DOI:
10.1039/D3SC00173C
(Edge Article)
Chem. Sci., 2023,
14, 3302-3310
Divergent total synthesis of the revised structures of marine anti-cancer meroterpenoids (+)-dysiherbols A–E†
Received
11th January 2023
, Accepted 28th February 2023
First published on 28th February 2023
Abstract
We report here a concise and divergent enantioselective total synthesis of the revised structures of marine anti-cancer sesquiterpene hydroquinone meroterpenoids (+)-dysiherbols A–E (6–10) using dimethyl predysiherbol 14 as a key common intermediate. Two different improved syntheses of dimethyl predysiherbol 14 were elaborated, one starting from Wieland–Miescher ketone derivative 21, which is regio- and diastereoselectively α-benzylated prior to establishing the 6/6/5/6-fused tetracyclic core structure through intramolecular Heck reaction. The second approach exploits an enantioselective 1,4-addition and a Au-catalyzed double cyclization to build-up the core ring system. (+)-Dysiherbol A (6) was prepared from dimethyl predysiherbol 14via direct cyclization, while (+)-dysiherbol E (10) was synthesized through allylic oxidation and subsequent cyclization of 14. Epoxidation of 14 afforded allylic alcohol 45 or unexpectedly rearranged homoallylic alcohol 44. By inverting the configuration of the hydroxy groups, exploiting a reversible 1,2-methyl shift and selectively trapping one of the intermediate carbenium ions through oxy-cyclization, we succeeded to complete the total synthesis of (+)-dysiherbols B–D (7–9). The total synthesis of (+)-dysiherbols A–E (6–10) was accomplished in a divergent manner starting from dimethyl predysiherbol 14, which led to the revision of their originally proposed structures.
Introduction
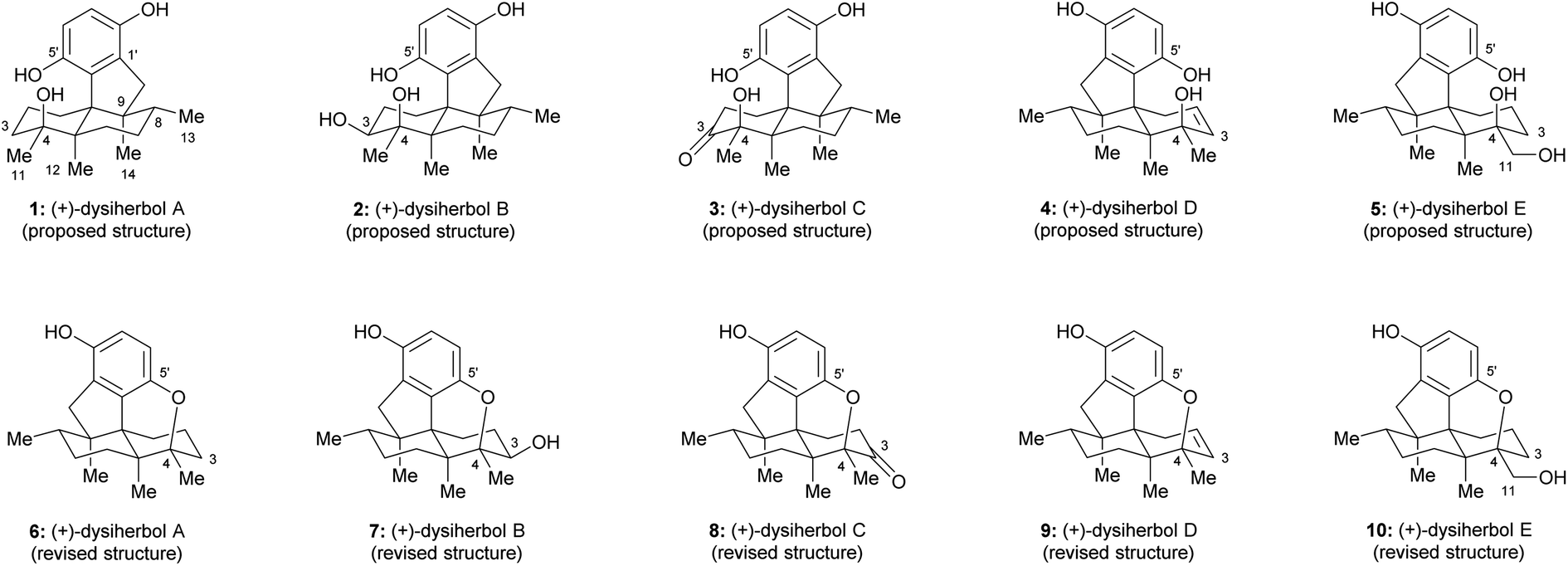
Sesquiterpene quinone/hydroquinone meroterpenoids are a class of mixed biosynthetic origin natural products mainly isolated from marine sources.1 In the past decade, a variety of sesquiterpene quinone/hydroquinone marine natural products with novel skeletons and anti-cancer activities were isolated and elucidated. Dysiherbols A–C (1–3, Fig. 1) are sesquiterpene hydroquinones isolated by Lin and colleagues from a Dysidea sp. marine sponge collected from the South China Sea in 2016.2 These three sesquiterpene hydroquinone natural products were reported to possess an intriguing 6/6/5/6-fused tetracyclic carbon skeleton.2 Preliminary bioactivity evaluation showed that dysiherbol A (1, Fig. 1) exhibited potent NF-κB inhibitory activity and cytotoxicity against human myeloma cancer cell line NCI H-929 with respective IC50 values of 0.49 and 0.58 μM, while dysiherbols B and C (2 and 3, Fig. 1) were about 10-fold and 20-fold less potent.2 In 2022, dysiherbols D and E (4 and 5, Fig. 1) were isolated by the same group from the marine sponge Dysidea avara collected from the South China Sea and were proposed to have the same carbon backbone but opposite absolute configuration than dysiherbols A–C (1–3).3 TNF-α-induced NF-κB activation evaluation in human HEK-293T cells showed that dysiherbols D and E (4 and 5) exhibited inhibitory activity with IC50 values of 10.2 and 8.6 μM, respectively.3
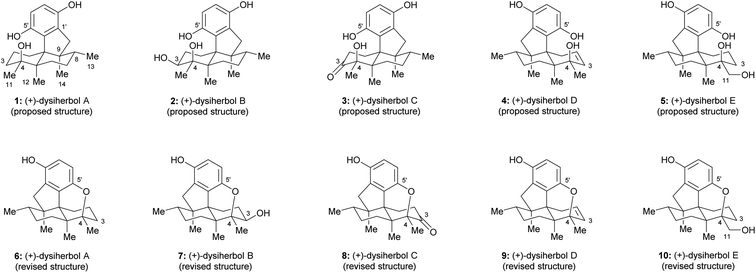 |
| | Fig. 1 Proposed and revised structures of (+)-dysiherbols A–E. | |
With their interesting structures, impressive bioactivities, and sparse availability from natural resources, dysiherbols A–E have attracted much attention from the synthetic community. In 2021, the Lu group disclosed the first total synthesis of (±)-dysiherbol A and revised the structure of dysiherbol A, which possesses an additional ether ring between C4 and C5′ instead of an alcohol group at C4 and a phenol group at C5′, as shown in Fig. 1.4 Only a few weeks later, the Schmalz group reported an enantioselective total synthesis of (−)-dysiherbol A, which not only confirmed the constitutional reassignment of dysiherbol A but also led to a revision of the absolute configuration of this natural product based on CD spectroscopic data.5 Last year, Liu et al. constructed the tetracyclic core structure of the dysiherbols through an intramolecular [2 + 2] cycloaddition, a Pd-catalyzed semipinacol rearrangement/Csp2−H arylation cascade and a visible-light-mediated ring-opening of a cyclopropyl silyl ether.6 Very recently, Tang and co-workers disclosed an elegant divergent total synthesis of dysiherbols A, C, and D by using a photo-induced quinone–alkene [2 + 2] cycloaddition and a tandem [1,2]-anionic rearrangement/cyclopropane fragmentation strategy.7
Due to the close relationship with dysiherbol A, it was proposed that the structures of dysiherbols B–E (2–5) also need to be revised as 7–10,4,5,7,8 as shown in Fig. 1. Here, we report a concise and divergent enantioselective total synthesis of the revised structures of (+)-dysiherbols A–E (6–10) in their natural absolute configuration.
