, 8659–8670 CrossRef CAS.
- S. Sato, T. Morikawa, T. Kajino and O. Ishitani, Angew. Chem., Int. Ed., 2013, 52, 988–992 CrossRef CAS.
- Y. Tamaki, K. Koike, T. Morimoto, Y. Yamazaki and O. Ishitani, Inorg. Chem., 2013, 52, 11902–11909 CrossRef CAS.
- P. Wang, R. Dong, S. Guo, J. Zhao, Z.-M. Zhang and T.-B. Lu, Natl. Sci. Rev., 2020, 7, 1459–1467 CrossRef CAS PubMed.
- X. Li, S. Kolemen, J. Yoon and E. U. Akkaya, Adv. Funct. Mater., 2017, 27, 1604053 CrossRef.
- V.-N. Nguyen, Y. Yan, J. Zhao and J. Yoon, Acc. Chem. Res., 2021, 54, 207–220 CrossRef CAS PubMed.
- J. Tian, J. Zhou, Z. Shen, L. Ding, J.-S. Yu and H. Ju, Chem. Sci., 2015, 6, 5969–5977 RSC.
- Q. Tang, W. Xiao, J. Li, D. Chen, Y. Zhang, J. Shao and X. Dong, J. Mater. Chem. B, 2018, 6, 2778–2784 RSC.
- M.-R. Ke, S.-L. Yeung, D. K. P. Ng, W.-P. Fong and P.-C. Lo, J. Med. Chem., 2013, 56, 8475–8483 CrossRef CAS.
- D. Chen, Z. Zhong, Q. Ma, J. Shao, W. Huang and X. Dong, ACS Appl. Mater. Interfaces, 2020, 12, 26914–26925 CrossRef CAS.
- J. Zhao, S. Ji and H. Guo, RSC Adv., 2011, 1, 937–950 RSC.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- C. Ye, L. Zhou, X. Wang and Z. Liang, Phys. Chem. Chem. Phys., 2016, 18, 10818–10835 RSC.
- N. Yanai and N. Kimizuka, Acc. Chem. Res., 2017, 50, 2487–2495 CrossRef CAS PubMed.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Principles of Molecular Photochemistry: An Introduction, University Science Books, Sausalito, CA, 2009 Search PubMed.
- K. Schmidt, S. Brovelli, V. Coropceanu, D. Beljonne, J. Cornil, C. Bazzini, T. Caronna, R. Tubino, F. Meinardi, Z. Shuai and J.-L. Brédas, J. Phys. Chem. A, 2007, 111, 10490–10499 CrossRef CAS.
- Z. Mahmood, A. A. Sukhanov, N. Rehmat, M. Hu, A. Elmali, Y. Xiao, J. Zhao, A. Karatay, B. Dick and V. K. Voronkova, J. Phys. Chem. B, 2021, 125, 9317–9332 CrossRef CAS PubMed.
- T. Yogo, Y. Urano, Y. Ishitsuka, F. Maniwa and T. Nagano, J. Am. Chem. Soc., 2005, 127, 12162–12163 CrossRef CAS PubMed.
- M. Nakashima, K. Iizuka, M. Karasawa, K. Ishii and Y. Kubo, J. Mater. Chem. C, 2018, 6, 6208–6215 RSC.
- W. Wu, H. Guo, W. Wu, S. Ji and J. Zhao, J. Org. Chem., 2011, 76, 7056–7064 CrossRef CAS PubMed.
- A. Gorman, J. Killoran, C. O'Shea, T. Kenna, W. M. Gallagher and D. F. O'Shea, J. Am. Chem. Soc., 2004, 126, 10619–10631 CrossRef CAS PubMed.
- N. Adarsh, R. R. Avirah and D. Ramaiah, Org. Lett., 2010, 12, 5720–5723 CrossRef CAS PubMed.
- V.-N. Nguyen, S. Qi, S. Kim, N. Kwon, G. Kim, Y. Yim, S. Park and J. Yoon, J. Am. Chem. Soc., 2019, 141, 16243–16248 CrossRef CAS.
- A. J. Tilley, R. D. Pensack, T. S. Lee, B. Djukic, G. D. Scholes and D. S. Seferos, J. Phys. Chem. C, 2014, 118, 9996–10004 CrossRef CAS.
