
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Site-selective radical reactions of kinetically stable open-shell singlet diradicaloid difluorenoheteroles with tributyltin hydride and azo-based radical initiators†
Naoki
Tabata
a,
Takumi
Uchino
a,
Chitoshi
Kitamura
a,
Kazunari
Yoshizawa
 b,
Yoshihito
Shiota
*b and
Shin-ichiro
Kato
*a
b,
Yoshihito
Shiota
*b and
Shin-ichiro
Kato
*a
aDepartment of Materials Chemistry, School of Engineering, The University of Shiga Prefecture, 2500 Hassaka-cho, Hikone, Shiga 522-8533, Japan. E-mail: kato.s@mat.usp.ac.jp
bInstitute for Materials Chemistry and Engineering (IMCE), Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: shiota@ms.ifoc.kyushu-u.ac.jp
First published on 2nd May 2023
Abstract
We have demonstrated site-selective radical reactions of the kinetically stable open-shell singlet diradicaloids difluoreno[3,4-b:4′,3′-d]thiophene (DFTh) and difluoreno[3,4-b:4′,3′-d]furan (DFFu) with tributyltin hydride (HSn(n-Bu)3) and azo-based radical initiators. Treatment of these diradicaloids with HSn(n-Bu)3 induces hydrogenation at the ipso-carbon in the five-membered rings, while treatment with 2,2′-azobis(isobutyronitrile) (AIBN) induces substitution at the carbon atoms in the peripheral six-membered rings. We have also developed one-pot substitution/hydrogenation reactions of DFTh/DFFu with various azo-based radical initiators and HSn(n-Bu)3. The resulting products can be converted into substituted DFTh/DFFu derivatives via dehydrogenation. Theoretical calculations unveiled a detailed mechanism of the radical reactions of DFTh/DFFu with HSn(n-Bu)3 and with AIBN, and that the site-selectivity of these radical reactions is controlled by the balance of the spin density and the steric hindrance in DFTh/DFFu.
Introduction
Open-shell singlet diradicaloids with a polycyclic π-conjugated backbone have attracted great attention due to their unique optoelectronic and magnetic properties.1–7 Scaffold moieties such as ortho- and para-quinodimethane (o-QDM and p-QDM, respectively), 2,6- and 1,5-naphtoquinodimethane (2,6-NQDM and 1,5-NQDM, respectively), and their π-extended derivatives play critical roles in inducing molecules to exhibit singlet diradical character.8 According to Clar's aromatic sextet rule, the benzenoid form of the open-shell contributor with two distinct radical centers is stabilized by an aromatic sextet. Installing bulky substituents into the aforementioned pro-aromatic quinoidal scaffolds has been a popular approach to synthesizing chemically and thermally stable open-shell polycyclic π-conjugated diradicaloids (PCDs).9,10 Elucidation of the inherent reactivity of PCDs is indispensable not only for understanding their diradical character,11–18 but also for the development of late-stage modifications19–21 and chemical transformations of PCDs into structurally and electronically interesting compounds that are difficult to access from the corresponding closed-shell compounds.22–31Unique reactivity that originates from diradical character has been sporadically reported in kinetically unstable PCDs without bulky substituents.32–37 For example, Haley's group has described the reaction of an in situ-formed PCD that possesses a 2,6-NQDM scaffold with the deuterium source tributyltin deuteride (DSn(n-Bu)3) at the ipso-carbons in the five-membered rings, which proceeded via a radical mechanism (Fig. 1a).34 More recently, Kubo's group has reported that a thermodynamically stable but kinetically unstable PCD that possesses an o-QDM scaffold undergoes dimerization via the [4 + 4] cycloaddition, which occurs through the radical stepwise mechanism (Fig. 1b);36 the σ-bonded dimer dissociates into a monomer upon heating or photoirradiation. A similar cyclodimerization, as well as the cyclotrimerization and higher oligomerization of a trimethylsilylethynyl-substituted indeno[1,2-b]fluorene derivative that incorporates a p-QDM scaffold, have previously been reported by Zhao's group despite the major closed-shell feature of the ground state (Scheme S1 in the ESI†).32 These reactions are rare examples that unveil the diradical contribution in the electronic ground state of PCDs from the viewpoint of their peculiar radical reactivity. It might, however, be argued that such radical reactions are found due to a lack of kinetic stability; in other words, in the stable PCDs synthesized so far, this unique reactivity is possibly masked due to the efficient kinetic protection. Moreover, kinetically stable PCDs usually involve relatively laborious synthetic routes, which may dissuade chemists from investigating their reactivity. Accordingly, the exploration of the reactivity of stable PCDs has been very limited to date and remains a challenging task.19–22,28
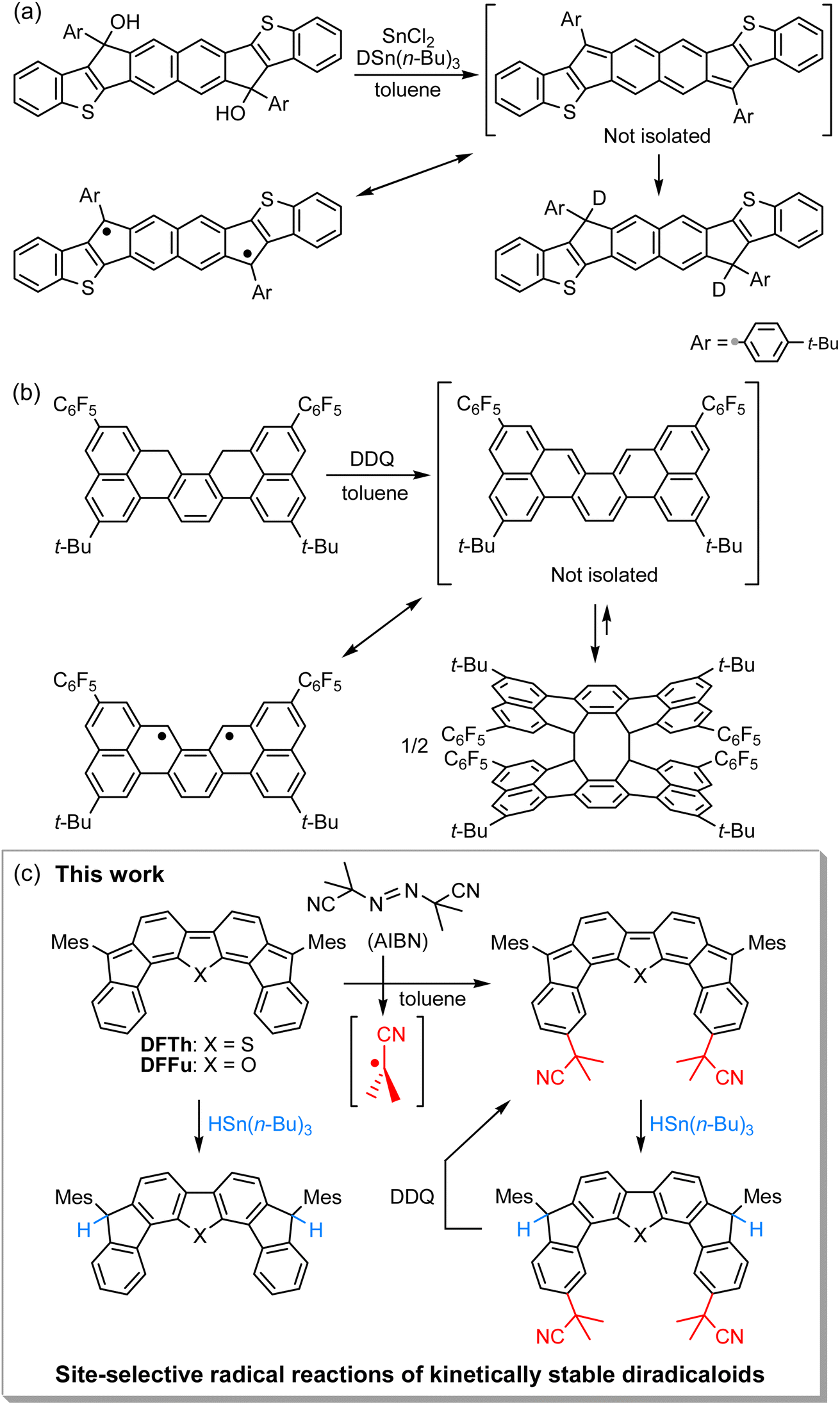 | ||
| Fig. 1 Reactions of recently reported kinetically unstable diradicaloids, in which (a) 2,6-NQDM or (b) o-QDM structures are embedded. (c) Site-selective radical reactions of difluoreno[4,3-b:3′,4′-d]thiophene (DFTh) and difluoreno[4,3-b:3′,4′-d]thiophene (DFFu; Mes = 2,4,6-trimethylphenyl) with AIBN and/or HSn(n-Bu)3, as well as dehydrogenation of the products. The resonance between the closed- and open-shell diradical structures of DFTh is shown in Fig. 3. | ||
We have recently designed and synthesized difluoreno[4,3-b:3′,4′-d]thiophene (DFTh) and difluoreno[4,3-b:3′,4′-d]furan (DFFu), which contain Tschitschibabin's hydrocarbon as a structural motif, and examined their diradical activity and functions (Fig. 1c; the open-shell contributors are shown in Fig. 3).38–40DFTh and DFFu are stable enough to be handled under ambient conditions without any special precautions due to the bulky Mes groups. They exhibit distinct diradical character with singlet–triplet energy gaps (ΔES–T) of −4.3 and −4.9 kcal mol−1 for DFTh and DFFu, respectively, which is reflected in their narrow optical/electrochemical gaps and ambipolar charge-transport behavior in organic field-effect transistor devices. Advantageously, these PCDs can be obtained in quantities exceeding 500 mg in a single sequence from commercially available materials (over five steps). The easy accessibility of DFTh and DFFu motivated us to explore their reactivity. Herein, we report that these PCDs react site-selectively with tributyltin hydride (HSn(n-Bu)3) and azo-based radical initiators such as 2,2′-azobis(isobutyronitrile) (AIBN) (Fig. 1c). Computational studies clearly demonstrated that DFTh/DFFu behave as π-diradicaloids in terms of reactivity; the site-selectivity observed in the reactions can be interpreted in terms of a balance between the diradical activity and the steric hindrance. We have developed a one-pot reaction of our PCDs with azo-based radical initiators and HSn(n-Bu)3; the products can be readily converted into substituted PCDs by oxidative dehydrogenation. This is, to the best of our knowledge, the first example of the synthesis of substituted diradicaloids through chemical transformations of the parent diradicaloids.
Results and discussion
Reaction of DFTh/DFFu with tributyltin hydride – radical hydrogenation
Initially, inspired by the radical hydrogenation of the PCD reported by Haley and co-workers (Fig. 1a),34 we investigated the hydrogenation of DFTh and DFFu (Scheme 1). Treating DFTh and DFFu with an excess of HSn(n-Bu)3 (10 equiv.) in toluene at 110 °C afforded the corresponding hydrogenated products DFThH (97%) and DFFuH (78%) as ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric mixtures. We then conducted a competitive-reaction experiment to investigate the reactivity of DFTh and DFFu in the hydrogenation. Upon heating a toluene-d8 solution of DFTh and DFFu (1:1) with HSn(n-Bu)3 at 90 °C for 0.5 h (for details, see the ESI†), DFTh was almost completely consumed, while ∼20% of the DFFu remained based on the 1H NMR spectrum (Fig. S1†). This finding suggests that DFTh is more reactive than DFFu, which might reflect the slightly smaller ΔES–T gap, i.e., a higher diradical activity of the former compared to that of the latter.
:1 diastereomeric mixtures. We then conducted a competitive-reaction experiment to investigate the reactivity of DFTh and DFFu in the hydrogenation. Upon heating a toluene-d8 solution of DFTh and DFFu (1:1) with HSn(n-Bu)3 at 90 °C for 0.5 h (for details, see the ESI†), DFTh was almost completely consumed, while ∼20% of the DFFu remained based on the 1H NMR spectrum (Fig. S1†). This finding suggests that DFTh is more reactive than DFFu, which might reflect the slightly smaller ΔES–T gap, i.e., a higher diradical activity of the former compared to that of the latter.
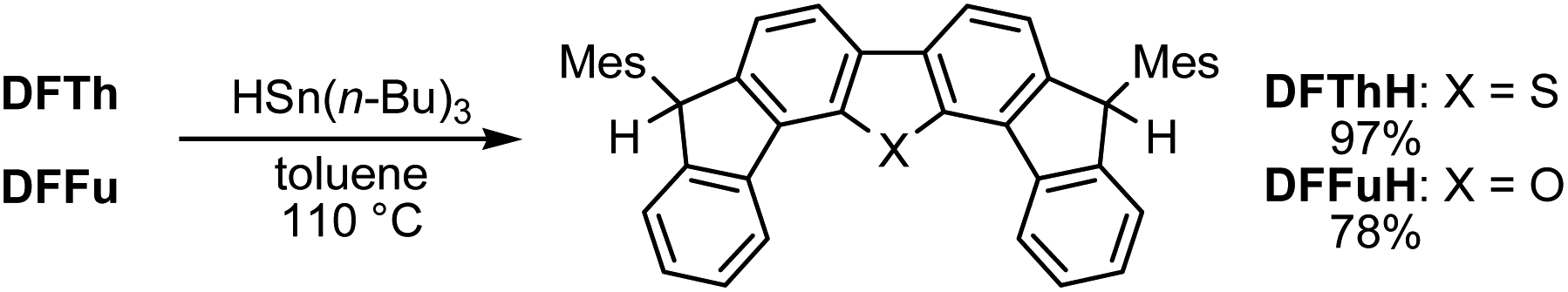 | ||
| Scheme 1 Hydrogenation of DFTh and DFFu with HSn(n-Bu)3 in toluene. | ||
Mechanistic insight into the hydrogenation of DFTh/DFFu using DFT calculations
The results of our DFT calculations at the B3LYP/6-31G** level suggest three forms of DFTh (A), corresponding to the closed-shell singlet state (CS), the open-shell singlet state (OSS), and the triplet state (T) (Fig. 2a)41 with calculated relative energies of +1.9 kcal mol−1 (ACS), 0.0 kcal mol−1 (AOSS), and +4.3 kcal mol−1 (AT). Of the three forms, the open-shell singlet is the ground state, which is diradical in nature and engages with HSn(n-Bu)3 in a hydrogenation. The calculated spin density in Fig. 3 clearly indicates a diradical state of AOSS, with contributions from the main form α, the minor form β, and the minor form γ.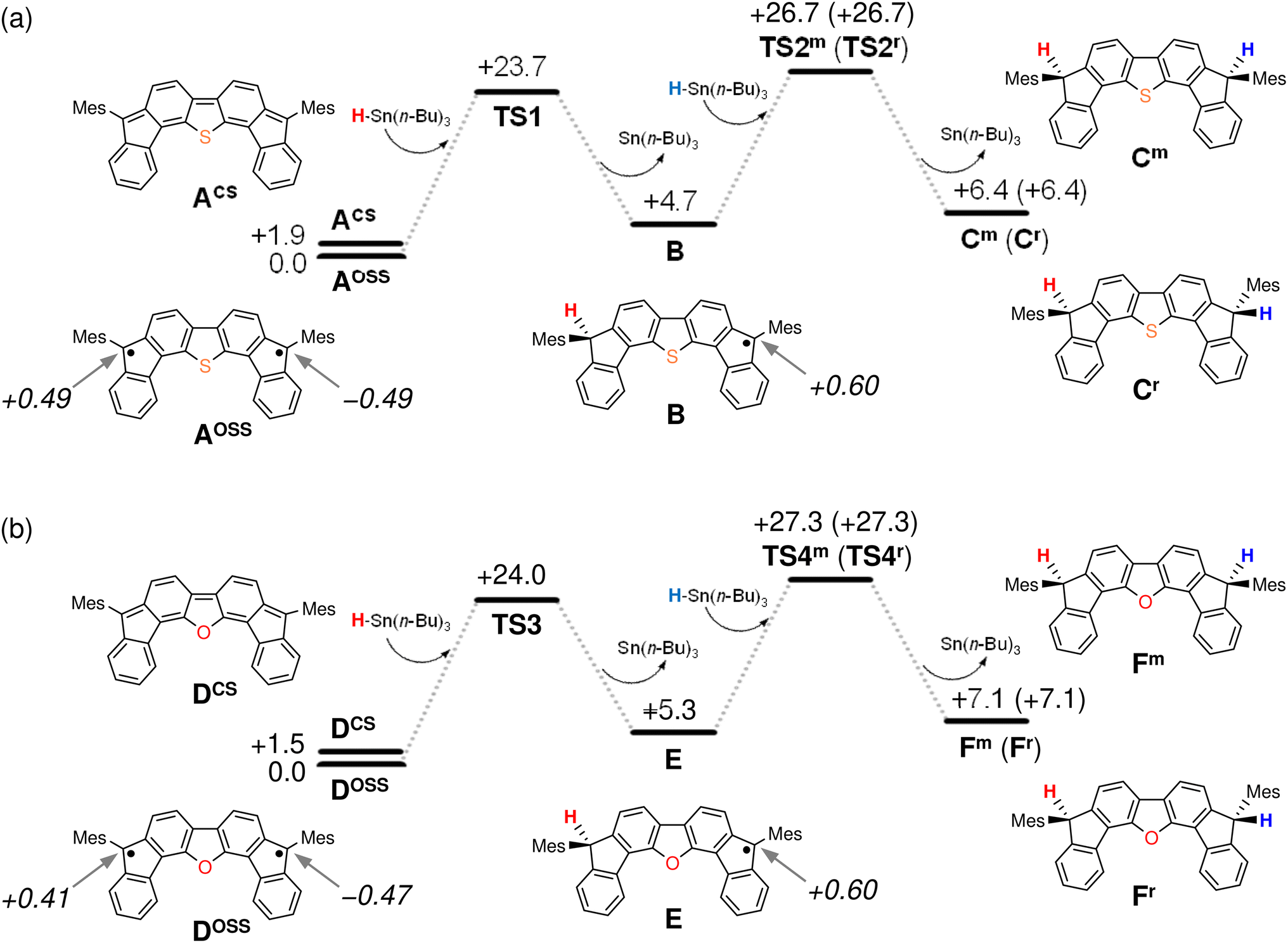 | ||
| Fig. 2 Potential energy diagrams for the hydrogenation of (a) DFTh and (b) DFFu calculated at the B3LYP/6-31G** level. Relative energies are given in kcal mol−1, and values in italics indicate spin density. | ||
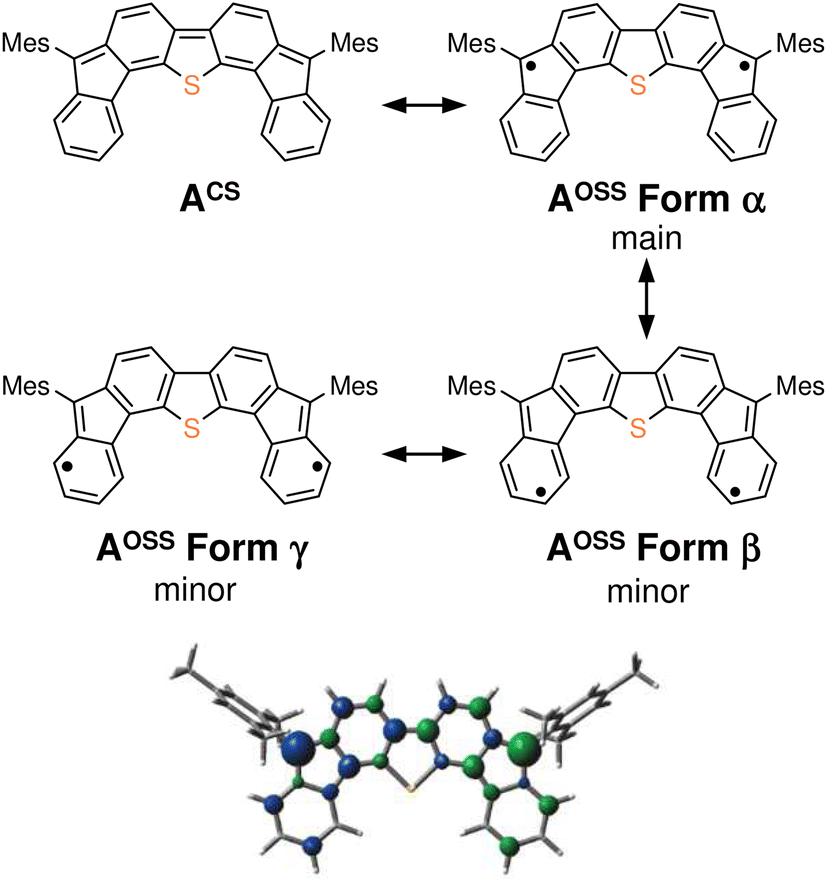 | ||
| Fig. 3 Kekulé resonance structures of ACS and AOSS, as well as the optimized structure of AOSS showing the spin density. In the spin density plot, blue is positive and green is negative (isovalue: 0.005). | ||
In Fig. 2a, the hydrogenation of DFTh proceeds via a two-step reaction with two transition states (TS1 and TS2) and one intermediate (B). The first H-atom migration occurs at a transition state with an activation energy of +23.7 kcal mol−1, resulting in the formation of a radical intermediate. The hydrogenation occurs at the ipso-carbon of one of the five-membered rings, which exhibits the highest spin density (+0.49) in the carbon framework (Fig. 2a). These results lead to the conclusion that the regioselective hydrogenation of DFTh is controlled by the diradical activity of the substrate, i.e., form α of DFTh (Fig. 3) is responsible for the hydrogenation with HSn(n-Bu)3. In the next step, the hydrogenation of radical intermediate B yields two types of products, i.e., a meso compound and a racemate. The activation energies of TS2m and TS2r, which correspond to the reactions to give the meso and racemic products (Cm and Cr), respectively, are both +22.0 kcal mol−1. Moreover, the calculated energies of Cm and Cr are also identical (+1.7 kcal mol−1). These computed results are consistent with the experimental observation that the ratio between the meso compound and the racemate is ∼1:1.
We next considered the hydrogenation of DFFu, as well as that of DFTh, as shown in Fig. 2b. In the initial structure (D) of DFFu, the ground state is the open-shell singlet state; the relative energies of the closed-shell singlet and the triplet state are +1.5 and +4.9 kcal mol−1, respectively. These results indicate that DFFu exhibits a diradical activity similar to that of DFTh and therefore undergoes hydrogenation. This reaction pathway is also a two-step reaction that involves two transition states (TS3 and TS4) and one radical intermediate (E). The reaction is initiated by the transfer of an H atom from D with an activation energy of +24.0 kcal mol−1. The activation energy of TS3 for DFFu is slightly higher (0.3 kcal mol−1) than that of TS1 for DFTh, which likely corresponds to the result of the aforementioned competitive-reaction experiment. The next step is the hydrogenation of the radical intermediate (E), which results in the formation of the meso compound and the racemate (Fm and Fr). The activation energies of TS4m and TS4r, which correspond to the reactions to give the meso and racemic products, respectively, were both calculated to be +22.0 kcal mol−1; this value is identical to the activation energies of TS2m and TS2r. The DFT calculations thus suggest that the hydrogenation of DFFu produces equal amounts of the meso compound and the racemate, as is the case with the hydrogenation of DFTh.
Reaction of DFTh with 2,2′-azobis(isobutyronitrile) – radical coupling and hydrogen abstraction
We chose AIBN for the reaction with DFTh, given that it is a representative azo-based radical initiator. We envisaged that the reaction between DFTh and the cyanoisopropyl radical (CIPR), which is formed upon heating AIBN, would occur in a site-selective manner; the sterically demanding CIPR can be expected to react at a carbon atom other than the ipso-carbons of the five-membered rings due to the steric hindrance between the CIPR and the mesityl group. Gratifyingly, treatment of DFTh with AIBN in toluene at 110 °C furnished 1 and 1H in 42% total yield as an inseparable 0.36:1 mixture within a few minutes (Scheme 2 and Fig. S2†). Notably, no regioisomers of 1 or 1H were detected. When the reaction time was prolonged, 1 almost disappeared, and 1H was obtained together with unidentified byproducts. No reaction was observed between pure 1H, which was prepared according to our reported procedures,39 and AIBN in toluene at 110 °C. These findings suggest that 1 is formed initially by the radical reaction between DFTh and CIPR, and subsequently hydrogenated to afford 1H.42 The use of benzene-d6 and toluene-d8 as reaction solvents also provided 1H, which suggests that H-atom transfer from CIPR to 1 affords 1H (Scheme S2†). The molecular structure of 1H was determined unambiguously via single-crystal X-ray diffraction analysis (Fig. S3†).
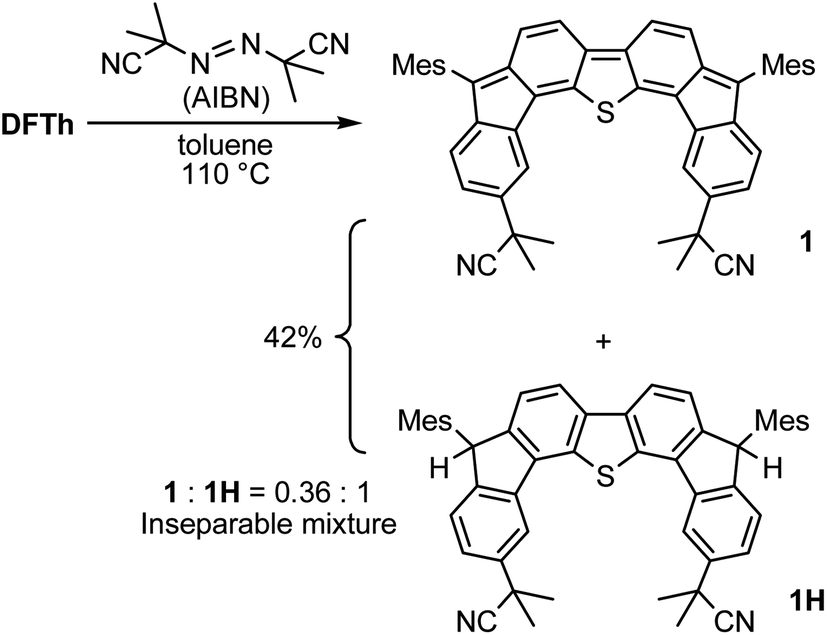 | ||
| Scheme 2 Reaction of DFTh with 2,2′-azobis(isobutyronitrile) (AIBN) and hydrogenation of the substituted product. | ||
Mechanistic insight into the reaction of DFTh/DFFu with AIBN using DFT calculations
We next considered the reaction of DFTh with AIBN to form a substituted product, as shown in Fig. 4. The decomposition of AIBN produces two CIPRs, which could induce either H-atom abstraction or radical C–C coupling. Thus, there are two possible reactions for the formation of 1 (Fig. 4a); one is the H-atom abstraction by CIPR, and the other is the C–C bond formation between CIPR and the carbon atom of the aromatic ring. H-atom abstraction from DFTh gives radical intermediate Int1 with an energy of +26.3 kcal mol−1. The C–C bond formation by radicals involves transition state TS5, which has an activation energy of +7.1 kcal mol−1 in the doublet state. Subsequently, Int2 with an energy of −15.5 kcal mol−1 in the doublet state is formed. These computational results indicate that the C–C bond formation is energetically favored over the H-atom abstraction in the first step. Thus, we can rule out the formation of Int1via direct H-atom abstraction from DFTh. In the next step, another CIPR reacts with Int2, resulting in the formation of Int3, which corresponds to monosubstituted DFTh.43,44 This process involves transition state TS6, in which the H-atom abstraction occurs with an activation energy of +8.2 kcal mol−1 in the open-shell singlet state, and therefore, this reaction is likely to proceed. Our DFT calculations furthermore suggest that the first substitution reaction is exothermic by 55 kcal mol−1 and that the transition state is low enough for the reaction to occur very easily.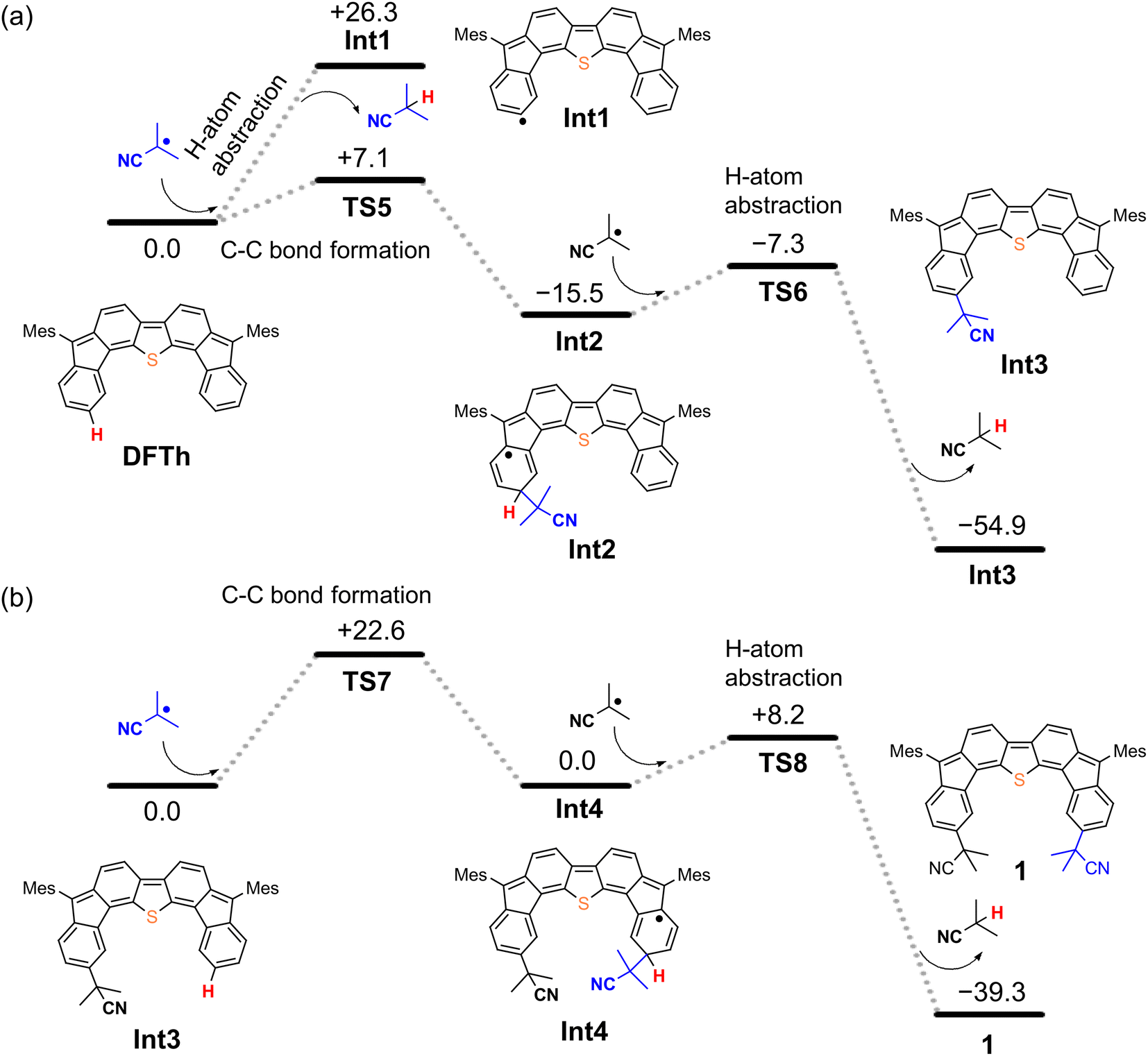 | ||
| Fig. 4 Potential energy diagrams for (a) the first substitution and (b) the second substitution of DFTh, calculated at the B3LYP/6-31G** level; units are given in kcal mol−1. | ||
As shown in Fig. 4b, the second substitution occurs via the same mechanism as the first substitution. The activation energy for the C–C bond formation in the first step of the second substitution via transition state TS7 requires +22.6 kcal mol−1. In the second step, H-atom abstraction occurs at TS8 with an activation energy of 8.2 kcal mol−1. These results suggest that the C–C bond formation in the second substitution is the rate-determining step. The CIPR plays a role in the formation of C–C bonds and the desorption of H atoms in the substitution reaction. The difference in activation energy between the first and the second C–C bond formation (+7.1 vs. +22.6 kcal mol−1) is quite large, whereas that between the first and the second H-atom abstraction is comparable (+8.2 kcal mol−1). An increase in the activation energy of the second C–C bond formation is likely due to the change in the electronic structure of the DFTh π-system resulting from the first cyanoisopropyl substitution. Based on the hydrogenation of DFTh, one might expect that the CIPRs would also react with the carbon atom at the highest spin density, as was observed in the hydrogenation with HSn(n-Bu)3. However, steric effects are also important for bulky radicals. In fact, the relative energy of the intermediate corresponding to the C–C bond formation at the ipso-carbons in the five-membered rings was calculated to be +9.7 kcal mol−1 at the B3LYP level. Therefore, we can assume that no such intermediate is present in the reaction with AIBN. As mentioned above, the reaction of DFTh with AIBN gives 1 and 1H. DFT calculations also suggest that the reaction between DFTh and CIPR is controlled by the steric hindrance rather than the diradical activity of DFTh. Since the major Form α of Aoss cannot contribute to the reaction due to steric hindrance, minor Form β plays an essential role in the substitution reaction.
One-pot reaction of DFTh/DFFu with azo-based radical initiators and HSn(n-Bu)3
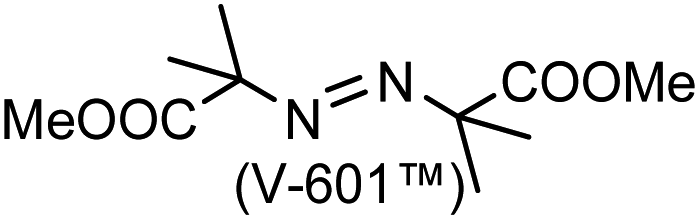
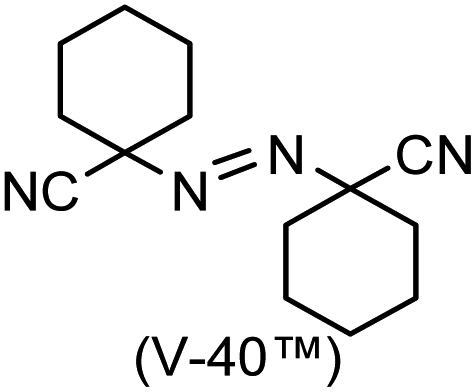
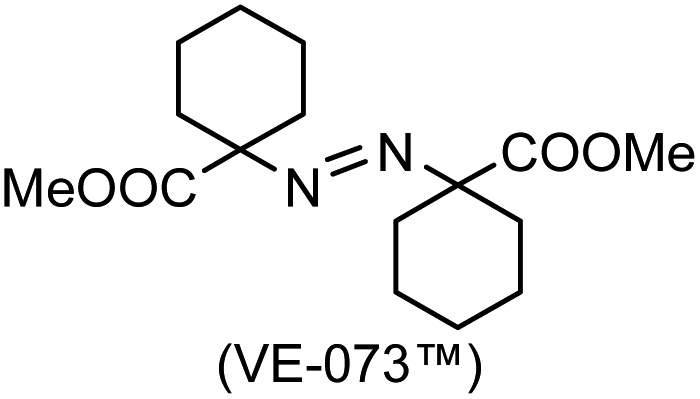
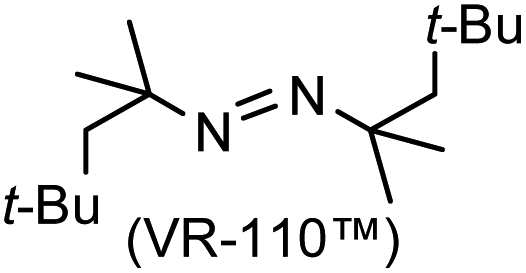
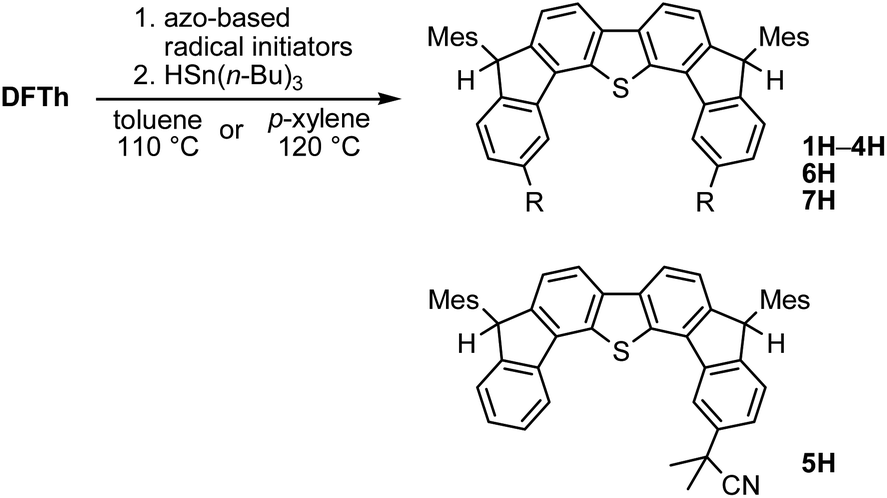
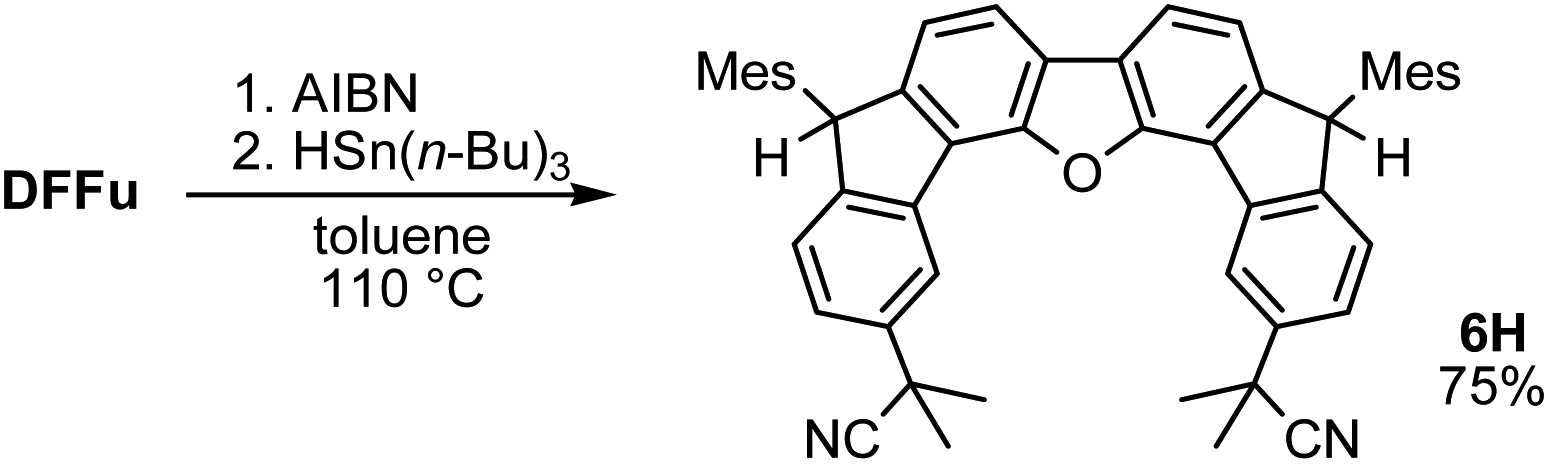
A prolonged reaction time of ca. 1 h for the reaction of DFTh with AIBN increased the amount of polar byproducts according to a TLC analysis, which renders the purification of 1H laborious. We therefore designed a one-pot reaction of DFTh with AIBN and HSn(n-Bu)3 (Table 1, entry 1); we expected that such a reaction could converge the product to 1H at a shorter reaction time and suppress the formation of byproducts. After DFTh was allowed to react with AIBN (5 equiv.) in toluene at 110 °C for 20 min, an excess of HSn(n-Bu)3 (10 equiv.) was added at once, which changed the color of the reaction solution from blue-green to pale yellow within 30 min. Fortunately, 1H was easily purified using column chromatography on silica gel and isolated in 65% yield. We then treated DFTh with various azo-based radical initiators under the one-pot conditions to clarify the reaction scope and limitations (Table 1). The use of 2,2′-azobis(2-methylpropionate) (V-601™), in which the cyano group of AIBN is replaced by a methoxycarbonyl group, and HSn(n-Bu)3 provided 2H in 51% yield after chromatographic separation of the reaction mixture (entry 2). The use of sterically more demanding 1,1′-azobis(cyclohexane-1-carbonitrile) (V-40™) and dimethyl 1,1′-azobis(1-cyclohexanecarboxylate) (VE-073™) also afforded 3H in 94% yield and 4H in 68% yield, respectively (entries 3 and 4). Decreasing the stoichiometry of AIBN from 5 equiv. to 1.5 equiv. provided monosubstituted 5H in 47% yield as the main product (entry 5). The one-pot reaction using AIBN and HSn(n-Bu)3 was also applicable to DFFu (Scheme 3), which furnished the disubstituted, hydrogenated product 6H in 75% yield. The reaction did not proceed for 2,2′-azobis(2,4,4-trimethylpentane) (VE-110™), not even after elevating the temperature to 140 °C in mesitylene (entry 6). The reaction with 2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile) (V-70™) gave a complex product mixture (entry 7).|
|
||||
|---|---|---|---|---|
| Entry | Azo-based radical initiator | Product | R | Yield [%] |
| a Azo-based radical initiator (5 equiv.) and HSn(n-Bu)3 (10 equiv.). b AIBN (1.5 equiv.) and HSn(n-Bu)3 (5 equiv.). c No reaction between DFTh and VR-110 was observed in mesitylene at 140 °C. d The reaction between DFTh and V-70 gave a complex product mixture. | ||||
| 1 |
|
1H |
|
65 |
| 2 |
|
2H |
|
51 |
| 3 |
|
3H |
|
94 |
| 4 |
|
4H |
|
68 |
| 5b | AIBN | 5H | 47 | |
| 6 |
|
7H |
|
N/Ac |
| 7 |
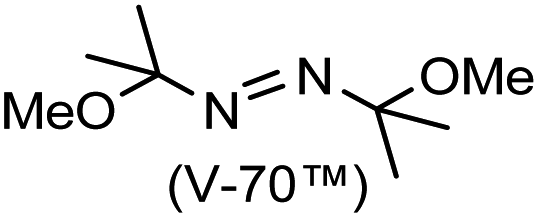
|
8H |
|
N/Ad |
 | ||
| Scheme 3 One-pot substitution and hydrogenation of DFFu with AIBN and tributyltin hydride. | ||
Oxidative dehydrogenation of 1H–6H – synthesis of substituted difluorenoheteroles
We then decided to convert 1H–6H into substituted DFTh and DFFu derivatives via oxidative dehydrogenation. Treatment of 1H–6H with 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) in toluene afforded the corresponding products (1–6) as deep-blue solids in good yield (Scheme 4). Similar to DFTh and DFFu, 1–6 were sufficiently stable under ambient conditions and could be handled without any special precautions.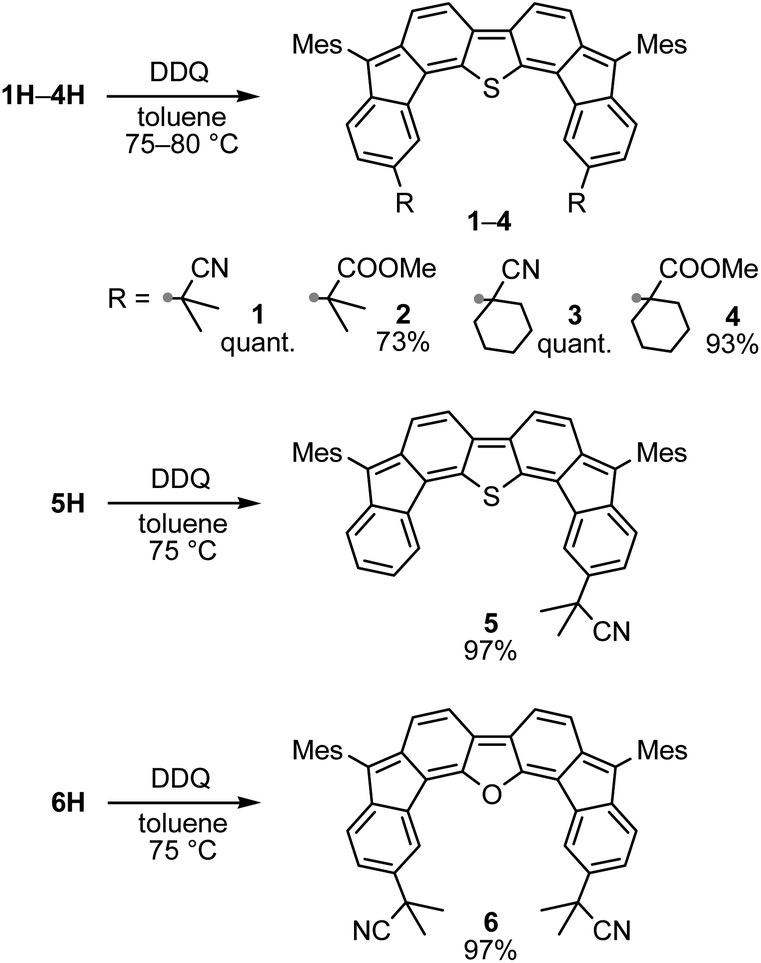 | ||
| Scheme 4 Oxidation of hydrogenated DFTh and DFFu derivatives 1H–6H with DDQ. | ||
Substituted difluorenoheteroles 1–6 are open-shell singlet diradicaloids from the parental DFTh and DFFu. The 1H NMR spectra of 1 and 6 at 25 °C in p-xylene-d10 display sharp signals in the aromatic region. Upon increasing the temperature to 110 °C, the aromatic resonances broaden (Fig. S5†). The signals are recovered to full height when the samples are cooled to 25 °C, which indicates that a thermally accessible triplet state is responsible for the signal broadening. The onset of peak broadening, implying population of the paramagnetic triplet state, is observed at ∼50 °C for 1 and ∼70 °C for 6, which are almost identical to those of DFTh and DFFu, respectively. Using the Yamaguchi method, the singlet diradical indices (y) of 1′, 6′, DFTh′, and DFFu′, in which the Mes groups of 1, 6, DFTh, and DFFu are replaced with protons, were estimated via DFT calculations at the LC-BLYP/6-311G*//M06-2X/6-31G* level (Table S1†). The y values of 1′ (0.50) and 6′ (0.46) are comparable to those of DFTh (0.50) and DFFu (0.45), respectively, which is in agreement with the experimental variable-temperature 1H NMR results.
The electrochemical properties of 1–6 were examined using cyclic voltammetry to gain insight into the electronic effects of the substituents (Fig. S6 and S7 and Table S2†). As is the case for DFTh and DFFu, 1–6 exhibit two reversible oxidation waves and two reversible reduction waves, thus indicating sufficient stability of these molecules toward redox reactions. The σ-inductive effects of the cyano residues of 1, 3, and 6 induce subtle but distinctive positive shifts of 0.06 V in the oxidation and reduction potentials relative to those of DFTh and DFFu. This indicates that the electrochemical properties of DFTh and DFFu can be tuned by functionalization of the outer six-membered rings. In the electronic absorption spectra (Fig. S8 and S9†), the absorption maxima of 1–6 are almost equal to the corresponding values for DFTh (609 nm) and DFFu (601 nm), and thus the optical gaps are retained after the introduction of substituents; however, substitution decreases the molar absorption coefficients in the long-wavelength region.
Conclusions
We have developed site-selective radical reactions of kinetically stabilized open-shell singlet diradicaloids, i.e., difluoreno[3,4-b:4′,3′-d]thiophene (DFTh) and difluoreno[3,4-b:4′,3′-d]furan (DFFu), with tributyltin hydride (HSn(n-Bu)3) and various azo-based radical initiators. The radical hydrogenation of DFTh/DFFu by HSn(n-Bu)3 occurs at the ipso-carbon of the five-membered rings, where the highest spin density is located. On the other hand, the radical coupling between DFTh/DFFu and the neutral radical derived from the decomposition of the azo-based radical initiators occurs at carbon atoms on the peripheral six-membered rings, where a relatively large spin density resides. This reactivity of the difluorenoheteroles demonstrates their open-shell π-radical nature,11 and is consistent with the singlet–triplet energy gaps of −4.3/−4.9 kcal mol−1, which indicate that DFTh/DFFu possess moderate diradical character. Theoretical calculations confirmed that the site-selectivity is controlled by the balance between the diradical activity of DFTh/DFFu and the steric hindrance. The one-pot reactions of DFTh/DFFu with azo-based radical initiators and HSn(n-Bu)3, followed by the dehydrogenation of the resulting products by DDQ, affords functionalized DFTh/DFFu derivatives. Although the substituents introduced in this way have marginal effects on the electronic properties, the present procedure provides a new approach to the late-stage functionalization of polycyclic conjugated diradicaloids. A strategy to access π-functionalized DFTh/DFFu derivatives is currently under investigation in our group.Data availability
The ESI† contains detailed description for the synthetic method and computational method. The supplementary spectroscopic and crystallographic data were also provided.Author contributions
N. Tabata synthesized the compounds and contributed to most of the experimental work. T. Uchino conducted additional complementary experiments. S.-i. Kato, Y. Shiota, and K. Yoshizawa performed the theoretical calculations. N. Tabata, C. Kitamura, and S.-i. Kato conducted the characterization of compounds. S.-i. Kato and Y. Shiota wrote the manuscript and played a critical role in the discussion of the experimental design, project direction, experiments and results, as well as the preparation of the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Grants-in-Aid for Scientific Research from the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT; 21K05042) and by the Cooperative Research Program ‘‘Network Joint Research Center for Materials and Devices’’ (Kyushu University). S.-i. K. gratefully acknowledges the Asahi Glass Foundation for financial support. The authors thank Mr Taisuke Matsumoto (Kyushu University) for carrying out a single-crystal X-ray diffraction analysis. Mass spectrometric data were collected at Hiroshima University (N-BARD: Ms Tomoko Amimoto). We thank Dr Shohei Ida (The University of Shiga Prefecture) for helpful comments.References
- M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed.
- T. Kubo, Chem. Lett., 2015, 44, 111–122 CrossRef.
- M. Nakano and B. Champagne, J. Phys. Chem. Lett., 2015, 6, 3236–3256 CrossRef CAS.
- J. Casado, Top. Curr. Chem., 2017, 375, 209–248 CrossRef PubMed.
- S. Dong and Z. Li, J. Mater. Chem. C, 2022, 10, 2431–2449 RSC.
- Z. X. Chen, Y. Li and F. Huang, Chem, 2021, 7, 288–332 CAS.
- Diradicaloids, ed. J. Wu, Jenny Stanford Publishing, New York, 2022 Search PubMed.
- Y. Tobe, Top. Curr. Chem., 2018, 376, 12 CrossRef PubMed.
- Z. Zeng, X. Shi, C. Chi, J. T. L. Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578–6596 RSC.
- S. Moles Quintero, M. M. Haley, M. Kertesz and J. Casado, Angew. Chem., Int. Ed., 2022, 61, e202209138 CrossRef CAS PubMed.
- T. Stuyver, B. Chen, T. Zeng, P. Geerlings, F. De Proft and R. Hoffmann, Chem. Rev., 2019, 119, 11291–11351 CrossRef CAS PubMed.
- J. Kolc and J. Michl, J. Am. Chem. Soc., 1970, 92, 4147–4148 CrossRef CAS.
- J. Kolc and J. Michl, J. Am. Chem. Soc., 1973, 95, 7391–7401 CrossRef CAS.
- C. R. Flynn and J. Michl, J. Am. Chem. Soc., 1974, 96, 3280–3288 CrossRef CAS.
- A. Castellan, J. Kolc and J. Michl, J. Am. Chem. Soc., 1978, 100, 6687–6692 CrossRef CAS.
- W. S. Trahanovsky and S. P. Lorimor, J. Org. Chem., 2006, 71, 1784–1794 CrossRef CAS PubMed.
- K. Uchida, S. Ito, M. Nakano, M. Abe and T. Kubo, J. Am. Chem. Soc., 2016, 138, 2399–2410 CrossRef CAS PubMed.
- T. Šolomek, P. Ravat, Z. Mou, M. Kertesz and M. Juríček, J. Org. Chem., 2018, 83, 4769–4774 CrossRef PubMed.
- J. J. Dressler, J. E. Barker, L. J. Karas, H. E. Hashimoto, R. Kishi, L. N. Zakharov, S. N. MacMillan, C. J. Gomez-Garcia, M. Nakano, J. I. Wu and M. M. Haley, J. Org. Chem., 2020, 85, 10846–10857 CrossRef CAS PubMed.
- J. Guo, Y. Yang, C. Dou and Y. Wang, J. Am. Chem. Soc., 2021, 143, 18272–18279 CrossRef CAS PubMed.
- X. Tian, J. Guo, W. Sun, L. Yuan, C. Dou and Y. Wang, Chem.–Eur. J., 2022, 28, e202200045 CAS.
- T. Kubo, M. Sakamoto, M. Akabane, Y. Fujiwara, K. Yamamoto, M. Akita, K. Inoue, T. Takui and K. Nakasuji, Angew. Chem., Int. Ed., 2004, 43, 6474–6479 CrossRef CAS PubMed.
- G. London, M. von Wantoch Rekowski, O. Dumele, W. B. Schweizer, J.-P. Gisselbrecht, C. Boudon and F. Diederich, Chem. Sci., 2014, 5, 965–972 RSC.
- H. Miyoshi, S. Nobusue, A. Shimizu, I. Hisaki, M. Miyata and Y. Tobe, Chem. Sci., 2014, 5, 163–168 RSC.
- P. Hu, S. Lee, T. S. Herng, N. Aratani, T. P. Gonçalves, Q. Qi, X. Shi, H. Yamada, K.-W. Huang, J. Ding, D. Kim and J. Wu, J. Am. Chem. Soc., 2016, 138, 1065–1077 CrossRef CAS PubMed.
- P. Hu, S. Lee, K. H. Park, S. Das, T. S. Herng, T. P. Gonçalves, K. W. Huang, J. Ding, D. Kim and J. Wu, J. Org. Chem., 2016, 81, 2911–2919 CrossRef CAS PubMed.
- S. Qiu, C. Wang, S. Xie, X. Huang, L. Chen, Y. Zhao and Z. Zeng, Chem. Commun., 2018, 54, 11383–11386 RSC.
- M. A. Majewski, P. J. Chmielewski, A. Chien, Y. Hong, T. Lis, M. Witwicki, D. Kim, P. M. Zimmerman and M. Stępień, Chem. Sci., 2019, 10, 3413–3420 RSC.
- H. Hayashi, J. E. Barker, A. Cárdenas Valdivia, R. Kishi, S. N. MacMillan, C. J. Gómez-García, H. Miyauchi, Y. Nakamura, M. Nakano, S.-i. Kato, M. M. Haley and J. Casado, J. Am. Chem. Soc., 2020, 142, 20444–20455 CrossRef CAS PubMed.
- C.-J. Zhen, S.-F. Lu, M.-H. Lin, J.-T. Wu, I. Chao and C.-H. Lin, Chem.–Eur. J., 2021, 27, 16682–16689 CrossRef CAS PubMed.
- J. E. Barker, T. W. Price, L. J. Karas, R. Kishi, S. N. MacMillan, L. N. Zakharov, C. J. Gómez-García, J. I. Wu, M. Nakano and M. M. Haley, Angew. Chem., Int. Ed., 2021, 60, 22385–22392 CrossRef CAS PubMed.
- X. Fu and D. Zhao, Org. Lett., 2015, 17, 5694–5697 CrossRef CAS PubMed.
- P. Ravat, T. Šolomek, M. Rickhaus, D. Häussinger, M. Neuburger, M. Baumgarten and M. Juríček, Angew. Chem., Int. Ed., 2016, 55, 1183–1186 CrossRef CAS PubMed.
- J. J. Dressler, M. Teraoka, G. L. Espejo, R. Kishi, S. Takamuku, C. J. Gómez-García, L. N. Zakharov, M. Nakano, J. Casado and M. M. Haley, Nat. Chem., 2018, 10, 1134–1140 CrossRef CAS PubMed.
- P. Ravat, T. Šolomek, D. Häussinger, O. Blacque and M. Juríček, J. Am. Chem. Soc., 2018, 140, 10839–10847 CrossRef CAS PubMed.
- K. Sahara, M. Abe, H. Zipse and T. Kubo, J. Am. Chem. Soc., 2020, 142, 5408–5418 CrossRef CAS PubMed.
- D. Čavlović, D. Häussinger, O. Blacque, P. Ravat and M. Juríček, JACS Au, 2022, 2, 1616–1626 CrossRef PubMed.
- S. Mori, M. Akita, S. Suzuki, M. S. Asano, M. Murata, T. Akiyama, T. Matsumoto, C. Kitamura and S.-i. Kato, Chem. Commun., 2020, 56, 5881–5884 RSC.
- S. Mori, S. Moles Quintero, N. Tabaka, R. Kishi, R. González Núñez, A. Harbuzaru, R. Ponce Ortiz, J. Marín-Beloqui, S. Suzuki, C. Kitamura, C. J. Gómez-García, Y. Dai, F. Negri, M. Nakano, S.-i. Kato and J. Casado, Angew. Chem., Int. Ed., 2022, 61, e202206680 CrossRef CAS PubMed.
- S.-i. Kato, Adv. Phys. Org. Chem., 2021, 55, 41–66 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 16, revision C.01, Gaussian, Inc., Wallingford, CT, 2016; for the full author list, see the ESI Search PubMed.
- We have also tested the reaction of DFTh with benzoyl peroxide (BPO), 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO), or (2,4,6-trimethylbenzoyl)diphenylphosphine oxide (TMDPO) (Scheme S3†). In the cases of BPO or TMDPO, only the decomposition of DFTh was observed, and unidentified polar materials were obtained; almost no reaction occurred between DFTh and TEMPO.
- The activation energy for the reaction of Int2 with AIBN was calculated to be +41.0 kcal mol−1, and that for the reaction of Int2 with AIBNH, which is possibly formed by the H-atom abstraction of Int2 by AIBN, was calculated to be +23.2 kcal mol−1 (Fig. S4 and Scheme S4†). Both activation energy values were found to be higher than the activation energy (+8.2 kcal mol−1) required for the reaction of Int2 with CIPR.
- According to DFT calculations at the B3LYP/6-31G** level, the dimerization of CIPR is an exothermic reaction (50.9 kcal mol−1) (Scheme S5†). This value is larger than that of the exothermic energy (39.4 kcal mol−1) required for the formation of Int3 from Int2, suggesting that the dimerization of CIPR competes with the H-atom abstraction of Int2 by CIPR.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, characterization, theoretical details, and supporting scheme, figures, and tables. CCDC 2231637. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00381g |
| This journal is © The Royal Society of Chemistry 2023 |