
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA rare isocyanide derived from an unprecedented neutral yttrium(II) bis(amide) complex†
Rashmi
Jena
 ,
Florian
Benner
,
Francis
Delano
IV
,
Daniel
Holmes
,
John
McCracken
*,
Selvan
Demir
* and
Aaron L.
Odom
*
,
Florian
Benner
,
Francis
Delano
IV
,
Daniel
Holmes
,
John
McCracken
*,
Selvan
Demir
* and
Aaron L.
Odom
*
Michigan State University, Department of Chemistry, 578 S. Shaw Ln, East Lansing, MI, USA 48824. E-mail: odoma@msu.edu; sdemir@chemistry.msu.edu; mccracke@msu.edu
First published on 21st March 2023
Abstract
A room temperature stable complex formulated as Y(NHAr*)2 has been prepared, where Ar* = 2,6-(2,4,6-(iPr)3C6H2)C6H3, by KC8 reduction of ClY(NHAr*)2. Based on EPR evidence, Y(NHAr*)2 is an example of a d1 Y(II) complex with significant delocalization of the unpaired electron density from the metal to the ligand. The isolation of molecular divalent metal complexes is challenging for rare earth elements such as yttrium. In fact, stabilization of the divalent state requires judicious ligand design that allows the metal center to be coordinatively saturated. Divalent rare earth elements tend to be reactive towards various substrates. Interestingly, Y(NHAr*)2 reacts as a radical donor towards tBuNC to generate an unusual yttrium isocyanide complex, CNY(NHAr*)2, based on spectroscopic evidence and single-crystal X-ray diffraction data.
Introduction
Stabilization of metal centers in unusual oxidation states provides opportunities to examine new electronic structures, metal–ligand interactions, and reactivity. While the vast majority of Group 3 transition metals and lanthanide compounds have the metal center in the +3 formal oxidation state, the oxidation states of many of the rare earth metals have been expanded to include examples where the formal oxidation state is +2, and Evans wrote in 2016 that, “Ln2+ ions are now known for all the lanthanides except {radioactive} Pm.”1The first Y(II) complex, {Y(C5H4SiMe3)3}−, was prepared by reduction of the neutral Y(III) complex (Chart 1), and the stability of these complexes is closely tied to the counterion used.2–4 Recently, an Y(II) complex using tris(aryloxide)mesitylene ligands was reported by Meyer and Evans that is stable for ∼48 h at room temperature in THF.5,6 In addition to cyclopentadienyl and alkoxide ligands, hexamethyldisilazides have been employed.7,8 In all cases, the currently known yttrium(II) complexes possess very bulky and strongly donating ligands in an approximately trigonal environment around the metal center, and all the ligands are retained on reduction to the anion.
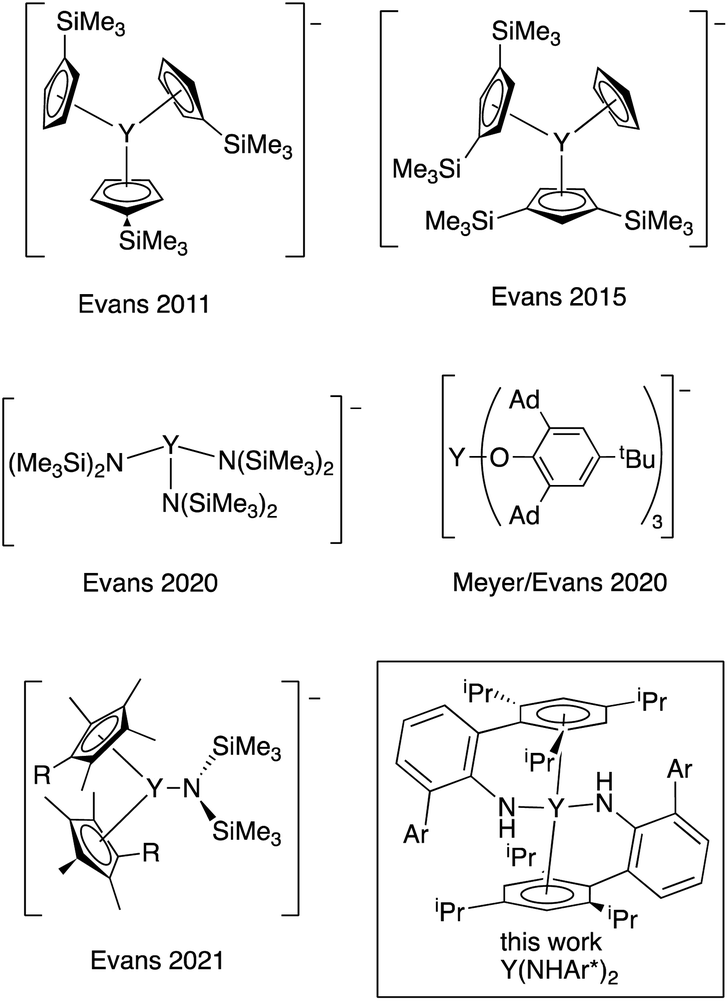 | ||
| Chart 1 Known Y(II) complexes2–7 and the complex described here (boxed). Ad = 1-adamantyl. Ar = 2,4,6-(iPr)3C6H2. | ||
The first fully characterized example of a rare earth η6-arene complex is Sm(η6-C6Me6)(μ2-AlCl4)3, reported by Cotton and Schwotzer in 1986.9 Cloke and coworkers prepared a host of interesting rare earth arene complexes in the apparent zero oxidation state.10 It is clear from these results that arenes are adept at stabilizing metals in low oxidation states. We wondered if an yttrium bis(η6-arene) complex in the formal oxidation state of Y(II) could be prepared using this arene coordination strategy to stabilize the low oxidation state. Here, we describe the synthesis and properties of a neutral Y(II) bis(amide) complex related to a recently reported U(II) system, U(NHAr*)2, where Ar* = 2,6-(2,4,6-iPr3C6H2)C6H3.11 In this uranium complex, there is evidence that reduction leads to increased bonding between the metal and the arene groups of the amide, stabilizing the neutral reduced complex. As a result, these ligands seem particularly well suited for the stabilization of highly reducing metal centers like yttrium(II), and Y(NHAr*)2 is relatively stable in diethyl ether at room temperature under inert atmosphere for days.
Based on EPR spectroscopy, the complex is formulated as d1 Y(II) with a significant portion of the unpaired electron density residing on the metal center. Despite possessing good thermal stability, the yttrium(II) complex is quite reactive, which we demonstrate through the synthesis and characterization of a new isocyanide CN–Y(NHAr*)2, where the CN ligand is only bound through nitrogen. The complex is prepared by presumed radical cleavage of a C–C or C–N bond. Evidence for this binding mode and discussion of when such a mode might be more favorable than the more common C-bound cyanide are included.