- M. Bröring, R. Krüger, S. Link, C. Kleeberg, S. Köhler, X. Xie, B. Ventura and L. Flamigni, Chem. – Eur. J., 2008, 14, 2976–2983 CrossRef PubMed.
- B. Ventura, G. Marconi, M. Bröring, R. Krüger and L. Flamigni, New J. Chem., 2009, 33, 428–438 RSC.
- Z. Zhu, X. Zhang, X. Guo, Q. Wu, Z. Li, C. Yu, E. Hao, L. Jiao and J. Zhao, Chem. Sci., 2021, 12, 14944–14951 RSC.
- W. Wu, J. Zhao, J. Sun and S. Guo, J. Org. Chem., 2012, 77, 5305–5312 CrossRef CAS PubMed.
- L. Huang, X. Yu, W. Wu and J. Zhao, Org. Lett., 2012, 14, 2594–2597 CrossRef CAS PubMed.
- Y. Wei, M. Zhou, Q. Zhou, X. Zhou, S. Liu, S. Zhang and B. Zhang, Phys. Chem. Chem. Phys., 2017, 19, 22049–22060 RSC.
- R. Ziessel, B. D. Allen, D. B. Rewinska and A. Harriman, Chem. – Eur. J., 2009, 15, 7382–7393 CrossRef CAS.
- J.-Y. Liu, M. E. El-Khouly, S. Fukuzumi and D. K. P. Ng, Chem. – Asian J., 2011, 6, 174–179 CrossRef CAS PubMed.
- C. B. KC, G. N. Lim, V. N. Nesterov, P. A. Karr and F. D'Souza, Chem. – Eur. J., 2014, 20, 17100–17112 CrossRef CAS PubMed.
- Z. Wang, J. Zhao, A. Barbon, A. Toffoletti, Y. Liu, Y. An, L. Xu, A. Karatay, H. G. Yaglioglu, E. A. Yildiz and M. Hayvali, J. Am. Chem. Soc., 2017, 139, 7831–7842 CrossRef CAS PubMed.
- Z. Wang, Y. Gao, M. Hussain, S. Kundu, V. Rane, M. Hayvali, E. A. Yildiz, J. Zhao, H. G. Yaglioglu, R. Das, L. Luo and J. Li, Chem. – Eur. J., 2018, 24, 18663–18675 CrossRef CAS PubMed.
- X. Zhang, A. A. Sukhanov, E. A. Yildiz, Y. E. Kandrashkin, J. Zhao, H. G. Yaglioglu and V. K. Voronkova, Chemphyschem, 2021, 22, 55–68 CrossRef CAS PubMed.
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, H. Savoie, K. J. Flanagan, C. Sy, E. Sitte, M. Telitchko, F. Laquai, R. W. Boyle and M. O. Senge, J. Am. Chem. Soc., 2017, 139, 6282–6285 CrossRef CAS PubMed.
- Z. Wang, A. A. Sukhanov, A. Toffoletti, F. Sadiq, J. Zhao, A. Barbon, V. K. Voronkova and B. Dick, J. Phys. Chem. C, 2019, 123, 265–274 CrossRef CAS.
- X. Zhang, Z. Wang, Y. Hou, Y. Yan, J. Zhao and B. Dick, J. Mater. Chem. C, 2021, 9, 11944–11973 RSC.
- E. Bassan, A. Gualandi, P. G. Cozzi and P. Ceroni, Chem. Sci., 2021, 12, 6607–6628 RSC.
- Y. Wu, Y. Zhen, Y. Ma, R. Zheng, Z. Wang and H. Fu, J. Phys. Chem. Lett., 2010, 1, 2499–2502 CrossRef CAS.
- H. Ito, H. Sakai, Y. Suzuki, J. Kawamata and T. Hasobe, Chem. – Eur. J., 2020, 26, 316–325 CrossRef CAS PubMed.
- K. Nagarajan, A. R. Mallia, K. Muraleedharan and M. Hariharan, Chem. Sci., 2017, 8, 1776–1782 RSC.
- Y. Dong, B. Dick and J. Zhao, Org. Lett., 2020, 22, 5535–5539 CrossRef CAS PubMed.
