
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Targeted proximity-labelling of protein tyrosines via flavin-dependent photoredox catalysis with mechanistic evidence for a radical–radical recombination pathway†
Taylor O.
Hope
a,
Tamara
Reyes-Robles
b,
Keun Ah
Ryu
b,
Steven
Mauries
a,
Nicole
Removski
a,
Jacinthe
Maisonneuve
a,
Rob C.
Oslund‡
 *b,
Olugbeminiyi O.
Fadeyi‡
*b and
Mathieu
Frenette
*a
*b,
Olugbeminiyi O.
Fadeyi‡
*b and
Mathieu
Frenette
*a
aDepartment of Chemistry, NanoQAM, Centre Québécois des Matériaux Fonctionnels (CQMF), Université du Québec à Montréal, Montréal, Québec H3C 3P8, Canada. E-mail: frenette.mathieu@uqam.ca
bExploratory Science Center, Merck & Co., Inc., Cambridge, MA, USA. E-mail: niyi@induprolabs.com; rob@induprolabs.com
First published on 17th May 2023
Abstract
Flavin-based photocatalysts such as riboflavin tetraacetate (RFT) serve as a robust platform for light-mediated protein labelling via phenoxy radical-mediated tyrosine–biotin phenol coupling on live cells. To gain insight into this coupling reaction, we conducted detailed mechanistic analysis for RFT-photomediated activation of phenols for tyrosine labelling. Contrary to previously proposed mechanisms, we find that the initial covalent binding step between the tag and tyrosine is not radical addition, but rather radical–radical recombination. The proposed mechanism may also explain the mecha-nism of other reported tyrosine-tagging approaches. Competitive kinetics experiments show that phenoxyl radicals are generated with several reactive intermediates in the proposed mechanism—primarily with the excited riboflavin-photocatalyst or singlet oxygen—and these multiple pathways for phenoxyl radical generation from phenols increase the likelihood of radical–radical recombination.
Introduction
Technologies that identify interacting proteins are crucial to understand fundamental biological processes and to enable new drug target discoveries. Visible light photocatalysis has emerged as an attractive platform for this purpose to achieve selective chemical transformations on and within biological materials with spatiotemporal control.1–11 A key feature is that visible light selectively excites a photocatalyst; the excited photocatalyst then activates chemical tags for covalent protein labelling. Tag molecules are usually activated to become reactive intermediates with short lifetimes, such as radicals,10,11 carbenes,8 or nitrenes,9 to limit the labelling radius within a complex biological environment. Importantly, a single photocatalyst can activate multiple tags resulting in substantial signal amplification for labelling with bioorthogonal handles such as biotin, azides, alkynes, or fluorophores for downstream protein analysis.1,12A major trend in proximity protein tagging is to exploit the reactivity of tyrosine at a protein's surface. Notably, peroxidase-enabled tyrosine labelling has been developed for profiling protein environments in numerous cellular contexts.13 In this system initiated by exogenous hydrogen peroxide, a phenol-containing tag is oxidized to phenoxyl radicals by heme-containing peroxidases resulting in the labelling of nearby proteins. Recently, we reported the use of a flavin-derived cofactor, riboflavin tetraacetate (RFT), as a photocatalyst for the generation of phenoxyl radical intermediates for tyrosine-based protein labelling (Fig. 1A).4 Blue-light activated RFT was shown to achieve proximity labelling of proteins, live cells, and cell–cell contact regions (Fig. 1B). Similar to other reported tyrosine tagging methods, radical addition onto a neutral tyrosine is traditionally proposed as a key step in the mechanism.10,11,14–16 However, to date, no direct evidence has been reported for this mechanism. Here, we examine the proposed radical addition mechanism and a competing radical–radical recombination pathway (Fig. 1C).
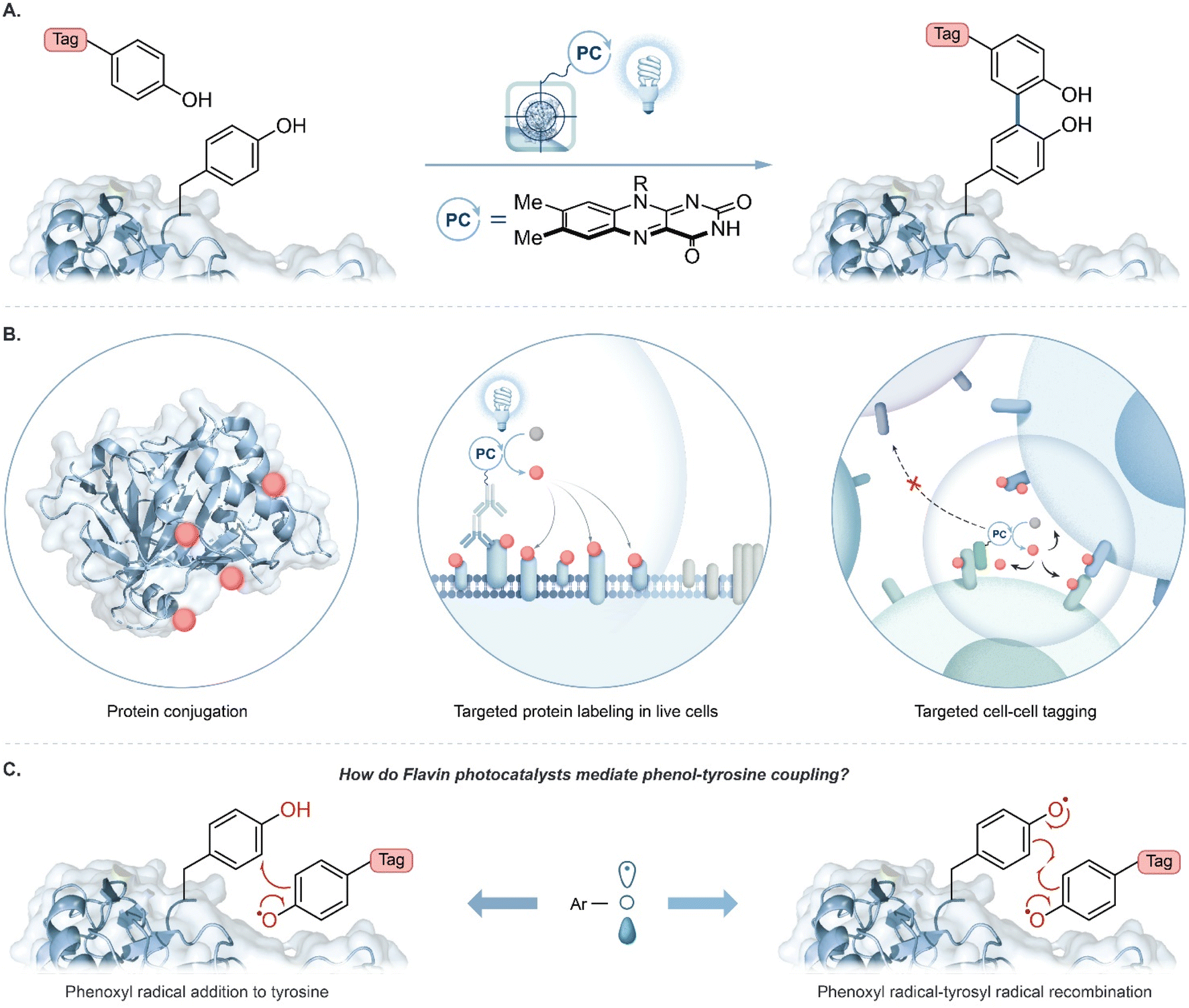 | ||
| Fig. 1 (A) Flavin-based photocatalytic tyrosine-tagging by phenol containing tags. (B) Biological applications of RFT-mediated photocatalysis in protein and cellular environments. (C) Competing mechanistic proposals for the labelling of tyrosine by phenoxyl radicals: radical addition to tyrosine versus radical recombination. | ||
Results and discussion
Prior to exploring the mechanistic basis of RFT-mediated phenol–phenol coupling, we developed a novel proteomic workflow to evaluate the effect of targeted activation of RFT in a localized microenvironment. Having previously demonstrated by flow cytometry and confocal imaging that we can achieve RFT-mediated protein labelling on live cells,4 this proteomic workflow focused on the evaluation of targeted versus untargeted activation of RFT localized to the cell surface (Fig. 2A). In these experiments, RFT was conjugated to a secondary antibody that can bind to a primary antibody recognizing cell surface proteins. We selected PTPRC (CD45), a T cell marker highly expressed on the cell surface,17 as our target.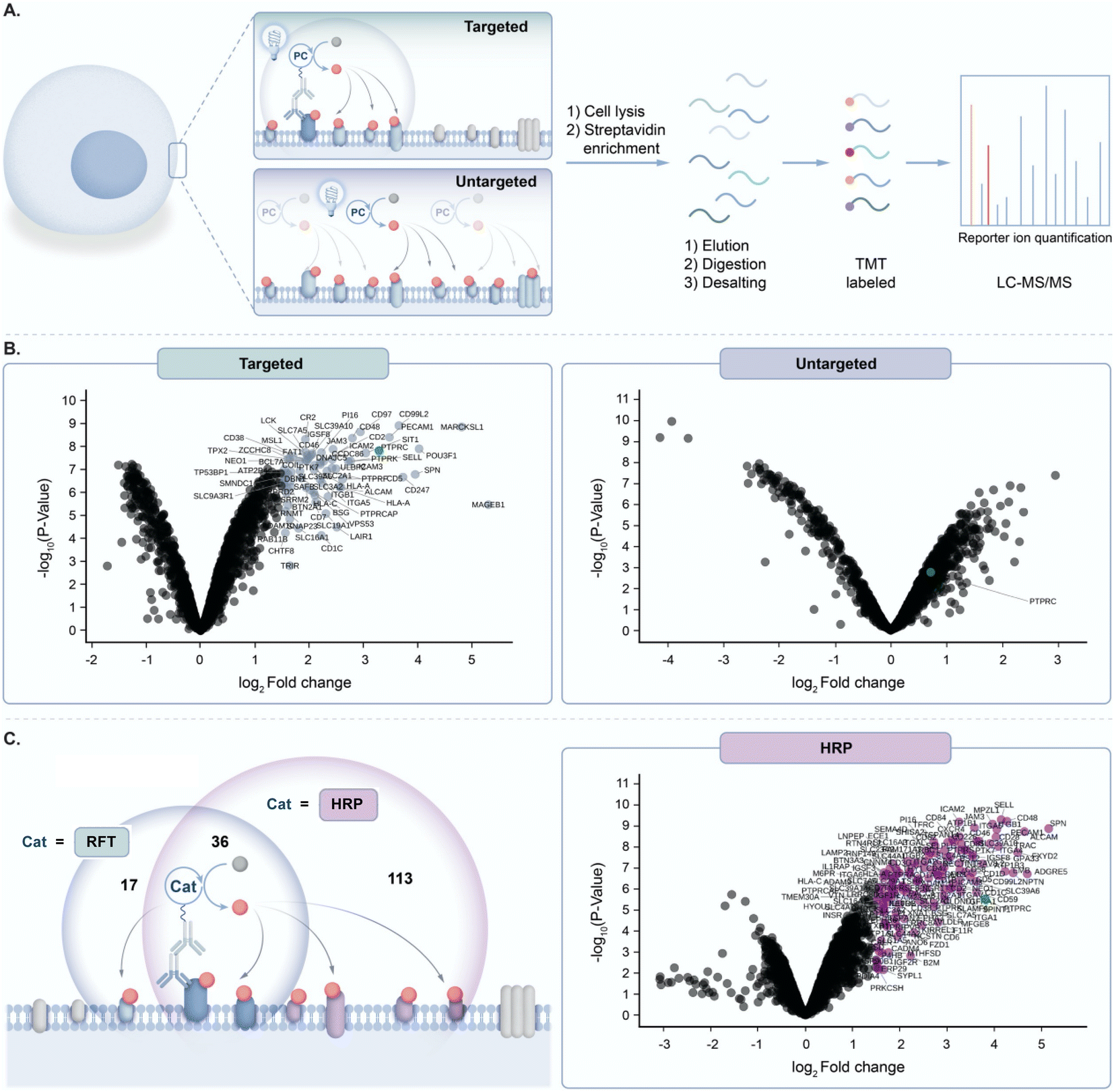 | ||
Fig. 2 (A) Schematic depicting targeted and untargeted labelling of CD45 (PTPRC) on the surface of Jurkat cells. Cells are labeled with an anti-CD45 primary antibody/secondary antibody RFT photocatalyst conjugate (targeted) or with free RFT photocatalyst (untargeted) followed by cell lysis, protein enrichment and digestion, and LC-MS/MS-based proteomic analysis. (B) Left, volcano plot of statistical significance vs. fold-enrichment for CD45-targeted vs. isotype-targeted biotinylation on Jurkat cells using an anti-CD45 primary antibody/secondary antibody RFT conjugate with 10 minutes of visible light activation in the presence of biotin tyramide. Significantly enriched proteins (p-value < 0.05 and log2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) FC > 1.5) are indicated in light blue and CD45 (PTPRC) is labeled in green (n = 3 experiments). Right, volcano plot of statistical significance vs. fold-enrichment of free RFT photocatalyst and biotin tyramide vs. biotin tyramide only on Jurkat cells after 10 minutes of visible light activation. CD45 (PTPRC) is labeled in green (n = 3 experiments). (C) Left, schematic depicting CD45 targeted labelling between RFT and peroxidase (HRP)-based methods. Circles reflect a Venn diagram of the significantly enriched proteins from targeted labelling