
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A donor-supported silavinylidene and silylium ylides: boroles as a flexible platform for versatile Si(II) chemistry†
Julijan
Sarcevic‡
a,
Tobias
Heitkemper‡
 a,
Paul Niklas
Ruth
b,
Leonard
Naß
b,
Maximilian
Kubis
c,
Dietmar
Stalke
b and
Christian P.
Sindlinger
*a
a,
Paul Niklas
Ruth
b,
Leonard
Naß
b,
Maximilian
Kubis
c,
Dietmar
Stalke
b and
Christian P.
Sindlinger
*a
aInstitut für Anorganische Chemie, Universität Stuttgart, Pfaffenwaldring 55, 70169 Stuttgart, Germany. E-mail: sindlinger@iac.uni-stuttgart.de
bInstitut für Anorganische Chemie, Georg-August-Universität Göttingen, Tammannstr. 4, 37077 Göttingen, Germany
cInstitut für Anorganische Chemie, RWTH Aachen University, Landoltweg 1a, 52074 Aachen, Germany
First published on 31st March 2023
Abstract
Electron-deficient, anti-aromatic 2,5-disilyl boroles are shown to be a flexibly adaptive molecular platform with regards to SiMe3 mobility in their reaction with the nucleophilic donor-stabilised precursor dichloro silylene SiCl2(IDipp). Depending on the substitution pattern, selective formation of two fundamentally different products of rivalling formation pathways is achieved. Formal addition of the dichlorosilylene gives the 5,5-dichloro-5-sila-6-borabicyclo[2.1.1]hex-2-ene derivatives. Under kinetically controlled conditions, SiCl2(IDipp) induces 1,3-trimethylsilyl migration and adds exocyclically to the generated carbene fragment giving an NHC-supported silylium ylide. In some cases interconversion between these compound classes was triggered by temperature or NHC-addition. Reduction of silaborabicyclo[2.1.1]hex-2-ene derivatives under forcing conditions gave clean access to recently described nido-type cluster Si(II) half-sandwich complexes of boroles. Reduction of a NHC-supported silylium ylide gave an unprecedented NHC-supported silavinylidene which rearranges to the nido-type cluster at elevated temperatures.
Introduction
Free 1H-boroles (boracyclopentadienes) fulfill the Breslow-requirements for anti-aromaticity.1 The extent of anti-aromatic destabilization is distinct, yet much weaker compared to its iso-electronic and utmost reactive carbon analog, the cyclopentadienyl cation.2 Therefore, along with cyclobutadienes free boroles allow preparative synthetic chemistry and experimental exploration of, albeit weak, anti-aromaticity which is a very rare feature for small localized π-systems within the chemical space.3 The thermodynamic driving force of the exceptional electronic situation associated with a relieve from anti-aromatic destabilization has manifested in a series of examples for the past five decades since the first experimental descriptions of an authentic sample of pentaphenyl borole.4–8 As such, boroles are easily reduced to form respective non-aromatic (single electron transfer) or aromatic (two-electron transfer) species,6,9–13 feature remarkable Lewis-acidity,5,14–17 but foremost reveal numerous rearrangements and ring-expansion reactions that ultimately result in removal of the cyclic conjugate 4 π-electron system.18–32 Most recently, Erker and coworkers elucidated an intriguing reversible isomerism between 2,5-disilyl-1H-boroles and respective Wade–Mingos-type clusters of borapyramidanes, which may also account for an ultimately apparent scrambling of the substitution pattern.33,34Migration of substituents around the central borole moiety in the course of chemical reactions has been previously noted in a few cases.35–37 We would now wish to report on our findings on a system, where depending on subtle electronic and steric changes in the substitution patterns, a borole proves to be a flexible platform allowing dynamic trimethylsilyl-migrations initially induced by a Si(II) donor-addition to generate in principle two products including an unexpected NHC-supported silylium ylide. Almost coinciding with this study, the Scheschkewitz and Nakamoto groups reported the highly related reaction involving trimethylsilyl migrations of a cyclobutadiene with Roesky's PhC(NtBu)2SiCl38 to give a donor-stabilized silene which can be reduced to give access to silapyramidane.39
We recently reported a synthetic route to a series of 2,5-disilyl-3,4-diaryl boroles [(Ar2TMS2C4)B–R, A/B–R] (backbone system A: Ar = Xyl = 3,5-Me2(C6H3); B: Ar = Ph* = 3,5-tBu2(C6H3), Chart 1) and successfully applied boroles as a dianionic, [Cp]−-mimicking ligand for E(+II) coordination chemistry.17,40–43
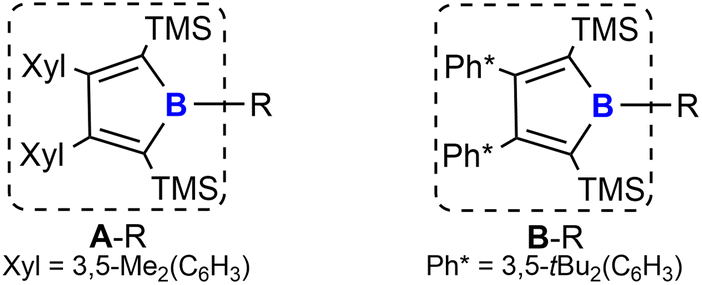 | ||
| Chart 1 Borole substitution patterns under investigation. | ||
In the following discussion of compounds, all product numbers will be accompanied by the borole identifier from which they derive. The Si(II) half-sandwich nido-type cluster compound 4B-Ph* has been reported to be formed by the reaction of the borolediide [B-Ph*]2− with two equivalents of Jutzi's [Cp*Si]+ as effective source of Si(II) in a salt metathesis reaction also affording silicocene Cp*2Si as a side product (Scheme 1).44–48 However, the necessity to provide [Cp*Si]+ with weakly coordinating anions rendered this approach rather costly and very atom inefficient and we were interested in studying alternative approaches. However, meaningful reagents that function as suitable sources of Si(II) for metathetic approaches are very scarce and mostly limited to said [Cp*Si]+, Cp*2Si,49 and Roesky's and Filippou's SiX2(IDipp) (X = Cl, Br, IDipp = 1,3-bis-2′,6′-iPr2(C6H3)-imidazole-2-ylidene).50–53
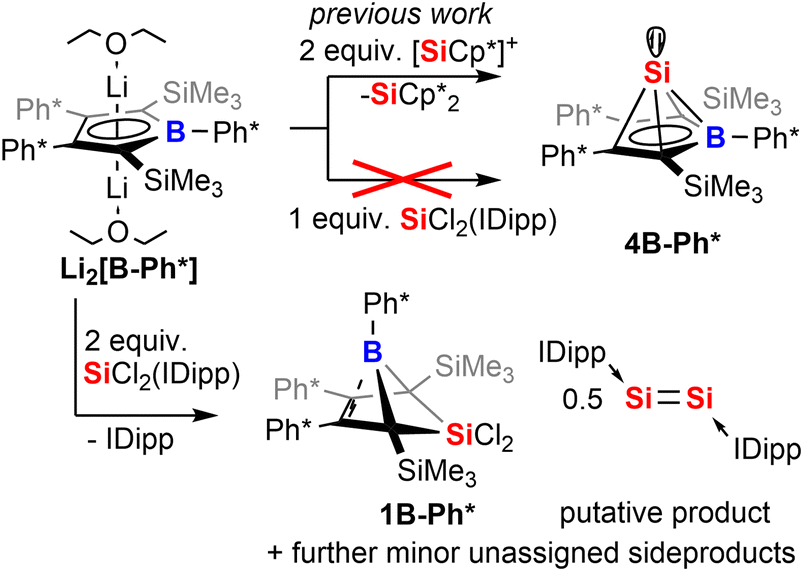 | ||
| Scheme 1 Reaction of borole dianion with SiCl2(IDipp). | ||
Results and discussion
Initial findings
When [B-Ph*]2− was respectively reacted with 1 equiv. of SiCl2(IDipp), the resulting mixture did not contain 4B-Ph* and revealed only ca. 50% conversion of [B-Ph*]2− along with full consumption of SiCl2(IDipp). Accordingly, with two equivalents SiCl2(IDipp), starting materials were fully converted to form a complex mixture of compounds of burgundy colour that reveal the NMR-spectroscopic fingerprints of free IDipp and 5,5-di-chloro-5-sila-6-borabicyclo[2.1.1]hex-2-ene 1B-Ph*, a formal oxidation (i.e. via chlorination) product of 4B-Ph* (Scheme 1).We reason that this product formation follows initial reduction of one equivalent of SiCl2(IDipp), putatively to Robinson's burgundy red Si0-species [Si2(IDipp)2]54 with the borolediide acting as a reducing agent (similar to the COT dianion (COT = 1,3,5,7-cyclooctatetraene)) rather than a salt metathesis precursor, regenerating the free borole B-Ph*.55,56 In a subsequent redox-process the free borole B-Ph* then reacts with SiCl2(IDipp) serving as a SiCl2 transfer reagent to form the 5-sila-6-borabicyclo [2.1.1]hex-2-ene 1B-Ph*. Formally, this formation can also be seen as a formal [4 + 1] cycloaddition reaction between the borole and dichloro silylene.
To corroborate this reasoning, we directly reacted the free borole B-Ph* with SiCl2(IDipp). NMR spectroscopic monitoring of the reaction mixture indeed revealed dominating (>95%) conversion to the anticipated 5,5-dichloro-5-sila-6-borabicyclo [2.1.1]hex-2-ene 1B-Ph* along with free IDipp and again a red-orange colour. This distinct colour could however not be routed back to [Si2(IDipp)2]. Upon crystallization from the crude mixture, we were able to identify this “coloured impurity” as a minor share of orange-red crystals aside to colourless specimen of 1B-Ph* (of which the structure is deposited and documented in the ESI†). X-ray diffractometric examination of these mediocrely diffracting orange-red crystals clearly revealed the unexpected connectivity pattern and molecular structure of an SiCl2(IDipp) adduct to a rearranged borole-scaffold which results from the migration of a SiMe3-group (2B-Ph*, Scheme 2). The determined molecular structure of 2B-Ph*(see ESI†) indicated a unique bonding situation. Unfortunately, further corroboration of the structure of 2B-Ph* by NMR remained impaired by low-concentrations in crude mixtures and broad, poorly defined signals sets resulting from steric congestion (vide infra).