- Y. Dong, P. Kumar, P. Maity, I. Kurganskii, S. Li, A. Elmali, J. Zhao, D. Escudero, H. Wu, A. Karatay, O. F. Mohammed and M. Fedin, Phys. Chem. Chem. Phys., 2021, 23, 8641–8652 RSC.
- Y. Yan, A. A. Sukhanov, M. H. E. Bousquet, Q. Guan, J. Zhao, V. K. Voronkova, D. Escudero, A. Barbon, Y. Xing, G. G. Gurzadyan and D. Jacquemin, J. Phys. Chem. B, 2021, 125, 6280–6295 CrossRef CAS PubMed.
- Z. Wang, L. Huang, Y. Yan, A. M. El-Zohry, A. Toffoletti, J. Zhao, A. Barbon, B. Dick, O. F. Mohammed and G. Han, Angew. Chem., Int. Ed., 2020, 59, 16114–16121 CrossRef CAS PubMed.
- L. Shi, Z. Liu, G. Dong, L. Duan, Y. Qiu, J. Jia, W. Guo, D. Zhao, D. Cui and X. Tao, Chem. – Eur. J., 2012, 18, 8092–8099 CrossRef CAS PubMed.
- T. Biet, K. Martin, J. Hankache, N. Hellou, A. Hauser, T. Bürgi, N. Vanthuyne, T. Aharon, M. Caricato, J. Crassous and N. Avarvari, Chem. – Eur. J., 2017, 23, 437–446 CrossRef CAS PubMed.
- Y. Dong, M. Taddei, S. Doria, L. Bussotti, J. Zhao, G. Mazzone and M. Di Donato, J. Phys. Chem. B, 2021, 125, 4779–4793 CrossRef CAS PubMed.
- Z. Wang, X. Zhang and J. Zhao, J. Phys. Chem. C, 2021, 125, 19097–19109 CrossRef CAS.
- S. V. K. Isukapalli and S. R. Vennapusa, J. Phys. Chem. A, 2022, 126, 7606–7612 CrossRef CAS PubMed.
- H. Kim, A. Burghart, M. B. Welch, J. Reibenspies and K. Burgess, Chem. Commun., 1999, 1889–1890 RSC.
- R. B. Alnoman, S. Rihn, D. C. O'Connor, F. A. Black, B. Costello, P. G. Waddell, W. Clegg, R. D. Peacock, W. Herrebout, J. G. Knight and M. J. Hall, Chem. – Eur. J., 2016, 22, 93–96 CrossRef CAS PubMed.
- M. Sapir and E. V. Donckt, Chem. Phys. Lett., 1975, 36, 108–110 CrossRef CAS.
- A. Loudet, R. Bandichhor, K. Burgess, A. Palma, S. O. McDonnell, M. J. Hall and D. F. O'Shea, Org. Lett., 2008, 10, 4771–4774 CrossRef CAS PubMed.
- S. Yamazawa, M. Nakashima, Y. Suda, R. Nishiyabu and Y. Kubo, J. Org. Chem., 2016, 81, 1310–1315 CrossRef CAS PubMed.
- S. Y. Leblebici, L. Catane, D. E. Barclay, T. Olson, T. L. Chen and B. Ma, ACS Appl. Mater. Interfaces, 2011, 3, 4469–4474 CrossRef CAS PubMed.
- S. Y. Leblebici, T. L. Chen, P. Olalde-Velasco, W. Yang and B. Ma, ACS Appl. Mater. Interfaces, 2013, 5, 10105–10110 CrossRef CAS PubMed.
- Y. Kubo, T. Shimada, K. Maeda and Y. Hashimoto, New J. Chem., 2020, 44, 29–37 RSC.
- M. Saikawa, T. Nakamura, J. Uchida, M. Yamamura and T. Nabeshima, Chem. Commun., 2016, 52, 10727–10730 RSC.
- R. D. Rieth, N. P. Mankad, E. Calimano and J. P. Sadighi, Org. Lett., 2004, 6, 3981–3983 CrossRef CAS PubMed.
- L. Copey, L. Jean-Gérard, E. Framery, G. Pilet and B. Andrioletti, Eur. J. Org. Chem., 2014, 2014, 4759–4766 CrossRef CAS.