Results and discussion
Biosynthetic hypothesis of dysiherbols A–E
From a biosynthetic point of view, it was postulated that dysiherbols A–E (6–10) might be generated from a common sesquiterpene hydroquinone natural product avarol (11),9 as shown in Scheme 1. Dehydrogenation of the C1–C10 single bond in avarol (11) gives rise to unconjugated diene 12 as a key intermediate. A 5-exo-trig cyclization of 12, connecting C6′ of hydroquinone moiety with C10 of the decalin unit, would give predysiherbol 13, which contains the core 6/6/5/6-fused tetracyclic framework of dysiherbols A–E (6–10). Direct cyclization of the C5′ phenol group with C4 of the double bond would give dysiherbol A (6). Oxidation of predysiherbol 13 and subsequent cyclization in a similar manner as in the case of dysiherbol A would afford dysiherbols B (7) and E (10). Further oxidation or elimination of the C3 hydroxy group of dysiherbol B (7) would render dysiherbols C (8) and D (9), respectively.
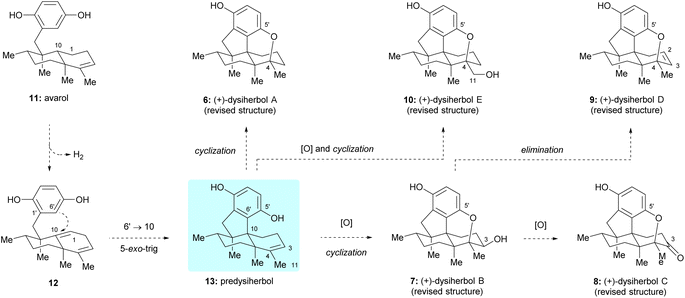 |
| | Scheme 1 Possible biosynthetic pathway of dysiherbols A–E. | |
Retrosynthetic analysis of dysiherbols A–E
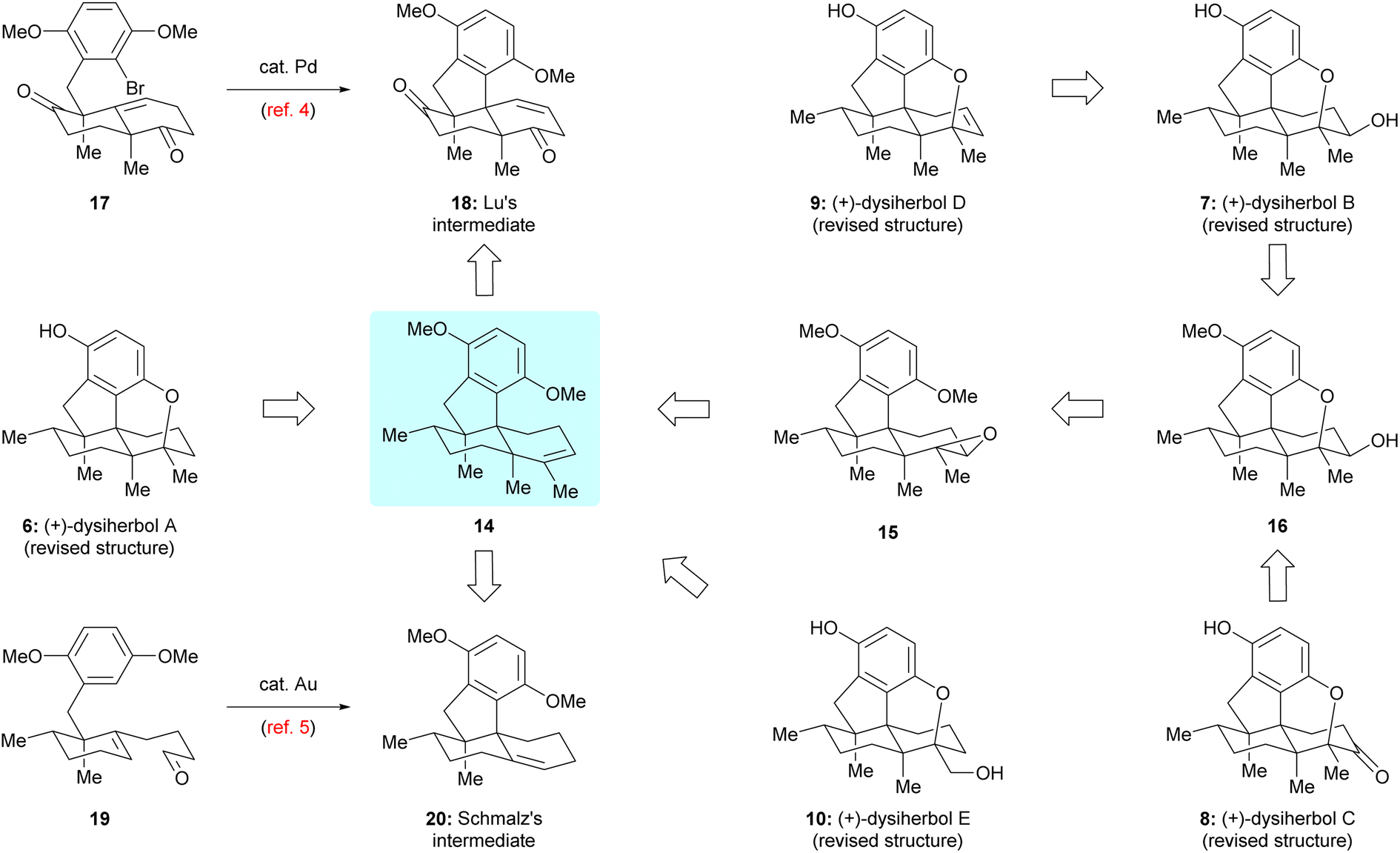
Guided by the above biosynthetic hypothesis, a retrosynthetic analysis of dysiherbols A–E (6–10) was conceived. To meet the challenge of synthesizing all dysiherbols in the natural absolute configuration, we envisioned using dimethyl predysiherbol 14 as a common late key precursor. As illustrated in Scheme 2, while this advanced key intermediate can be readily converted to dysiherbol A (6) by treatment with BBr3 as described previously,4,5 dysiherbol E (10) could be synthesized through allylic oxidation and subsequent cyclization of dimethyl predysiherbol 14.
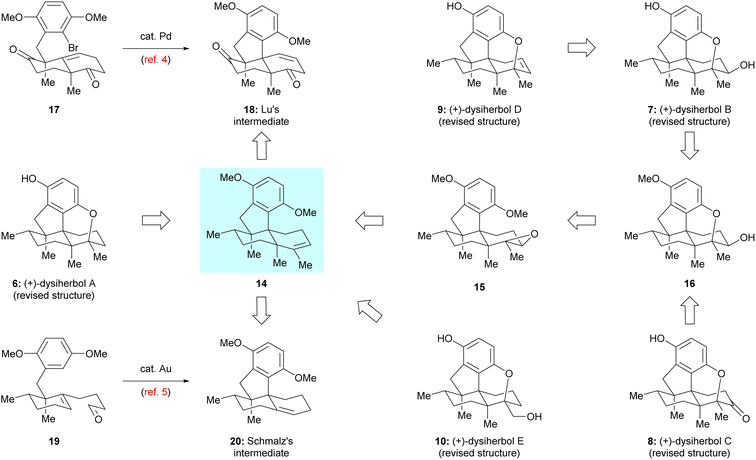 |
| | Scheme 2 Retrosynthetic analysis of dysiherbols A–E by two different routes. | |
We also reasoned that the synthesis of dysiherbols B–D (7–9) could be achieved by epoxidation of the double bond within 14 to give 15 followed by cyclization to an advanced intermediate such as pentacyclic alcohol 16. We report here the implementation of this concept, which proved to be surprisingly challenging due to the high tendency of the systems to undergo Wagner–Meerwein rearrangements. In addition, we elaborated two different efficient routes to dimethyl predysiherbol 14 by adapting, shortening, and optimizing our previously developed synthetic sequences.4,5 Noteworthy, both strategies exploit a metal-catalyzed cyclization step to set up the tetracyclic carbon skeleton. While the Lu's synthesis uses an intramolecular Heck reaction for the conversion of bromoalkene 17 into tetracyclic diketone 18, the Schmalz's synthesis features a gold-catalyzed double cyclization of aldehyde 19 to give tetracyclic olefin 20. Thus, besides confirming the proposed structures of the dysiherbols by total synthesis, the present work also demonstrates the power of modern synthetic methodology for the construction of complex polycyclic molecules displaying a high density of quaternary carbon centers.10
Synthesis of dimethyl predysiherbol 14
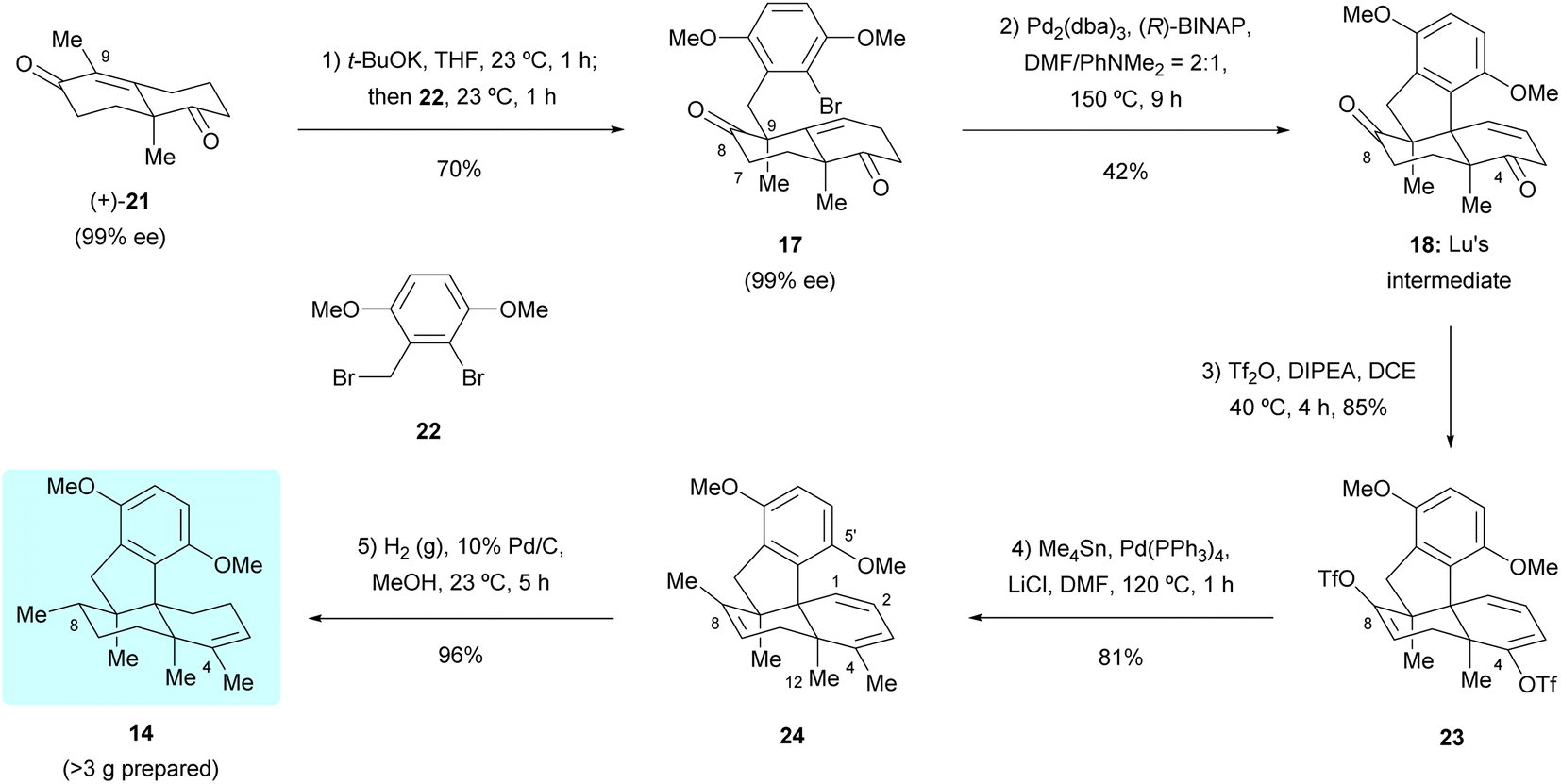
Our first synthetic route towards dimethyl predysiherbol 14 started from the union of Wieland–Miescher ketone derivative 21 and benzyl bromide 22. As shown in Scheme 3, treatment of enantioenriched Wieland–Miescher ketone derivative 21 (99% ee)11 with benzyl bromide 22 in the presence of t-BuOK proceeded smoothly on a gram scale to give bromoalkene 17 in high isolated yield (70%). The enantiomeric purity of bromoalkene 17 retained well (99% ee). However, the following intramolecular Heck reaction of 17 to construct tetracyclic diketone 18 proved to be unexpectedly challenging due to a competing arylation reaction12 at C7 and Grignard-type nucleophilic addition13 of the arylpalladium intermediate to the keto group at C8. After extensive exploration of catalysts, ligands, bases, and solvents,14 we were pleased to find that the intramolecular Heck reaction of 17 with Pd2(dba)3 as catalyst, (R)-BINAP as ligand and PhNMe2 as base rendered the cyclized product 18 in acceptable isolated yield (42%). All our attempts to perform a reductive Heck reaction15 of 17 under various conditions only led to trace amounts of the desired product. The tetracyclic diketone 18 was then converted to the bistriflate 23, the Stille coupling of which with Me4Sn afforded the triene 24 in 69% overall yield over both steps.16 The chemo- and diastereoselective reduction of triene 24 was perfectly achieved by heterogeneous hydrogenation, giving dimethyl predysiherbol 14 in almost quantitative yield (96%). The excellent chemo- and diastereoselective hydrogenation occurs at C8 is attributed to steric hindrances. We found that the disubstituted olefin at C1 and C2 was first reduced. For the hydrogenation of these two trisubstituted olefins, the C12 methyl group blocks the approach of H–Pd species to C3![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C4 and C7C8 double bonds from the bottom face and the methoxyl group on C5′ blocks the approach of H–Pd species to C3C4 double bond from the top face, which also explains the easy formation of the bridged ether ring between C5′ and C4 (vide infra). Thus, the hydrogenation of 24 occurs at C8 from the top face, giving 14 in excellent yield and diastereoselectivity. Noteworthy, a total of more than 3 grams of 14 were prepared, which ensured the material supply for the synthesis of (+)-dysiherbols A–E (6–10) and their designed analogs for further bioactivity evaluation.
C4 and C7C8 double bonds from the bottom face and the methoxyl group on C5′ blocks the approach of H–Pd species to C3C4 double bond from the top face, which also explains the easy formation of the bridged ether ring between C5′ and C4 (vide infra). Thus, the hydrogenation of 24 occurs at C8 from the top face, giving 14 in excellent yield and diastereoselectivity. Noteworthy, a total of more than 3 grams of 14 were prepared, which ensured the material supply for the synthesis of (+)-dysiherbols A–E (6–10) and their designed analogs for further bioactivity evaluation.
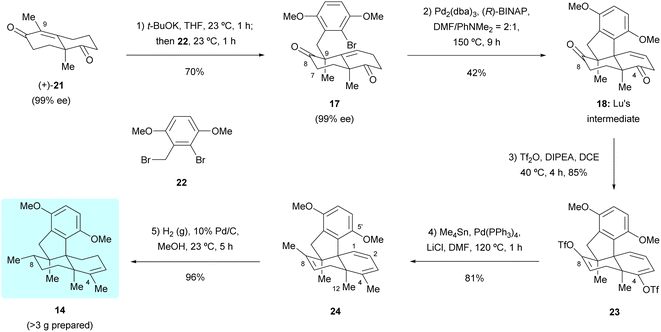 |
| | Scheme 3 Synthesis of dimethyl predysiherbol 14 by Lu's intermediate 18. (R)-BINAP = (R)-2,2′-bis(diphenylphosphino)-1,1′-dinaphthalene, DCE = 1,2-dichloroethane, DIPEA = N,N-diisopropylethylamine, DMF = N,N-dimethylformamide, Pd2(dba)3 = tris(dibenzylideneacetone)dipalladium (0), Tf2O = trifluoromethanesulfonic anhydride. | |
The second route towards dimethyl predysiherbol 14 was developed based on the previously reported synthesis of Schmalz's intermediate 20,5 as shown in Scheme 4. Now, we employed the enantiomeric phosphoramidate ligand L* in the chirogenic opening step (Cu-catalyzed 1,4-addition/enolate trapping) to ensure the required absolute configuration. Starting from 2-methylcyclohexenone 25 and benzyl iodide 26, the triple substituted cyclohexanone 27 was obtained with high enantioselectivity (96% ee). While the conversion of 27 into tetracyclic ketone 28via intermediate 19 and its Au-catalyzed tandem cyclization to Schmalz's intermediate 20 succeeded smoothly under reported conditions,5 we spend some effort to optimize the remaining steps of the synthesis of dimethyl predysiherbol 14. The challenging α-methylation of ketone 28 at the bridgehead carbon to give 30 was achieved by (1) regioselective deprotonation/O-methylation (LiH, TPPA, 160 °C; then MeI), (2) Simmons–Smith cyclopropanation of the resulting enol ether, and (3) proteolytic cleavage of the cyclopropane 29. This way, ketone 30 was obtained in an improved overall yield of 57% (3 steps) on a 120 mg scale. The final conversion of ketone 30 into dimethyl predysiherbol 14 was then achieved in 73% overall yield through enol triflate formation and subsequent Stille coupling.
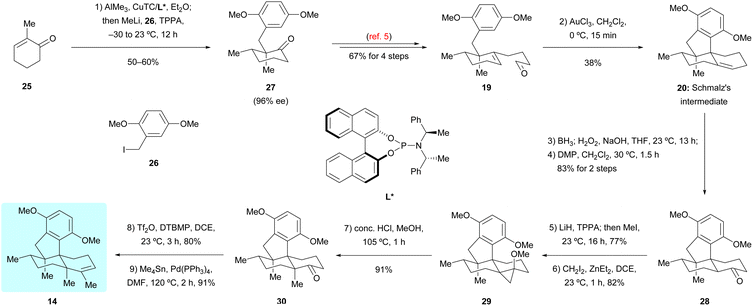 |
| | Scheme 4 Synthesis of dimethyl predysiherbol 14 by Schmalz's intermediate 20. CuTC = copper(I) 2-thiophenecarboxylate, DTBMP = 2,6-bis(1,1-dimethylethyl)-4-methylpyridine, TPPA = tripyrrolidinophosphoric acid triamide. | |
Total synthesis of (+)-dysiherbols A and E
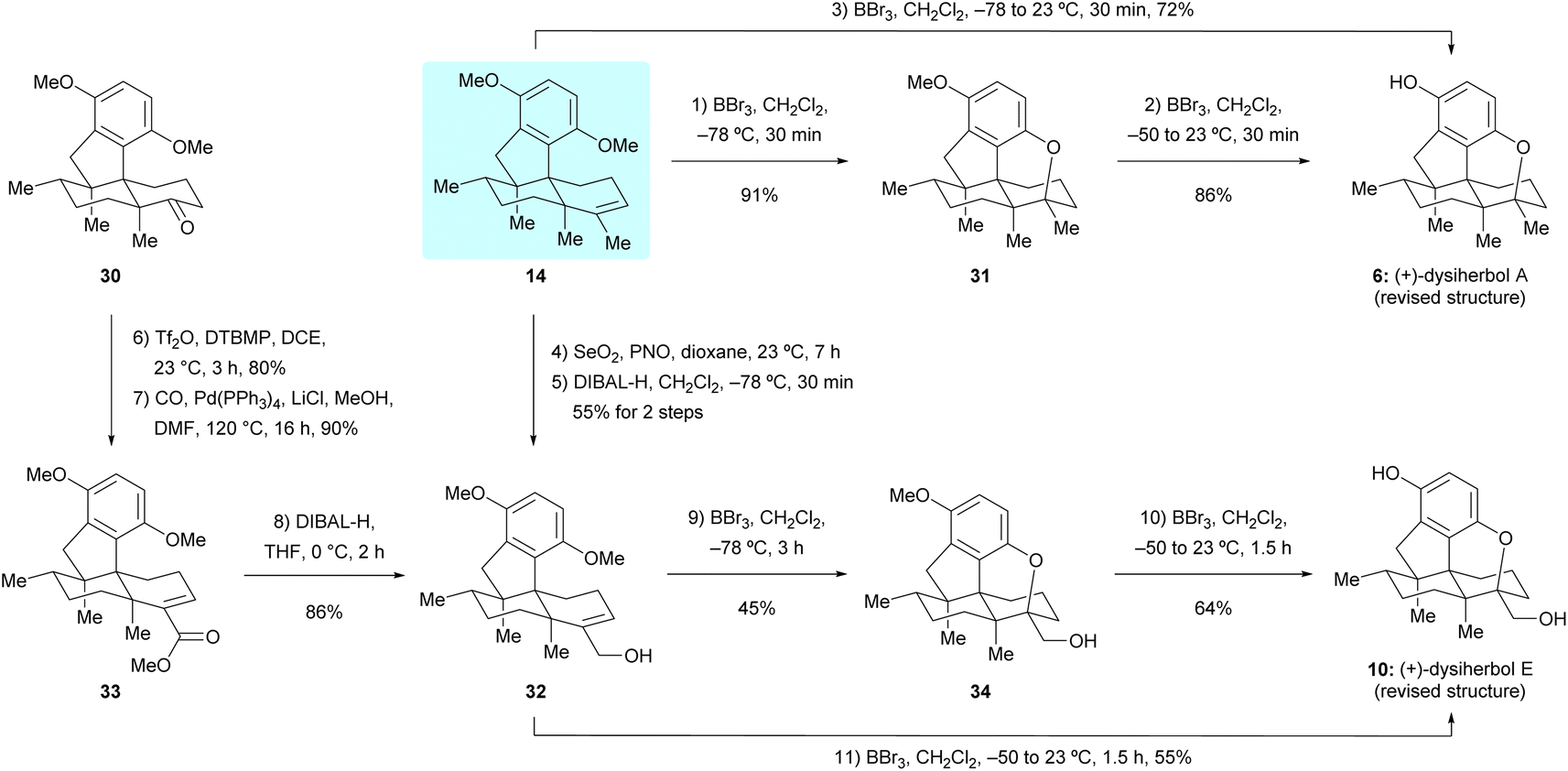
With sufficient quantities of dimethyl predysiherbol 14 in hand, we entered the final stage of its transformation to target natural products (+)-dysiherbols A and E (6 and 10). As shown in Scheme 5, exposure of dimethyl predysiherbol 14 with BBr3 at −78 °C gave cyclized compound 31 in 91% yield under the previously developed reaction conditions.4 Deprotection of the remaining O-methyl group of 31 with BBr3 at 23 °C afforded (+)-dysiherbol A in 86% yield, which also could be prepared from dimethyl predysiherbol 14 directly in 72% overall yield. The spectroscopic and optical rotation data of synthetic (+)-dysiherbol A (6) matched well with those for natural (+)-dysiherbol A.
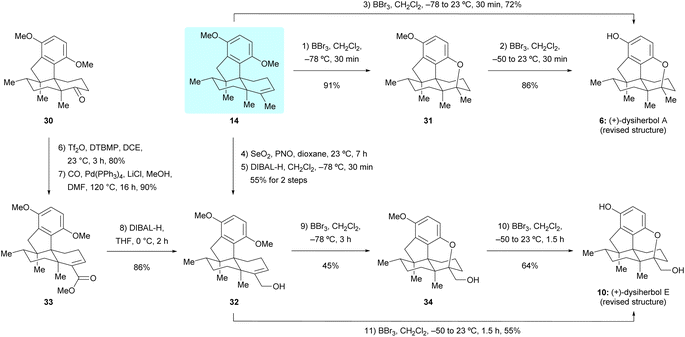 |
| | Scheme 5 Total synthesis of the revised structures of (+)-dysiherbols A and E. DIBAL-H = diisobutylaluminium hydride, PNO = pyridine N-oxide. | |
We then turned our attention to the synthesis of (+)-dysiherbol E. Allylic oxidation of dimethyl predysiherbol 14 to alcohol 32 proved to be challenging due to the existence of different oxidizable positions. SeO2 and other tested oxidation reagents only led to poor yields of 32.17 After extensive screening of oxidating reagents and reaction conditions, we were pleased to find that the combination of SeO2 and pyridine N-oxide (PNO)18 gave the best combined yield of allylic alcohol 32 and the corresponding aldehyde resulting from overoxidation. Treatment of the crude product mixture with diisobutylaluminium hydride (DIBAL-H) afforded allylic alcohol 32 in 55% yield for the two steps. As an alternative access to the alcohol 32, we found that the enol triflate derived from ketone 30 can be efficiently methoxycarbonylated by reaction with CO and MeOH in the presence of Pd(PPh3)4 as a catalyst and LiCl as an additive. Reduction of the resulting ester 33 using DIBAL-H then afforded 32 in 77% yield over two steps. Under similar cyclization conditions as used in the synthesis of (+)-dysiherbol A (6), exposure of allylic alcohol 32 to BBr3 at −78 °C for 3 h gave the cyclized intermediate 34 in 45% yield besides a small amount of unreacted starting material 32 and the O-demethylated product, namely (+)-dysiherbol E (10). Deprotection of the methyl group of 34 with BBr3 at 23 °C gave (+)-dysiherbol E (10) in 64% yield. Direct treatment of allylic alcohol 34 with BBr3 at 23 °C for 1.5 h also gave (+)-dysiherbol E (10) in 55% yield. The spectroscopic and optical rotation data of synthetic (+)-dysiherbol E (10) matched well with those reported for natural (+)-dysiherbol E. The calculated electronic circular dichroisms (ECDs) of (+)-dysiherbols A (6) and E (10) matched well with experimental ECDs (see Fig. 2b).
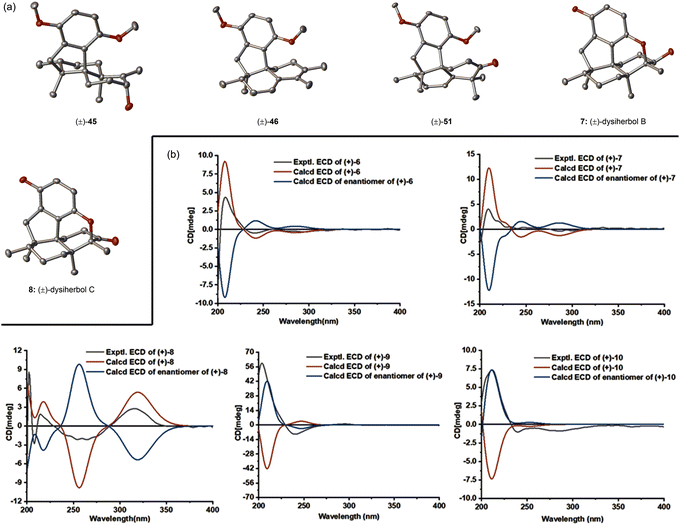 |
| | Fig. 2 (a) ORTEP drawing of the structures of key intermediates and the revised structures of dysiherbols B and C. (b) Experimental and calculated ECDs for (+)-dysiherbols A–E. Thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms have been omitted for clarity. | |
1,2-Methyl shift processes in dysiherbol-related compounds
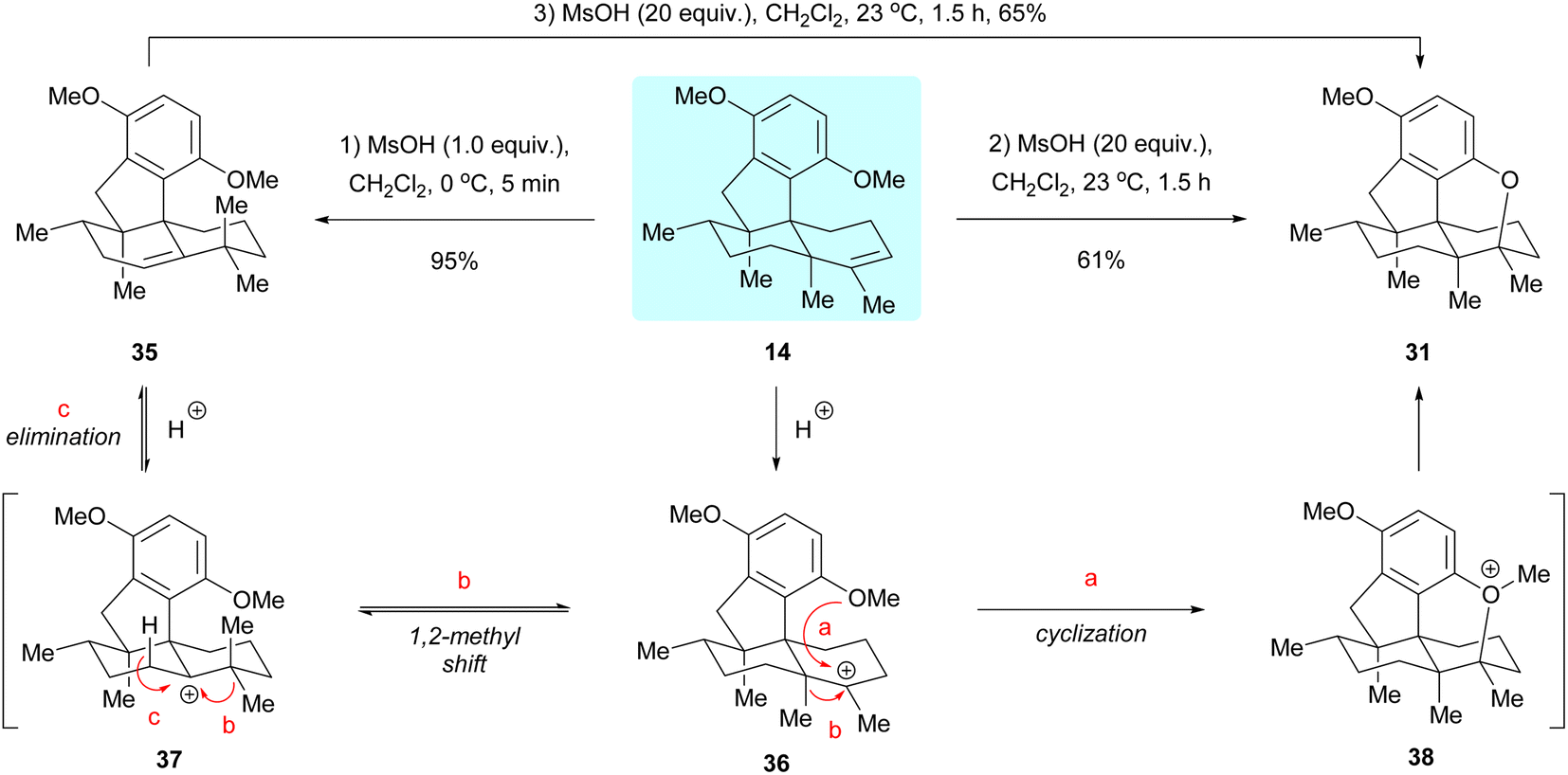
In studying the cyclization reaction of dimethyl predysiherbol 14, we found that treatment of this compound with 1.0 equivalent of methanesulfonic acid (MsOH) at 0 °C gave the rearranged product 35 in 95% yield, while treatment of 14 with 20 equivalents of MsOH at 23 °C afforded the cyclized ether 31 in 61% yield, as shown in Scheme 6.5 Further experiments showed that the rearranged product 35 could also be converted to the cyclized ether 31 in 65% yield by treatment with 20 equivalents of MsOH at 23 °C. We rationalized that the carbenium ion 36 is first generated from dimethyl predysiherbol 14 under acidic conditions. Then reversible methyl migration (Wagner–Meerwein rearrangement) gives the cation 37, from which the rearranged product 35 is formed by proton elimination (pathway c). Under more drastic conditions, however, the carbenium ion 36 is trapped by the adjacent methoxy group to irreversibly yield the cyclized product 31via the oxonium intermediate 38 (pathway a). This proves the reversibility of such 1,2-methyl shift processes in dysiherbol-related compounds.5
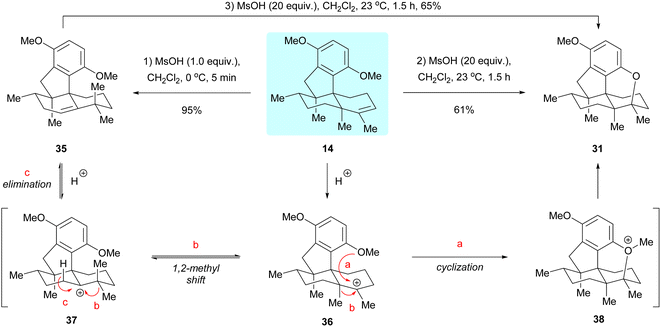 |
| | Scheme 6 1,2-Methyl shift processes in dysiherbol-related compounds. MsOH = methanesulfonic acid. | |
Oxidation and cyclization investigation of dimethyl predysiherbol 14
Having successfully accomplished the total synthesis of (+)-dysiherbols A (6) and E (10), we switched our attention to the synthesis of (+)-dysiherbols B–D (7–9). However, the transformation of dimethyl predysiherbol 14 to (+)-dysiherbols B–D (7–9) proved to be surprisingly challenging and could not be achieved in the envisioned straightforward manner. Aiming at the synthesis of (+)-dysiherbols B–D (7–9), we envisioned that epoxidation of the double bond of dimethyl predysiherbol 14 and subsequent cyclization would give pentacyclic alcohol 43via intermediates 39, 40, 41 and 42 (pathway a), as depicted in Scheme 7. For this purpose, dimethyl predysiherbol 14 was treated with m-chloroperbenzoic acid (m-CPBA). Surprisingly, only the rearranged homoallylic alcohol 44 was obtained in 80% yield when the reaction was quenched at −78 °C, presumably by direct proton elimination of carbenium 40 (pathway c). The desired cyclization did not occur when the reaction mixture was warmed to 23 °C. Instead, the allylic alcohol 45 was isolated in 64% yield presumably through a similar rearrangement (1,2-methyl migration) of the carbenium ion 40 to 41 and proton elimination of the latter (pathway d). The structure of allylic alcohol 45 and the orientation of the newly generated hydroxy group were verified by X-ray crystallographic analysis (see Fig. 2a).
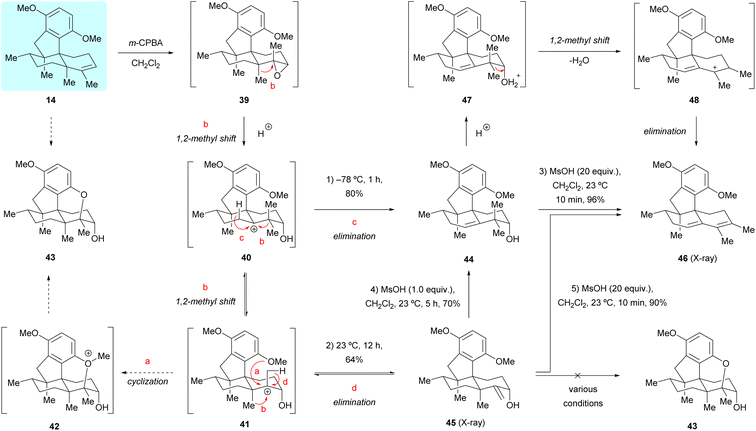 |
| | Scheme 7 Oxidation and cyclization investigation of dimethyl predysiherbol 14. | |
We assumed that the homoallylic alcohol 44 could be transformed to cyclized ether 43 through the 1,2-methyl shift of 40 to the cation 41 and subsequent cyclization. However, when homoallylic alcohol 44 was treated with various Brønsted acids (e.g., TsOH and MsOH) or Lewis acids (e.g., BBr3 and BF3·OEt2) the expected cyclized product was not observed. In addition, upon treatment of 44 with excess MsOH, the tetracyclic diene 46 was isolated in near quantitative yield (96%), presumably via the cationic intermediates 47 and 48. The transformation of 45 to pentacyclic alcohol 43 also met with failure under various tested conditions. Indeed, treatment of allylic alcohol 45 with various Brønsted acids or Lewis acids also led to the elimination product 46 in high yield probably via the homoallylic alcohol 44.19 The structure of 46 was secured by X-ray crystallographic analysis (see Fig. 2a). We would like to note that homoallylic alcohol 44 could be prepared in high yield (67–80%) from dimethyl predysiherbol 14 either using magnesium monoperoxyphthalate (MMPP)20 or trifluoroacetone-oxone-Na2EDTA21 and the isomeric, non-rearranged allylic alcohol 45 could also be prepared from dimethyl predysiherbol 14 through 1O2 ene reaction22 under photochemical conditions in 62% yield (see ESI† for details).
Total synthesis of (+)-dysiherbols B–D
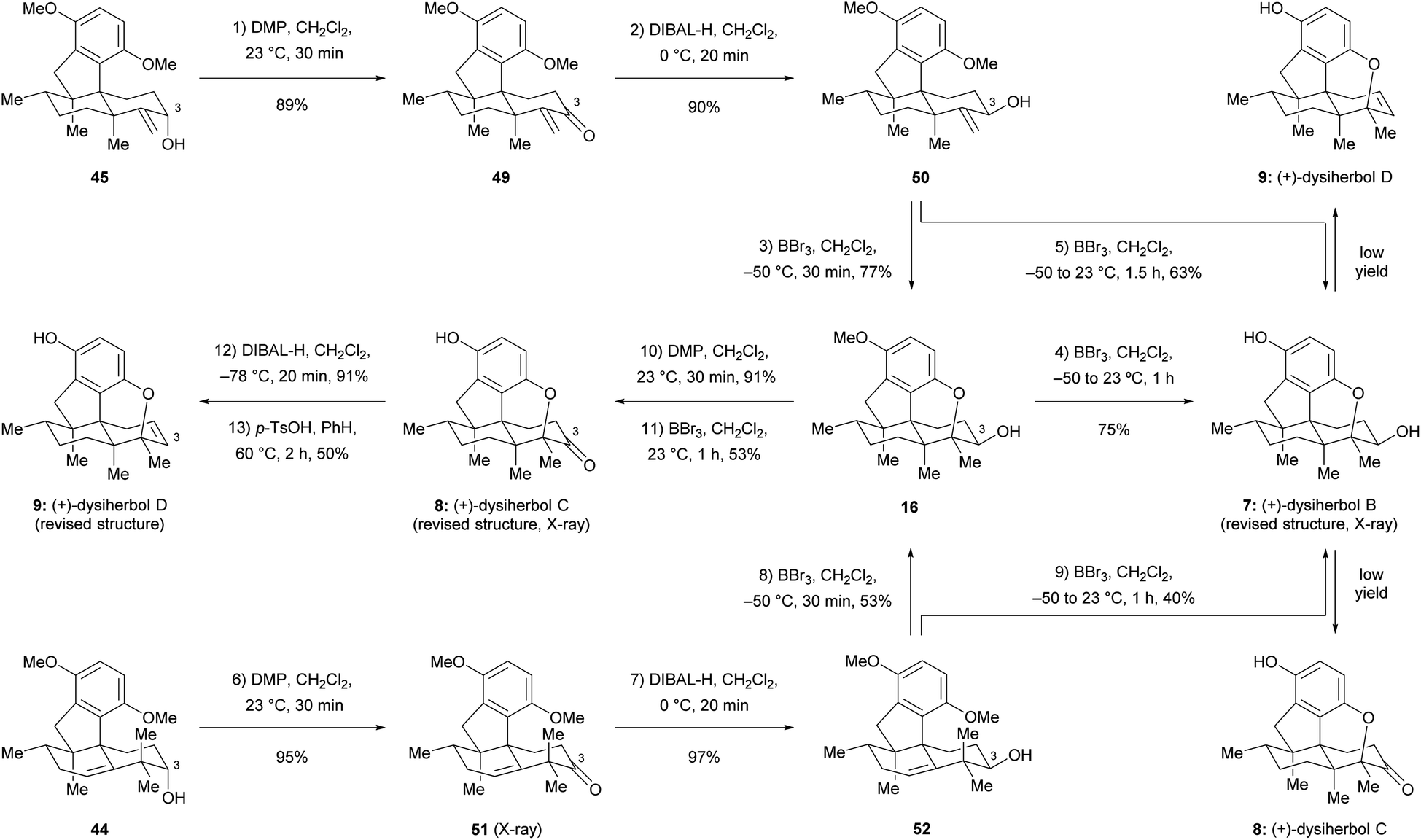
Since the direct cyclization of homoallylic alcohol 44 or allylic alcohol 45 to the cyclized ether 43 failed, we anticipated that the configuration of the C3 hydroxy group of these two compounds might affect the formation of the additional ether bridge by changing the conformational bias of the carbenium ion intermediates. To this end, we inverted the configuration of the C3 hydroxy group prior to the cyclization. As shown in Scheme 8, Dess–Martin periodinane (DMP) oxidation of allylic alcohol 45 (89% yield) and subsequent DIBAL-H reduction of the resulting ketone 49 (90% yield) afforded allylic alcohol 50, which possesses the correct configuration of the C3 hydroxy group, as found in (+)-dysiherbol B. Much to our delight, the ether ring formation reaction now proceeded smoothly when allylic alcohol 50 was treated with BBr3 at −50 °C for 30 min, affording cyclized ether 16 in 77% yield. Removal of the remaining methyl group by the same reagent but at 23 °C for 1 h resulted in (+)-dysiherbol B (7) in 75% yield. In addition, (+)-dysiherbol B (7) could be directly prepared from 50 through a one-pot procedure in 63% yield. The synthetic (+)-dysiherbol B (7) exhibited identical spectroscopic and optical rotation data as those reported for the natural product. X-ray crystallographic analysis of synthetic dysiherbol B (7) clearly showed the additional ether bridge between C4 and C5′, thus confirming the revised structure of this natural product (see Fig. 2a).
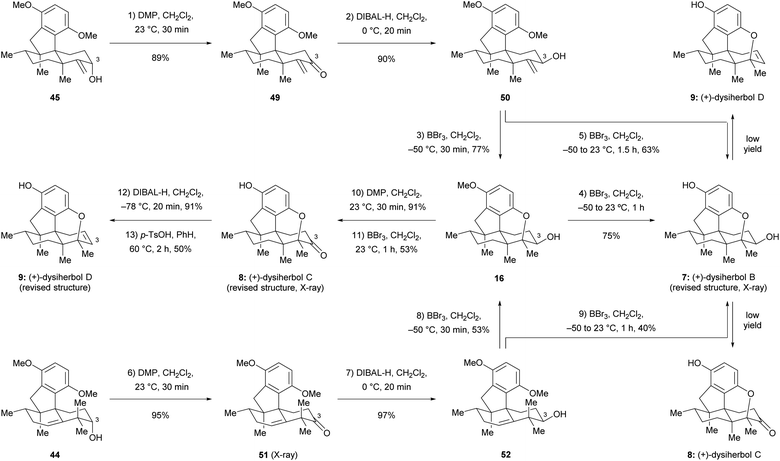 |
| | Scheme 8 Total synthesis of the revised structures of (+)-dysiherbols B–D. DMP = Dess–Martin periodinane, p-TsOH = p-toluenesulfonic acid. | |
Prompted by the above results, we anticipated that homoallylic alcohol 44 also could be transformed to (+)-dysiherbol B (7) after inversion of the configuration at C3 and subsequent cyclization. Thus, homoallylic alcohol 44 was converted to its epimer 52 through DMP oxidation and subsequent DIBAL-H reduction in 92% yield over two steps. The structure of ketone 51 was also confirmed by X-ray crystallographic analysis (see Fig. 2a). As we expected, the subjection of homoallylic alcohol 52 to BBr3 also gave the cyclized ether 16 in 53% yield, probably via above-mentioned methyl migration and subsequent ether ring formation, along with a significant amount of elimination byproduct 46 (43%). Homoallylic alcohol 52 was also converted to (+)-dysiherbol B in a one-pot procedure in 40% yield, besides 40% of 46.
With (+)-dysiherbol B (7) readily available, we anticipated that oxidation and elimination of the C3 hydroxy group could afford (+)-dysiherbols C (8) and D (9), respectively. However, under various tested oxidation and elimination reagents and conditions, very low yields of (+)-dysiherbols C (8) or D (9) were obtained presumably due to the instability of the free phenol functionality. (+)-Dysiherbols C (8) and D (9) could be alternatively synthesized from 16 avoiding exposure of the sensitive free phenol to oxidation and elimination conditions. To this end, DMP oxidation of the C3 hydroxy group (91%) and deprotection of the remaining methyl group of 16 resulted in (+)-dysiherbol C (8) in 53% yield. Attempts to directly eliminate the equatorial OH group in 16 proved to be fruitless. After inversion of the configuration of the hydroxy group by DIBAL-H reduction (91% yield) of (+)-dysiherbol C (8), acid-mediated elimination afforded (+)-dysiherbol D (9) in acceptable yield (50%). The spectroscopic and optical rotation data of synthetic (+)-dysiherbols C (8) and D (9) matched with those reported for natural products. The revised structure of dysiherbol C (8) was also confirmed by X-ray crystallographic analysis (see Fig. 2a). The calculated ECDs of (+)-dysiherbols B–D (7–9) matched well with experimental ECDs (see Fig. 2b).
Conclusions
In summary, we have elaborated a concise and divergent total synthesis of the revised structures of marine anti-cancer sesquiterpene hydroquinone meroterpenoids (+)-dysiherbols A–E (6–10) in their natural absolute configuration via the common intermediate dimethyl predysiherbol 14. The key common intermediate dimethyl predysiherbol 14 was efficiently synthesized in non-racemic form by two different routes, both exploiting a transition metal-catalyzed cyclization reaction to construct the tetracyclic carbon skeleton. The synthesis of (+)-dysiherbols A (6) and E (10) was accomplished through direct cyclization and allylic oxidation and subsequent cyclization of dimethyl predysiherbol 14, respectively. The synthesis of (+)-dysiherbols B–D (7–9) was achieved by epoxidation of dimethyl predysiherbol 14 and cationic cyclization, overcoming unexpected difficulties resulting from the tendency of the systems to undergo reversible 1,2-methyl migration (“methyl shuttle”). Importantly, the synthesis of (+)-dysiherbols A–E (6–10) confirmed their revised structures. The work described here also paves the way for the synthesis and evaluation of the anti-cancer activity of a wide variety of analogs of (+)-dysiherbols A–E (6–10), which is currently underway in our laboratories and will be reported in due course.
Data availability
All experimental and characterization data in this manuscript are available in the ESI.† Crystallographic data for compounds 45, 46, 51, 7, and 8 have been deposited at the Cambridge Crystallographic Data Center and assigned numbers 2124205, 2192301, 2192302, 2141304, and 2141326.
Author contributions
Z. L. and H.-G. S. conceived and supervised the project. C. C., L. C., I. G., Q. Z., Y. K., B. W., J. K., and J. B. carried out the experimental work, collected the data, and analyzed the results. W. L. and Y. G. conducted the DFT calculations of ECDs. Z. L. and H.-G. S. wrote the paper.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 22171146, 21971121, and 22188101 to ZL) and the China Postdoctoral Science Foundation (No. 2021M701775 to CC). We thank the Jürgen Manchot Foundation (doctoral stipend to IG) and the Fonds der Chemischen Industrie (doctoral stipend to JB). We are grateful to Dr Ruocheng (Ronnie) Yu (Rice University) for his kind help during the preparation of this manuscript. We thank Dr Qingxin Cui (Nankai University) for NMR spectroscopic assistance and Dr Quanwen Li (Nankai University) for X-ray crystallographic analysis.
Notes and references
-
(a) P. A. García, Á. P. Hernández, A. San Feliciano and M. Á. Castro, Mar. Drugs, 2018, 16, 292 CrossRef PubMed;
(b) M. Gordaliza, Mar. Drugs, 2010, 8, 2849 CrossRef CAS PubMed;
(c) I. S. Marcos, A. Conde, R. F. Moro, P. Basabe, D. Diez and J. G. Urones, Mini-Rev. Org. Chem., 2010, 7, 230 CrossRef CAS.
- W.-H. Jiao, G.-H. Shi, T.-T. Xu, G.-D. Chen, B.-B. Gu, Z. Wang, S. Peng, S.-P. Wang, J. Li, B.-N. Han, W. Zhang and H.-W. Lin, J. Nat. Prod., 2016, 79, 406 CrossRef CAS PubMed.
- H.-Y. Liu, M. Zhou, R.-Y. Shang, L.-L. Hong, G.-H. Wang, W.-J. Tian, W.-H. Jiao, H.-F. Chen and H.-W. Lin, Chin. J. Nat. Med., 2022, 20, 148 CAS.
- C. Chong, Q. Zhang, J. Ke, H. Zhang, X. Yang, B. Wang, W. Ding and Z. Lu, Angew. Chem., Int. Ed., 2021, 60, 13807 CrossRef CAS PubMed.
- J. Baars, I. Grimm, D. Blunk, J.-M. Neudörfl and H.-G. Schmalz, Angew. Chem., Int. Ed., 2021, 60, 14915 CrossRef CAS PubMed.
- R. Liu, M. Xia, C. Ling, S. Fu and B. Liu, Org. Lett., 2022, 24, 1642 CrossRef CAS PubMed.
- S. Hu and Y. Tang, J. Am. Chem. Soc., 2022, 144, 19521 CrossRef CAS PubMed.
- C. Chong and Z. Lu, Synlett, 2021, 32, 1777 CrossRef CAS.
-
(a) R. Puliti, S. De Rosa and C. A. Mattia, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1994, 50, 830 CrossRef;
(b) S. De Rosa, L. Minale, R. Riccio and G. Sodano, J. Chem. Soc., Perkin Trans. 1, 1976, 1408 RSC;
(c) L. Minale, R. Riccio and G. Sodano, Tetrahedron Lett., 1974, 15, 3401 CrossRef.
-
(a) Z. Xin, H. Wang, H. He and S. Gao, Tetrahedron Lett., 2021, 71, 153029 CrossRef CAS;
(b) M. Büschleb, S. Dorich, S. Hanessian, D. Tao, K. B. Schenthal and L. E. Overman, Angew. Chem., Int. Ed., 2016, 55, 4156 CrossRef PubMed.
-
(a) K. K. Wan, K. Iwasaki, J. C. Umotoy, D. W. Wolan and R. A. Shenvi, Angew. Chem., Int. Ed., 2015, 54, 2410 CrossRef CAS PubMed;
(b) T. Ling, J. Xu, R. Smith, A. Ali, C. L. Cantrell and E. A. Theodorakis, Tetrahedron, 2011, 67, 3023 CrossRef CAS PubMed;
(c) H. Hagiwara and H. Uda, J. Org. Chem., 1988, 53, 2308 CrossRef CAS;
(d) A. B. Smith III and R. Mewshaw, J. Org. Chem., 1984, 49, 3685 CrossRef;
(e) K. Hiroi and S.-I. Yamada, Chem. Pharm. Bull., 1975, 23, 1103 CrossRef CAS;
(f) W. Acklin, V. Prelog and A. P. Prieto, Helv. Chim. Acta, 1958, 61, 1416 CrossRef;
(g) P. Wieland and K. Miescher, Helv. Chim. Acta, 1950, 33, 2215 CrossRef CAS.
-
(a) B. C. Hamann and J. F. Hartwig, J. Am. Chem. Soc., 1997, 119, 12382 CrossRef CAS;
(b) M. Palucki and S. L. Buchwald, J. Am. Chem. Soc., 1997, 119, 11108 CrossRef CAS.
-
(a) D. Solé, L. Vallverdú, X. Solans, M. Font-Bardía and J. Bonjoch, J. Am. Chem. Soc., 2003, 125, 1587 CrossRef PubMed;
(b) L. G. Quan, M. Lamrani and Y. Yamamoto, J. Am. Chem. Soc., 2000, 122, 4827 CrossRef CAS;
(c) J. Vicente, J.-A. Abad and J. Gil-Rubio, J. Organomet. Chem., 1992, 436, C9 CrossRef CAS.
-
(a) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed;
(b) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 CrossRef CAS PubMed;
(c) A. Endo, A. Yanagisawa, M. Abe, S. Tohma, T. Kan and T. Fukuyama, J. Am. Chem. Soc., 2002, 124, 6552 CrossRef CAS PubMed;
(d) S. P. Govek and L. E. Overman, J. Am. Chem. Soc., 2001, 123, 9468 CrossRef CAS PubMed;
(e) L. E. Overman, Pure Appl. Chem., 1994, 66, 1423 CrossRef CAS;
(f) C. Y. Hong, N. Kado and L. E. Overman, J. Am. Chem. Soc., 1993, 115, 11028 CrossRef CAS.
-
(a) T. Ghosh, ChemistrySelect, 2019, 4, 4747 CrossRef CAS;
(b) A. Amorese, A. Arcadi, E. Bernocchi, S. Cacchi, S. Cerrini, W. Fedeli and G. Ortar, Tetrahedron, 1989, 45, 813 CrossRef CAS;
(c) S. Cacchi and A. Arcadi, J. Org. Chem., 1983, 48, 4236 CrossRef CAS.
- H. Zhang, S. Ma, Z. Yuan, P. Chen, X. Xie, X. Wang and X. She, Org. Lett., 2017, 19, 3478 CrossRef CAS PubMed.
- A. Nakamura and M. Nakada, Synthesis, 2013, 45, 1421 CrossRef CAS.
- K. A. Parker and A. Dermatakis, J. Org. Chem., 1997, 62, 6692 CrossRef CAS.
- H. Ageta, K. Shiojima and Y. Arai, Chem. Pharm. Bull., 1987, 35, 2705 CrossRef CAS.
- S. Meninno, R. Villano and A. Lattanzi, Eur. J. Org. Chem., 2021, 2021, 1758 CrossRef CAS.
- P. O'Brien and C. D. Pilgram, Tetrahedron Lett., 1999, 40, 8427 CrossRef.
-
(a) Y.-J. Hu, C.-C. Gu, X.-F. Wang, L. Min and C.-C. Li, J. Am. Chem. Soc., 2021, 143, 17862 CrossRef CAS PubMed;
(b) L. Min, X. Lin and C.-C. Li, J. Am. Chem. Soc., 2019, 141, 15773 CrossRef CAS PubMed;
(c) X. Yu, L. Xiao, Z. Wang and T. Luo, J. Am. Chem. Soc., 2019, 141, 3440 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Le
Chang‡
a,
Isabelle
Grimm‡
b,
Qunlong
Zhang
a,
Le
Chang‡
a,
Isabelle
Grimm‡
b,
Qunlong
Zhang