with HRP (purple circle, (C) right panel) or RFT (light blue circle, Fig. S2†). Right, volcano plot of statistical significance vs. fold-enrichment for CD45-targeted vs. isotype-targeted biotinylation on Jurkat cells using an anti-CD45 primary antibody/secondary antibody peroxidase conjugate (HRP) or isotype/secondary antibody peroxidase conjugate with 1 minute of labelling in the presence of biotin tyramide and hydrogen peroxide. Significantly enriched proteins (p-value < 0.05 and log2FC > 1.5) are indicated in purple and CD45 (PTPRC) is labeled in green (n = 3 experiments). FC > 1.5) are indicated in light blue and CD45 (PTPRC) is labeled in green (n = 3 experiments). Right, volcano plot of statistical significance vs. fold-enrichment of free RFT photocatalyst and biotin tyramide vs. biotin tyramide only on Jurkat cells after 10 minutes of visible light activation. CD45 (PTPRC) is labeled in green (n = 3 experiments). (C) Left, schematic depicting CD45 targeted labelling between RFT and peroxidase (HRP)-based methods. Circles reflect a Venn diagram of the significantly enriched proteins from targeted labelling with HRP (purple circle, (C) right panel) or RFT (light blue circle, Fig. S2†). Right, volcano plot of statistical significance vs. fold-enrichment for CD45-targeted vs. isotype-targeted biotinylation on Jurkat cells using an anti-CD45 primary antibody/secondary antibody peroxidase conjugate (HRP) or isotype/secondary antibody peroxidase conjugate with 1 minute of labelling in the presence of biotin tyramide and hydrogen peroxide. Significantly enriched proteins (p-value < 0.05 and log2FC > 1.5) are indicated in purple and CD45 (PTPRC) is labeled in green (n = 3 experiments). | ||
Accordingly, targeted delivery of RFT to PTPRC via the primary/secondary antibody system, followed by irradiation with blue light in the presence of biotin tyramide led to detection of PTPRC among the highest enriched proteins (Fig. 2B). Known PTPRC interactors such as the PTPRC associated protein (PTPRCAP), CD2, and LCK were also highly co-enriched with PTPRC (Fig. 2B and S1†). In contrast, untargeted delivery of free RFT catalysts to live cells (Fig. 2A) followed by irradiation with blue light in the presence of biotin tyramide failed to enrich PTPRC (Fig. 2B). Since the peroxidase enzyme similarly leverages biotin tyramide to achieve protein labelling,13 we compared our RFT protein system to peroxidase-based labelling of CD45 on live cells followed by mass spectrometry based proteomic analysis. We observed a nearly three-fold increase in total protein enrichment using peroxidase-based labelling compared to RFT (Fig. 2C and S2†) suggesting a much larger effective labelling radius achieved by the enzyme-based method. Collectively these results highlight the ability of targeted activation of RFT to achieve localized phenoxyl radical tagging within complex cellular systems.
Likewise, tyrosine radical addition onto a neutral aromatic system has been proposed (Fig. 1C).10,14 In both cases, prohibitively large activation energies are calculated for these proposed mechanisms (Tables S12 and S15†). A major contribution of this study is to bring convincing evidence against radical addition to tyrosine; the thermodynamic cost of this radical addition step is prohibitively unfavorable (Fig. 3A, ΔGDFT > 34 kcal mol−1, Table S10†). The recombination of phenoxyl radicals, however, is much more probable (Fig. 3B, ΔGDFT = −1 to +3 kcal mol−1, Table S9†). The slight endergonic recombination is a testament to phenoxyl radical stability—of course, the initial recombination product will rapidly tautomerize to regenerate aromaticity for a largely favorable end-product (ΔGDFT = −36 kcal mol−1, Table S9†). Photocatalysis often employs metal-based complexes due to their long-lived excited state lifetimes and tunable redox potentials,18–20 however, more bio-compatible photocatalysts such as RFT are desirable in protein labelling.21 We explored RFT photochemistry and found several pathways by which phenoxyl radicals, from tags and tyrosine, could be generated. The multiple pathways that generate phenoxyl radicals support evidence of radical–radical recombination as a likely mechanism in these systems.
 | ||
| Fig. 3 (A) The addition of a phenoxyl radical tag to tyrosine is highly unfavorable (Table S10†). (B) Radical–radical recombination of phenoxyl radicals is more favorable, and rearromatization leads to a strong covalent bond (Table S9†). The relative rate constants were estimated according to Eyring's equation (see ESI† for details). | ||
The photocatalytic cycle begins with blue light absorption by RFT followed by formation of a triplet excited state, 3[RFT]*. We observed 3[RFT]* to display a relatively long excited state lifetime of 12 μs in deoxygenated solution, as determined by following its characteristic absorption at 700 nm in laser-flash photolysis experiments (Fig. 4A).
 | ||
| Fig. 4 (A) Transient absorption of RFT (0.06 mM) in deoxygenated acetonitrile:water (1:1) following 355 nm laser excitation. (B) Stern–Volmer quenching of 3[RFT]* with 2,6-dimethoxyphenol (grey), biotin tyramide (orange), Ac–Tyr–NHMe (yellow) and BSA (blue). (C) Transient absorption of phenoxyl radicals from biotin tyramide and fit to second order decay kinetics (see ESI and Fig. S17† for details). | ||
Due to their oxidizing nature, flavin-based triplet excited states can initiate radical chemistry.22 In our system, phenol-containing tags or tyrosines in a protein will be rapidly oxidized by 3[RFT]* to generate phenoxyl radicals. The rate constant (kq) for this bimolecular process was determined by Stern–Volmer kinetics (Fig. 4B), which shows the oxidation of phenols with 3[RFT]* to be fast in measured examples (108 to 109 M−1 s−1) (Table S2†). When generated in laser flash photolysis experiments, phenoxyl radicals display a typical 2nd order decay as shown in Fig. 4C since they undergo a radical–radical recombination as their main decay pathway.
In the absence of oxygen, 3[RFT]* will quickly be reduced by phenols, either from a tag or tyrosine group (Fig. 5A, Table S5†). The transient absorption of 3[RFT]* at 700 nm rapidly decays in the presence of phenols to form a relatively stable intermediate with an absorption centered at 600 nm (Fig. 5B). We identify this peak as the semi reduced form, H-RFT˙, with supporting evidence provided by TD-DFT predicted spectra (Fig. 5D). Semi-reduced flavins with similar chemical structures absorb in the same region.22 Both transient absorption peaks associated with 3[RFT]* and H-RFT˙ are strongly attenuated by the introduction of oxygen (Fig. 5C).
 | ||
| Fig. 5 (A) Reactions observed by laser-flash photolysis. (B) Transient absorption of RFT (0.06 mM) and biotin tyramide (0.3 mM) in the absence of oxygen after a 355 nm laser pulse. (C) Transient absorption of the same RFT (0.06 mM) and biotin tyramide (0.3 mM) solution open to air. (D) TD-DFT predicted absorption spectra of ground state RFT (blue), 3[RFT]* (black), H-RFT˙ (orange) and H2-RFT (dashed grey). | ||
We applied this photocatalytic coupling method on nucleophilic phenol substrates and tyrosine containing peptides where we observed tyrosine–phenol cross-coupling in moderate yields (Fig. S3 and Table S4†). During the optimization of our synthetic method, we found oxygen to be crucially important in maximizing the conversion efficiency (Table S4†). While the synthetic and protein labelling conditions differ in phenol type and concentration, this observation led us to investigative the role oxygen plays in the overall reaction mechanism.
Flavins are well-known singlet oxygen (1O2) sensitizers22 and 1O2 can oxidize phenols to phenoxyl radicals.24–27 Using competitive kinetics with the probe 9,10-diphenylanthracene (DPA),23 we measured the rate constant for the reaction of 1O2 with electron rich phenols and a tyrosine analog to be ∼107 M−1 s−1 (Fig. S24†). The H-abstraction from phenols by 1O2 is calculated to be favorable with a ΔGDFT from −12.3 to −18.0 kcal mol−1 (Table S7†).
Oxygen also plays a role in the photocatalytic turnover of RFT. Following excitation, 3[RFT]* will react with phenols to form phenoxyl radicals and H-RFT˙. Two pathways to regenerate RFT from H-RFT˙ are considered. As often proposed in the literature for flavins, H-RFT˙ can be further reduced to H2-RFT,11,22,28 however, we calculate this reduction to be significantly unfavorable in our system. Phenoxyl radical generation from the reaction between phenols and H-RFT˙ gave ΔGDFT values in the range of +19.4 to +25.2 kcal mol−1 (Table S6†). A second pathway to regenerate RFT from H-RFT˙ bypasses the need to generate H2-RFT. In the proposed mechanism (Fig. 6A), oxygen will directly oxidize H-RFT˙ back to RFT with the formation of HOO˙. This pathway has a more favorable ΔGDFT of +4.99 kcal mol−1 than the full reduction (ΔGDFT > +17 kcal mol−1).
 | ||
| Fig. 6 (A) Proposed photocatalytic reaction mechanism for the generation of phenoxyl radicals from tyrosine-containing protein and phenol-containing tag molecules. (B) Phenoxyl radical are persistent radicals, and as such will recombine followed by rearomatization, as the preferred mechanism for phenol–phenol coupling. ΔGDFT, shown in kcal mol−1, were calculated using B3LYP-D3BJ/6-311+G(2d,2p)//CPCM(H2O) with various phenols (see ESI† for details). | ||
The detection of H2O2 as a by-product has been used as evidence for the catalytic cycle to involve H2-RFT.16 Indeed, the reduction of H2-RFT, in a presumably multistep process, could favorably generate H2O2 in the presence of oxygen (ΔGDFT = −23.4 kcal mol−1). However, H2O2 can also be generated in pathways that do not include H2-RFT. The reaction of 1O2 with phenols will generate HOO˙ as could the reaction of 3O2 with H-RFT˙. Both H-abstraction by HOO˙ and recombination of two HOO˙ can explain the presence of H2O2.
Finally, as proposed in Fig. 6B the phenoxyl radicals will recombine and favourably tautomerize (ΔGDFT = −1 to +3 kcal mol−1 and ∼−36 kcal mol−1, for each step respectively. See Table S9†).
Traditionally, the need for two radicals to recombine in a synthetically useful transformation is rare—the concentration of transient radicals is often too low to explain high yielding reactions. In this case, however, the persistence of phenoxyl radicals is well-documented—several antioxidants are based on phenoxyl radical's lack of side-reactions.29 In biological systems, phenoxyl radicals are relatively persistent due to their low reactivity with oxygen and their inability to perform H-abstraction with most biomolecules—their estimated lifetime is ∼0.1 ms in cell media.8
The proposed mechanism described herein helps explain the success of phenoxyl radical labelling methods. Radical–radical recombination follows bimolecular kinetics, and the efficiency of this process will dramatically decrease as the distance increases away from the photochemical source of phenoxyl radicals. Another important factor in the success of this protein tagging method is the strength of the bonds formed. Radical–radical recombination becomes more predominant with increasing radical persistence; however, the recombination of persistent radicals can lead to weak and reversible bonds as is the case for certain carbon-centered radicals.30 The recombination of phenoxyl radical is followed by tautomeric aromatization leading to bond strengthening. Altogether, these mechanistic insights will be an important consideration as different tag motifs are considered in photochemical protein tagging. Furthermore, the radical–radical recombination mechanism described here might be a potential pathway that occurs in the case of peroxidase-based labelling systems.31,32
Conclusion
We envision that the insights gained from our mechanistic analysis will lead to improved catalytic performance of these flavin-based systems and further encourage the development of new and effective light-mediated labelling strategies in the field of chemical biology.Data availability
Computational data is resumed in the ESI.† No other data in data repositories.Author contributions
The manuscript was written by T. O. H., R. C. O., O. O. F. and M. F. All authors have given approval to the final version of the manuscript.Conflicts of interest
T. R. R., K. A. R., R. C. O., and O. O. F. were employed by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Rah-way, NJ, USA during the experimental planning, execution and/or preparation of this manuscript.Acknowledgements
We gratefully acknowledge NSERC for funding in the form of a Discovery Grant to M. F. (RGPIN-2016-06773). We are grateful to NanoQAM and to Compute Canada and Gaussian for computational resources.Notes and references
- K. A. Ryu, C. M. Kaszuba, N. B. Bissonnette, R. C. Oslund and O. O. Fadeyi, Interrogating biological systems using visible-light-powered catalysis, Nat. Rev. Chem., 2021, 5, 322–337 CrossRef CAS PubMed.
- M. Müller, F. Gräbnitz, N. Barandun, Y. Shen, F. Wendt, S. N. Steiner, Y. Severin, S. U. Vetterli, M. Mondal, J. R. Prudent and R. Hofmann, Light-mediated discovery of surfaceome nanoscale organization and intercellular receptor interaction networks, Nat. Commun., 2021, 12, 7036 CrossRef PubMed.
- B. F. Buksh, S. D. Knutson, J. V. Oakley, N. B. Bissonnette, D. G. Oblinsky, M. P. Schwoerer, C. P. Seath, J. B. Geri, F. P. Rodriguez-Rivera, D. L. Parker and G. D. Scholes, μMap-Red: Proximity Labelling by Red Light Photocatalysis, J. Am. Chem. Soc., 2022, 144, 6154–6162 CrossRef CAS PubMed.
- R. C. Oslund, T. Reyes-Robles, C. H. White, J. H. Tomlinson, K. A. Crotty, E. P. Bowman, D. Chang, V. M. Peterson, L. Li, S. Frutos, M. Vila-Perello, D. Vlerick, K. Cromie, D. H. Perlman, S. Ingale, S. D. O’Hara, L. R. Roberts, G. Piizzi, E. C. Hett, D. J. Hazuda and O. O. Fadeyi, Detection of Cell-Cell Interactions via Photocatalytic Cell Tagging, Nat. Chem. Biol., 2022, 18, 850–858 CrossRef CAS PubMed.
- H. Wang, Y. Zhang, K. Zeng, J. Qiang, Y. Cao, Y. Li, Y. Fang, Y. Zhang and Y. Chen, Selective Mitochondrial Protein Labelling Enabled by Biocompatible Photocatalytic Reactions inside Live Cells, JACS Au, 2021, 1, 1066–1075 CrossRef CAS PubMed.
- T. Tamura, M. Takato, K. Shiono and I. Hamachi, Development of a photoactivatable proximity labelling method for the identification of nuclear proteins, Chem. Lett., 2020, 49, 145–148 CrossRef CAS.
- A. D. Trowbridge, C. P. Seath, F. P. Rodriguez-Rivera, B. X. Li, B. E. Dul, A. G. Schwaid, B. F. Buksh, J. B. Geri, J. V. Oakley, O. O. Fadeyi, R. C. Oslund, K. A. Ryu, C. White, T. Reyes-Robles, P. Tawa, D. L. Parker Jr and D. W. C. MacMillan, Small molecule photocatalysis enables drug target identification via energy transfer, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2208077119 CrossRef CAS PubMed.
- J. B. Geri, J. V. Oakley, T. Reyes-Robles, T. Wang, S. J. McCarver, C. H. White, F. P. Rodriguez-Rivera, D. L. Parker, E. C. Hett, O. O. Fadeyi, R. C. Oslund and D. W. C. MacMillan, Microenvironment mapping via Dexter energy transfer on immune cells, Science, 2020, 367, 1091–1097 CrossRef CAS PubMed.
- N. Tay, K. A. Ryu, J. Weber, A. Olow, D. Reichman, R. Oslund, O. O. Fadeyi and T. Rovis, Targeted Activation in Localized Protein Environments via Deep Red Photoredox Catalysis, ChemRxiv, 2021, preprint, DOI:10.26434/chemrxiv-2021-x9bjv, accessed 2022-07-28.
- S. Sato and H. Nakamura, Ligand-Directed Selective Protein Modification Based on Local Single-Electron-Transfer Catalysis, Angew. Chem., Int. Ed., 2013, 52, 8681–8684 CrossRef CAS PubMed.
- B. X. Li, D. K. Kim, S. Bloom, R. Y. C. Huang, J. X. Qiao, W. R. Ewing, D. G. Oblinsky, G. D. Scholes and D. W. C. MacMillan, Site-selective tyrosine bioconjugation via photoredox catalysis for native-to-bioorthogonal protein transformation, Nat. Chem., 2021, 13, 902–908 CrossRef CAS PubMed.
- N. Stephanopoulos and M. B. Francis, Choosing an effective protein bioconjugation strategy, Nat. Chem. Biol., 2011, 7, 876–884 CrossRef CAS PubMed.
- V. Hung, N. D. Udeshi, S. S. Lam, K. H. Loh, K. J. Cox, K. Pedram, S. A. Carr and A. Y. Ting, Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2, Nat. Protoc., 2016, 11, 456–475 CrossRef CAS PubMed.
- M. Tsushima, S. Sato and H. Nakamura, Selective Purification and Chemical Labelling of a Target Protein on Ruthenium Photocatalyst Functionalized Affinity Beads, Chem. Commun., 2017, 53, 4838–4841 RSC.
- D. A. Fancy and T. Kodadek, Chemistry for the Analysis of Protein-Protein Interactions: Rapid and Efficient Cross-Linking Triggered by Long Wavelength Light, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6020–6024 CrossRef CAS PubMed.
- K. A. Niederer, P. H. Gilmartin and M. C. Kozlowski, Oxidative Photocatalytic Homo-and Cross-Coupling of Phenols: Nonenzymatic; Catalytic Method for Coupling Tyrosine, ACS Catal., 2020, 10, 14615–14623 CrossRef CAS PubMed.
- P. Danaher, S. Warren, L. Dennis, L. D'Amico, A. White, M. L. Disis, M. A. Geller, K. Odunsi, J. Beechem and S. P. Fling, Gene expression markers of tumor infiltrating leukocytes, J. Immunother. Cancer, 2017, 5, 1–15 CrossRef PubMed.
- A. Juneau, T. O. Hope, J. Malenfant, M. Mesko, J. McNeill and M. Frenette, Methods to Predict Potential Reagents in Iridium-Based Photoredox Catalysis Calibrated with Stern–Volmer Quenching Rate Constants, ACS Catal., 2022, 12, 2348–2356 CrossRef CAS.
- A. C. Hernandez-Perez and S. K. Collins, Heteroleptic Cu-based sensitizers in photoredox catalysis, Acc. Chem. Res., 2016, 49, 1557–1565 CrossRef CAS PubMed.
- F. Glaser and O. S. Wenger, Recent progress in the development of transition-metal based photoredox catalysts, Coord. Chem. Rev., 2020, 405, 213129 CrossRef CAS.
- Y. B. Lee, S. Lim, Y. Lee, C. H. Park and H. J. Lee, Green Chemistry for Crosslinking Biopolymers: Recent Advances in Riboflavin-Mediated Photochemistry, Materials, 2023, 16, 1218 CrossRef CAS PubMed.
- (a) B. Zhuang, U. Liebl and M. H. Vos, Flavoprotein Photochemistry: Fundamental Processes and Photocatalytic Perspectives, J. Phys. Chem. B, 2022, 126, 3199–3207 CrossRef CAS PubMed; (b) D. Grosheva and T. K. Hyster, Light-driven flavin-based biocatalysis, Flavin-Based Catalysis: Principles and Applications, ed. R. Cibulka and M. Fraaije, Wiley-VCH Verlag GmbH & Co. KGaA, 2021, pp. 291–313, DOI:10.1002/9783527830138.ch12.
- S. P. Pitre, C. D. McTiernan, W. Vine, R. DiPucchio, M. Grenier and J. C. Scaiano, Visible-light actinometry and intermittent illumination as convenient tools to study Ru(bpy)3Cl2 mediated photoredox transformations, Sci. Rep., 2015, 5, 1–10 CrossRef PubMed.
- M. C. DeRosa and R. J. Crutchley, Photosensitized singlet oxygen and its applications, Coord. Chem. Rev., 2002, 233, 351–371 CrossRef.
- M. J. Thomas and C. S. Foote, Chemistry of singlet oxygen—XXVI. Photooxygenation of phenolsy, Photochem. Photobiol., 1978, 27, 683–693 CrossRef CAS.
- F. E. Scully Jr and J. Hoigné, Rate constants for reactions of singlet oxygen with phenols and other compounds in water, Chemosphere, 1987, 16, 681–694 CrossRef.
- J. Al-Nu'airat, B. Z. Dlugogorski, X. Gao, N. Zeinali, J. Skut, P. R. Westmoreland, I. Oluwoye and M. Altarawneh, Reaction of phenol with singlet oxygen, Phys. Chem. Chem. Phys., 2019, 21, 171–183 RSC.
- W. Chen, J. J. Chen, R. Lu, C. Qian, W. W. Li and H. Q. Yu, Redox reaction characteristics of riboflavin: a fluorescence spectroelectrochemical analysis and density functional theory calculation, Bioelectrochemistry, 2014, 98, 103–108 CrossRef CAS PubMed.
- G. W. Burton and K. U. Ingold, Vitamin E: application of the principles of physical organic chemistry to the exploration of its structure and function, Acc. Chem. Res., 1986, 19, 194–201 CrossRef CAS.
- M. Frenette, C. Aliaga, E. Font-Sanchis and J. C. Scaiano, Bond dissociation energies for radical dimers derived from highly stabilized carbon-centered radicals, Org. Lett., 2004, 6, 2579, DOI:10.1021/ol049111j.
- T. Michon, M. Chenu, N. Kellershon, M. Desmadril and J. Guéguen, Horseradish peroxidase oxidation of tyrosine-containing peptides and their subsequent polymerization: a kinetic study, Biochemistry, 1997, 36, 8504–8513 CrossRef CAS PubMed.
- J. V. Oakley, B. F. Buksh, D. F. Fernández, D. G. Oblinsky, C. P. Seath, J. B. Geri, G. D. Scholes and D. W. C. MacMillan, Radius measurement via super-resolution microscopy enables the development of a variable radii proximity labelling platform, Proc. Natl. Acad. Sci. U. S. A., 2022, 19, e2203027119 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00638g |
| ‡ Current address: InduPro, Cambridge, Massachusetts, USA. |
| This journal is © The Royal Society of Chemistry 2023 |