- X.-Y. Zhu, H. Wu, X.-F. Guo and H. Wang, Dyes Pigm., 2019, 165, 400–407 CrossRef CAS.
- M. A. Filatov, Org. Biomol. Chem., 2020, 18, 10–27 RSC.
- D. J. Gibbons, A. Farawar, P. Mazzella, S. Leroy-Lhez and R. M. Williams, Photochem. Photobiol. Sci., 2020, 19, 136–158 CrossRef CAS PubMed.
- Y. Hou, X. Zhang, K. Chen, D. Liu, Z. Wang, Q. Liu, J. Zhao and A. Barbon, J. Mater. Chem. C, 2019, 7, 12048–12074 RSC.
- W. Yang, J. Zhao, C. Sonn, D. Escudero, A. Karatay, H. G. Yaglioglu, B. Küçüköz, M. Hayvali, C. Li and D. Jacquemin, J. Phys. Chem. C, 2016, 120, 10162–10175 CrossRef CAS.
- Z. Yu, Y. Wu, Q. Peng, C. Sun, J. Chen, J. Yao and H. Fu, Chem. – Eur. J., 2016, 22, 4717–4722 CrossRef CAS PubMed.
- Y. Tomimori, T. Okujima, T. Yano, S. Mori, N. Ono, H. Yamada and H. Uno, Tetrahedron, 2011, 67, 3187–3193 CrossRef CAS.
- K. Chen, M. Taddei, L. Bussotti, P. Foggi, J. Zhao and M. Di Donato, ChemPhotoChem, 2020, 4, 487–501 CrossRef CAS.
- S. Sasaki, K. Hattori, K. Igawa and G.-i. Konishi, J. Phys. Chem. A, 2015, 119, 4898–4906 CrossRef CAS PubMed.
- Z. Wang and J. Zhao, Org. Lett., 2017, 19, 4492–4495 CrossRef CAS PubMed.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed.
- H. Lu, J. Mack, Y. Yang and Z. Shen, Chem. Soc. Rev., 2014, 43, 4778–4823 RSC.
- F. Tanaka and J. Osugi, Chem. Phys. Lett., 1974, 27, 133–137 CrossRef CAS.
- K. Chen, I. V. Kurganskii, X. Zhang, A. Elmali, J. Zhao, A. Karatay and M. V. Fedin, Chem. – Eur. J., 2021, 27, 7572–7587 CrossRef CAS PubMed.
- Y. Hou, T. Biskup, S. Rein, Z. Wang, L. Bussotti, N. Russo, P. Foggi, J. Zhao, M. Di Donato, G. Mazzone and S. Weber, J. Phys. Chem. C, 2018, 122, 27850–27865 CrossRef CAS.
- V. Bandi, H. B. Gobeze, V. Lakshmi, M. Ravikanth and F. D'Souza, J. Phys. Chem. C, 2015, 119, 8095–8102 CrossRef CAS.
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, S. Callaghan, K. J. Flanagan, M. Telitchko, T. Wiesner, F. Laquai and M. O. Senge, Phys. Chem. Chem. Phys., 2018, 20, 8016–8031 RSC.
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, S. Callaghan, K. J. Flanagan, T. Wiesner, F. Laquai and M. O. Senge, ChemPhotoChem, 2018, 2, 606–615 CrossRef CAS.
- M. Jurczok, P. Plaza, M. M. Martin, Y. H. Meyer and W. Rettig, Chem. Phys., 2000, 253, 339–349 CrossRef CAS.
- K. Xu, J. Zhao, D. Escudero, Z. Mahmood and D. Jacquemin, J. Phys. Chem. C, 2015, 119, 23801–23812 CrossRef CAS.
- K. Chen, M. Hussain, S. S. Razi, Y. Hou, E. A. Yildiz, J. Zhao, H. G. Yaglioglu and M. D. Donato, Inorg. Chem., 2020, 59, 14731–14745 CrossRef CAS PubMed.
- Z. Lou, Y. Hou, K. Chen, J. Zhao, S. Ji, F. Zhong, Y. Dede and B. Dick, J. Phys. Chem. C, 2018, 122, 185–193 CrossRef CAS.
- Y. Dong, A. A. Sukhanov, J. Zhao, A. Elmali, X. Li, B. Dick, A. Karatay and V. K. Voronkova, J. Phys. Chem. C, 2019, 123, 22793–22811 CrossRef CAS.