Results and discussion
Synthesis and properties of Y(NHAr*)2
Unsolvated KNHAr* was prepared in 64% yield by addition of solid KCH2SiMe3 to H2NAr* in n-hexane.11–13 Salt metathesis of YCl3 in diethyl ether with 2 equiv. of KNHAr* resulted in the formation of ClY(NHAr*)2 (1) (Fig. 1). Single-crystal X-ray diffraction on 1 revealed an η6-interaction (Ar1 in Fig. 1, red) between one of the arene substituents of an NHAr* ligand and yttrium. The η6-arene centroid to Y distance was found to be 2.493(3) Å, whereas the next closest Y-(arene centroid) distance is 3.699(3) Å. However, 1H and 13C NMR spectroscopy suggests all the arene rings are equivalent on the timescale of these experiments, indicating that fast exchange occurs between the bound and unbound aromatic groups of 1 in solution.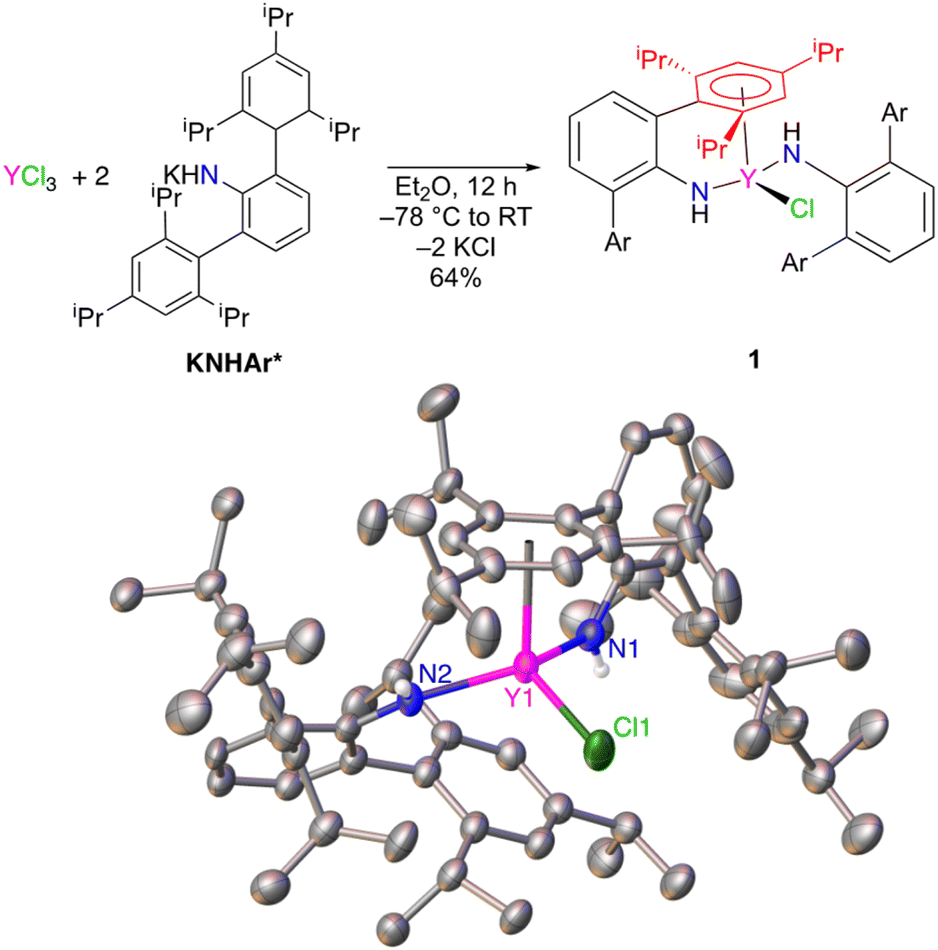 | ||
| Fig. 1 Synthesis of ClY(NHAr*)2 (1) (top) and (bottom) the structure of 1 from single-crystal X-ray diffraction (thermal ellipsoids drawn at 50%), where Ar is 2,4,6-(iPr)3C6H2. Hydrogen atoms on carbon and solvent molecules (2 n-hexane) are not shown for clarity. The η6-arene (red) is discussed as Ar1 in the text. Some selected structural parameters: Y–N distances: 2.249(2) Å, 2.210(2) Å. Y–Ar1(centroid): 2.493(3) Å. | ||
The centroid-(Ar1)–Y1–N1 angle is 94.9(7)°, and the centroid (Ar1)–Y1–N2 angle is 101.8(6)°. The Y1–N1 distance is 2.249(2) Å, and Y1–N2 is 2.210(2) Å for the two inequivalent amides. The Y1–N1–C1(ipso) angle is 131.5(2)°, and Y1–N–C36(ipso) is 144.0(2)°, with the large disparity caused by the η6-arene coordination to only one ligand.
Treatment of ClY(NHAr*)2 (1) with excess KC8 results in a color change from light yellow to a very dark yellow within an hour (Fig. 2). The product formed, Y(NHAr*)2 (2), retains its color as a clear yellow diethyl ether solution at room temperature for at least 1 week. We observed dilute ether solutions at room temperature for >100 h, and decomposition seemed have a rate of ∼3% per day under these conditions (see the ESI† for more details.)
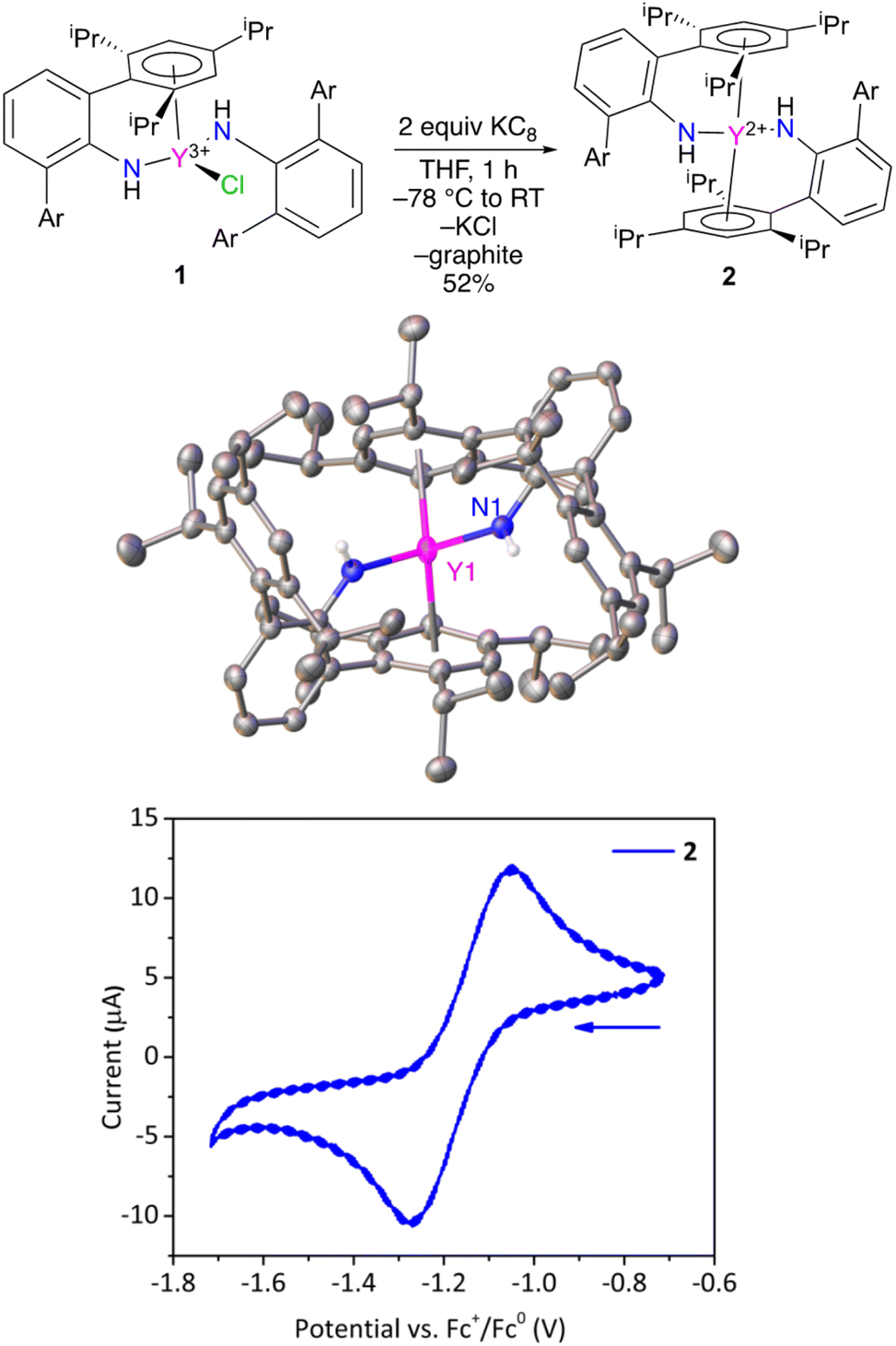 | ||
| Fig. 2 (top) Synthesis of 2 and (middle) structure of Y(NHAr*)2 from single-crystal X-ray diffraction (thermal ellipsoids drawn at 50%), where Ar is 2,4,6-(iPr)3C6H2. Hydrogen atoms on carbon and solvent molecule (THF) in the crystal lattice are not shown for clarity. The molecule resides on a crystallographic 2-fold axis. Some selected parameters: Y–N: 2.261(1) Å. Y–Ar(centroid): 2.468(8) Å. (bottom) Cyclic voltammogram of 2 (1.5 mM) in Et2O with 0.1 M NBu4+ B(3,5-(CF3)2C6H3)4− as electrolyte, 100 mV s−1 scan rate, with a glassy carbon working electrode. | ||
X-ray diffraction quality crystals of 2 were obtained from n-hexane, and the geometries of 1 and 2 vary significantly in the solid-state. Complex 2 crystallizes on a 2-fold rotation axis bisecting the N–Y–N angle, Fig. 2 and S28.† In this case, ortho-aromatic rings from both amide groups are η6-coordinated to the metal center, similar to previously reported U(NHAr*)2.11
In this lower oxidation state, the Y1–Ar1(centroid) distance shortened slightly (but not significantly relative to three times the e.s.d.) relative to 1 from 2.493(3) Å to 2.468(8) Å in 2. Naturally, the expectation is that the radius of the metal will increase on reduction, which is observed in the Y–N distance in 1vs.2 of 2.230(2) vs. 2.261(1) Å, respectively. The apparent contraction in Y–Ar(centroid) distance in 2vs.1 is consistent with additional yttrium to arene bonding between the formally reduced metal center to the arene in the reduced complex, an effect observed in the related U(NHAr*)2 complex as well.11
Y(II) complex 2 was examined by cyclic voltammetry (Fig. 2) using NBu4+ B(3,5-(CF3)2C6H3)4− as the electrolyte in diethyl ether with a glassy carbon working electrode. A reversible feature was observed at E1/2 = −1.16 ± 0.01 V in the voltammogram (see the ESI†) vs. FeCp2+1/0 under these conditions, which is assigned to the Y(NHAr*)2+1/0 couple (Fig. 2, bottom). For comparison, a recent report listed the quasi-reversible Y(C5H4SiMe3)3−/0 potential as E1/2 = −3.06 V in THF with NBu4+ BPh4− as electrolyte.14 One can readily see the extreme difference in the two metal environments. Complex 2 contains aromatic groups that we believe act as acceptors, making the complex more stable and much more readily reduced, while Y(C5H4SiMe3)3 contains anionic (presumably strongly donating) cyclopentadienyls that make the metal quite difficult to reduce.
EPR spectra collected for Y(NHAr*)2 (2) in Et2O at 60 K and 295 K are shown in Fig. 3. The frozen solution spectrum (top, black trace) shows an axial lineshape with hyperfine splitting that arises from an I = 1/2 nucleus, consistent with an 89Y-centered paramagnet. Analysis of this spectrum shows that it can be well-simulated (top, red trace) using an axial g-tensor with g‖ = 1.985 and g⊥ = 2.004, an axial 89Y hyperfine coupling with A‖ = 38.9 MHz and A⊥ = 40.9 MHz, and an intrinsic Gaussian lineshape with an average width of 1.06 mT (FWHM). The solution spectrum of 2 in Et2O (bottom, black trace) features a broad intrinsic lineshape with a modest inflection, also consistent with hyperfine splitting from an I = 1/2 nucleus. This spectrum was simulated using both fast- and slow-motion calculation models (bottom, red trace). The results of both approaches yield isotropic g- and 89Y-hyperfine coupling values of 1.995 and 49 MHz, respectively.15 For these simulations, a broad Lorentzian intrinsic lineshape of 2.5 mT was required, casting greater uncertainty on the spin Hamiltonian values obtained from the solution spectrum.
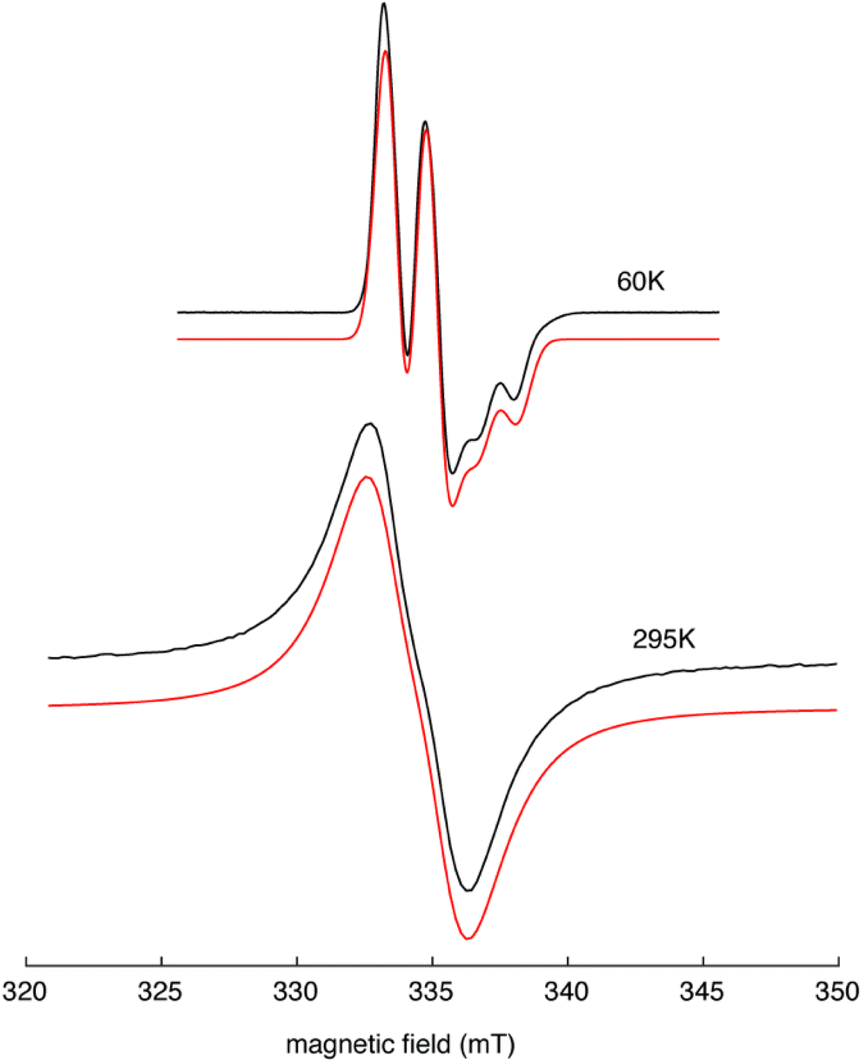 | ||
| Fig. 3 EPR spectra of Y(NHAr*)2 (2) in Et2O at 60 K (top) and 295 K (bottom). The black traces are the experimental spectra collected using the following conditions: microwave frequency, 9.3972 GHz (top), 9.3270 GHz (bottom); microwave power, 5 mW (top), 0.125 mW (bottom); field modulation amplitude, 0.1 mT (top), 0.4 mT (bottom). The red traces are spectra simulations offset for visualization purposes. Simulations were done with EasySpin15 using the spin Hamiltonian parameters given in the text. | ||
The spin Hamiltonian values obtained from analysis of the solid state spectrum of 2 are most similar to those reported for the Y(C5H4SiMe3)3− complexes reported by Evans and coworkers.4 These complexes also showed a modest g-anisotropy, g‖ = 2.00 and g⊥ = 1.99, but with g‖ > g⊥. For 2, our analysis shows g‖ < g⊥, most likely reflecting the different coordination geometry of the ligands about the metal center.16 For both 2 and Y(C5H4SiMe3)3−, the presence of at least one g-value below 2.0023 and a resolved I = 1/2 isotropic hyperfine coupling is indicative of a d1-centered paramagnet. The isotropic hyperfine coupling attributed to 89Y of 2 (40.2 MHz) is 40% of that reported for Y(C5H4SiMe3)3−, consistent with more of the unpaired spin being delocalized onto the NHAr* ligands. This picture of 2's electronic structure is supported by the lack of detectable 14N hyperfine coupling in both liquid and solid state EPR spectra. Still, it is worth noting that in single crystal EPR studies of Y(II) in SrCl2, 89Y isotropic hyperfine couplings of 80.8 MHz were resolved.17 DFT calculations on 2 are consistent with this EPR analysis, showing extensive unpaired spin on the metal and coordinated rings with little on the nitrogen atoms (see the ESI† for more details).
The compound was examined in the solid-state using SQUID magnetometry (Fig. 4 and additional information in the ESI†). The fits of the χMT vs. T data engendered slightly lower g values than the expected g value of 2.0023 for an unpaired electron that is unaffected by spin–orbit coupling. Hence, the isothermal field-dependent magnetization (M vs. H) data were collected between 2 and 10 K up and at fields up to 7 T. The resulting experimental data was fit to a set of Brillouin functions to afford g values near the expected value (1.9938(19)–2.3163(36)), which are in excellent agreement with the values attained from EPR spectroscopy. The magnetic properties of 2 were also probed through measuring a toluene solution of 2 employing Evans's method between temperatures of 183 and 298 K, Fig. S17.† Similar to the determined χMT values on the solid sample, a higher magnetic moment μeff = 2.39μB was obtained relative to the spin-only value of 1.73μB for an unpaired electron, which is likely ascribed to a TIP contribution.
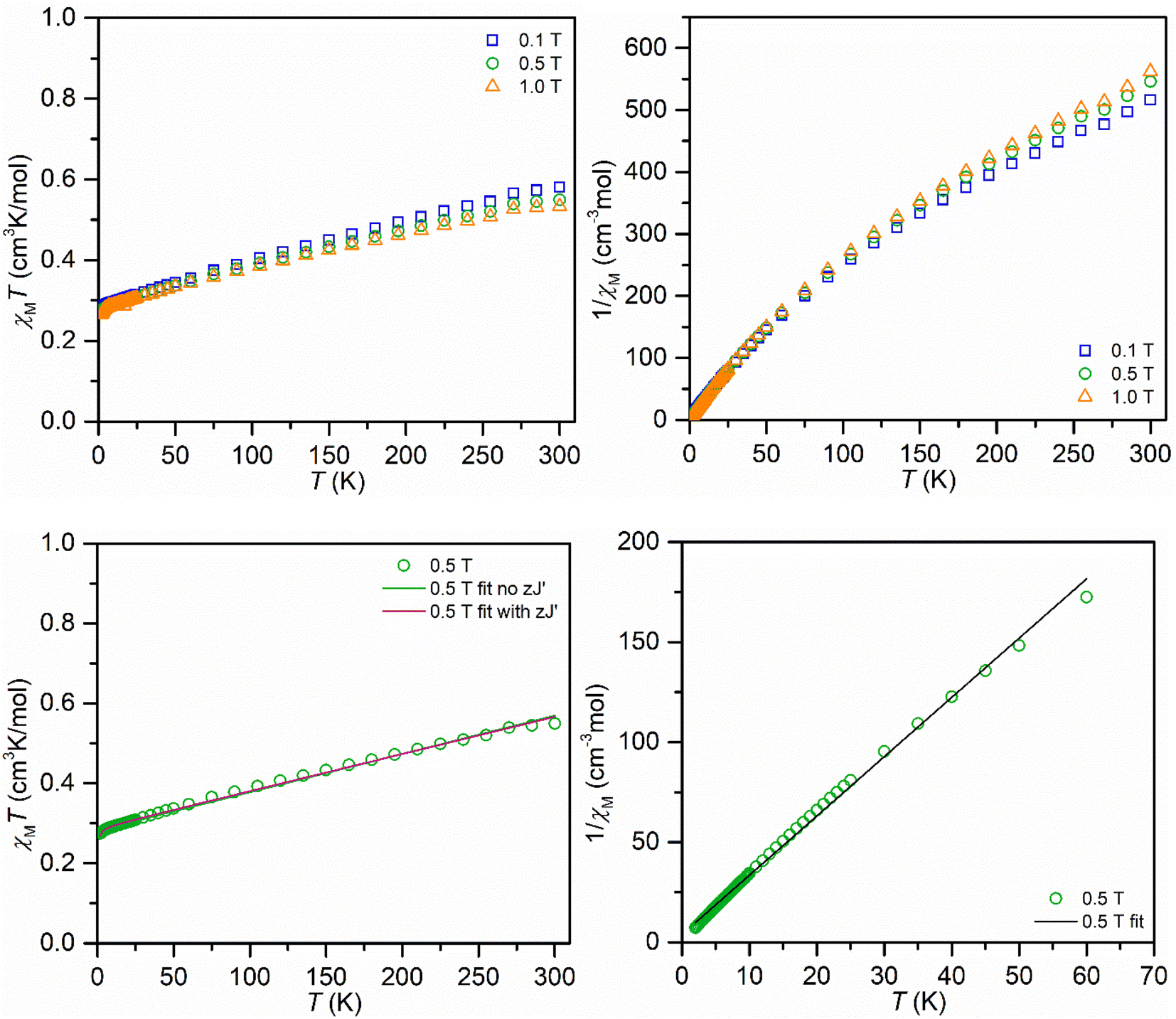 | ||
| Fig. 4 Variable-temperature dc magnetic susceptibility data for a restrained polycrystalline sample of Y(NHAr*)2 (2) collected under 0.1 T, 0.5 T and 1.0 T applied dc fields and at temperatures from 2 to 300 K (top left). Corresponding Curie–Weiss plots (1/χMvs. T) for 2 at 0.1 T, 0.5 T and 1.0 T (top right). Explementary fits for χMT vs. T (bottom left) and 1/χMvs. T plots at 0.5 T. Parameters for the χMvs. T fit: g = 1.7334(14), TIP = 9.586(51) × 10−4 and g = 1.7521(19), TIP = 9.297(45) × 10−4, zJ′ = −0.1195(102) cm−1. Parameters for the 1/χMvs. T fit: C = 0.338(22) cm3 K mol−1, Θ = −1.298 K. | ||
The absorption spectrum of 2 in the UV-Vis-NIR was obtained in diethyl ether. The yellow–brown complex has large, intense bands at 752 nm (ε = 605 cm−1 M−1), 244 nm (ε = 7300 cm−1 M−1), and 297 nm (ε = 1040 cm−1 M−1), all assigned to charge transfer transitions. Similar absorptions occur in the previously reported U(II)(NHAr*)2 (ref. 11) (see the ESI† for more details.)
An yttrium isocyanide: CN–Y(NHAr*)2
The reactivity of Y(NHAr*)2 (2) was explored as well. The compound is expected to be a high energy metallaradical, and it was anticipated that the complex would react with CNtBu to liberate (presumably) a tert-butyl radical. This was found to be the case; however, instead of producing the more common cyanide (M–CN) the isocyanide (M–NC) complex is isolated (Fig. 5).18,19 The same reaction with NCtBu provided the same product by 1H NMR spectroscopy.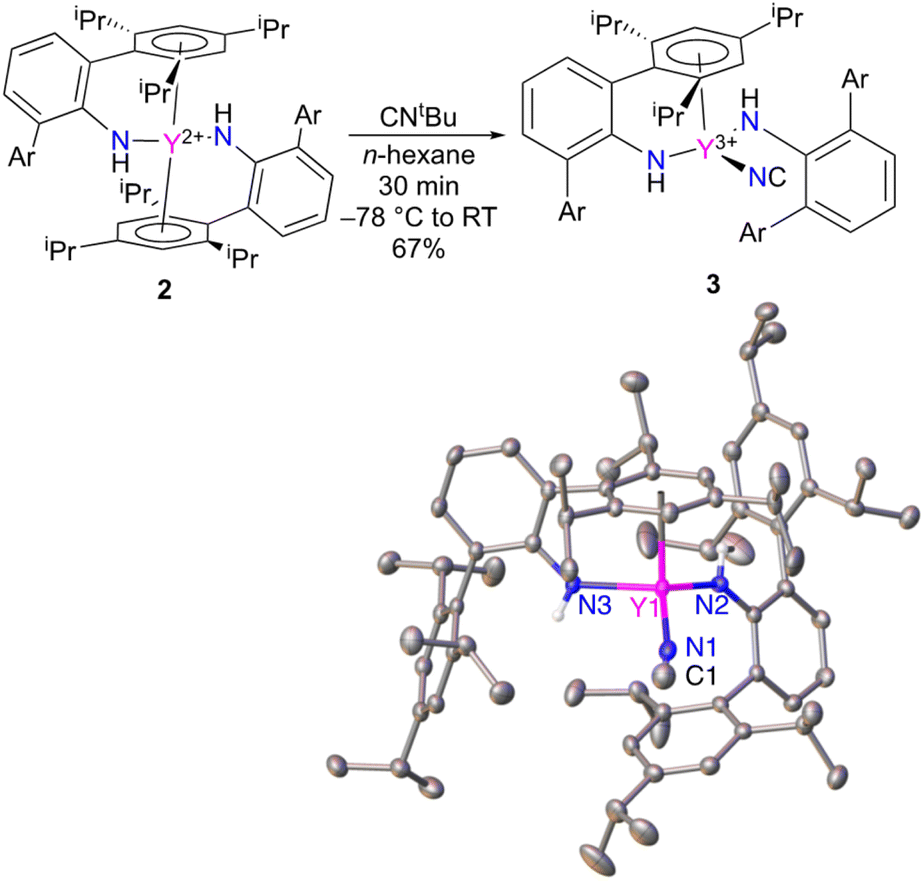 | ||
| Fig. 5 Synthesis of CN–Y(NHAr*)2 (3) (top) and structure from single crystal X-ray diffraction (thermal ellipsoids at 50%), where Ar is 2,4,6-(iPr)3C6H2. (bottom) Hydrogen atoms on the carbon atoms and solvent molecules in the crystal lattice are not shown for clarity. Selected bond distances (Å) and angles (°): Y1–N1: 2.348(2), Y1–N2: 2.221(2), Y1–N3: 2.221(2), C1–N1: 1.039(4), Y1–N1–C1: 170.4(2)°. | ||
Within the rich chemistry of isonitrile (CNR) reactions with metal complexes,20 there are a few other instances where nitriles and isonitriles have been used to add a CN (cyanide or isocyanide) ligand to a metal center. Carmona, Andersen, and coworkers showed that tris(cyclopentadienyl)uranium(III) complexes will react with CNtBu to give cyanide uranium(IV).21 Jones and coworkers have extensively studied the isonitrile (and related nitrile) cleavage reactions with nickel(0), ruthenium(0), and iron(0).22–27 Co(I),28,29 Sm(II),30,31 and V(II)32 complexes have exhibited such reactions, as well. Maron, Arnold, and coworkers used the same reaction to add a CN ligand to a low oxidation state thorium complex.33
A couple of different avenues were readily available to determine experimentally that 3 is indeed an isocyanide. The first is to examine the X-ray diffraction data (Fig. 5) modelled as both the isocyanide and cyanide to see which configuration gives better statistics for the structure. The assigned isocyanide Y–NC structure has R1 = 4.11% and wR2 = 9.53%, while the cyanide has R1 = 4.37% and wR2 = 10.61%. In addition, the isocyanide structure has improved thermal parameters over the cyanide isomer. More details can be found in the ESI.†
As a second method of determining the relative position of the carbon in the CN ligand, we turned to NMR spectroscopy. First, we were able to account for every observable proton and carbon in the 1H and 13C spectra, which was consistent with the structure shown in Fig. 5. There were no observable couplings between the protons and the carbon assigned to the CN ligand, which argues against a “Y–NCH” structure. The 89Y–13C coupling constant was found to be 9.8 Hz in the 13C NMR spectrum (Fig. 6). Perhaps the best method for relating this to cyanide vs. isocyanide couplings is to look at the 89Y–13C coupling constants for the α- and β-carbon atoms of alkynyls, and a few examples are shown in Fig. 6.34–40 The typical range for the one-bond coupling constant to the α-carbon atom of an alkynyl is 50–75 Hz. While the two-bond couplings to the β-carbon atom of an alkynyl are typically 5–15 Hz. As a result, 3 falls in the middle of the two-bond coupling range, consistent with an isocyanide and is quite distinct from the typical one-bond coupling.
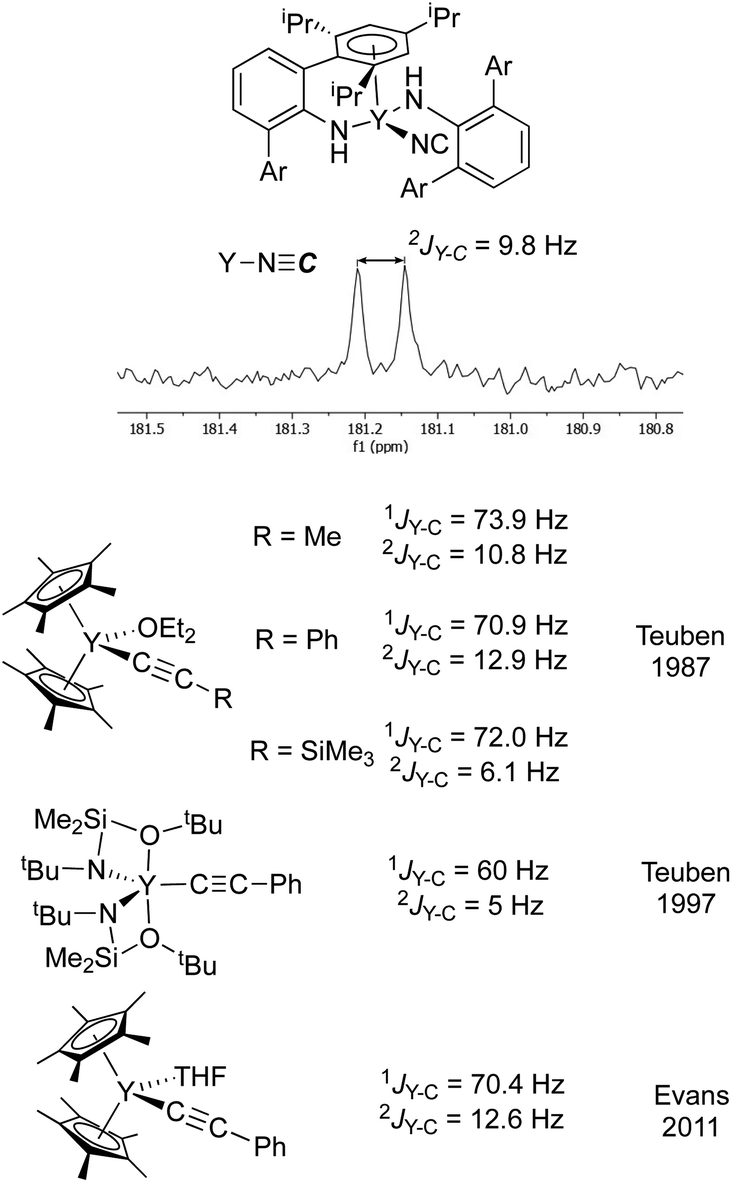 | ||
| Fig. 6 (top) Excerpt of the 13C NMR spectrum for 3 showing the isocyanide resonance coupled to yttrium. (bottom) The isocyanide resonance is compared with Y–C one- and two-bond coupling constants for some representative alkynyl complexes from Teuben and Evans.34,36,37 The coupling constant observed for 3 is in the typical range for a two-bond coupling, much smaller than a typical one-bond coupling constant. Ar = 2,4,6-(iPr)3C6H2. | ||
Yttrium features 100% abundance of an NMR active nucleus, 89Y with I = 1/2, and 89Y NMR chemical shifts can be obtained for 1 and 3. Experimental determination of the chemical shift of 2 was inhibited, as would be expected, by the paramagnetic nature of the Y(II) ion. The 89Y NMR spectra were recorded in toluene-d8 solutions for 1 and 3 (Fig. S3, S8 and S9†). While ClY(NHAr*)2 (1) has a chemical shift for the metal center at +427.7 ppm, the resonance in CN–Y(NHAr*)2 (3) is observed at +350.0 ppm. For comparison, the 89Y NMR chemical shifts for the metal centers in Y{N(SiMe3)2}3 and Y(C5H4Me)3(THF) were found at +570 (CDCl3) and −371 ppm (THF), respectively.41,42
As an addition to the experimental investigations, we examined the cyanide and isocyanide computationally using DFT. It was found that the N-bound isocyanide is indeed ∼4 kcal mol−1 lower in energy than the cyanide for this Y(II) system (see ESI† for more details.)
While rare, isocyanides are known, especially for the main group and f-block elements. For example, there is the well-characterized Mg(dipyrrolylmethene)isocyanide reported by Harder and coworkers in 2019,43 and a report of a thallium porphyrin complex.44 In the f-block, thorium and uranium isocyanide complexes have been well-established.33,45–49 One obvious conclusion from the extensive work of Ephritikhine and coworkers on uranium CN complexes is that small changes in structure can change a cyanide ligand into an isocyanide.47 Indeed calculations on a large number of cyanide vs. isocyanide energies suggest that increased covalency favors the cyanide M–CN conformation and ionic structures can slightly favor the isocyanide M–NC structure (vide infra).43 Well-characterized examples of isocyanides of the transition metals seem rarer than of the f-block metals and main group. Structurally characterized, terminal cyanides of Group-3 elements seem to be quite rare in general.
The IR spectrum for 3 shows an intense band at 2053 cm−1 in n-hexane at room temperature assigned to the isocyanide stretch. The CN-triple bond stretches in the CNtBu and NCtBu starting materials appears at 2125 and 2250 cm−1, respectively. The cyanide stretch in NBu4+ CN− is found at 2050 cm−1.47 The isocyanide stretch here is quite similar to other reported metal isocyanides such as Arnold and coworkers' thorium complex (2046 cm−1),33 along with Ephritikhine and coworkers' CN–U{N(SiMe3)2}3 (2044 cm−1) and NEt4+ (CN)2U{N(SiMe3)2}3− (2058 cm−1).47 In Harder's magnesium complex, both cyanide and isocyanide are observable in equilibrium with stretching frequencies of 2162 and 2085 cm−1, respectively.43
The ∼100 cm−1 difference in stretching frequency when a substituent is bound to the carbon vs. nitrogen atom of the CN triple bond fragment is likely due to the expected difference in alleviating lone pair bond weakening on those atoms.50 In CN−, both atoms contain a lone pair of electrons. A lone pair will require more s-character than the only other orbital using s-character in the ligand—the C–N σ-bond (Bent's Rule).51,52 However, the more electronegative nitrogen atom will be able to support the lone pair of electrons with less s-character than the carbon atom. All of this is borne out by Natural Bond Orbital calculations on the cyanide anion (M06L, def2-tzvp, NBO7), which gives that the nitrogen and carbon lone pairs are held by sp1.1- and sp0.55-hybrids, respectively. The C–N σ-bond is calculated by NBO to be comprised of overlapping carbon-based sp1.5- and nitrogen-based sp0.9 orbitals. When another atom binds to the lone pair, some or all the lone pair bond weakening is alleviated, and more s-character can be used in the C–N σ-bond, often strengthening the bond.
There is very little difference in hybridization between the lone pair and σ-bonding orbital on the nitrogen atom (sp0.9vs. sp1.1), while there is a larger difference for the carbon atom (sp1.5vs. sp0.55). Consequently, very little additional C–N bonding is gained by the metal bonding to the nitrogen atom, and the CN stretching frequency does not change a great deal from free CN− for isocyanide systems (assuming no significant backbonding). In contrast, the CN bond can strengthen more significantly when a non-backbonding metal is attached to the carbon atom, and the stretching frequency increases over free cyanide anion. This effect to increase the bond strength and stretching frequency, which occurs in CO chemistry as well, is sometimes referred to as “nonclassical” bonding for the ligand since the CO (or CN−) increases in stretching frequency and bond strength instead of weakening, as would be expected in systems where backbonding to the diatomic occurs.53–56 As a result, the slightly lower frequency of the isocyanide stretch relative to related cyanide complexes is not likely to be due to increased backbonding but is instead due to differences in lone pair bond weakening in the two bonding modes of the ambidentate ligand.
Conclusions
Using a triarylamide ligand (NHAr*), we were able to prepare a thermally stable Y(II) complex, Y(NHAr*)2 (2). The compound exhibits a geometry similar to previously reported U(NHAr*)2 with an aryl ring of each amide η6-bound to the metal center to give a sandwich structure (Fig. 2). The complex was prepared by chemical reduction of ClY(NHAr*)2 (1), a half-sandwich complex, with the strong reductant KC8, potassium graphite. Cyclic voltammetry on 2 shows a reversible feature at −1.16 ± 0.01 V relative to FeCp2+/0 in Et2O assigned to the Y(NHAr*)2+/0 couple. By EPR, the unpaired electron-density in 2 is delocalized with significant yttrium character, and we assign the complex as Y(II) d1.The strongly reducing radical 2 is quite reactive despite being relatively stable under inert atmosphere at room temperature in diethyl ether. Reaction of 2 with CNtBu or NCtBu results in supposed loss of tert-butyl radical and formation of the unusual isocyanide, CN–Y(NHAr*)2 (3), a half-sandwich complex. The isocyanide structure was assigned based on single-crystal X-ray diffraction and the 89Y–13C coupling constant in the 13C spectrum, which is consistent with a two-bond coupling in relation to known yttrium acetylides (Fig. 6). Further, the IR stretching frequency for isocyanide metal complexes are expected to be somewhat lower (∼100 cm−1) than cyanide stretching frequencies (negligible backbonding in all cases), and 3 is similar to other known isocyanide complexes in CN stretching frequency. DFT calculations on CN–Y(NHAr*)2 (3) and hypothetical NC–Y(NHAr*)2 suggest the isocyanide is more stable by ∼4 kcal mol−1.
As mentioned, well-characterized examples of transition metal isocyanides seem to be quite rare; however, there are well established examples for the main group and f-block elements. Their existence anywhere in the periodic table as stable species is somewhat surprising based on a molecular orbital analysis of the cyanide ion, which has both the HOMO and LUMO predominantly on carbon (Fig. 7), something that is obvious from the orbital pictures (M06/def2-tzvp). However, Natural Population Analysis gives that about 75% of the anionic charge of cyanide resides on nitrogen. Consequently, metal centers that are very electropositive and with little or no ability to backbond to CN− may favor the isocyanide structure. Put another way, highly polar systems may give the charge-controlled structure (as opposed to frontier orbital controlled structure) with the nitrogen bound to the metal center.63–66
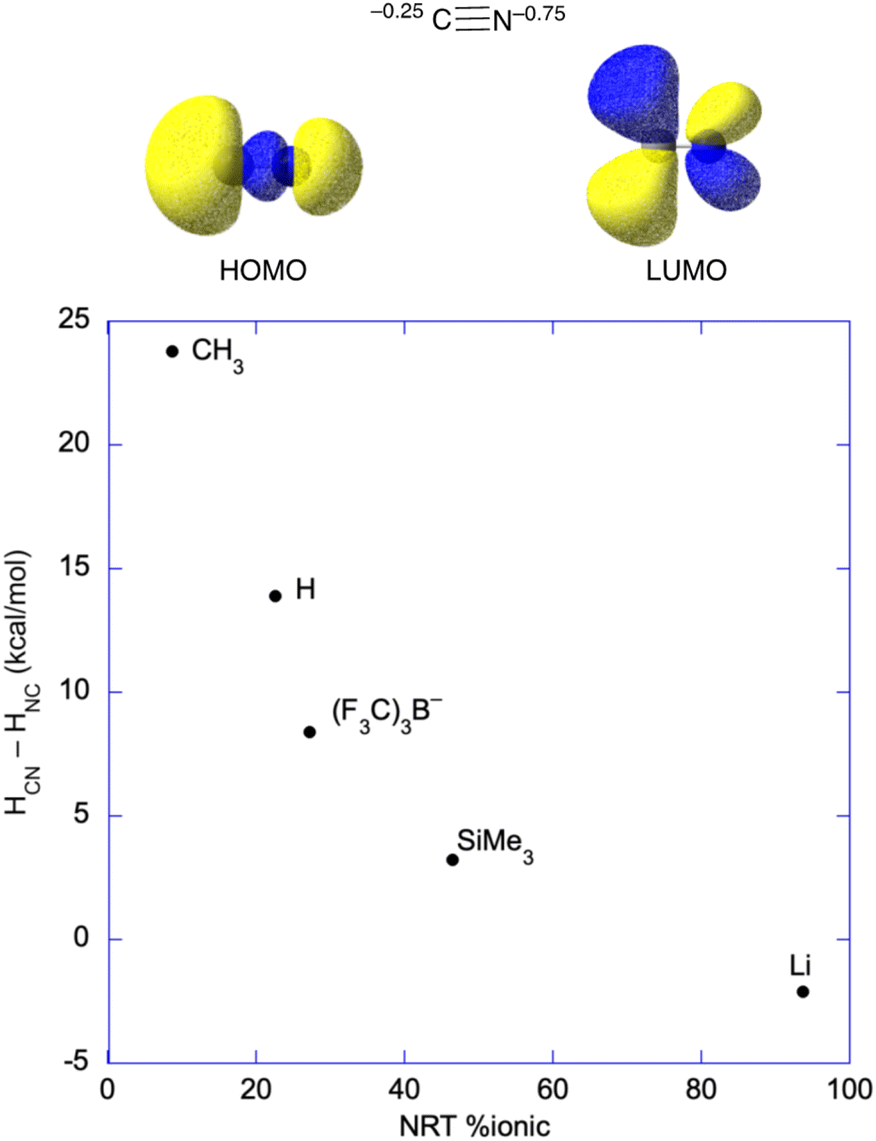 | ||
| Fig. 7 (top) Natural population analysis gives the anion charge which is 75% on the nitrogen atom, and MO analysis gives that both the HOMO and the LUMO are centered on the carbon. In covalent systems and/or systems with significant backbonding, binding through the carbon is expected based on the MOs; however, very polar systems may bind to the nitrogen of cyanide. (bottom) Plot of the enthalpy difference57–61 between the cyanide (e.g., HCN) and isocyanide (e.g., HNC) structures vs. the percentage of ionic bonding from Natural Resonance Theory (NRT).62 | ||
The structural effects of a highly polar bond were discussed by Harder and coworkers on their article regarding a Mg–CN complex, where they pointed out that more X–CN (where X–CN = MeCN, HCN, (F3C)3BCN−, Me3SiCN, and LiCN)57–61 covalency leads to thermodynamically favored carbon-bonding, as opposed to nitrogen in the cyanide unit, and higher barriers to generating the isocyanide.43 Natural Resonance Theory (NRT) can be used to determine the ionic character in these main group systems, which is plotted against the enthalpic favorability of the ground state cyanide (X–CN) structure in Fig. 7.62 Clearly, the more covalent systems, e.g., H3C–CN, favor binding through carbon, while bonding to nitrogen only becomes favorable for very polar systems like LiCN, where HCN–HNC = −2 kcal mol−1.
Currently, we are examining related f-block systems using these intriguing ligands.
Data availability
The ESI† contains all spectroscopy, characterization data, and computational data. Structures are available on CCDC.Author contributions
RJ prepared all the compounds in the paper and aided with characterization. FB and FD did the initial preparation of 1. FB and RJ collected and analyzed the CV data. DH was instrumental in NMR characterization of 3. JM collected and analyzed the EPR spectroscopic data. SD and ALO conceived the project, provided resources, and support. ALO wrote the initial draft of the article, and all authors aided in editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
ALO would like to thank the National Science Foundation (CHE-1953254) and the American Chemical Society Petroleum Research Fund (65702-ND3) for support of their research. S. D. is grateful to the Department of Chemistry at Michigan State University (MSU) for generous start-up funds. We extend our thanks to Prof. James K. McCusker for providing access to the UV-Vis-NIR spectrophotometer and Dr Richard J. Staples for his assistance in the interpretation of the collected X-ray diffraction data. Funding for the single-crystal X-ray diffractometer was provided through the MRI program of the National Science Foundation under Grant No. MRI-1919565.References
- W. J. Evans, Organometallics, 2016, 35, 3088–3100 CrossRef CAS.
- M. R. MacDonald, J. E. Bates, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2013, 135, 9857–9868 CrossRef CAS PubMed.
- M. R. MacDonald, J. W. Ziller and W. J. Evans, J. Am. Chem. Soc., 2011, 133, 15914–15917 CrossRef CAS PubMed.
- W. N. G. Moore, J. W. Ziller and W. J. Evans, Organometallics, 2021, 40, 3170–3176 CrossRef CAS.
- S. A. Moehring, M. Miehlich, C. J. Hoerger, K. Meyer, J. W. Ziller and W. J. Evans, Inorg. Chem., 2020, 59, 3207–3214 CrossRef CAS PubMed.
- C. T. Palumbo, D. P. Halter, V. K. Voora, G. P. Chen, J. W. Ziller, M. Gembicky, A. L. Rheingold, F. Furche, K. Meyer and W. J. Evans, Inorg. Chem., 2018, 57, 12876–12884 CrossRef CAS PubMed.
- A. J. J. Ryan, J. W. Ziller and W. J. Evans, Chem. Sci., 2020, 11, 2006–2014 RSC.
- T. F. Jenkins, S. Bekoe, J. W. Ziller, F. Furche and W. J. Evans, Organometallics, 2021, 40, 3917–3925 CrossRef CAS.
- F. A. Cotton and W. Schwotzer, J. Am. Chem. Soc., 1986, 108, 4657–4658 CrossRef CAS.
- F. G. N. Cloke, Chem. Soc. Rev., 1993, 22, 17–24 RSC.
- B. S. Billow, B. N. Livesay, C. C. Mokhtarzadeh, J. McCracken, M. P. Shores, J. M. Boncella and A. L. Odom, J. Am. Chem. Soc., 2018, 140, 17369–17373 CrossRef CAS PubMed.
- B. Twamley, C. S. Hwang, N. J. Hardman and P. P. Power, J. Organomet. Chem., 2000, 609, 152–160 CrossRef CAS.
- P. L. Arnold and S. T. Liddle, C. R. Chim., 2008, 11, 603–611 CrossRef CAS.
- M. T. Trinh, J. C. Wedal and W. J. Evans, Dalton Trans., 2021, 50, 14384–14389 RSC.
- S. Stoll and A. Schweiger, J Mag. Res., 2006, 178, 42–55 CrossRef CAS PubMed.
- J. R. Pilbrow, Transition Ion Electron Paramagnetic Resonance, Oxford University Press, 1990 Search PubMed.
- J. R. Herrington, T. L. Estle and L. A. Boatner, Phys. Rev. B: Solid State, 1973, 7, 3003–3013 CrossRef CAS.
- G. P. Moss, P. A. S. Smith and D. Tavernier, Pure Appl. Chem., 1995, 67, 1307–1375 Search PubMed.
- In 1995, IUPAC guidelines suggested the use of “cyanide” for salts of CN• and C-organyl derivatives of hydrogen cyanide, HCN, e.g., CH3CN as “methyl cyanide” rather than the more common “acetonitrile”. It would have been less confusing to call metal-CN complexes “cyanides” and the organic compounds nitriles, following common usage. In addition, the same recommendations described the term “isonitrile” as “An obsolete term, which should not be used, for isocyanides.” In other words, they describe RNC compounds as “isocyanides” and disposed of “isonitrile” as archaic. Unfortunately, these recommendations cause great confusion when trying to describe complexes of the type M–NC. These N-bound cyanides should be rightfully called “isocyanide complexes” but are conflated with RNC–M compounds of the same name, where the latter should have been uniquely named “isonitrile complexes”. All this confusion aside, we will use the term that could have been reserved for M–NC species here, “isocyanides”.
- V. P. Boyarskiy, N. A. Bokach, K. V. Luzyanin and V. Y. Kukushkin, Chem. Rev., 2015, 115, 2698–2779 CrossRef CAS PubMed.
- M. D. Conejo, J. S. Parry, E. Carmona, M. Schultz, J. G. Brennann, S. M. Beshouri, R. A. Andersen, R. D. Rogers, S. Coles and M. Hursthouse, Chem.–Eur. J., 1999, 5, 3000–3009 CrossRef.
- M. E. Evans, T. Li and W. D. Jones, J. Am. Chem. Soc., 2010, 132, 16278–16284 CrossRef CAS PubMed.
- C. L. Tennent and W. D. Jones, Can. J. Chem., 2005, 83, 626–633 CrossRef CAS.
- J. J. Garcia, A. Arevalo, N. M. Brunkan and W. D. Jones, Organometallics, 2004, 23, 3997–4002 CrossRef CAS.
- J. J. Garcia, N. M. Brunkan and W. D. Jones, J. Am. Chem. Soc., 2002, 124, 9547–9555 CrossRef CAS PubMed.
- J. J. Garcia and W. D. Jones, Organometallics, 2000, 19, 5544–5545 CrossRef CAS.
- W. D. Jones and W. P. Kosar, Organometallics, 1986, 5, 1823–1829 CrossRef CAS.
- X. Y. Li, H. J. Sun, F. L. Yu, U. Florke and H. F. Klein, Organometallics, 2006, 25, 4695–4697 CrossRef CAS.
- H. W. Xu, P. G. Williard and W. H. Bernskoetter, Organometallics, 2012, 31, 1588–1590 CrossRef CAS.
- W. J. Evans and D. K. Drummond, Organometallics, 1988, 7, 797–802 CrossRef CAS.
- M. G. Gardiner, A. N. James, C. Jones and C. Schulten, Dalton Trans., 2010, 39, 6864–6870 RSC.
- S. Hasegawa, Y. Ishida and H. Kawaguchi, Chem. Commun., 2021, 57, 8296–8299 RSC.
- M. E. Garner, S. Hohloch, L. Maron and J. Arnold, Angew. Chem., Int. Ed., 2016, 55, 13789–13792 CrossRef CAS PubMed.
- I. J. Casely, J. W. Ziller and W. J. Evans, Organometallics, 2011, 30, 4873–4881 CrossRef CAS.
- B. J. Deelman, W. M. Stevels, J. H. Teuben, M. T. Lakin and A. L. Spek, Organometallics, 1994, 13, 3881–3891 CrossRef CAS.
- K. H. Denhaan, Y. Wielstra and J. H. Teuben, Organometallics, 1987, 6, 2053–2060 CrossRef CAS.
- R. Duchateau, E. A. C. Brussee, A. Meetsma and J. H. Teuben, Organometallics, 1997, 16, 5506–5516 CrossRef CAS.
- A. V. Karpov, A. S. Shavyrin, A. V. Cherkasov, G. K. Fukin and A. A. Trifonov, Organometallics, 2012, 31, 5349–5357 CrossRef CAS.
- D. Robert, P. Voth, T. P. Spaniol and J. Okuda, Eur. J. Inorg. Chem., 2008, 2810–2819 CrossRef CAS.
- J. Sun, D. J. Berg and B. Twamley, Organometallics, 2008, 27, 683–690 CrossRef CAS.
- P. S. Coan, L. G. Hubertpfalzgraf and K. G. Caulton, Inorg. Chem., 1992, 31, 1262–1267 CrossRef CAS.
- W. J. Evans, J. H. Meadows, A. G. Kostka and G. L. Closs, Organometallics, 1985, 4, 324–326 CrossRef CAS.
- G. Ballmann, H. Elsen and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 15736–15741 CrossRef CAS PubMed.
- A. G. Coutsolelos, A. Tsapara, D. Daphnomili and D. L. Ward, J. Chem. Soc., Dalton Trans., 1991, 3413–3417 RSC.
- Y. Bouzidi, L. Belkhiri, M. Ephritikhine, J. F. Halet and A. Boucekkine, J. Organomet. Chem., 2017, 847, 82–89 CrossRef CAS.
- X. T. Chen, Q. N. Li, Y. Gong, L. Andrews, B. K. Liebov, Z. T. Fang and D. A. Dixon, Inorg. Chem., 2017, 56, 5060–5068 CrossRef CAS PubMed.
- A. Herve, Y. Bouzidi, J. C. Berthet, L. Belkhiri, P. Thuery, A. Boucekkine and M. Ephritikhine, Inorg. Chem., 2015, 54, 2474–2490 CrossRef CAS PubMed.
- J. C. Berthet, P. Thuery and M. Ephritikhine, Dalton Trans., 2015, 44, 7727–7742 RSC.
- A. Herve, Y. Bouzidi, J. C. Berthet, L. Belkhiri, P. Thuery, A. Boucekkine and M. Ephritikhine, Inorg. Chem., 2014, 53, 6995–7013 CrossRef CAS PubMed.
- D. Lauvergnat, P. Maitre, P. C. Hiberty and F. Volatron, J. Phys. Chem., 1996, 100, 6463–6468 CrossRef CAS.
- H. A. Bent, Chem. Rev., 1961, 61, 275–311 CrossRef CAS.
- F. Weinhold and C. R. Landis, Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective, Cambridge University Press, Cambridge, 2005 Search PubMed.
- A. L. Odom, in Comprehensive Organometallic Chemistry IV, ed. P. Holland, Elsevier, 2022, vol. 1, pp. 2–30 Search PubMed.
- P. K. Hurlburt, J. J. Rack, J. S. Luck, S. F. Dec, J. D. Webb, O. P. Anderson and S. H. Strauss, J. Am. Chem. Soc., 1994, 116, 10003–10014 CrossRef CAS.
- A. J. Lupinetti, S. H. Strauss and G. Frenking, in Prog. Inorg. Chem., ed. K. D. Karlin, 2001, vol. 49, pp. 1–112 Search PubMed.
- S. H. Strauss, J. Chem. Soc., Dalton Trans., 2000, 1–6 RSC.
- M. Finze, E. Bernhardt, H. Willner and C. W. Lehmann, J. Am. Chem. Soc., 2005, 127, 10712–10722 CrossRef CAS PubMed.
- V. S. Rao, A. Vijay and A. K. Chandra, Can. J. Chem., 1996, 74, 1072–1077 CrossRef CAS.
- M. H. Baghal-Vayjooee, J. L. Collister and H. O. Pritchard, Can. J. Chem., 1977, 55, 2634–2636 CrossRef CAS.
- M. R. Booth and S. G. Frankiss, Spectrochim. Acta, Part A, 1970, 26, 859 CrossRef CAS.
- M. R. Booth and S. G. Frankiss, Chem. Commun., 1968, 1347 RSC.
- Here, we are using the ionic bond order divided by the total bond order from the NRT times 100 to give % ionic for the bond.
- T. Stuyver and S. Shaik, J. Am. Chem. Soc., 2020, 142, 20002–20013 CrossRef CAS PubMed.
- J. S. M. Anderson, J. Melin and P. W. Ayers, J. Mol. Model., 2016, 22, 1–11 CrossRef CAS PubMed.
- J. S. M. Anderson, J. Melin and P. W. Ayers, J. Chem. Theor. Comput., 2007, 3, 358–374 CrossRef CAS PubMed.
- J. S. M. Anderson, J. Melin and P. W. Ayers, J. Chem. Theor. Comput., 2007, 3, 375–389 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and additional characterization data. X-ray crystallographic data has been deposited. CCDC 2207616–2207618. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00171g |
| This journal is © The Royal Society of Chemistry 2023 |