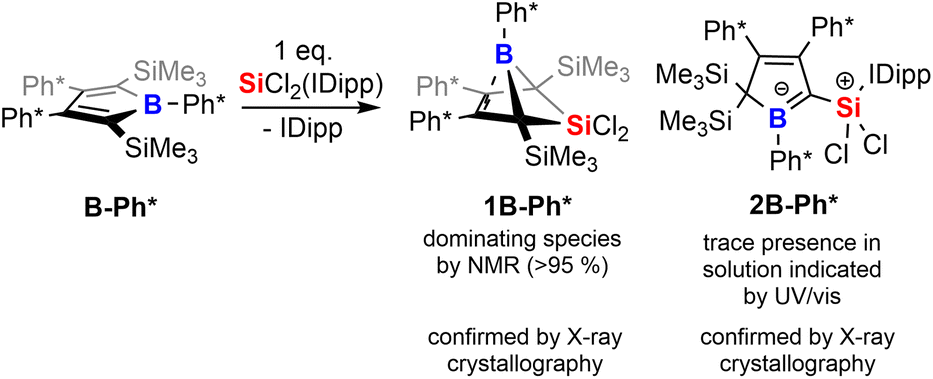 | ||
| Scheme 2 Reaction of free borole with SiCl2(IDipp). | ||
Substitution-dependent selectivity and mechanistic proposal
The intriguing electronic bonding situation of the NHC-supported silylium ylide 2B-Ph* prompted us to investigate synthetic strategies for the selective formation of such species. We therefore probed the outcome of the reaction of SiCl2(IDipp) with a series of differently substituted free 2,5-disilylboroles iteratively tuning both the B-bound residue R (R = Cl, Me, Xyl [3,5-Me2(C6H3)], p-Xyl [2,5-Me2(C6H3)], Ph*) and the 3,4-bound aryls (Xyl, vs. Ph*) and thus changing electronic and steric profiles of the respective free boroles. The results of this screening are tabulated in Scheme 3.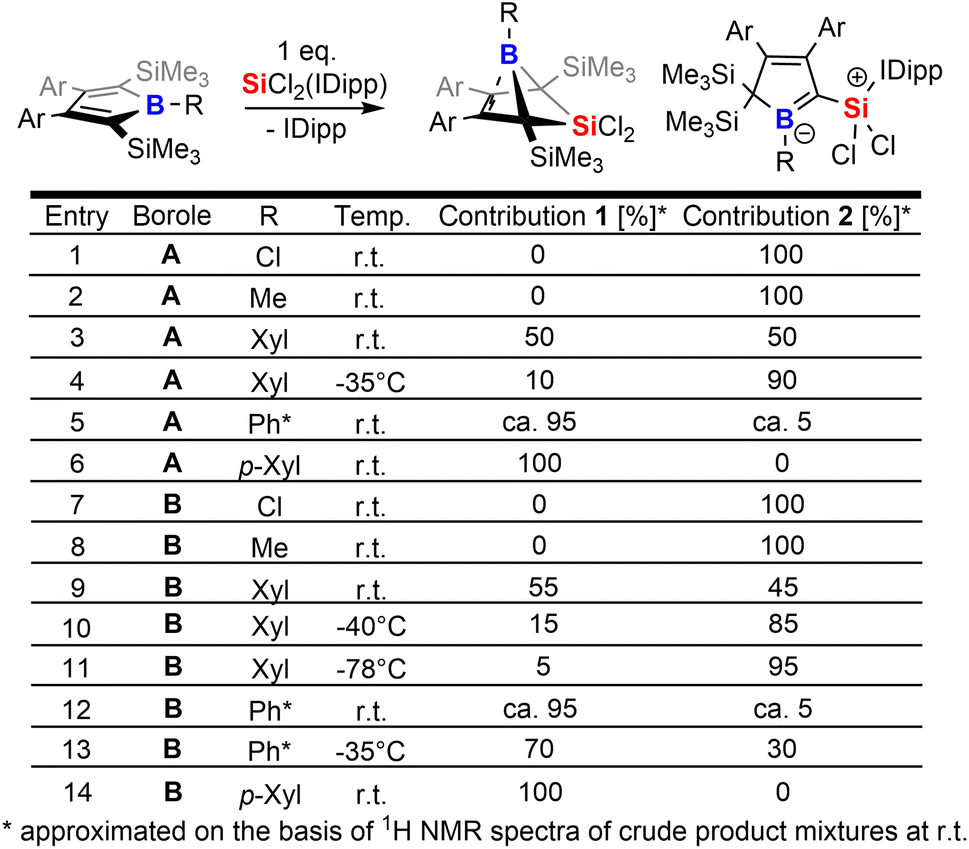 | ||
| Scheme 3 Tabulated summary for the product ratios between 1 and 2 at r.t. (20 °C). | ||
From this experimental insight a few observations can be summarized: (1) the steric profile of the borole backbone, i. e. system A or B, has no major influence on the selectivity; (2) for small substituents at the boron-atom (Cl, Me), the NHC-supported silylium ylide 2 are obtained selectively (entries 1, 2 and 7, 8); (3) only very bulky substituents such as Ph* result in clear favourisation of 1 (entries 5, 12) making our initially probed system a particularly unfortunate case; (4) boron-bound aryl-substituents with one ortho-methyl group (such as pXyl) partially blocking one hemisphere of the borole plane completely circumvent silyl migration and formation of the silylium ylide 2 in favor of 1 (entries 6, 14); (5) systems with a similar steric profile but non-π-plane interfering B-bound aryl group (such as m-Xyl vs. p-Xyl) are suitably balanced to give rise to both products 1 and 2 in almost equal shares at room temperature (20 °C, entries 3, 9); (6) conducting these unselective reactions at low temperatures notably increases the formation of NHC supported silylium ylide 2 in the obtained reaction mixtures indicating 2 to be a kinetically favored product (entries 9–11); (7) potential π-donation interactions from Cl atoms in chloroboroles do not influence the selectivity.
Taking these observations into account we propose the following rivalling reaction pathways for the formation of 1 and 2, summarized in Scheme 4.
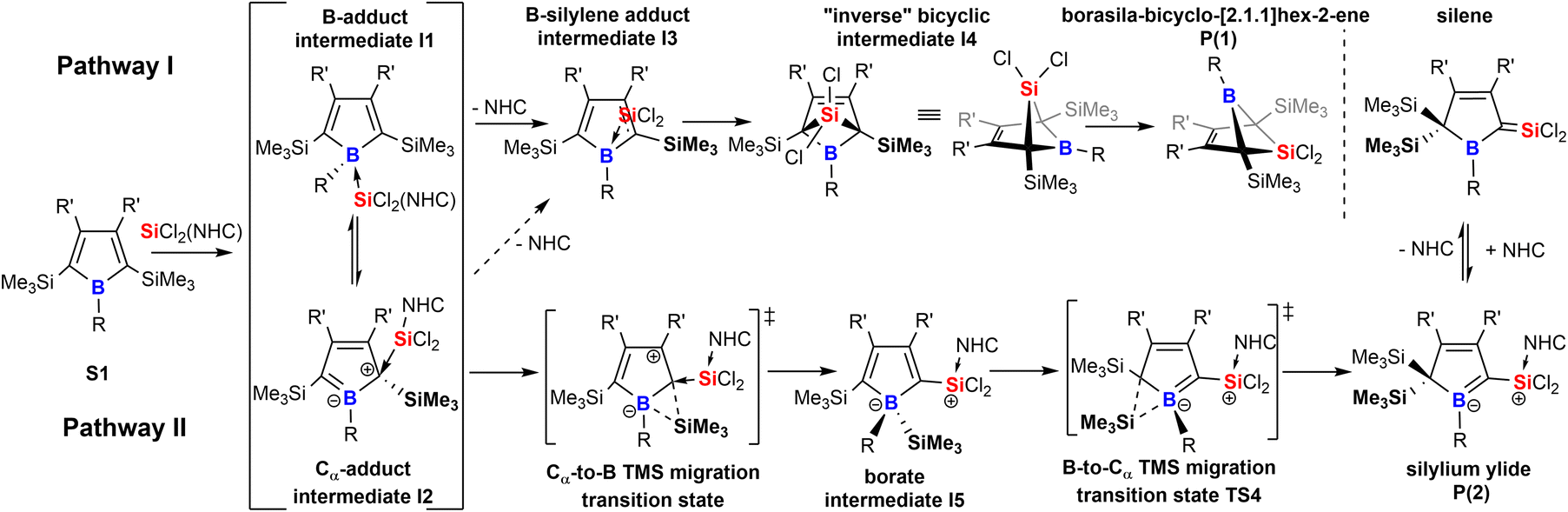 | ||
| Scheme 4 Proposed mechanistic steps for the formation of products 1 and 2. | ||
A more detailed account on the computational assessment of this mechanistic proposal including transition states for a sterically reduced model system (S1: R,R′ = Me; NHC: 1,3-Me2-imidazol-2-ylidene, Scheme 4) and a graphical depiction of relative energies of proposed species in the course of the reaction is documented in the ESI.† However, as sterics are most likely to play an important role, only relative free energies (BP86/def2-TZVPP and benzene solvation model) of intermediates with experimentally probed substitution patterns are given here. The anticipated initial step is an adduct formation of the Lewis-base SiCl2(IDipp) with free borole S1. This would intuitively add to the Lewis-acidic boron atom to give I1. So reported an example of a silylene-donor adduct to pentaphenyl borole.57 An alternative target for a nucleophilic attack at boroles is the Cα atom which reveals a vinylogous connection to the boron-site to result in a zwitter-ionic boratabutadiene-type adduct I2.36 For example, Braunschweig previously reported on a reversible B-/Cα-atom adduct formation of 2,6-lutidine to boroles.58,59 Formation of both I1 (ca. −17 kcal mol−1) and I2 (ca. −2 kcal mol−1) would be exergonic. However, these putative intermediates I1 and I2 have not been experimentally observed in the course of the reaction, suggesting that subsequent processes must have low barriers. We propose the decisive forking of the pathway to occur at this stage.
Pathway I (Scheme 4) assumes an energetically uphill dissociation of the NHC. Computational NHC removal from both adducts I1 and I2 affords the identical structure I3 after optimization. I3 can be described as an adduct of an ambiphilic dichlorosilylene to the equally ambiphilic borole. The dissociation free energy at 298 K of the NHC from I1 is high for A-Xyl (18 kcal mol−1) and almost prohibitively high for A/B-Me (ca. 24 kcal mol−1). For our model system low barriers (ca. 1 kcal mol−1) for the formation of the bicyclic I4 from I3 and conversion/isomerization of I4 to the bicyclic product P1 are suggested. The overall free energies of the formation of P1 are exergonic by ca. −10 kcal mol−1.
We propose the pathway II to originate from the Cα-adduct I2 from which a 1,2-SiMe3 migration from Cα to the B-atom occurs forming the silyl borate intermediate I5. Experimentally, no direct evidence for the involvement of a species of the I5-type were observed. However, the complete suppression of trimethylsilyl migration in systems with ortho-methyl substituted aryls at boron (A/B-pXyl) is considered as a strong indirect proof for a migration via the boron atom. Subsequent 1,2-SiMe3-migration provides silylium ylide product class P2 and again small barriers (ca. 1 kcal mol−1) are suggested for the reduced model system for this step.
An example for comparable sigmatropic 1,2- and 1,3-shifts of hydrogen in borole-NHC adducts have been described by Braunschweig and coworkers.35 For all substitution variants probed, despite considerable steric bulk and thus repulsive interactions involved, the NHC-supported silylium ylides P2 were the global energy minimum (ca. −30 kcal mol−1vs.S1) of all species investigated and much more favoured than the bicyclic species P1 (−10 kcal mol−1vs.S1). For further details, see the ESI.† A key experimental finding is that mixing the starting materials in the cold favors the formation of the NHC-supported silylium ylides, suggesting that a decisive energy barrier at an early stage after bifurcation must be lower on the path towards P2. It seems reasonable that the NHC dissociation to form the I3 is the key barrier process.
The seemingly ideal test system, A-Xyl allowed us to develop protocols toward separation of either product from the respective mixtures. However, achieving rigorous separation to yield pure isolated products 1/2 from mixtures often remained a tedious task affecting the yields. The NHC supported silylium ylides 2 reveal considerable dipolar moment and are less soluble in hexane. Careful extraction of crude product mixtures with hexane thus resulted in removal of the more soluble 1. Removal of remaining free IDipp can be achieved by addition of ZnCl2 to solutions of 1 in hexane and precipitation of IDipp(ZnCl2). This protocol can also be applied for further purification and isolation of the other product.
However, we note the clean and irreversible thermal conversion of the supposedly thermodynamically favoured NHC-supported silylium ylides 2A-Xyl or 2B-Xyl into the respective bicyclic species 1A-Xyl or 1B-Xyl under NHC elimination at ca. 50–80 °C. However, when we treated the readily isolable bicyclic species 1A-pXyl and 1B-pXyl with the much smaller 2,3,4,5-tetramethylimidazol-2-ylidene (MeNHC), immediate colorization to yellow and essentially quantitative formation of the MeNHC-supported silylium ylides 3(A,B)-pXyl are formed (Scheme 5).
 | ||
| Scheme 5 NHC-induced silyl-migration of 1. | ||
We assume that this product formation is enabled by the much less bulky NHC that can attack the SiCl2 moiety of the bicyclic compound 1 to proceed to a I2-type Cα-adduct intermediate via cleavage of a Si–Cbridgehead bond. The X-ray structure of 1A-pXyl reveals the ortho-methyl group of the boron-bound pXyl-group to point toward the SiCl2 bridge (Fig. 1). We assume that a SiMe3 migration from this so generated I2-type intermediate is now possible, because the SiCl2(NHC) donor and the respective ortho-methyl group of the boron-bound aryl are located on the same half sphere of the borole plane allowing the SiMe3 group to migrate on the opposite, unblocked side. In the reactions of free pXyl-bearing boroles with the SiCl2(NHC) nucleophile, the SiCl2(NHC) adduct would, for steric reasons, always attack from the unprotected side resulting in an anti-periplanar type arrangement of the ortho-methyl group and the SiCl2(NHC) fragment.
 | ||
| Fig. 1 ORTEP of the solid state molecular structure of 5-sila-6-borabicyclo[2.1.1]hex-2-ene 1A-pXyl. A second molecule in the asymmetric unit and H-atoms are omitted for the sake of clarity. Anisotropic displacement parameters are drawn at 50% probability. Selected interatomic distances [Å] and angles [°]: B1–(C2/C3)centr 1.673; (C2/C3)centr–(C1/C4)centr–B1 82.7°, (C2/C3)centr–(C1/C4)centr–Si1 130.8°. Further details are listed in Table 1. | ||
Structural and spectroscopic properties of 1 and 2
Almost all derivatives based on the 3,5-tBu2(C6H3)-substituted borole system B revealed very poor crystallinity and severe disorder and thus only allowed for collection of poor or mediocre data from X-ray diffraction. Derivatives of borole A however crystallized reliably. Structural features of substitution derivatives of 1 and 2, respectively, did not differ much within the series of compounds. Exemplarily representing their substitution derivatives, the solid state molecular structures of 1A-pXyl (Fig. 1) and 2A-Me (Fig. 2) are depicted.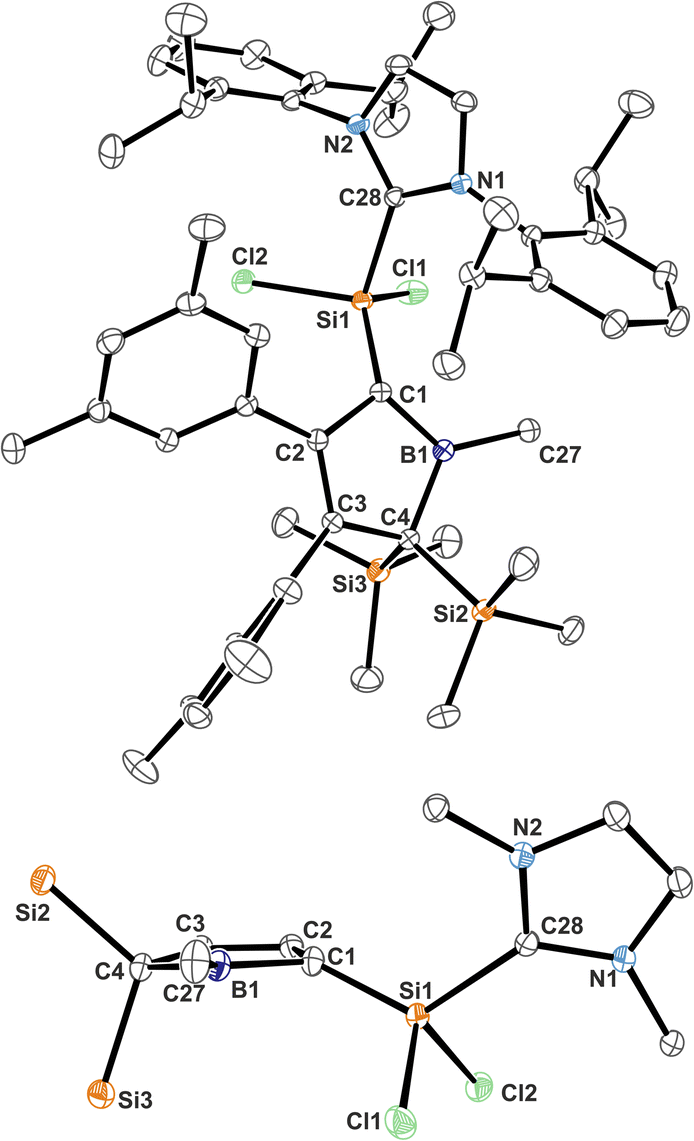 | ||
| Fig. 2 ORTEP of the solid state molecular structure of NHC-supported silylium ylide 2A-Me. H-atoms are omitted for the sake of clarity. Anisotropic displacement parameters are drawn at 50% probability. Selected interatomic distances are listed in Table 1. | ||
Compared to the starting material of a free borole with a localised 1,3-butadiene backbone of short C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds (ca. 1.36 Å) and a long C–C bond (1.54 Å), the 5-sila-6-borabicyclo[2.1.1]hex-2-ene derivatives 1 all feature a short C2–C3 distance (ca. 1.4 Å) and elongated C1–C2/C3–C4 contacts (ca. 1.50 Å) indicative of a 2-butene moiety. This is in line with a description of 1 as the product of a formal [4 + 1] cycloaddition of the butadiene moiety of the borole with a [SiCl2] fragment.
C bonds (ca. 1.36 Å) and a long C–C bond (1.54 Å), the 5-sila-6-borabicyclo[2.1.1]hex-2-ene derivatives 1 all feature a short C2–C3 distance (ca. 1.4 Å) and elongated C1–C2/C3–C4 contacts (ca. 1.50 Å) indicative of a 2-butene moiety. This is in line with a description of 1 as the product of a formal [4 + 1] cycloaddition of the butadiene moiety of the borole with a [SiCl2] fragment.
The borane moiety stands almost perpendicular on the C4-plane [(C2/C3)centr–(C1/C4)centr–B1 82.7°] indicating clear interaction of the Lewis-acidic borane with the π-bond of the 2-butene moiety, corroborated by short C2/3–B1 contacts (B1–(C2/C3)centr 1.673 Å). Such an arrangement has been observed previously in related products of pericyclic Diels–Alder reactions of free boroles with alkynes and alkenes and a cationic bicyclic hydrosilylium cation.18,41,61 Jutzi described a related η4-(Cp*R′)BR′ (R′ = Cp*(Br)2Si–) as an arachno-tetracarbaborane cluster.62
NHC-supported silylium ylide derivatives 2 reveal a rather unusual structural motif. Structural features are summarized in Table 1. The silyl group at C1 migrated to C4 and was replaced by the SiCl2(IDipp) moiety – an electronic equivalent of a formally neutral silyl group. This rearrangement goes along with a shortening of the C2–C3 contact to 1.371(1) Å indicative of a double-bond. The short C1–B1 contact (1.513(1) Å) lies between typical boron–carbon bonds (ca. 1.55–1.60 Å) and authentic boron–carbon double bonds (ca. 1.44–1.46 Å).63,64 Most notably, the Si1–C1 contact is rather short (1.7590(9) Å). Si–C single bonds usually amount to 1.85 Å while selected examples for SiC have been reported at 1.702(5) Å (in silenes)65 or 1.774(2) Å (in NHC@Si-adducts to R2CSiSiCR2 heterocumulenes).66 The Si1–C1 bond vector is significantly deviating by 21° from the (B1–C1–C2)-plane in line with a deviation from (trigonal) planarity at C1 (sum of basal angles ca. 354°). A related motif of a slightly trans-bent geometry at the carbon atom was described by Roesky and coworkers for a cyclic amidinate-stabilised silene.67 This hints at a rather unusual electronic situation at carbon-atom C1 as, to the best of our knowledge, known NHC supported silylium ylides (or silenes) only reveal planar C-atoms.
| Entry | B1–C1, C1–C2, C2–C3, C3–C4, C4–B1a | C1/(C4)–Si1a | Si1–Cl1/2a | Si1–CNHCa |
|
λ max exp/comp | 29Sic(Si1) | 11B |
|---|---|---|---|---|---|---|---|---|
| a In Å, following the numbering scheme shown in Fig. 1 and 2. b In nm (toluene solutions) – TDDFT calculations using RI-CAM-B3LYP/def2SVP/J. c In ppm. d Crystals diffracted poorly and structure could not be determined to allow extensive discussion. e The species is only present in solution in very small quantities with broad ill-defined spectra potentially due to dynamic equilibria which do not allow unambiguous identification by NMR spectroscopy. f Two molecules in the asymmetric unit. g Shoulder. | ||||||||
| 1A-Xyl | No X-ray structure | 1.8 | −26.7 | |||||
| 1A-pXyl | 1.693(2), 1.494(3), 1.398(2), 1.502(3), 1.674(2) | 1.830(2), 1.837(2) | 2.0633(5), 2.050(1) | 3.0 | −22.0 | |||
| 1B-Xyl | No X-ray structure | 1.0 | −26.7 | |||||
| 1B-Ph* | 1.694(4), 1.496(3), 1.411(3), 1.493(3), 1.702(3) | 1.842(2), 1.836(2) | 2.072(1), 2.0485(8) | 1.5 | −25.3 | |||
| 1B-pXyl | No X-ray structure | 3.0 | −22.4 | |||||
| 2A-Me | 1.513(1), 1.477(1), 1.371(1), 1.530(1), 1.610(1) | 1.7590(9) | 2.0772(6), 2.0691(5) | 1.947(1) | 21° | 548/535 | −26.2 | 61.8 |
| 2A-Cl | 1.491(2), 1.483(2), 1.369(2), 1.536(2), 1.591(2) | 1.765(1) | 2.0673(6), 2.0667(5) | 1.939(1) | 21.6° | 500/498 | −24.8 | 53.8 |
| 2A-Xyl | 1.513(3), 1.476(3), 1.368(3), 1.525(3), 1.619(4) | 1.773(2) | 2.069(1), 2.059(1) | 1.957(3) | 13.8° | 514/497 | −23.4 | 54.7 |
| 2B-Me | 1.50(1), 1.480(8), 1.37(1), 1.536(9), 1.618(9)d | 1.753(8)d | 2.070(2), 2.074(3)d | 1.931(7)d | 28.2°d | 560/511 | −28.0 | 62.9 |
| 2B-Cl | 1.49(2), 1.48(1), 1.37(1), 1.54(1), 1.63(1) d | 1.77(1)d | 2.072(3)d 2.073(5)d | 1.926(9)d | 32.2°/16.4°df | 500/496 | −24.1 | 52.4 |
| 2B-Xyl | No X-ray structure | 487 | −25.2 | 57.4 | ||||
| 2B-Ph* | 1.515(4), 1.481(4), 1.365(4), 1.531(4), 1.616(4) | 1.776(3) | 2.055(1), 2.072(1) | 1.963(3) | 9.2° | 493/444 | —e | —e |
| 3A-pXyl | No X-ray structure | 410g | −19.9 | 54.8 | ||||
| 3B-pXyl | 1.499(3), 1.466(3), 1.355(3), 1.538(3), 1.633(3) | 1.765(2) | 2.0772(8), 2.0851(9) | 1.908(2) | 0.2° | 415g/398 | −20.0 | 54.7 |
| 5B-Me | 1.537(3), 1.475(4), 1.360(3), 1.539(3), 1.603(4) | 1.784(2) | 1.975(3) | 0° | 496/456 | 226.3 | 61.3 | |

The molecular structure of 3B-pXyl is shown in Fig. 3 and details are listed in Table 1. Key difference to IDipp adducts 2 is the lack of distortion of the [SiCl2(NHC)]-fragment from the borole plane and a planar C1-atom suggesting that the IDipp sterics impose this pyramidalisation of C1 in derivatives of 2. It is to note that the orientation of the Si–CNHC vector towards the borole-plane differs. In 2 the NHC stands syn-periplanar to a lone pair at C1 while in 3B-pXyl a chlorine atom adopts this position.
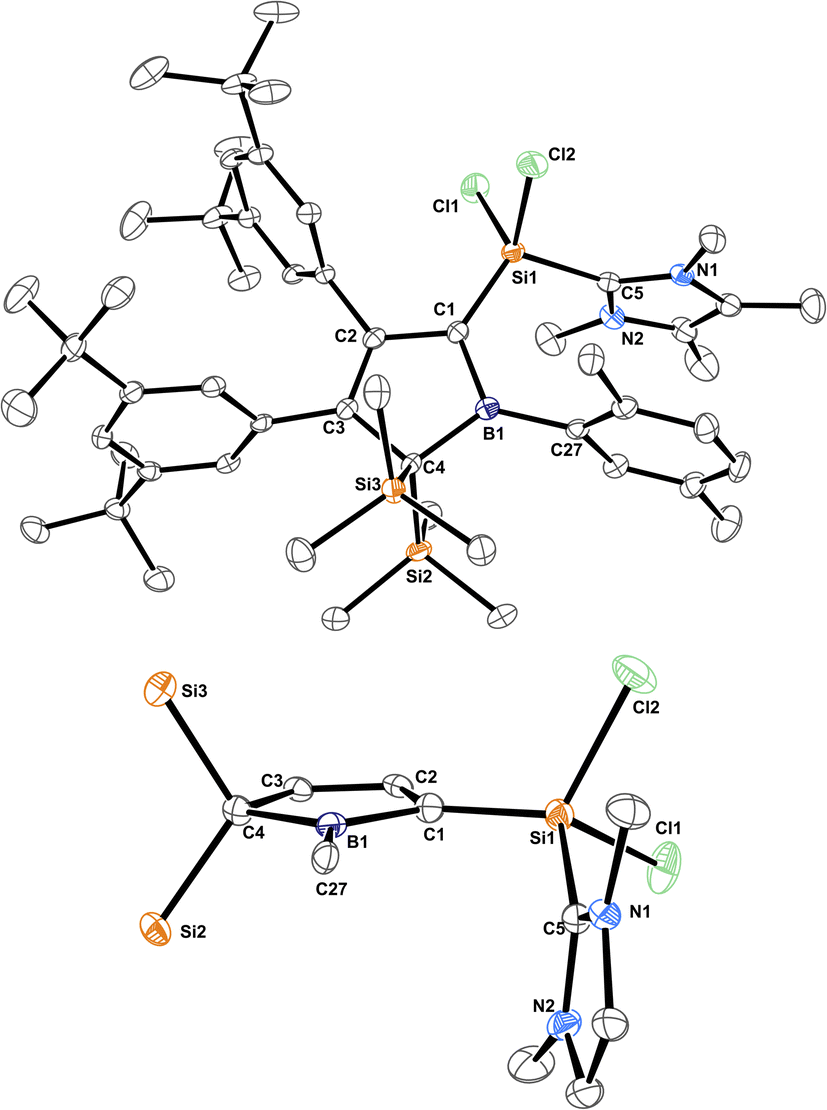 | ||
| Fig. 3 ORTEP of the solid state molecular structure of NHC-supported silylium ylide 3B-pXyl. H-atoms are omitted for the sake of clarity. Anisotropic displacement parameters are drawn at 50% probability. Interatomic distances are listed in Table 1. | ||
Trends for NMR spectroscopic observables will be discussed as summary for the two compound classes 1 and 2. Individual details for each compound are listed in Table 1. The most notable NMR spectroscopic property of compound class 1 is the considerably highfield shifted, narrow 11B NMR signal (FWHM between ca. 350–450 Hz), which is observed at δ(11B) around −22 ppm. 11B NMR signals of 2 are much broader (FWHM ca. 1400–1600 Hz). This is a characteristic region (ca. −5–−50 ppm) for boron atoms intramolecularly stabilised by π-donor interactions.36,61,62,68–72 The 29Si-resonance of the bridging SiCl2 lies in the regular region around 0 ppm. The bridgehead 13C signals are found around 54 ppm and the π-bonded carbon atoms in the backbone of the [2.1.1]-cyclohexene framework are found around 127 ppm.
A very notable feature of the 1H-NMR spectra of the B–Cl/Me derivatives of compound class 2 is the time-averaged presence of a mirror-plane symmetry (e.g. only one resonance for both SiMe3-groups) through the borole-plane, despite the solid-state structure suggests otherwise (Fig. 2). This could be rationalised by either a putative free rotation around the C1–Si1 bond or a dynamic NHC-coordination and dissociation fast enough within the timescale of the NMR experiment. The more bulky B–Ar derivatives (Ar = Xyl, Ph*) of compound class 2 show significantly broadened and seemingly ill-defined signal patterns particularly for the most bulky aryl group Ph*, which indicates presence of reduced symmetry due to a putative sterics-impaired rotation around the C1–Si1 bond. Heating these samples to ca. 60–75 °C causes sharpening of these signals.
Compounds 2 reveal a low-field shifted 11B-NMR signal around δ(11B) = 60 ppm. The B-chloro derivatives reveal resonances shifted to lower frequencies (ca. 53 ppm) in line with an increased occupation of the empty p-orbital at the boron atom due to π-donation from the chlorine atom or increased CB π-interaction resulting from higher Lewis-acidity caused by the electron withdrawing chlorine atom. The observed 11B resonances lie in between the shift observed for the respective free boroles (δ(11B) = ca. 80 ppm, tricoordinate B-atom) and a more highfield shifted boratafulvene derivative that features authentic BC double bonds (δ(11B) = ca. 40 ppm). This indicates the B-atom in 2 to be involved in π-bonding to C1 with a partially populated p-orbital, however not to the extent of an authentic double bond.
Spectroscopic characteristics of 3 except for the perceived colour and the respective absorption in the UV-vis spectra barely deviate.
We reason that the mild pyramidalization at C1 stems in part from the localisation of a lone pair of electrons, which is only partially delocalised into the accepting p-orbital at the boron atom to form a B1 = C1 double bond. This is corroborated by the respective notably shortened B–C contact. The lone pair of electrons at C1 is attracted by both π-accepting moieties: the borane as well as the adjacent vinyl moiety. (Allylic) delocalisation into the vinyl moiety explains the rather short C1–C2 contact. Feasible deviation from planarity to account for steric pressure of the C–Si vector is only reasonable if no SiC double-bond character of the ylide vs. ylene is assumed, which is in line with the coordination number at Si. A similar mildly pyramidalised carbon atom with a lone pair of electrons delocalised between two accepting boron-atoms was recently described by Erker.73
The experimental properties advocate for a description of 2 as NHC-supported silylium ylide in which the ylide is further internally stabilised by adjacent π-accepting moieties. The cationic charge is mostly localised on the imidazolium moiety. Suitable mesomeric descriptions (I.–III.) for the Lewis-structure of 2 are summarised in Scheme 6. The short Si–C contact is thus a consequence of coulombic attraction rather than π-bonding.
 | ||
| Scheme 6 Mesomeric descriptions of NHC-supported silylium ylides. | ||
A to some extent comparable electronic situation has been previously described by Berndt and coworkers for their true (i.e. unsupported) germene/stannene derivatives of cyclic diboryl-substituted methylene fragments or respective phosphine adducts.74–78
A somewhat related 1,3-silyl migration was described by Erker and coworkers who noted the formation of an unusual ketene derivative from reaction of 2,5-disilylboroles with carbon monoxide.36 However, compared to the essentially purely σ-donating SiCl2(IDipp), CO is a potent π-acceptor readily forming an electronically trivial, authentic CC double-bond.
Electronic structure of silylium ylides 2
To further elucidate the electronic structure of the silylium-ylides 2, these structures were probed computationally using DFT.79,80 Structure optimisation with RI-BP86-D3(BJ)/def2-TZVP model chemistry reproduced the experimentally found features very well, including the deviation from planarity at C1.81–85The results for 2A-Me are presented and discussed exemplarily for all experimentally accessed derivatives as deviations are only marginal. NBO analyses86,87 on 2A-Me corroborated electronic structure II. (Scheme 6) with a cyclic borata-butadiene moiety as the leading Lewis-structure. However, the CB double bond is strongly polarised towards the C-atom, both in its σ- (72%) and even more in its π-contribution (82%), the latter NBO being only occupied by 1.6 electrons. Second order perturbation theory finds no significant delocalisation toward the Si-atom (a potential silene resonance structure) but a strong (28 kcal mol−1) delocalisation of the CB π-electron density into the π*-orbital of the CC doublebond in the boratabutadiene moiety. This is in line with an allyl-type delocalisation of a C1-centered lone pair of electrons (as in resonance structure III., Scheme 6) in line with structural parameters. The respective intrinsic bonding orbital (IBO)88 essentially represents a lone pair of electrons in a C1-centered p-orbital mildly polarised into the three adjacent atoms (Fig. 4).
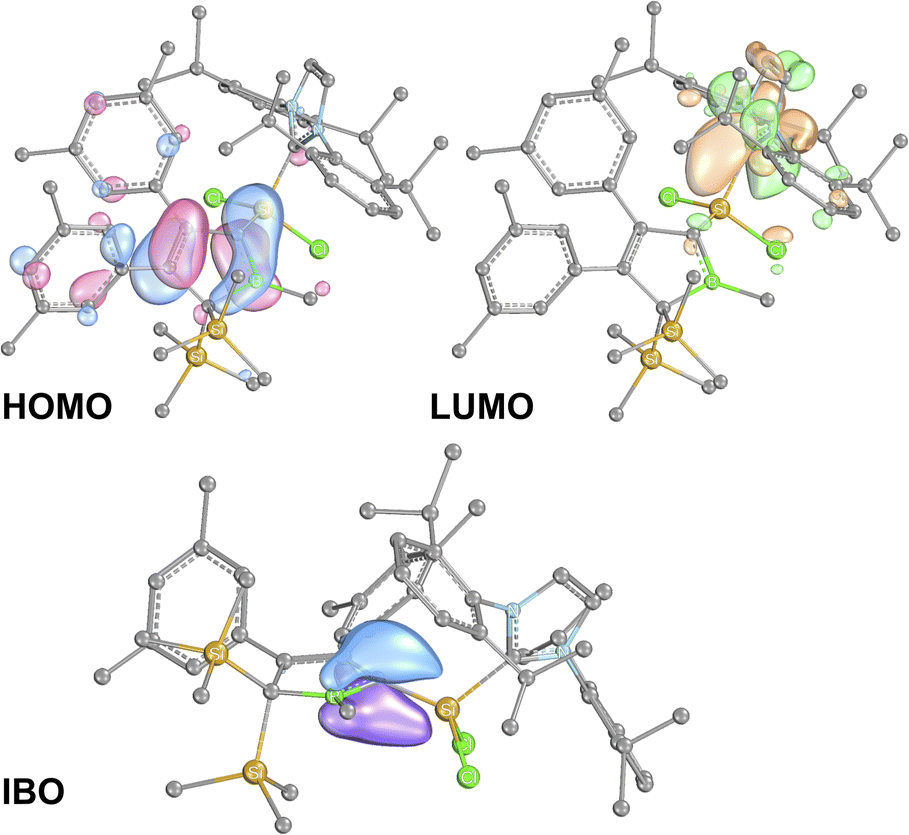 | ||
| Fig. 4 Frontier molecular orbitals of 2A-Me. (BP86/def2-TZVP). Iso-surfaces at 60% probability. | ||
The canonical frontier molecular orbitals reveal the boratabutadiene π-system to be the HOMO with the LUMO located on the imidazolium NCN π*-orbital with a fairly small energy gap (0.86 eV for 2A-Me, Fig. 4). The intense red to purple colour observed for these NHC-supported silylium ylides 2 goes back to respective π/π* HOMO to LUMO transitions as corroborated by TD-DFT calculations that reproduce these electronic spectra in the Vis range. An alternative description of compounds 2 other than NHC-supported silylium ylides would be an internally π-accepting boryl-substituted carbene fragment that is stabilized by a nucleophilic silylene donor.
Contrary to conventional widely applied nucleophilic carbenes such as NHC, this putative intermediate carbene fragment would be electrophilic an thus readily stabilised by the nucleophilic [:SiCl2(IDipp)] donor. Similar descriptions were previously suggested by Berndt.78In situ generation of a σ-donating cAAC by 1,2-H migration in a cyclic alkene induced by a borane was observed previously by Kinjo and coworkers for a 2,3-dihydro-1H-1,2-azaborole derivative to finally form an adduct of the carbene to the borane.89
Reduction of compounds 1 and 2
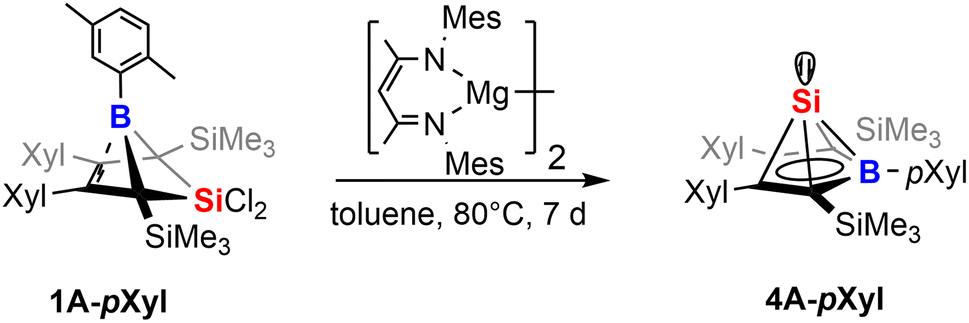
Our initial investigations aimed at more efficient approaches toward the synthesis of neutral Si(II) half-sandwich compounds (such as 4B-Ph*, Scheme 1) circumventing the necessity of two equivalents of [Cp*Si]+ reagents.41 We therefore attempted the reduction of the now straightforwardly available dichlorosilylene addition derivatives 1 and 2.While our attempts to achieve successful reductions of 1 with common reducing agents (Li(naph), 5% Na on NaCl, KC8) revealed only unsatisfyingly poor selectivity, a rather clean (as monitored by NMR spectroscopy) reliable conversion was achieved by 1 equiv. of Jones' and Stasch's [MesNacnac(MgI)]2 reagent.90,91 Starting from 1B-Ph* we were able to selectively form the previously described Si(II) borole compound 4B-Ph*. The reverse reaction, an oxidative conversion of the half-sandwich to a bicyclic 5-sila-6-borabicyclo[2.1.1]hex-2-ene was previously observed by formal protonation of the Si-cluster 4B-Ph*.41
Accordingly, we converted 1A-pXyl into the respective new Si(II) cluster 4A-pXyl by reduction with 1 equiv. of [MesNacnac(MgI)]2 reagent (Scheme 7). NMR monitoring suggested clean conversions, however due to the high solubility of the resulting clusters, poor isolated yields of crystallized compounds 4 (ca. 20%) were obtained. The Si(II) cluster 4A-pXyl was obtained as a colorless compound revealing a mixture of two conformers (with regards to rotation around the B-pXyl bond) in a ratio of ca. 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85 as observed by NMR spectroscopy. The X-ray structure of 4A-pXyl is shown in Fig. 5 and only reveals the trans-type conformer where the apical Si-atom and the ortho-methyl group occupy different half-spheres of the borole plane. The key bond lengths of the nido-clusters [C4B]Si moiety are, within the error, identical to those previously described for 4B-Ph* and feature the essential bond length homologisation that is to be expected when a free borole (with localized single- and double bonds) is reduced to a borole dianion, isoelectronic to Cp−.
:85 as observed by NMR spectroscopy. The X-ray structure of 4A-pXyl is shown in Fig. 5 and only reveals the trans-type conformer where the apical Si-atom and the ortho-methyl group occupy different half-spheres of the borole plane. The key bond lengths of the nido-clusters [C4B]Si moiety are, within the error, identical to those previously described for 4B-Ph* and feature the essential bond length homologisation that is to be expected when a free borole (with localized single- and double bonds) is reduced to a borole dianion, isoelectronic to Cp−.
 | ||
| Scheme 7 Reduction of 1 to give Si(II) half-sandwich compounds. | ||
 | ||
| Fig. 5 ORTEP of the solid state molecular structure of Si(II) borole half-sandwich compound 4A-pXyl. H-atoms are omitted for the sake of clarity. Anisotropic displacement parameters are drawn at 50% probability. Selected interatomic distances [Å]: B1–C1 1.549(3), C1–C2 1.462(3), C2–C3 1.429(3), C3–C4 1.460(3), C4–B1 1.547(3), B1–Si1 2.190(2), C1–Si1 2.100(2), C2–Si1 2.094(2), C3–Si1 2.109(2), C4–Si1 2.102(2). (BC4-borole)centr–Si1 1.700. | ||
The 11B resonance is found at δ(11B) = 30.6 ppm (31.5 ppm, in 4B-Ph*) and the characteristically high field shifted 29Si resonances are found at δ(29Si) = −354.8 ppm (minor conformer) and −355.7 ppm (major conformer). The latter also clearly reveals the lowfield-shifted shoulder at −355.6 ppm assigned to the 10B–29Si isotopologue.
Strikingly, when we attempted the reduction of derivatives of 2 we found rather unselective formations of intractable mixtures however, for 2B-Me a rather clean reduction was observed with lithium naphthalenide (Scheme 8).
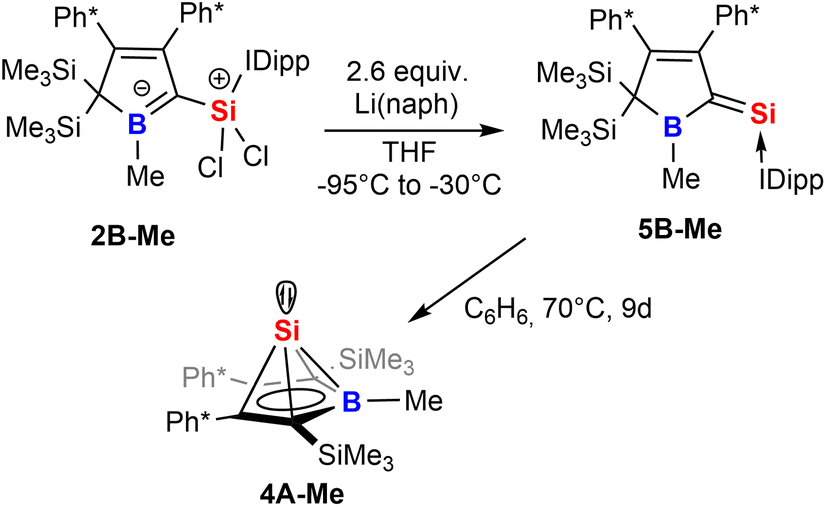 | ||
| Scheme 8 Reduction of 2 to give an NHC-supported silavinylidene 5 that can be thermally converted to a stable Si(II) half-sandwich compound. | ||
Carefully allowing to warm the coloured reaction mixture from −95 °C to ca. −30 °C led to an intensely blood red colourisation and we were able to isolate the unprecedented NHC-supported silavinylidene 5B-Me featuring an authentic CSi: double bond. A number of donor-stabilised heavier homologs of vinylidenes have been reported in the last decade (R2Ge = Ge*,92,93 R2Si = Ge*,94,95 R2Ge = Si*,96 R2Si = Si*;97,98 asterisks denote further donor-stabilisation of the atom). To the best of our knowledge, silavinylidene 5B-Me is the first example for the lightest heavier congener of (donor-stabilised) vinylidenes. Filippou recently reported a NHC- and isonitrile donor-stabilised Si0 atom which reveals the structural motif of a NHC-supported :Si=CNR fragment.99 The molecular structure of 5B-Me is shown in Fig. 6.
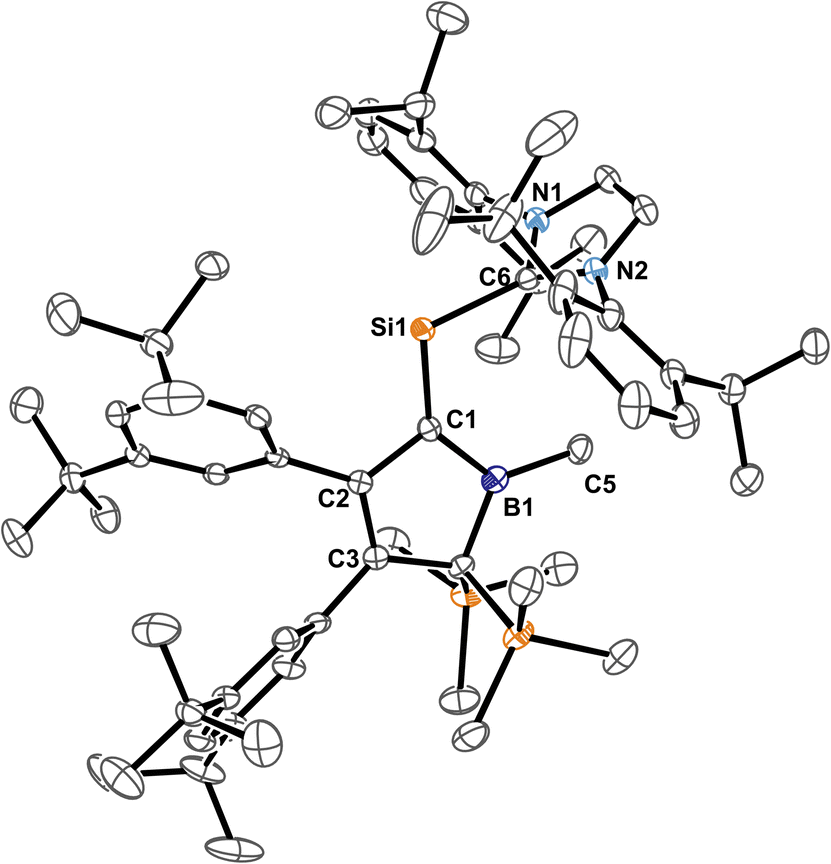 | ||
| Fig. 6 ORTEP of the solid state molecular structure of NHC-supported silavinylidene 5B-Me. H-atoms and partially occupied ether molecule are omitted for the sake of clarity. Anisotropic displacement parameters are drawn at 50% probability. Details on interatomic distances are summarized in Table 1. | ||
Direct comparison of bond length in 5B-Me with the set of structural features of the silylium ylides reveals only minor changes with a slight elongation of the C1–B1, C1–Si1 and Si1–CNHC distances being most notable. The elongation of the Si1 distances may be a result of the larger covalent radius of the reduced Si-atom. The Si1C1 distance found is identical to what Filippou reported.99 The C1–B1 elongation may hint at a reduced π-interaction of the formerly ylidic lone-pair with the boron atom as π-bonding to the Si1 atom can now be established. The 11B-NMR signal of 5B-Me is found at δ(11B) = 61.3 ppm, barely shifted from 2B-Me. The structural data are in line with a Lewis-structure as depicted in Scheme 8 which is also corroborated by NBO analysis. The comparably short C1–C2 and C1–B1 can be rationalised with some delocalisation of the C1Si1 π-density into the adjacent accepting vinyl and boryl moieties. This is corroborated by second order perturbation theory in NBO that finds donation from the C1Si1 NBO (populated by 1.58e) into the boron p-orbital (11 kcal mol−1) and the C3C4 π*-NBO (29 kcal mol−1). The lone-pair at Si is predicted to be high in s-orbital character (72%). The canonical HOMO-1 represents the Si-based lone-pair, while the HOMO is located on the silabutadiene moiety. The LUMO is a π-type orbital on the Si–NHC-vector (Fig. 7).
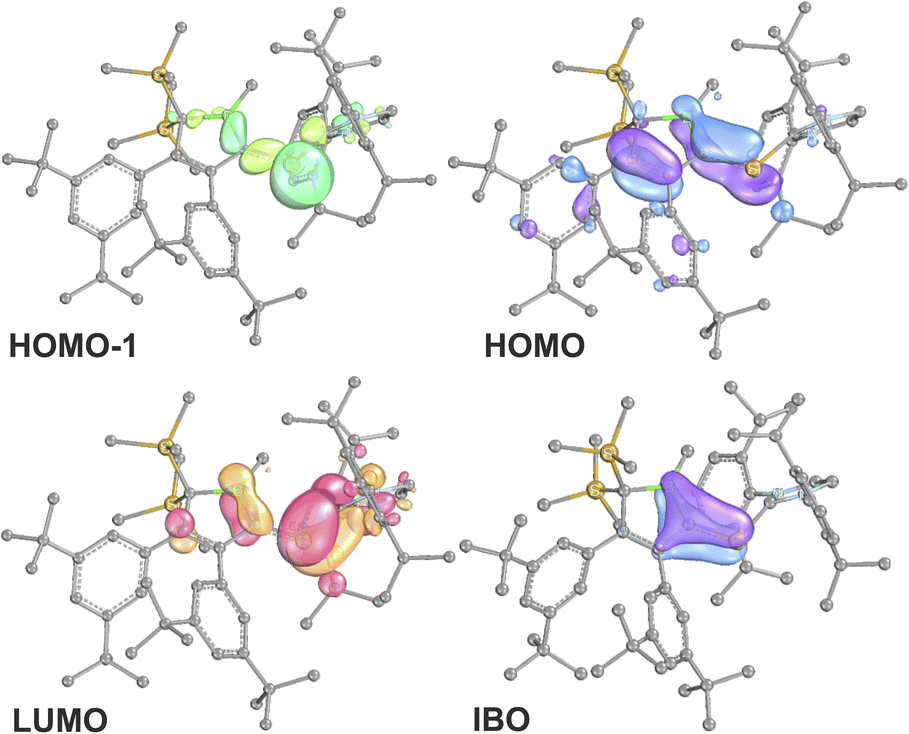 | ||
| Fig. 7 Frontier molecular orbitals of 5B-Me (BP86/def2-TZVP). Iso-surfaces at 60% probability. | ||
The most obvious change is observed for the 29Si-resonance that shifts drastically to higher frequencies and is found at δ(29Si) = 226.3 ppm. The value is reproduced by computational predictions (GIAO:PBE0/def2-TZVP δ(29Si)calc = 228 ppm). This value deviates drastically from other silavinylidene derivatives such as Filippou's reported value for (NHC): Si = CNR (δ(29Si) = −142 ppm)99 or Wesemann's germasila vinylidene (δ(29Si) = −48 ppm)96 which have been discussed as masked Si0 compounds and is closer to Robinson's (IDipp)SiSi(IDipp) (δ(29Si) = 225 ppm)54 or Filippou's (IDipp)Si = PMes* (δ(29Si) = 267 ppm).100 The intensely blood-red 5B-Me reveals a respective absorption band at 496 nm with a shoulder at 436 nm.
When solutions of 5B-Me were heated at ca. 70 °C for several days, we observed a very clean and complete conversion of the deep red silavinylidene to the colourless Si(II) half-sandwich compound 4B-Me and free IDipp which becomes unequivocally clear from the respective characteristic heteronuclear NMR pattern with δ(29Si) = −347.6 ppm and δ(11B) = 31.4 ppm. The isomerisation of NHC-supported silavinylidene 5B-Me to 4B-Me and IDipp was computationally approximated to be endergonic at 298 K (4.3 kcal mol−1). The conversion of the silavinylidene 5 into the half-sandwich compounds including a remigration of the silyl groups further highlights the mobility of silicon atoms on the borole-platform. Further inspection revealed that the half-sandwich compound was also present as a minor side product in the crude reduction reaction mixture of 5.
Therefore, reductions of both products of the reaction of free boroles with SiCl2(IDipp) ultimately allow access to the Si(II)-centered, nucleophilic nido-cluster 4 allowing for the development of more straightforward synthetic procedures than introducing Si(II) via [Cp*Si]+.
Conclusions
This study gives a detailed account on the reactivity of antiaromatic free boroles with SiCl2(IDipp) as a low-valent silicon precursor. They either react in a formal [4 + 1] cycloaddition reaction to give sila-borabicyclo[2.1.1]hex-2-ene derivatives or induce silyl migration to generate NHC-supported silylium ylides. Mechanistic proposals corroborated by computational assessment and experimental derivatizations have been presented for the diverging product formation pathways. We have been able to show, that the borole platform allows for facile and reversible migration of silyl groups. Depending on the substitution pattern and reaction conditions selective formation of either of these products was achieved. It was shown that the NHC-supported silylium ylides can be thermally converted into the bicyclic products but addition of small NHC to the bicycles can induce trimethyl silyl migration leading to new NHC-supported silylium ylides. Finally examples for both species, sila-borabicyclo[2.1.1]hex-2-ene and NHC-supported silylium ylides were successfully reduced to borole-based half-sandwich compounds of Si(II), our initial target structure. In the case of the reduction of silylium ylides, an unprecedented silavinylidene was isolated and identified as a key intermediate in the formation of the Si(II) half-sandwich cluster. This study perfectly complements to most recent findings by the groups of Nakamoto and Scheschkewitz that found virtually identical silyl migration in anti-aromatic cyclobutadienes by addition of nucleophilic silylenes to give donor-supported silenes.39 Reduction of such silenes gave Si(II) nido-clusters (silapyramidanes). Our work suggests that silavinylidenes could also be relevant intermediates in the formation of silapyramidanes.Data availability
Synthetic details and analytical data, including depictions of all spectra and detailed accounts on the methods applied are documented in the ESI.† Crystallographic data is made available via the CCDC. Coordinate data of all computationally optimised species is provided as a separate xyz-file as accompanying ESI.†Author contributions
JS, TH, LN and MK conducted and validated the experiments. TH, JS and CPS conceptualised the project. PNR and CPS acquired and analysed X-ray data. DS and CPS acquired funding and provided resources and supervision. CPS managed the project, performed computations, wrote the original draft. The manuscript was further reviewed and edited from contributions of JS, TH and PNR.Conflicts of interest
There are no conflicts to declare.Acknowledgements
CPS is grateful to excellent support, colleagueship and conditions at the Institute für Anorganische Chemie at Göttingen, Aachen and Stuttgart. The Fonds der chemischen Industrie is acknowledged for funding (Liebig-Fellowship to CPS/TH). Hans Gildenast and Steven van Terwingen (Aachen) are gratefully acknowledged for technical assistance with X-ray measurements. Dr Gerhard Fink (Aachen) for NMR measurements. The Boehringer Ingelheim Foundation (BIS) is acknowledged for support by an Exploration Grant. This research was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 389479699/GRK2455 and 496024290/SI2721.Notes and references
- R. Breslow, Acc. Chem. Res., 1973, 6, 393–398 CrossRef CAS.
- P. v. R. Schleyer, H. Jiao, B. Goldfuss and P. K. Freeman, Angew. Chem., Int. Ed. Engl., 1995, 34, 337–340 CrossRef CAS.
- H. Braunschweig, I. Krummenacher and J. Wahler, in Advances in Organometallic Chemistry, ed. A. F. Hill and M. J. Fink, Academic Press, 2013, vol. 61, pp. 1–53 Search PubMed.
- J. J. Eisch, N. K. Hota and S. Kozima, J. Am. Chem. Soc., 1969, 91, 4575–4577 CrossRef CAS.
- J. J. Eisch, J. E. Galle and S. Kozima, J. Am. Chem. Soc., 1986, 108, 379–385 CrossRef CAS PubMed.
- G. E. Herberich, B. Buller, B. Hessner and W. Oschmann, J. Organomet. Chem., 1980, 195, 253–259 CrossRef CAS.
- H. Braunschweig, I. Fernández, G. Frenking and T. Kupfer, Angew. Chem., Int. Ed., 2008, 47, 1951–1954 CrossRef CAS PubMed.
- C. Fan, W. E. Piers and M. Parvez, Angew. Chem., Int. Ed., 2009, 48, 2955–2958 CrossRef CAS PubMed.
- H. Braunschweig, F. Breher, C.-W. Chiu, D. Gamon, D. Nied and K. Radacki, Angew. Chem., Int. Ed., 2010, 49, 8975–8978 CrossRef CAS PubMed.
- H. Braunschweig, V. Dyakonov, J. O. C. Jimenez-Halla, K. Kraft, I. Krummenacher, K. Radacki, A. Sperlich and J. Wahler, Angew. Chem., Int. Ed., 2012, 51, 2977–2980 CrossRef CAS PubMed.
- R. Bertermann, H. Braunschweig, R. D. Dewhurst, C. Hörl, T. Kramer and I. Krummenacher, Angew. Chem., Int. Ed., 2014, 53, 5453–5457 CrossRef CAS PubMed.
- P. Bissinger, H. Braunschweig, A. Damme, C. Hörl, I. Krummenacher and T. Kupfer, Angew. Chem., Int. Ed., 2015, 54, 359–362 CrossRef CAS PubMed.
- C.-W. So, D. Watanabe, A. Wakamiya and S. Yamaguchi, Organometallics, 2008, 27, 3496–3501 CrossRef CAS.
- H. Braunschweig and T. Kupfer, Chem. Commun., 2008, 4487–4489 RSC.
- A. Fukazawa, J. L. Dutton, C. Fan, L. G. Mercier, A. Y. Houghton, Q. Wu, W. E. Piers and M. Parvez, Chem. Sci., 2012, 3, 1814–1818 RSC.
- A. Y. Houghton, J. Hurmalainen, A. Mansikkamäki, W. E. Piers and H. M. Tuononen, Nat. Chem., 2014, 6, 983–988 CrossRef CAS PubMed.
- T. Heitkemper and C. P. Sindlinger, Chem. - Eur. J., 2020, 26, 11684–11689 CrossRef CAS PubMed.
- C. Fan, W. E. Piers, M. Parvez and R. McDonald, Organometallics, 2010, 29, 5132–5139 CrossRef CAS.
- C. Fan, L. G. Mercier, W. E. Piers, H. M. Tuononen and M. Parvez, J. Am. Chem. Soc., 2010, 132, 9604–9606 CrossRef CAS PubMed.
- A. Y. Houghton, V. A. Karttunen, C. Fan, W. E. Piers and H. M. Tuononen, J. Am. Chem. Soc., 2013, 135, 941–947 CrossRef CAS PubMed.
- H. Braunschweig, C. Hörl, L. Mailänder, K. Radacki and J. Wahler, Chem.–Eur. J., 2014, 20, 9858–9861 CrossRef CAS PubMed.
- H. Braunschweig, F. Hupp, I. Krummenacher, L. Mailänder and F. Rauch, Chem.–Eur. J., 2015, 21, 17844–17849 CrossRef CAS PubMed.
- H. Braunschweig, I. Krummenacher, L. Mailänder and F. Rauch, Chem. Commun., 2015, 51, 14513–14515 RSC.
- H. Braunschweig, M. A. Celik, F. Hupp, I. Krummenacher and L. Mailänder, Angew. Chem., Int. Ed., 2015, 54, 6347–6351 CrossRef CAS PubMed.
- F. Lindl, S. Lin, I. Krummenacher, C. Lenczyk, A. Stoy, M. Müller, Z. Lin and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 338–342 CrossRef CAS PubMed.
- F. Lindl, S. Lin, I. Krummenacher, C. Lenczyk, A. Stoy, M. Müller, Z. Lin and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 338–342 CrossRef CAS PubMed.
- S. A. Couchman, T. K. Thompson, D. J. D. Wilson, J. L. Dutton and C. D. Martin, Chem. Commun., 2014, 50, 11724–11726 RSC.
- K. Huang, S. A. Couchman, D. J. D. Wilson, J. L. Dutton and C. D. Martin, Inorg. Chem., 2015, 54, 8957–8968 CrossRef CAS PubMed.
- K. Huang and C. D. Martin, Inorg. Chem., 2015, 54, 1869–1875 CrossRef CAS PubMed.
- S. Yruegas, K. Huang, D. J. D. Wilson, J. L. Dutton and C. D. Martin, Dalton Trans., 2016, 45, 9902–9911 RSC.
- J. H. Barnard, S. Yruegas, S. A. Couchman, D. J. D. Wilson, J. L. Dutton and C. D. Martin, Organometallics, 2016, 35, 929–931 CrossRef CAS.
- S. Yruegas, C. Wilson, J. L. Dutton and C. D. Martin, Organometallics, 2017, 36, 2581–2587 CrossRef CAS.
- Q. Sun, C. G. Daniliuc, X. Yu, C. Mück-Lichtenfeld, G. Kehr and G. Erker, J. Am. Chem. Soc., 2022, 144, 7815–7821 CrossRef CAS PubMed.
- V. Y. Lee, H. Sugasawa, O. A. Gapurenko, R. M. Minyaev, V. I. Minkin, H. Gornitzka and A. Sekiguchi, J. Am. Chem. Soc., 2018, 140, 6053–6056 CrossRef CAS PubMed.
- H. Braunschweig, C.-W. Chiu, T. Kupfer and K. Radacki, Inorg. Chem., 2011, 50, 4247–4249 CrossRef CAS PubMed.
- F. Ge, G. Kehr, C. G. Daniliuc and G. Erker, Organometallics, 2015, 34, 229–235 CrossRef CAS.
- Q. Sun, C. G. Daniliuc, C. Mück-Lichtenfeld, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2022, 61, e202205565 CAS.
- C.-W. So, H. W. Roesky, J. Magull and R. B. Oswald, Angew. Chem., Int. Ed., 2006, 45, 3948–3950 CrossRef CAS PubMed.
- T. Imagawa, L. Giarrana, D. M. Andrada, B. Morgenstern, M. Nakamoto and D. Scheschkewitz, J. Am. Chem. Soc., 2023, 145, 4757–4764 CrossRef CAS PubMed.
- T. Heitkemper, L. Naß and C. P. Sindlinger, Dalton Trans., 2020, 49, 2706–2714 RSC.
- T. Heitkemper, J. Sarcevic and C. P. Sindlinger, J. Am. Chem. Soc., 2020, 142, 21304–21309 CrossRef CAS PubMed.
- J. Sarcevic, T. Heitkemper and C. P. Sindlinger, Chem. Commun., 2022, 58, 246–249 RSC.
- P. Tholen, Z. Dong, M. Schmidtmann, L. Albers and T. Müller, Angew. Chem., Int. Ed., 2018, 57, 13319–13324 CrossRef CAS PubMed.
- P. Jutzi, Chem.–Eur. J., 2014, 20, 9192–9207 CrossRef CAS PubMed.
- K. Leszczyńska, K. Abersfelder, M. Majumdar, B. Neumann, H.-G. Stammler, H. S. Rzepa, P. Jutzi and D. Scheschkewitz, Chem. Commun., 2012, 48, 7820 RSC.
- P. Jutzi, A. Mix, B. Rummel, W. W. Schoeller, B. Neumann and H.-G. Stammler, Science, 2004, 305, 849–851 CrossRef CAS PubMed.
- P. Jutzi, U. Holtmann, D. Kanne, C. Krüger, R. Blom, R. Gleiter and I. Hyla-Kryspin, Chem. Ber., 1989, 122, 1629–1639 CrossRef CAS.
- P. Jutzi, D. Kanne and C. Krüger, Angew. Chem., Int. Ed. Engl., 1986, 25, 164 CrossRef.
- K. I. Leszczyńska, V. Huch, C. Präsang, J. Schwabedissen, R. J. F. Berger and D. Scheschkewitz, Angew. Chem., Int. Ed., 2019, 58, 5124–5128 CrossRef PubMed.
- R. S. Ghadwal, R. Azhakar and H. W. Roesky, Acc. Chem. Res., 2013, 46, 444–456 CrossRef CAS PubMed.
- R. S. Ghadwal, H. W. Roesky, S. Merkel, J. Henn and D. Stalke, Angew. Chem., Int. Ed., 2009, 48, 5683–5686 CrossRef CAS PubMed.
- A. C. Filippou, Y. N. Lebedev, O. Chernov, M. Straßmann and G. Schnakenburg, Angew. Chem., Int. Ed., 2013, 52, 6974–6978 CrossRef CAS PubMed.
- A. C. Filippou, O. Chernov and G. Schnakenburg, Angew. Chem., Int. Ed., 2009, 48, 5687–5690 CrossRef CAS PubMed.
- Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer, P. v. R. Schleyer and G. H. Robinson, Science, 2008, 321, 1069–1071 CrossRef CAS PubMed.
- G. E. Herberich and H. Ohst, Chem. Ber., 1985, 118, 4303–4313 CrossRef CAS.
- P. E. Romero, W. E. Piers, S. A. Decker, D. Chau, T. K. Woo and M. Parvez, Organometallics, 2003, 22, 1266–1274 CrossRef CAS.
- Y. Li, R. K. Siwatch, T. Mondal, Y. Li, R. Ganguly, D. Koley and C.-W. So, Inorg. Chem., 2017, 56, 4112–4120 CrossRef CAS PubMed.
- H. Braunschweig, C.-W. Chiu, D. Gamon, K. Gruß, C. Hörl, T. Kupfer, K. Radacki and J. Wahler, Eur. J. Inorg. Chem., 2013, 2013, 1525–1530 CrossRef CAS.
- K. Ansorg, H. Braunschweig, C. W. Chiu, B. Engels, D. Gamon, M. Hügel, T. Kupfer and K. Radacki, Angew. Chem., Int. Ed., 2011, 50, 2833–2836 CrossRef CAS PubMed.
- Deposition numbers 2233795 (for A-Me), 2233796 (for A-Xyl), 2233797 (for B-Xyl), 2233798 (for 1A-pXyl), 2233799 (for 1B-Ph*), 2233800 (for 2A-Me), 2233801 (for 2A-Cl), 2233802 (2A-Xyl), 2233803 (for 2B-Ph*), 2233804 (for 3B-pXyl), 2233805 (for 4A-pXyl), and 2239382 (for 5B-Me) contain the supplementary crystallographic data for this paper.
- P. J. Fagan, E. G. Burns and J. C. Calabrese, J. Am. Chem. Soc., 1988, 110, 2979–2981 CrossRef CAS.
- U. Holtmann, P. Jutzi, T. Kühler, B. Neumann and H.-G. Stammler, Organometallics, 1999, 18, 5531–5538 CrossRef CAS.
- M. M. Olmstead, P. P. Power, K. J. Weese and R. J. Doedens, J. Am. Chem. Soc., 1987, 109, 2541–2542 CrossRef CAS.
- T. Heitkemper, L. Naß and C. P. Sindlinger, Angew. Chem., Int. Ed., 2021, 60, 20055–20060 CrossRef CAS PubMed.
- N. Wiberg, G. Wagner and G. Müller, Angew. Chem., Int. Ed. Engl., 1985, 24, 229–230 CrossRef.
- Z. Wang, J. Zhang, J. Li and C. Cui, J. Am. Chem. Soc., 2016, 138, 10421–10424 CrossRef CAS PubMed.
- X. Sun, A. Hinz, H. Kucher, M. T. Gamer and P. W. Roesky, Chem.–Eur. J., 2022, 28, e202201963 CAS.
- C. Balzereit, H.-J. Winkler, W. Massa and A. Berndt, Angew. Chem., Int. Ed., 1994, 33, 2306 CrossRef.
- F. Ge, G. Kehr, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2014, 136, 68–71 CrossRef CAS PubMed.
- X. Su, J. J. Baker and C. D. Martin, Chem. Sci., 2020, 11, 126–131 RSC.
- C.-T. Shen, Y.-H. Liu, S.-M. Peng and C.-W. Chiu, Angew. Chem., Int. Ed., 2013, 52, 13293–13297 CrossRef CAS PubMed.
- H.-C. Tseng, C.-T. Shen, K. Matsumoto, D.-N. Shih, Y.-H. Liu, S.-M. Peng, S. Yamaguchi, Y.-F. Lin and C.-W. Chiu, Organometallics, 2019, 38, 4516–4521 CrossRef CAS.
- Q. Sun, C. G. Daniliuc, C. Mück-Lichtenfeld, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2022, 61, e202205565 CAS.
- H. Meyer, G. Baum, W. Massa, S. Berger and A. Berndt, Angew. Chem., Int. Ed. Engl., 1987, 26, 546–548 CrossRef.
- H. Meyer, G. Baum, W. Massa and A. Berndt, Angew. Chem., Int. Ed. Engl., 1987, 26, 798–799 CrossRef.
- M. Stürmann, W. Saak, M. Weidenbruch, A. Berndt and D. Scheschkewitz, Heteroat. Chem., 1999, 10, 554–558 CrossRef.
- M. Weidenbruch, H. Kilian, M. Stürmann, S. Pohl, W. Saak, H. Marsmann, D. Steiner and A. Berndt, J. Organomet. Chem., 1997, 530, 255–257 CrossRef CAS.
- R. Wehrmann, H. Klusik and A. Berndt, Angew. Chem., Int. Ed. Engl., 1984, 23, 826–827 CrossRef.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew and W. Yue, Phys. Rev. B, 1986, 33, 8800–8802 CrossRef PubMed.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- E. D. Glendening, C. R. Landis and F. Weinhold, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 1–42 CAS.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO7.0, 2018 Search PubMed.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS PubMed.
- W. Lu, Y. Li, R. Ganguly and R. Kinjo, J. Am. Chem. Soc., 2017, 139, 5047–5050 CrossRef CAS PubMed.
- S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754–1757 CrossRef CAS PubMed.
- S. J. Bonyhady, C. Jones, S. Nembenna, A. Stasch, A. J. Edwards and G. J. McIntyre, Chem.–Eur. J., 2010, 16, 938–955 CrossRef CAS PubMed.
- A. Rit, J. Campos, H. Niu and S. Aldridge, Nat. Chem., 2016, 8, 1022–1026 CrossRef CAS PubMed.
- K. M. Krebs, D. Hanselmann, H. Schubert, K. Wurst, M. Scheele and L. Wesemann, J. Am. Chem. Soc., 2019, 141, 3424–3429 CrossRef CAS PubMed.
- A. Jana, V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed., 2013, 52, 12179–12182 CrossRef CAS PubMed.
- A. Jana, M. Majumdar, V. Huch, M. Zimmer and D. Scheschkewitz, Dalton Trans., 2014, 43, 5175–5181 RSC.
- C. Wilhelm, D. Raiser, H. Schubert, C. P. Sindlinger and L. Wesemann, Inorg. Chem., 2021, 60, 9268–9272 CrossRef CAS PubMed.
- P. Ghana, M. I. Arz, U. Das, G. Schnakenburg and A. C. Filippou, Angew. Chem., Int. Ed., 2015, 54, 9980–9985 CrossRef CAS PubMed.
- R. Kobayashi, S. Ishida and T. Iwamoto, Organometallics, 2021, 40, 843–847 CrossRef CAS.
- S. Karwasara, L. R. Maurer, B. Peerless, G. Schnakenburg, U. Das and A. C. Filippou, J. Am. Chem. Soc., 2021, 143, 14780–14794 CrossRef CAS PubMed.
- D. Geiß, M. I. Arz, M. Straßmann, G. Schnakenburg and A. C. Filippou, Angew. Chem., Int. Ed., 2015, 54, 2739–2744 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, analytical and spectroscopic data and spectra depictions, crystallographic and computational details. CCDC 2233795–2233805 and 2239382. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00808h |
| ‡ These authors contributed equally and should be allowed to name themselves first in personal references of this work. |
| This journal is © The Royal Society of Chemistry 2023 |