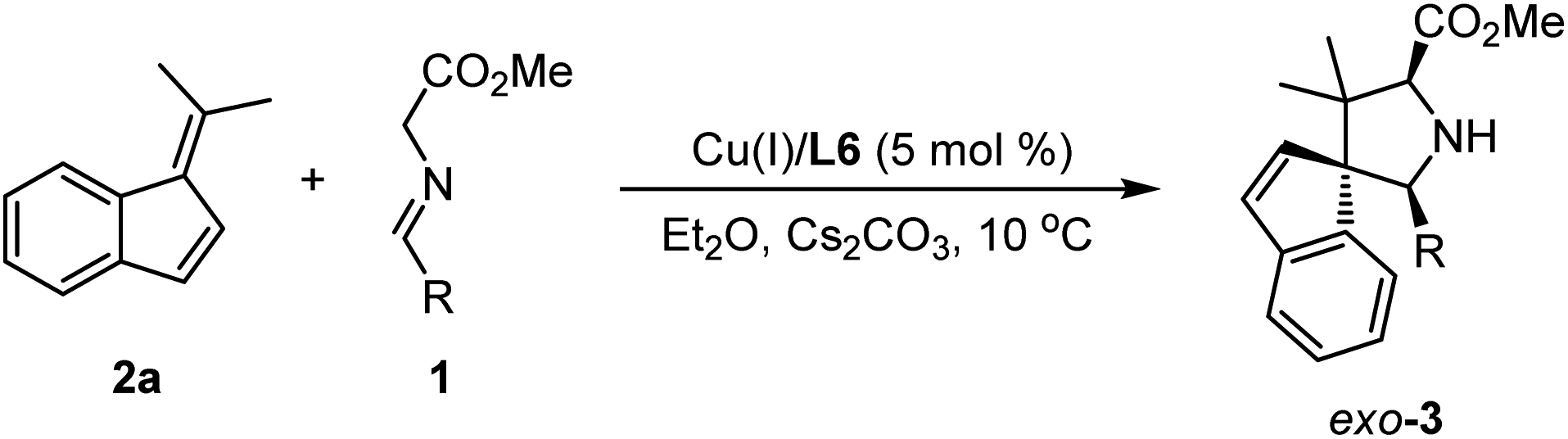DOI:
10.1039/D3SC00435J
(Edge Article)
Chem. Sci., 2023,
14, 5460-5469
Electron-rich benzofulvenes as effective dipolarophiles in copper(I)-catalyzed asymmetric 1,3-dipolar cycloaddition of azomethine ylides†
Received
26th January 2023
, Accepted 25th April 2023
First published on 25th April 2023
Abstract
A series of benzofulvenes without any electron-withdrawing substituents were employed as 2π-type dipolarophiles for the first time to participate in Cu(I)-catalyzed asymmetric 1,3-dipolar cycloaddition (1,3-DC) reactions of azomethine ylides. An intrinsic non-benzenoid aromatic characteristic from benzofulvenes serves as a key driving force for activation of the electron-rich benzofulvenes. Utilizing the current methodology, a wide range of multi-substituted chiral spiro-pyrrolidine derivatives containing two contiguous all-carbon quaternary centers were formed in good yield with exclusive chemo-/regioselectivity and high to excellent stereoselectivity. Computational mechanistic studies elucidate the origin of the stereochemical outcome and the chemoselectivity, in which the thermostability of these cycloaddition products is the major factor.
Introduction
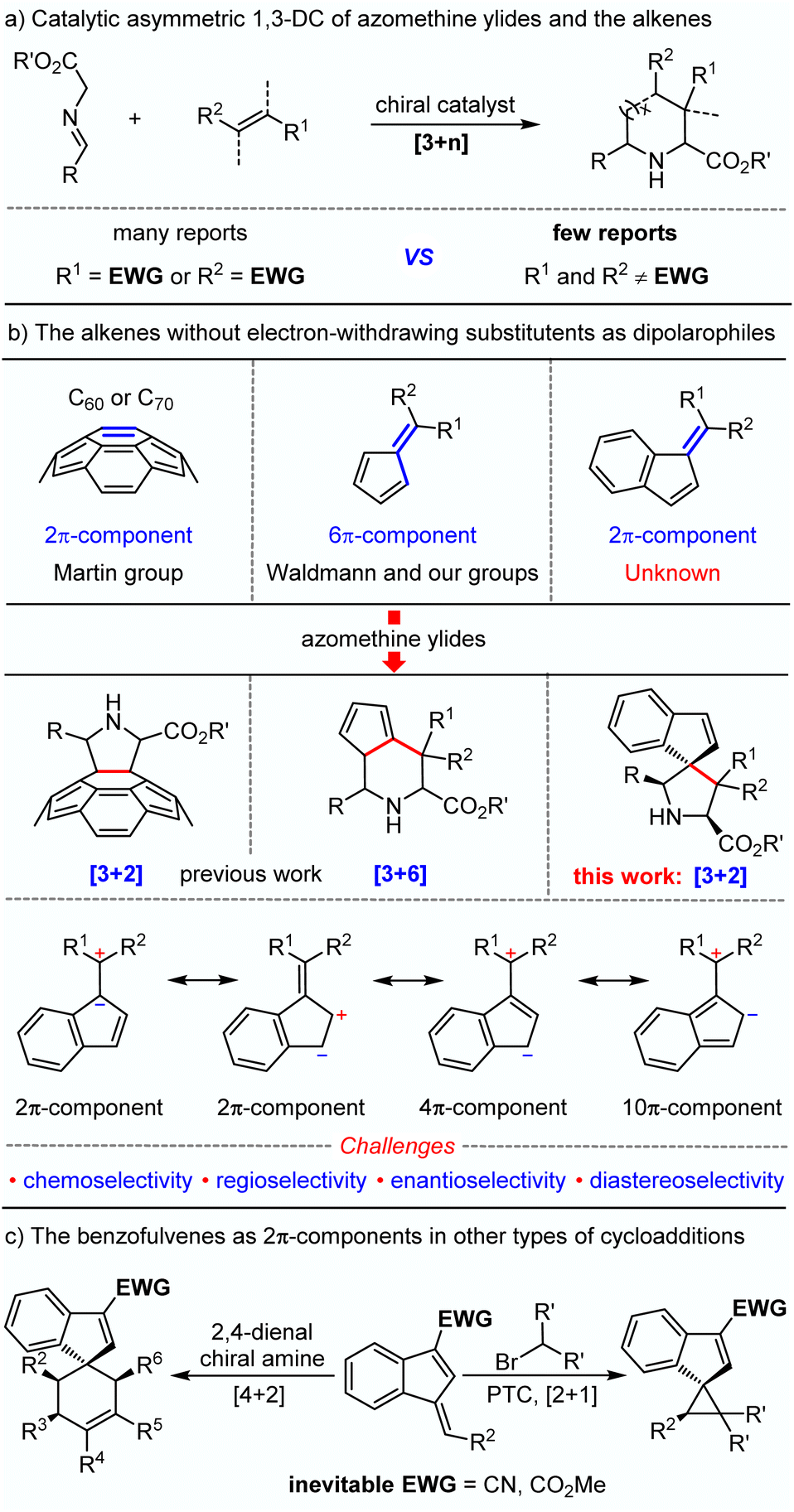
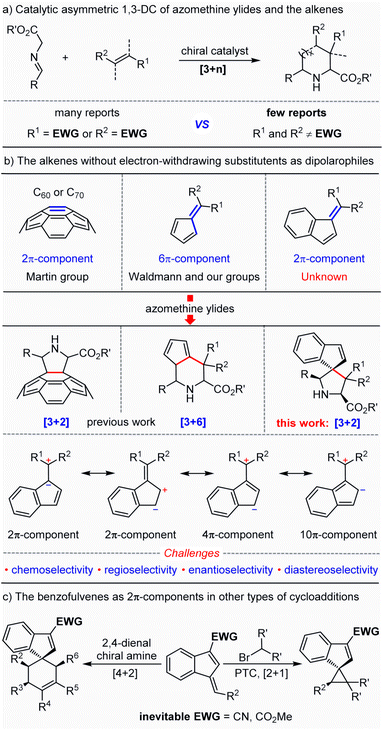
The catalytic asymmetric 1,3-dipolar cycloaddition (1,3-DC) reactions of azomethine ylides are one of the most powerful and versatile strategies for the construction of enantioenriched nitrogen-containing heterocyclic compounds,1,2 which serve as the privileged skeletons widely distributed in a range of bioactive natural products and pharmaceutical molecules.3 As a consequence, great efforts have been devoted to developing various novel catalytic systems for further extension of the applicable range of this methodology.4 Among the currently reported examples, alkenes bearing at least one electron-withdrawing group to activate alkenes (dipolarophiles) have been commonly and generally used as dipolarophiles. In sharp contrast, alkenes without any electron-withdrawing substituents rarely play a role of dipolarophiles in the catalytic asymmetric 1,3-dipolar cycloaddition reaction with azomethine ylides,5 which may be ascribed to the absence of LUMO activation of alkenes (Scheme 1a). It was not until 2009 that this challenge was first attempted by the Martin group.6 They reported a seminal asymmetric version of transition metal-catalyzed 1,3-dipolar cycloaddition reactions between fullerene C60 and azomethine ylides, successfully constructing an array of enantioenriched pyrrolidinofullerenes in high yields with excellent enantio-selectivities under mild reaction conditions (Scheme 1b). The strain of the curved double bond in C60 serving as the 2π-component provides the crucial driving force for the event of 1,3-dipolar cycloaddition.7 Later, Waldmann8 and our group9 independently developed fulvene-involved catalytic asymmetric higher-order [3 + 6] cycloaddition reactions of azomethine ylides, in which electron-rich fulvenes acted as 6π-components and the intrinsic nonbenzenoid aromatic character10 of fulvenes offers the major driving force. Despite these advances, such catalytic synthetic transformations with electron-rich olefines are still of great challenge and have remained in an infant stage. Therefore, further investigation of other types of olefines without any electron-withdrawing groups as dipolarophiles in azomethine ylide-involved catalytic asymmetric 1,3-dipolar cycloadditions was in high demand and desirable.
 |
| | Scheme 1 (a and b) Catalytic asymmetric 1,3-DC of azomethine ylides and alkenes. (c) Other cycloaddition reactions with benzofulvene derivatives incorporating electron-withdrawing substituents. | |
Benzofulvenes, a kind of synthon having semi-aromatic characteristics, have been mainly employed in the synthesis of polymeric compounds and transition-metal-complexes.11 In comparison, the utilization of these synthons to construct small molecules was rarely reported, particularly in a catalytic asymmetric manner,12 which may be ascribed to the low activity of the diene in the benzofulvene. In 2016, Jørgensen introduced an electron-withdrawing group at the 3-position of the benzofulvene to enhance the reactivity of the exocyclic double bond, which served as a 2π-component to react with 2,4-dienals or dimethyl bromomalonate in the presence of chiral catalysts.12a,b With the aid of electron-withdrawing groups, a series of spiroindenes bearing an all-carbon quaternary stereocenter were smoothly formed in excellent yield and diastereoselectivity (Scheme 1c). In continuation of our research interest in azomethine ylide-involved catalytic asymmetric 1,3-dipolar cycloaddition reactions,2i,j,13 we envisioned that benzofulvenes without any electron-withdrawing substituents may serve as 2π type dipolarophiles to participate in azomethine ylide-involved catalytic asymmetric 1,3-dipolar cycloadditions to enable the modular assembly of enantioenriched spiro-pyrrolidine derivatives. Nevertheless, several challenges may exist in the design: (1) whether reactivity driven by the intrinsic nonbenzenoid aromatic characteristic is enough for the novel dipolarophiles (benzofulvenes) because electron-withdrawing groups were inevitable in the previous [2 + 1] and [4 + 2] cycloadditions promoted by organocatalysts; (2) the control of chemo- and regioselectivity due to the existence of diverse electronic resonance structures in benzofulvene; (3) the construction of two contiguous all carbon quaternary centers including a challenging spiro chirality in a catalytic asymmetric manner.
Herein, we reported an unprecedented catalytic asymmetric 1,3-dipolar cycloaddition reaction of azomethine ylides and benzofulvenes, in which benzofulvenes without any electron-withdrawing substituents were capable to act as efficient 2π dipolarophiles for the high-efficiency synthesis of a series of multi-substituted enantioenriched pyrrolidine derivatives bearing a spiroindene molecular architecture which is prevalent in diverse active pharmaceutical ingredients and natural products.14
Results and discussion
Reaction development and optimization
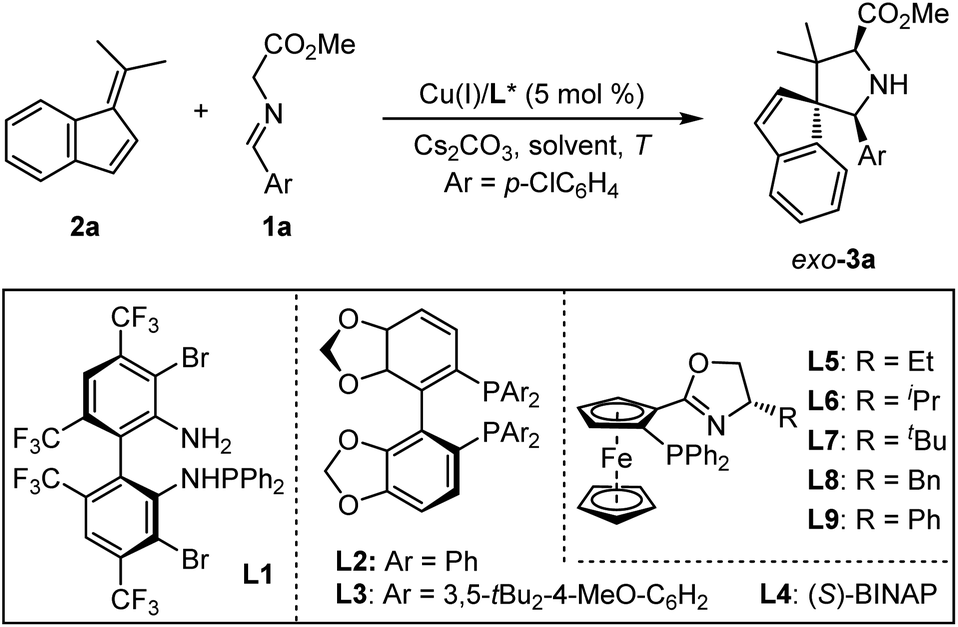
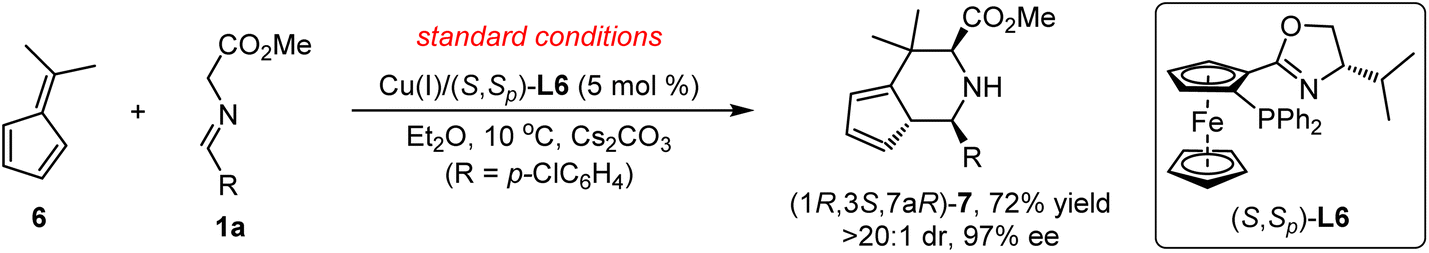
Initially, we choose readily available benzofulvene 2a and aldimine ester 1a as the model substrate and Cs2CO3 as the base to examine the feasibility of this design. As shown in Table 1, the optimal chiral ligand TF-BiphamPhos9,15 in the fulvene-involved catalytic asymmetric 1,3-dipolar [3 + 6] cycloaddition was first examined in dichloromethane at room temperature. Unfortunately, no desired spiroheterocyclic product was observed (Table 1, entry 1). We turned our attention to the investigation of other types of chiral ligands. Chiral bisphosphine ligands (L2, L3, L4) did make the corresponding reaction happen, but with unsatisfactory yield and stereoselectivity (Table 1, entries 2–4). Subsequently, a series of chiral N–P ligands Phosferrox L5–L9 were further evaluated (Table 1, entries 5–9). These experimental results suggested that the chiral ligand L6 derived from L-valine represents a better performance, delivering the desired cycloadduct 3a in 98% yield with 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and 87% ee (Table 1, entry 6). To further improve the stereoselectivity, we explored other experimental conditions. Switching Cu(I) with Ag(I) was proved to be ineffective and can be detrimental to diastereoselectivity (Table 1, entry 10). Reducing the reaction temperature to 10 °C led to an obvious increase in the enantioselectivity at the expense of diastereoselectivity (Table 1, entry 11). The reaction became very sluggish when the temperature was reduced further (Table 1, entry 12). Solvent-screening (Table 1, entries 13–19) revealed that Et2O was the best choice, affording a satisfactory outcome (81% yield, 18:1 dr, 96% ee; Table 1, entry 15), which was determined as the optimal reaction condition in this study.
:1 dr and 87% ee (Table 1, entry 6). To further improve the stereoselectivity, we explored other experimental conditions. Switching Cu(I) with Ag(I) was proved to be ineffective and can be detrimental to diastereoselectivity (Table 1, entry 10). Reducing the reaction temperature to 10 °C led to an obvious increase in the enantioselectivity at the expense of diastereoselectivity (Table 1, entry 11). The reaction became very sluggish when the temperature was reduced further (Table 1, entry 12). Solvent-screening (Table 1, entries 13–19) revealed that Et2O was the best choice, affording a satisfactory outcome (81% yield, 18:1 dr, 96% ee; Table 1, entry 15), which was determined as the optimal reaction condition in this study.
Table 1 Optimization of reaction conditionsa
Substrate scope
With the optimal reaction conditions in hand, we commenced examining the generality of this catalytic asymmetric 1,3-dipolar cycloaddition by treating benzofulvene 2a with different aldimine esters, 1a–1n. As shown in Table 2, a variety of aldimine esters bearing electron-withdrawing, electron-neutral and electron-donating substituents at the ortho-, meta- or para-position of the aromatic ring could react successfully with 2a, affording the corresponding cycloadducts 3a–3i in good yields (67–98%) with excellent stereoselectivity (10:1–20:1 dr, 91–98% ee; entries 1–9). With further extension from the phenyl ring to a fused aromatic ring (bulky 1-naphthyl 1j and 2-naphthyl 1k), an excellent level of enantioselectivity (96 and 95% ee) could still be achieved with moderate to good yield (56 and 98% yield) and diastereoselectivity (7:1 and 10:1 dr; entries 10 and 11). In addition, subjecting heteroaromatic aldehyde-derived imine ester 1l and 1m to this catalytic system, the desired cycloadducts 3l and 3m were isolated in acceptable yields with high to excellent enantioselectivity (entries 12 and 13). It is worth noting that the challenging and less reactive aldimine ester 1n derived from cyclohexylaldehyde was proven to be a suitable precursor of azomethine ylide, furnishing the cycloadduct 3n in 72% yield with 20:1 dr and 97% ee (entry 14). The absolute configuration of 3j was unambiguously determined as (1R,2′S,5′S) by the X-ray diffraction analysis (Fig. 1).16
Table 2 Substrate scope of imine estersa
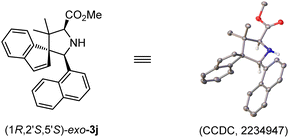 |
| | Fig. 1 X-Ray Structure of (1R,2′S,5′S)-3j. | |
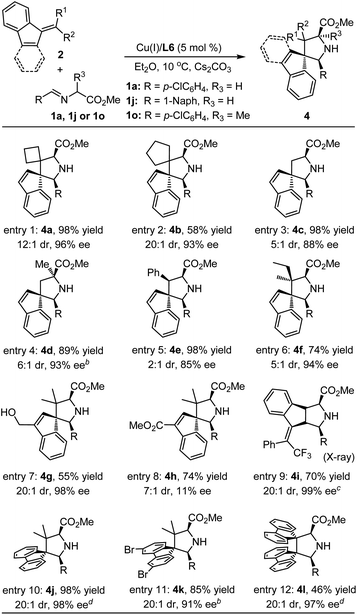
Having established the scope of aldimine esters, we then set out to evaluate the scope of benzofulvenes 2 with aldimine ester 1a. As outlined in Table 3, employing symmetrical cyclobutanone and cyclopentanone-derived benzofulvenes 2b and 2c as dipolarophiles, the corresponding cycloadducts 4a and 4b containing two contiguously spirocyclic skeletons could be formed smoothly in good to excellent yield with good stereoselectivity(entries 1–2). Notably, when the highly active benzofulvene 2d bearing a terminal alkene moiety was selected as the dipolarophile, the corresponding cycloaddition could occur in a satisfactory result, even when the more challenging α-Me-substituted aldimine ester 1o was employed in this system (entries 3 and 4). Switching the symmetrical ketone/aldehyde with unsymmetrical 2-butanone or benzaldehyde, a similar reactivity and enantiocontrol were also achieved with acceptable diastereoselectivity (entries 5 and 6). To further explore the compatibility of this reaction, a series of substituent groups possessing different electronic properties on the benzofulvene were further investigated. The corresponding experimental results indicated that the electron-donating hydroxylmethyl group positioned at the endocyclic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond had little impact on the reactivity and selectivity (entry 7). The electron-withdrawing CO2Me group located at the endocyclic CC bond indeed enhanced the polarization of the exocyclic CC bond, affording the corresponding spiro heterocyclic adduct 4h in good yield albeit with unsatisfactory stereoselectivity (7:1 dr and 11% ee) and yield (74%) (entry 8). Switching the electron-withdrawing group from the endocyclic double bond to the exocyclic double bond would directly result in the change of regioselectivity. The endocyclic double bond reacted with azomethine ylides, giving rise to the corresponding fused cycloadduct 4i in moderate yield (70%) with excellent stereoselectivity (20:1 dr, 99% ee), which revealed that the inductive effect of the CF3 group was stronger than the driving force from the reduced intrinsic nonbenzenoid aromatic character of the benzofulvene 2i (entry 9). The absolute configuration of 4i was unambiguously determined as (1R,3S,3aR,8aS,E) by the X-ray diffraction analysis.16 In addition to indene-derived benzofulvenes, more bulky 9-fluorenone-derived olefins proved to be viable substrates for this catalytic asymmetric cycloaddition reaction, producing the desired cycloadducts 4j and 4k in high yield (98%, 85%) with excellent diastereoselectivity (20:1 dr) and enantiomeric excess (98%, 91%, entries 10 and 11). Encouraged by these results, a more challenging dipolarophile 2l bearing double bulkier fluorene moieties was investigated to further determine the compatibility of this catalytic system. Gratifyingly, the corresponding cycloaddition reactions still underwent smoothly to deliver the desired adducts 4l in 46% yield with exclusive diastereoselectivity and excellent enantioselectivity (entry 12).
C bond had little impact on the reactivity and selectivity (entry 7). The electron-withdrawing CO2Me group located at the endocyclic CC bond indeed enhanced the polarization of the exocyclic CC bond, affording the corresponding spiro heterocyclic adduct 4h in good yield albeit with unsatisfactory stereoselectivity (7:1 dr and 11% ee) and yield (74%) (entry 8). Switching the electron-withdrawing group from the endocyclic double bond to the exocyclic double bond would directly result in the change of regioselectivity. The endocyclic double bond reacted with azomethine ylides, giving rise to the corresponding fused cycloadduct 4i in moderate yield (70%) with excellent stereoselectivity (20:1 dr, 99% ee), which revealed that the inductive effect of the CF3 group was stronger than the driving force from the reduced intrinsic nonbenzenoid aromatic character of the benzofulvene 2i (entry 9). The absolute configuration of 4i was unambiguously determined as (1R,3S,3aR,8aS,E) by the X-ray diffraction analysis.16 In addition to indene-derived benzofulvenes, more bulky 9-fluorenone-derived olefins proved to be viable substrates for this catalytic asymmetric cycloaddition reaction, producing the desired cycloadducts 4j and 4k in high yield (98%, 85%) with excellent diastereoselectivity (20:1 dr) and enantiomeric excess (98%, 91%, entries 10 and 11). Encouraged by these results, a more challenging dipolarophile 2l bearing double bulkier fluorene moieties was investigated to further determine the compatibility of this catalytic system. Gratifyingly, the corresponding cycloaddition reactions still underwent smoothly to deliver the desired adducts 4l in 46% yield with exclusive diastereoselectivity and excellent enantioselectivity (entry 12).
Table 3 Substrate scope of benzofulvenesa
| All reactions were carried out with 0.4 mmol 1 and 0.2 mmol 2, 0.01 mmol Cu/L6 and 0.2 mmol of Cs2CO3 in 2 mL of Et2O for 16 h, Cu(I) = Cu(CH3CN)4BF4. Isolated yield. The dr value was determined by the crude 1H NMR, and the ee value was determined by HPLC analysis. The reaction was run at 0 °C. R = 1 − Naph. The reaction was run at 20 °C. |
 |
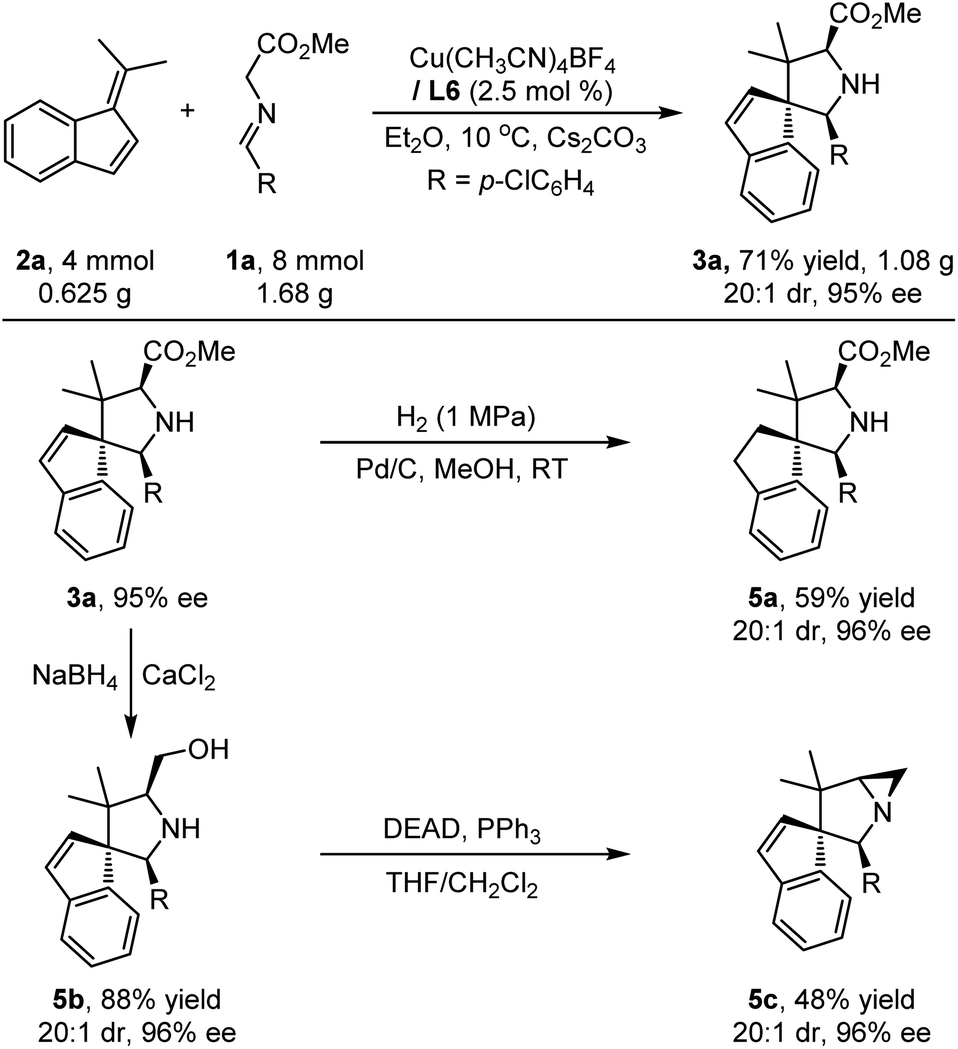
Scale-up experiments and synthetic application
To demonstrate the practicability and synthetic utility of the current methodology, we performed a gram–scale reaction of 1a and 2a under slightly modified reaction conditions with reduced catalyst loading (2.5 mol%). As shown in Scheme 2, a comparable yield (71%) and stereoselectivity (20:1 dr, 95% ee) were observed. Under 1 MPa of H2, Pd/C-catalyzed reduction of the endocyclic double bond of the product 3a could be realized, providing the target compound 5a in 59% yield with maintained stereoselectivity (20:1 dr, 96% ee). Treating cycloadduct 3a with NaBH4 and CaCl2 could selectively reduce the ester group of 3a efficiently, and the corresponding amino alcohol 5b was isolated in high yield with excellent stereoselectivity, which could be further stereospecifically transformed into more complex product 5c bearing a biologically important 1-azabicyclo[3.1.0]hexane heterocyclic moiety.17
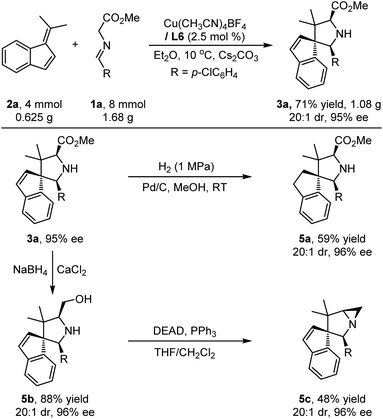 |
| | Scheme 2 Scale-up experiments and synthetic elaborations. | |
Computational study of the reaction mechanism for the formation of 3a
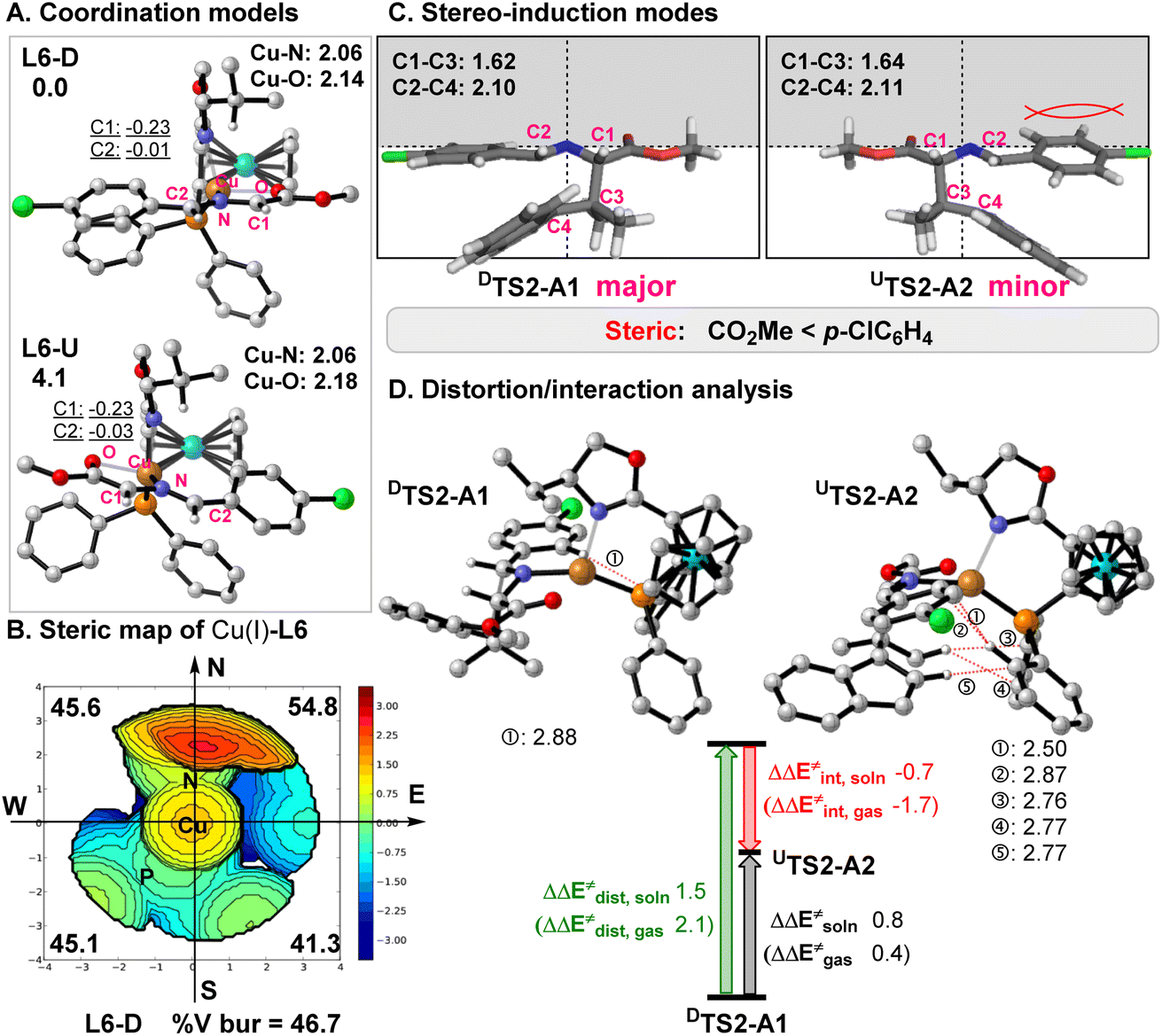
To understand the origin of the stereo-, chemo-, and regioselective 1,3-dipolar cycloaddition reaction of benzofulvenes catalyzed by Cu(I)-L6, systematic DFT calculations (the PCM M06-L//M06-L method mainly) were carried out.18–20 As shown in Fig. 2A, there are two possible coordination models (L6-U and L6-D) for a four-coordinate intermediate containing Cu(I)-L6 and the deprotonated 1a (dipole). L6-D was computed to be more stable than L6-U by 4.1 kcal mol−1 in the solution by the PCM M06-L//M06-L method, due to more steric congestion between the ferrocene group in L6 and a larger aryl substituent in the dipole (more C–H repulsion in L6-U, see Fig. 2A and S3†). In addition, the steric map21 (Fig. 2B) shows that the i-Pr group in L6 occupies the upper coordination sphere and should exclude the cycloaddition of benzofulvene substrates on the top side. Therefore, our results show that the dipolarophile substrate preferentially attacks the dipole from the bottom side (Fig. 2C).
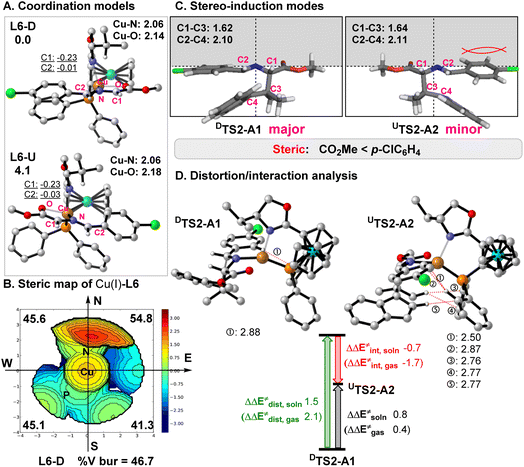 |
| | Fig. 2 (A) Two optimized coordination modes (L6-D and L6-U) of the active four-coordinate Cu(I) intermediate in the gas phase by the M06-L method with the key bond distances. The relative free energies and NPA charge for the key atoms in diethylether solution by the PCM M06-L//M06-L method are given. (B) Steric map of the Cu(I)-L6 part from the optimized L6-D with the computed buried volumes. (C) Stereo-induction modes for the enantioselectivity. (D) Distortion/interaction analysis for the Cu-catalyzed [3 + 2] cycloaddition of 2a with the key repulsions between substrates and L6 (in angstrom). | |
Fig. 2 and 3 further depict two energetically favorable [3 + 2] cycloaddition pathways for the substrate 2a. Our DFT calculations show that the formation of these favorable major and minor cycloaddition products (A1 and A2) is a two-step process. Addition of 2a to the four-coordinate intermediate L6-D (or L6-U) forms a less stable intermediate DIN1-A1 (or UIN1-A2; ΔGsoln = 5.2–5.8 kcal mol−1). Then, the first C1–C3 bond addition from an electron-rich C1 of the dipole (q(C1): −0.23, Fig. 2A) to the C3 site of the dipolarophile occurs viaDTS1-A1 or UTS1-A2 with a barrier of 16.5 or 19.9 kcal mol−1 to afford a less stable zwitterionic intermediate DIN2-A1 or UIN2-A2 (ΔGsoln = 14.1 or 18.9 kcal mol−1), respectively. Such regioselective addition to the C3 position is kinetically and thermodynamically much more favorable than the addition to another C4 position of the dipolarophile by at least 1.6 kcal mol−1 (Fig. 3 and S1†), due to stabilization of the formal resultant carbanion by π-conjugation interaction with the aromatic ring (c.f.DIN2-A1).
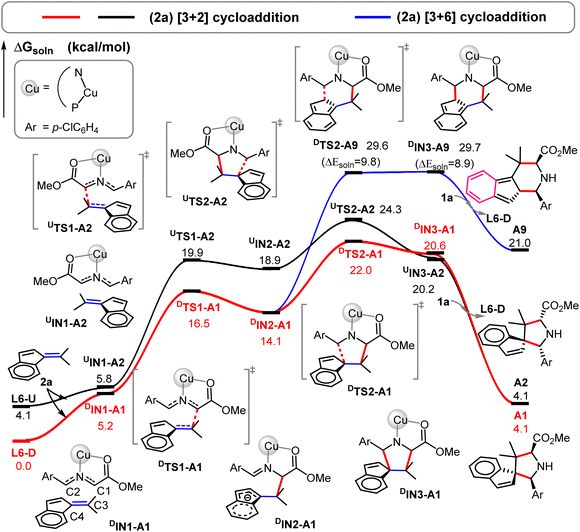 |
| | Fig. 3 Free energy profiles for the Cu-catalyzed [3 + 2] (in red and black solid lines) and [3 + 6] (in the blue solid line) cycloadditions of 2a in the solution by the PCM M06-L//M06-L method. | |
Afterward, DIN2-A1 and UIN2-A2 undergo the second C2–C4 bond addition to form the major cycloadduct intermediate DIN3-A1 and the minor cycloadduct UIN3-A2viaDTS2-A1 and UTS2-A2, respectively. This final cyclization step in both [3 + 2] pathways is suggested to be the rate-determining step with a barrier of 22.0–24.3 kcal mol−1. DTS2-A1 leading to the major product DIN3-A1 has a lower barrier than UTS2-A2 forming UIN3-A2 (ΔΔGsoln± = ∼2.3 kcal mol−1 and ΔΔEsoln± = ∼0.8 kcal mol; Fig. 2 and 3), which qualitatively explains the observed stereo- and regiochemistry. The entropy effect (∼1.1 kcal mol−1) and a smaller distortion energy22 (ΔΔEdist,soln = ∼1.5 kcal mol; Fig. 2d) play the key roles in controlling the enantioselectivity, whereas the solvation also slightly favors the major-type cyclization TS (∼0.4 kcal mol−1). In addition, more congestion between the substrates and ligand were found in UTS2-A2 than DTS2-A1 (Fig. 2d). Finally, ligand exchange of DIN3-A1 (or UIN3-A2) with another 1a substrate can regenerate the active species (L6-D) as well as release the cycloaddition product A1 (or A2) and energy.
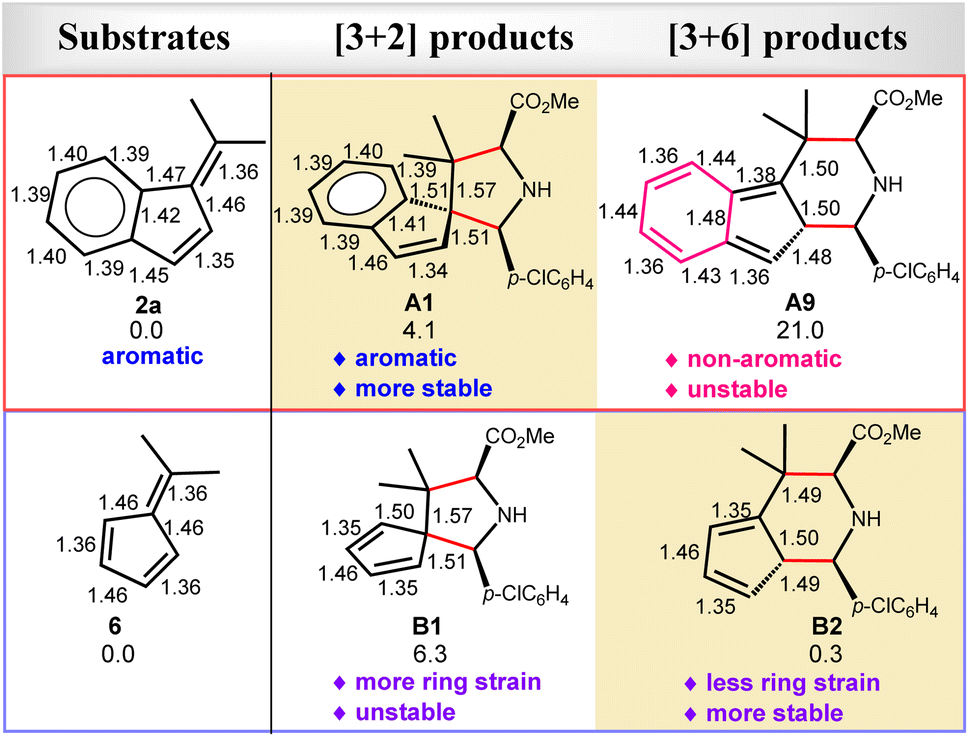
In comparison, our calculations reveal that DIN2-A1 requires a much larger barrier (29.6 kcal mol−1) for the [3 + 6] cycloaddition (branching) pathway viaDTS2-A9 by 7.6 kcal mol−1, compared to the most-favorable major pathway viaDTS2-A1. Remarkably, the [3 + 6] cycloadduct intermediate DIN3-A9 is found to be highly unstable (ΔGsoln = 29.7 kcal mol−1). It should be noted that the [3 + 6] cycloaddition induces π-bond shifting and, thus, transforms the aromatic ring of 2a into the non-aromatic ring in the highly unstable [3 + 6] cycloaddition product A9 (ΔGsoln = 21.0 kcal mol−1). As A9 is thermodynamically much unstable than the [3 + 2] cycloaddition products A1 and A2 (ΔGsoln = 4.1 kcal mol−1), thermostability of these cycloaddition products is the major factor in the chemoselective [3 + 2] cycloaddition and excludes the formation of the unstable [3 + 6] cycloadducts with benzofulvenes (Fig. 3–5).
Interestingly, our DFT calculations predict that the same cycloaddition reaction with the simplest fulvene substrate 6 favors the [3 + 6] cycloaddition in this catalytic system (Scheme 3), instead of the [3 + 2] cycloaddition (Fig. 4 and 5). Its reaction barrier for the [3 + 2] cycloaddition (25.5 kcal mol−1viaDTS2-B1) is much higher than that for the [3 + 6] cycloaddition (16.9 kcal mol−1viaDTS2-B2) by 8.6 kcal mol−1. Notably, the [3 + 2] cycloadduct intermediate DIN3-B1 becomes thermodynamically less stable than the [3 + 6] cycloadduct intermediate DIN3-B2 by 12.4 kcal mol−1. Likewise, the [3 + 2] product B1 (5/5-spiro-ring; ΔGsoln = 6.3 kcal mol−1) is also less stable than the [3 + 6] product B2 (5/6-fused-ring; ΔGsoln = 0.3 kcal mol−1) by 6.0 kcal mol−1. Apparently, less ring strain and more substituted alkene part should gain more thermostability in the 5/6-fused ring ([3 + 6] cycloadduct) than the 5/5-spiro ring ([3 + 2] cycloadduct). Therefore, the higher thermostability in the simple 5/6-fused ring should be the key factor in dictating chemoselective [3 + 6] cycloaddition with fulvenes. In comparison, the [3 + 6] product (6/5/6-fused-ring) for benzofulvene 2a loses the aromatic stability and has more steric congestion between one methyl and phenyl groups (due to the roughly-planar geometry, Fig. S3†), which renders highly unstable [3 + 6] cycloadduct as well as raises its reaction barrier (29.6 kcal mol−1). In contrast, thermochemistry of the [3 + 2] product for 2a is slightly more favorable than that for 6, which promotes the [3 + 2] cycloaddition with 2a. These combined experimental and computational results demonstrate interesting substrate-dependent/switching chemoselective 1,3-dipolar cycloadditions.
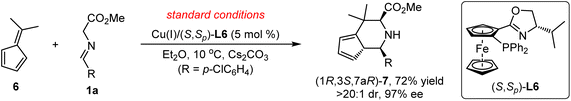 |
| | Scheme 3 Experimental proof of Cu(I)/(S,Sp)-L6-catalyzed [3 + 6] cycloaddition of aldimine ester 1a with fulvene 6 as the dipolarophile. | |
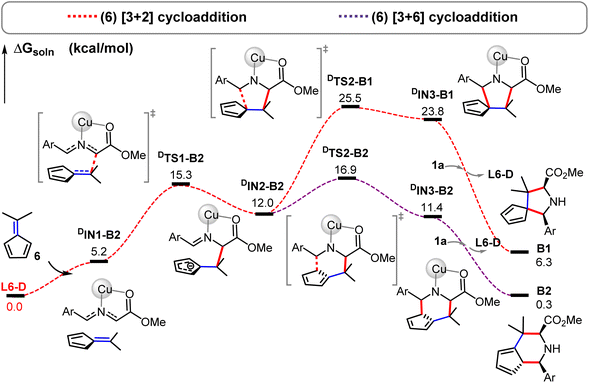 |
| | Fig. 4 Free energy profiles for the Cu-catalyzed [3 + 2] (in the red dashed line) and [3 + 6] (in the purple dashed line) cycloaddition reactions of substrate 6 in the solution by the PCM M06-L//M06-L method. | |
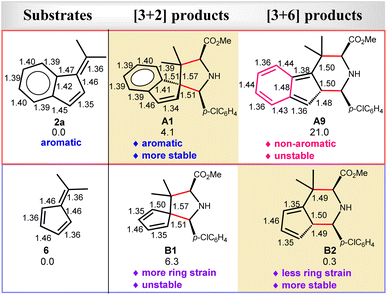 |
| | Fig. 5 The key bond distances (in angstrom) of the optimized substrates (2a and 6) and their corresponding cycloaddition products by the M06-L method (those optimized by the M06-2X method given in Fig. S4†). The relative free energies (kcal mol−1) of these cycloaddition products in the solution by the PCM M06-L//M06-L method are also given. | |
Conclusions
In conclusion, combining the intrinsic nonbenzenoid aromatic characteristic of benzofulvenes, we have successfully developed catalytic asymmetric [3 + 2] cycloaddition between azomethine ylides and electron-rich benzofulvenes without any electron-withdrawing substituents for the first time. Such efficient reaction gives access to a variety of multi-substituted chiral spiropyrrolidine derivatives containing two contiguous all carbon quaternary centers in high yields with exclusive chemo-/regioselectivity and high to excellent stereoselectivity. Extensive density functional theory (DFT) calculations not only elucidated the reaction mechanism of the formation of the cycloadducts, but also demonstrated that the excellent chemo-/regioselectivity is dependent on the thermostability of the corresponding cycloaddition products.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†
Author contributions
C.-J. W. conceived and designed the research. C.-J. W. and X. C. directed the project. X. C. and X.-T. L. performed the research. F. L., Y. Y., L. W. C. performed the DFT calculations. X. C., L. W. C. and C.-J. W. co-wrote the paper. All authors analysed the data, discussed the results, commented on the manuscript, collected data and prepared the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the NSFC (22071186, 22101216, 22073067, 21933003, 22193020, and 22193023), the Hubei Province Natural Science Foundation (2020CFA036), China Postdoctoral Science Foundation (2021M702514), the Fundamental Research Funds for the Central Universities (2042022kf1180), the Shenzhen Nobel Prize Scientists Laboratory Project (C17783101) and Guangdong Provincial Key Laboratory of Catalytic Chemistry (2020B121201002). The authors thank Dr Ran Zhang from the Core Facility of Wuhan University for his generous support in the X-ray structure analysis and the Center for Computational Science and Engineering at the Southern University of Science and Technology and CHEM HPC at SUSTech for partly supporting this work.
Notes and references
-
(a) R. Huisgen, Angew. Chem., Int. Ed., 1963, 2, 565–598 CrossRef
 ;
(b) S. L. Schreiber, Science, 2000, 287, 1964–1969 CrossRef CAS PubMed ;
(c) A. Padwa and W. H. Pearson, Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, ed. H. Feuer, Wiley-VCH, 2002 CrossRef .
;
(b) S. L. Schreiber, Science, 2000, 287, 1964–1969 CrossRef CAS PubMed ;
(c) A. Padwa and W. H. Pearson, Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, ed. H. Feuer, Wiley-VCH, 2002 CrossRef . - For selected reviews on catalytic asymmetric 1,3-dipolar cycloadditions, see:
(a) I. Coldham and R. Hufton, Chem. Rev., 2005, 105, 2765–2810 CrossRef CAS PubMed ;
(b) C. Nájera and J. M. Sansano, Angew. Chem., Int. Ed., 2005, 44, 6272–6276 CrossRef PubMed ;
(c) G. Pandey, P. Banerjee and S. R. Gadre, Chem. Rev., 2006, 106, 4484–4517 CrossRef CAS PubMed ;
(d) H. Pellissier, Tetrahedron, 2007, 63, 3235–3285 CrossRef CAS ;
(e) M. Álvarez-Corral, M. Muñoz-Dorado and I. Rodríguez-García, Chem. Rev., 2008, 108, 3174–3198 CrossRef PubMed ;
(f) L. M. Stanley and M. P. Sibi, Chem. Rev., 2008, 108, 2887–2902 CrossRef CAS PubMed ;
(g) J. Adrio and J. C. Carretero, Chem. Commun., 2011, 47, 6784–6794 RSC ;
(h) J. Adrio and J. C. Carretero, Chem. Commun., 2014, 50, 12434–12446 RSC ;
(i) T. Hashimoto and K. Maruoka, Chem. Rev., 2015, 115, 5366–5412 CrossRef CAS PubMed ;
(j) X. Fang and C.-J. Wang, Org. Biomol. Chem., 2018, 16, 2591–2601 RSC ;
(k) L. Wei, X. Chang and C.-J. Wang, Acc. Chem. Res., 2020, 53, 1084–1100 CrossRef CAS PubMed .
-
(a) K. Ding, Y. Lu, Z. Nikolovska-Coleska, S. Qiu, Y. Ding, W. Gao, J. Stuckey, K. Krajewski, P. P. Roller, Y. Tomita, D. A. Parrish, J. R. Deschamps and S. Wang, J. Am. Chem. Soc., 2005, 127, 10130–10131 CrossRef CAS PubMed ;
(b) J. P. Michael, Nat. Prod. Rep., 2008, 25, 139–165 RSC ;
(c) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed ;
(d) M. Setoguchi, S. Iimura, Y. Sugimoto, Y. Yoneda, J. Chiba, T. Watanabe, F. Muro, Y. Iigo, G. Takayama, M. Yokoyama, T. Taira, M. Aonuma, T. Takashi, A. Nakayama and N. Machinaga, Bioorg. Med. Chem., 2013, 21, 42–61 CrossRef CAS PubMed ;
(e) Y. Zhao, L. Liu, W. Sun, J. Lu, D. McEachern, X. Li, S. Yu, D. Bernard, P. Ochsenbein, V. Ferey, J.-C. Carry, J. R. Deschamps, D. Sun and S. Wang, J. Am. Chem. Soc., 2013, 135, 7223–7234 CrossRef CAS PubMed ;
(f) M. Kuhnert, A. Blum, H. Steuber and W. E. Diederich, J. Med. Chem., 2015, 58, 4845–4850 CrossRef CAS PubMed ;
(g) Y. Zhao, A. Aguilar, D. Bernard and S. Wang, J. Med. Chem., 2015, 58, 1038–1052 CrossRef CAS PubMed ;
(h) L. Shu, C. Gu, D. Fishlock and Z. Li, Org. Process Res. Dev., 2016, 20, 2050–2056 CrossRef CAS ;
(i) J. Hartung, S. N. Greszler, R. C. Klix and J. M. Kallemeyn, Org. Process Res. Dev., 2019, 23, 2532–2537 CrossRef CAS ;
(j) T. Ince, R. Serttas, B. Demir, H. Atabey, N. Seferoglu, S. Erdogan, E. Sahin, S. Erat and Y. Nural, J. Mol. Struct., 2020, 1217, 128400 CrossRef CAS ;
(k) K. V. Kudryavtsev, M. N. Sokolov, E. E. Varpetyan, A. A. Kirsanova, N. I. Fedotcheva, N. L. Shimanovskii and T. A. Fedotcheva, ChemistrySelect, 2020, 5, 11467–11470 CrossRef CAS ;
(l) X.-J. Zou, W.-L. Yang, J.-Y. Zhu and W.-P. Deng, Chin. J. Chem., 2020, 38, 435–438 CrossRef CAS .
- For the most recent examples of transition-metal-catalyzed asymmetric 1,3-dipolar cycloadditions, see:
(a) I. N. Chaithanya Kiran, K. Fujita, S. Tanaka and M. Kitamura, ChemCatChem, 2020, 12, 5613–5617 CrossRef CAS ;
(b) X. Cheng, D. Yan, X.-Q. Dong and C.-J. Wang, Asian J. Org. Chem., 2020, 9, 1567–1570 CrossRef CAS ;
(c) H. Cui, K. Li, Y. Wang, M. Song, C. Wang, D. Wei, E.-Q. Li, Z. Duan and F. Mathey, Org. Biomol. Chem., 2020, 18, 3740–3746 RSC ;
(d) S. Furuya, K. Kanemoto and S.-i. Fukuzawa, J. Org. Chem., 2020, 85, 8142–8148 CrossRef CAS PubMed ;
(e) E. Garcia-Mingueens, V. Selva, O. Larranaga, C. Najera, J. M. Sansano and A. de Cozar, ChemCatChem, 2020, 12, 2014–2021 CrossRef CAS ;
(f) J.-P. Tan, X. Li, Y. Chen, X. Rong, L. Zhu, C. Jiang, K. Xiao and T. Wang, Sci. China: Chem., 2020, 63, 1091–1099 CrossRef CAS ;
(g) D. Zhang, Z. Su, Q. He, Z. Wu, Y. Zhou, C. Pan, X. Liu and X. Feng, J. Am. Chem. Soc., 2020, 142, 15975–15985 CrossRef CAS PubMed ;
(h) R. G. Biswas, S. K. Ray, V. K. Kannaujiya, R. A. Unhale and V. K. Singh, Org. Biomol. Chem., 2021, 19, 4685–4690 RSC ;
(i) S. J. Kalita, F. Cheng, Q.-H. Fan, N. Shibata and Y.-Y. Huang, J. Org. Chem., 2021, 86, 8695–8705 CrossRef CAS PubMed ;
(j) I. N. C. Kiran, K. Fujita, K. Kobayashi, S. Tanaka and M. Kitamura, Bull. Chem. Soc. Jpn., 2021, 94, 295–308 CrossRef CAS ;
(k) Y.-N. Li, X. Chang, Q. Xiong, X.-Q. Dong and C.-J. Wang, Chin. Chem. Lett., 2021, 32, 4029–4032 CrossRef CAS ;
(l) P. Mahto, K. Shukla, A. Das and V. K. Singh, Tetrahedron, 2021, 87, 132115 CrossRef CAS ;
(m) V. A. Motornov, A. A. Tabolin, Y. V. Nelyubina, V. G. Nenajdenko and S. L. Ioffe, Org. Biomol. Chem., 2021, 19, 3413–3427 RSC ;
(n) Y. Suzuki, K. Kanemoto, A. Inoue, K. Imae and S.-i. Fukuzawa, J. Org. Chem., 2021, 86, 14586–14596 CrossRef CAS PubMed ;
(o) H. Wang, C. Gong, C. Zhang, X. Zheng, Q. Hou, Q. Zhou, G. Zhou and Y. Chen, Synlett, 2021, 32, 1437–1446 CrossRef CAS ;
(p) O. Yildirim, M. Grigalunas, L. Brieger, C. Strohmann, A. P. Antonchick and H. Waldmann, Angew. Chem., Int. Ed., 2021, 60, 20012–20020 CrossRef CAS PubMed ;
(q) G.-J. Wang, L. Wang, G.-D. Zhu, J. Zhou, H.-Y. Bai and S.-Y. Zhang, Org. Lett., 2021, 23, 8434–8438 CrossRef CAS PubMed ;
(r) J. Corpas, A. Ponce, J. Adrio and J. C. Carretero, Org. Lett., 2018, 20, 3179–3182 CrossRef CAS PubMed .
-
(a) K. Popandova-Yambolieva, Collect. Czech. Chem. Commun., 1994, 59, 489–494 CrossRef ;
(b) M. Maggini, G. Scorrano and M. Prato, J. Am. Chem. Soc., 2002, 115, 9798–9799 CrossRef ;
(c) B. C. Hong, A. K. Gupta, M. F. Wu, J. H. Liao and G. H. Lee, Org. Lett., 2003, 5, 1689–1692 CrossRef CAS PubMed ;
(d) J. Jayashankaran, R. D. R. S. Manian and R. Raghunathan, Tetrahedron Lett., 2004, 45, 7303–7305 CrossRef CAS ;
(e) R. D. R. S. Manian, J. Jayashankaran and R. Raghunathan, Tetrahedron, 2006, 62, 12357–12362 CrossRef CAS ;
(f) E. R. Zaitseva, A. Y. Smirnov, N. S. Baleeva and M. S. Baranov, Chem. Heterocycl. Compd., 2019, 55, 676–678 CrossRef CAS .
-
(a) S. Filippone, E. E. Maroto, A. Martin-Domenech, M. Suarez and N. Martin, Nat. Chem., 2009, 1, 578–582 CrossRef CAS PubMed ;
(b) E. E. Maroto, S. Filippone, A. Martin-Domenech, M. Suarez and N. Martin, J. Am. Chem. Soc., 2012, 134, 12936–12938 CrossRef CAS PubMed ;
(c) E. E. Maroto, S. Filippone, M. Suarez, R. Martinez-Alvarez, A. de Cozar, F. P. Cossio and N. Martin, J. Am. Chem. Soc., 2014, 136, 705–712 CrossRef CAS PubMed ;
(d) E. E. Maroto, J. Mateos, M. Garcia-Borras, S. Osuna, S. Filippone, M. A. Herranz, Y. Murata, M. Sola and N. Martin, J. Am. Chem. Soc., 2015, 137, 1190–1197 CrossRef CAS PubMed .
-
(a) R. Taylor and D. R. M. Walton, Nature, 1993, 363, 685–693 CrossRef CAS ;
(b) F. Wudl, Acc. Chem. Res., 2002, 25, 157–161 CrossRef .
-
(a) M. Potowski, J. O. Bauer, C. Strohmann, A. P. Antonchick and H. Waldmann, Angew. Chem., Int. Ed., 2012, 51, 9512–9516 CrossRef CAS PubMed ;
(b) M. Potowski, A. P. Antonchick and H. Waldmann, Chem. Commun., 2013, 49, 7800–7802 RSC .
- Z. L. He, H. L. Teng and C. J. Wang, Angew. Chem., Int. Ed., 2013, 52, 2934–2938 CrossRef CAS PubMed .
-
(a) M. J. S. Dewar, Nature, 1945, 155, 50–51 CrossRef CAS ;
(b) T. Nakajima and S. Katagiri, Bull. Chem. Soc. Jpn., 1962, 35, 910–916 CrossRef CAS ;
(c) J. Griffiths and M. Lockwood, J. Chem. Soc., Perkin Trans. 1, 1976, 48–54 RSC .
- For selected examples, see:
(a) M. Diekmann, G. Bockstiegel, A. Lützen, M. Friedemann, W. Saak, D. Haase and R. Beckhaus, Organometallics, 2006, 25, 339–348 CrossRef CAS ;
(b) A. Cappelli, S. Galeazzi, I. Zanardi, V. Travagli, M. Anzini, R. Mendichi, S. Petralito, A. Memoli, E. Paccagnini, W. Peris, A. Giordani, F. Makovec, M. Fresta and S. Vomero, J. Nanopart. Res., 2010, 12, 895–903 CrossRef CAS ;
(c) Y. Kosaka, K. Kitazawa, S. Inomata and T. Ishizone, ACS Macro Lett., 2013, 2, 164–167 CrossRef CAS PubMed ;
(d) Y. Kosaka, S. Kawauchi, R. Goseki and T. Ishizone, Macromolecules, 2015, 48, 4421–4430 CrossRef CAS .
-
(a) B. S. Donslund, N. I. Jessen, J. B. Jakobsen, A. Monleon, R. P. Nielsen and K. A. Jørgensen, Chem. Commun., 2016, 52, 12474–12477 RSC ;
(b) B. S. Donslund, R. P. Nielsen, S. M. Monsted and K. A. Jorgensen, Angew. Chem., Int. Ed., 2016, 55, 11124–11128 CrossRef CAS PubMed ;
(c) J.-F. Yue, G.-Y. Ran, X.-X. Yang, W. Du and Y.-C. Chen, Org. Chem. Front., 2018, 5, 2676–2679 RSC .
-
(a) M.-C. Tong, X. Chen, H.-Y. Tao and C.-J. Wang, Angew. Chem., Int. Ed., 2013, 52, 12377–12380 CrossRef CAS PubMed ;
(b) Q.-H. Li, L. Wei and C.-J. Wang, J. Am. Chem. Soc., 2014, 136, 8685–8692 CrossRef CAS PubMed ;
(c) H. L. Teng, L. Yao and C. J. Wang, J. Am. Chem. Soc., 2014, 136, 4075–4080 CrossRef CAS PubMed ;
(d) X. Chang, X.-S. Sun, C. Che, Y.-Z. Hu, H.-Y. Tao and C.-J. Wang, Org. Lett., 2019, 21, 1191–1196 CrossRef CAS PubMed ;
(e) C. Shen, Y. Yang, L. Wei, W.-W. Dong, L. W. Chung and C.-J. Wang, iScience, 2019, 11, 146–159 CrossRef CAS PubMed ;
(f) S.-M. Xu, L. Wei, C. Shen, L. Xiao, H.-Y. Tao and C.-J. Wang, Nat. Commun., 2019, 10, 5553 CrossRef PubMed ;
(g) X. Chang, Y. Yang, C. Shen, K.-S. Xue, Z.-F. Wang, H. Cong, H.-Y. Tao, L. W. Chung and C.-J. Wang, J. Am. Chem. Soc., 2021, 143, 3519–3535 CrossRef CAS PubMed ;
(h) X. Chang, X. Cheng and C.-J. Wang, Chem. Sci., 2022, 13, 4041–4049 RSC ;
(i) C. S. Huachao Liu, Xin Chang, Chunjiang Wang, Chin. J. Org. Chem., 2022, 42, 3322–3334 CrossRef ;
(j) L. Xiao, B. Li, F. Xiao, C. Fu, L. Wei, Y. Dang, X.-Q. Dong and C.-J. Wang, Chem. Sci., 2022, 13, 4801–4812 RSC ;
(k) X. Xu, L. Bao, L. Ran, Z. Yang, D. Yan, C.-J. Wang and H. Teng, Chem. Sci., 2022, 13, 1398–1407 RSC .
-
(a) R. Misra, R. C. Pandey and J. V. Silverton, J. Am. Chem. Soc., 1982, 104, 4478–4479 CrossRef CAS ;
(b) P. Jimonet, A. Boireau, M. Chevé, D. Damour, A. Genevois-Borella, A. Imperato, J. Pratt, J. C. R. Randle, Y. Ribeill, J.-M. Stutzmann and S. Mignani, Bioorg. Med. Chem. Lett., 1999, 9, 2921–2926 CrossRef CAS PubMed ;
(c) P. de Almeida Leone, A. R. Carroll, L. Towerzey, G. King, B. M. McArdle, G. Kern, S. Fisher, J. N. A. Hooper and R. J. Quinn, Org. Lett., 2008, 10, 2585–2588 CrossRef CAS PubMed ;
(d) A. C. Wei, M. A. Ali, Y. K. Yoon, R. Ismail, T. S. Choon, R. S. Kumar, N. Arumugam, A. I. Almansour and H. Osman, Bioorg. Med. Chem. Lett., 2012, 22, 4930–4933 CrossRef CAS PubMed ;
(e) Y. Zheng, C. M. Tice and S. B. Singh, Bioorg. Med. Chem. Lett., 2014, 24, 3673–3682 CrossRef CAS PubMed ;
(f) Y.-J. Zheng and C. M. Tice, Expert Opin. Drug Discovery, 2016, 11, 831–834 CrossRef PubMed .
-
(a) C. J. Wang, G. Liang, Z. Y. Xue and F. Gao, J. Am. Chem. Soc., 2008, 130, 17250–17251 CrossRef CAS PubMed ;
(b) Z.-Y. Xue, Q.-H. Li, H.-Y. Tao and C.-J. Wang, J. Am. Chem. Soc., 2011, 133, 11757–11765 CrossRef CAS PubMed .
- CCDC 2234947 ((1R,2'S,5'S)-3j), 2256257 (1R,3S,3aR,8aS,E)-4i contain the supplementary crystallographic data for this paper.
- M. Hashimoto and S. Terashima, Tetrahedron Lett., 1994, 35, 9409–9412 CrossRef CAS .
-
(a) See computational details in the ESI.†.;
(b) Y.-h. Lam, M. N. Grayson, M. C. Holland, A. Simon and K. N. Houk, Acc. Chem. Res., 2016, 49, 750–762 CrossRef CAS PubMed ;
(c) Q. Peng and R. S. Paton, Acc. Chem. Res., 2016, 49, 1042–1051 CrossRef CAS PubMed ;
(d) R. B. Sunoj, Acc. Chem. Res., 2016, 49, 1019–1028 CrossRef CAS PubMed ;
(e) D. J. Tantillo, Acc. Chem. Res., 2016, 49, 741–749 CrossRef CAS PubMed ;
(f) X. Zhang, L. W. Chung and Y.-D. Wu, Acc. Chem. Res., 2016, 49, 1302–1310 CrossRef CAS PubMed ;
(g) S. Ahn, M. Hong, M. Sundararajan, D. H. Ess and M.-H. Baik, Chem. Rev., 2019, 119, 6509–6560 CrossRef CAS PubMed ;
(h) J. N. Harvey, F. Himo, F. Maseras and L. Perrin, ACS Catal., 2019, 9, 6803–6813 CrossRef CAS ;
(i) J. Lan, X. Li, Y. Yang, X. Zhang and L. W. Chung, Acc. Chem. Res., 2022, 55, 1109–1123 CrossRef CAS PubMed .
- Selected related computational studies:
(a) D. H. Ess and K. N. Houk, J. Am. Chem. Soc., 2008, 130, 10187–10198 CrossRef CAS PubMed ;
(b) M. Wang, C.-J. Wang and Z. Lin, Organometallics, 2012, 31, 7870–7876 CrossRef CAS ;
(c) A. Pascual-Escudero, A. de Cózar, F. P. Cossío, J. Adrio and J. C. Carretero, Angew. Chem., Int. Ed., 2016, 55, 15334–15338 CrossRef CAS PubMed ;
(d) L. R. Domingo, M. Ríos-Gutiérrez and P. Pérez, J. Org. Chem., 2018, 83, 10959–10973 CrossRef CAS PubMed ;
(e) F. Cheng, S. J. Kalita, Z.-N. Zhao, X. Yang, Y. Zhao, U. Schneider, N. Shibata and Y.-Y. Huang, Angew. Chem., Int. Ed., 2019, 58, 16637–16643 CrossRef CAS PubMed ;
(f) Y. Xiong, Z. Du, H. Chen, Z. Yang, Q. Tan, C. Zhang, L. Zhu, Y. Lan and M. Zhang, J. Am. Chem. Soc., 2019, 141, 961–971 CrossRef CAS PubMed ;
(g) S. V. Kumar and P. J. Guiry, Angew. Chem., Int. Ed., 2022, 61, e202205516 CAS ;
(h) B. Li, H. Xu, Y. Dang and K. N. Houk, J. Am. Chem. Soc., 2022, 144, 1971–1985 CrossRef CAS PubMed .
- Our computational studies on first-row transition-metal catalysis:
(a) L. Xu, L. W. Chung and Y.-D. Wu, ACS Catal., 2016, 6, 483–493 CrossRef CAS ;
(b) W. Gao, H. Lv, T. Zhang, Y. Yang, L. W. Chung, Y.-D. Wu and X. Zhang, Chem. Sci., 2017, 8, 6419–6422 RSC ;
(c) J. Lan, T. Liao, T. Zhang and L. W. Chung, Inorg. Chem., 2017, 56, 6809–6819 CrossRef CAS PubMed ;
(d) X. Zhang and L. W. Chung, Chem. Eur. J., 2017, 23, 3623–3630 CrossRef CAS PubMed ;
(e) S.-B. Wu, T. Zhang, L. W. Chung and Y.-D. Wu, Org. Lett., 2019, 21, 360–364 CrossRef CAS PubMed ;
(f) Y. Yang, X. Zhang, L.-P. Zhong, J. Lan, X. Li, C.-C. Li and L. W. Chung, Nat. Commun., 2020, 11, 1850 CrossRef CAS PubMed ;
(g) X. Du, Y. Xiao, Y. Yang, Y.-N. Duan, F. Li, Q. Hu, L. W. Chung, G.-Q. Chen and X. Zhang, Angew. Chem., Int. Ed., 2021, 60, 11384–11390 CrossRef CAS PubMed ;
(h) J. Lan, T. Zhang, Y. Yang, X. Li and L. W. Chung, Inorg. Chem., 2022, 61, 18019–18032 CrossRef CAS PubMed .
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed .
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed .
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2234947 and 2256257. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00435j |
| ‡ These three authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c and
Chun-Jiang Wang
*c and
Chun-Jiang Wang