- L. Huang, J. Zhao, S. Guo, C. Zhang and J. Ma, J. Org. Chem., 2013, 78, 5627–5637 CrossRef CAS PubMed.
- L. Huang, X. Cui, B. Therrien and J. Zhao, Chem. – Eur. J., 2013, 19, 17472–17482 CrossRef CAS PubMed.
- Y. Hou, Q. Liu and J. Zhao, Chem. Commun., 2020, 56, 1721–1724 RSC.
- X. Zhang, M. Ivanov, Z. Wang, M. H. E. Bousquet, X. Liu, Y. Wan, J. Zhao, A. Barbon, D. Escudero, D. Jacquemin and M. Fedin, Angew. Chem., Int. Ed., 2022, 61, e202210419 CAS.
- Z. Wang, A. Toffoletti, Y. Hou, J. Zhao, A. Barbon and B. Dick, Chem. Sci., 2021, 12, 2829–2840 RSC.
- W. E. Ford and P. V. Kamat, J. Phys. Chem., 1987, 91, 6373–6380 CrossRef CAS.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolf, Handbook of PhotochemistryCRC Press, Boca Raton, 2006 Search PubMed.
- T. Zhang, X. Ma and H. Tian, Chem. Sci., 2020, 11, 482–487 RSC.
- Z. Wang, J. Zhao, M. Di Donato and G. Mazzone, Chem. Commun., 2019, 55, 1510–1513 RSC.
- L. Franco, A. Toffoletti, M. Ruzzi, L. Montanari, C. Carati, L. Bonoldi and R. Po’, J. Phys. Chem. C, 2013, 117, 1554–1560 CrossRef CAS.
- S. Väth, K. Tvingstedt, A. Baumann, M. C. Heiber, A. Sperlich, J. A. Love, T.-Q. Nguyen and V. Dyakonov, Adv. Energy Mater., 2017, 7, 1602016 CrossRef.
- Z. E. X. Dance, S. M. Mickley, T. M. Wilson, A. B. Ricks, A. M. Scott, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. A, 2008, 112, 4194–4201 CrossRef CAS PubMed.
- S. Weber, eMagRes, 2017, 6, 255–270 CAS.
- S. Richert, C. E. Tait and C. R. Timmel, J. Magn. Reson., 2017, 280, 103–116 CrossRef CAS PubMed.
- J. W. Verhoeven, J. Photochem. Photobiol., C, 2006, 7, 40–60 CrossRef CAS.
- A. Toffoletti, Z. Wang, J. Zhao, M. Tommasini and A. Barbon, Phys. Chem. Chem. Phys., 2018, 20, 20497–20503 RSC.
- M. Imran, X. Zhang, Z. Wang, X. Chen, J. Zhao, A. Barbon and V. K. Voronkova, Phys. Chem. Chem. Phys., 2021, 23, 15835–15868 RSC.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- B. de Souza, G. Farias, F. Neese and R. Izsák, J. Chem. Theory Comput., 2019, 15, 1896–1904 CrossRef CAS PubMed.
- A. Baiardi, J. Bloino and V. Barone, J. Chem. Theory Comput., 2013, 9, 4097–4115 CrossRef CAS PubMed.
- B. d. Souza, F. Neese and R. Izsák, J. Chem. Phys., 2018, 148, 034104 CrossRef PubMed.
- C. M. Marian, Annu. Rev. Phys. Chem., 2021, 72, 617–640 CrossRef CAS PubMed.
- E. R. Menzel, Chem. Phys. Lett., 1974, 26, 45–49 CrossRef CAS.
- A. Kirch, M. Gmelch and S. Reineke, J. Phys. Chem. Lett., 2019, 10, 310–315 CrossRef CAS PubMed.
- J. J. Snellenburg, S. Laptenok, R. Seger, K. M. Mullen, and I. H. M. Van Stokkum, Glotaran: A Java-based graphical user interface for the R package TIMP, 2012 Search PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, i. G. Scalman, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. J. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, M. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision A.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental methods, synthesis of compounds, molecular structure characterization, X-ray crystallographic data, computational details and additional spectra. CCDC 2210911. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00854a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |