
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBlue-emitting InP quantum dots participate in an efficient resonance energy transfer process in water†
Pradyut
Roy
 ,
Mishika
Virmani
and
Pramod P.
Pillai
*
,
Mishika
Virmani
and
Pramod P.
Pillai
*
Department of Chemistry, Indian Institute of Science Education and Research (Pune), Dr Homi Bhabha Road, Pashan, Pune – 411008, India. E-mail: pramod.pillai@iiserpune.ac.in
First published on 20th April 2023
Abstract
Development of stable blue-emitting materials has always been a challenging task because of the necessity of high crystal quality and good optical properties. We have developed a highly efficient blue-emitter, based on environmentally friendly indium phosphide/zinc sulphide quantum dots (InP/ZnS QDs) in water, by controlling the growth kinetics of the core as well as the shell. A rational combination of less-reactive metal-halides, phosphorus, and sulphur precursors is the key for achieving the uniform growth of the InP core and ZnS shell. The InP/ZnS QDs showed long-term stable photoluminescence (PL) in the pure-blue region (∼462 nm), with an absolute PL quantum yield of ∼50% and a colour purity of ∼80% in water. Cytotoxicity studies revealed that the cells can withstand up to ∼2 micromolar concentration of pure-blue emitting InP/ZnS QDs (∼120 μg mL−1). Multicolour imaging studies show that the PL of InP/ZnS QDs was well-retained inside the cells as well, without interfering with the fluorescence signal of commercially available biomarkers. Moreover, the ability of InP based pure-blue emitters to participate in an efficient Förster resonance energy transfer (FRET) process is demonstrated. Installing a favorable electrostatic interaction turned out to be crucial in achieving an efficient FRET process (E ∼75%) from blue-emitting InP/ZnS QDs to rhodamine B dye (Rh B) in water. The quenching dynamics fits well with the Perrin formalism and the distance-dependent quenching (DDQ) model, which confirms an electrostatically driven multi-layer assembly of Rh B acceptor molecules around the InP/ZnS QD donor. Furthermore, the process of FRET was successfully translated into the solid state, proving their suitability for device-level studies as well. In short, our study expands the spectrum of aqueous QDs based on InP towards the blue region for future biological and light harvesting studies.
Introduction
Luminescent materials find applications in broad areas of science and technology including full-color display devices, photovoltaics, bioimaging and targeting, sensing, and so on.1–3 The diversity of modern science demands for the need of luminescent materials that emit in all regions of the visible and near-infrared spectra.1–5 But, it is challenging to prepare ‘pure’ blue-emitting luminescent materials as they often fail to meet the stringent requirement of colour purity, high and stable quantum yield, and long-term photostability.1,2 This has been an obstacle in realizing the full use of luminescent materials in different areas of energy and medical research. Quantum dots (QDs) can be one class of materials that could solve this challenge, as they possess unique optoelectronic properties, such as large absorption cross-section, tunable and narrow emission bandwidth, high photoluminescence quantum yield (PL QY), and enhanced photostability.3–14 In this direction, indium phosphide (InP) based QDs are emerging as one of the luminescent materials of choice because of their intrinsically lower toxicity over traditional Cd- and Pb-based QDs.15–27 However, the creation of highly luminescent blue-emitting InP QDs is not a straight-forward task in QD research because of the necessity of an ultra-small sized QD core (<2 nm).6,20 The ultra-small QDs are prone to contain more crystal defects that could hamper their luminescent properties, and this demands for improved synthetic strategies. Moreover, the high covalent nature of InP QDs makes them more vulnerable to oxidizing environments.28 A few protocols are available to prepare blue-emitting InP QDs in organic medium, wherein the growth kinetics of QDs is slowed down by controlling the reaction temperature, precursor reactivity, surface etching, shell growth, competitive secondary nucleation process, etc.29–36 In one example, stable blue-emitting InP/ZnS/ZnS QDs (λem ∼475 nm) were prepared by fine-tuning the inherent reactivity between phosphorus and indium precursors, thereby achieving a control over nucleation and growth processes.33 This has led to the development of efficient blue-emitting light emitting diodes (LEDs) based on InP/ZnS/ZnS core/shell/shell QDs. The use of a double layer of a very thick ZnS shell was crucial in this study for stabilizing the QDs and achieving high PL QY. The presence of a thick ZnS shell in this study (∼8 monolayers) eventually increases the overall size of the QDs to ∼6.8 nm, which might have inherent limitations in light-harvesting (poor energy transfer efficiency because of the thick shell)37 as well as in biological studies (poor renal clearance because of the large size).38 In a very recent study, Douhal and co-workers were successful in preparing smaller sized InP/ZnS/ZnS QDs (∼4 nm) with the PL centered in the deep-blue region in organic medium, by fine-tuning the growth kinetics and ZnS shell-thickness.36 The other demand in the area is to retain the high PL of blue-emitting InP/ZnS QDs in aqueous medium to expand their scope in diverse areas of science. Therefore, there is a necessity to optimize the synthetic protocol for preparing stable pure-blue emitting InP/ZnS QDs in the aqueous medium, along with demonstrating their versatile applied properties in water.Here, we report the preparation of highly luminescent pure-blue emitting InP/ZnS QDs (∼462 nm) in organic medium and their successful transfer to the aqueous phase, with an absolute PL quantum yield of ∼50% and a colour purity of ∼80% (CIE 0.16,0.15) in water. These InP QDs contain a thin shell of ZnS compared to previous reports (3 vs. 8 monolayers), which motivated us to test their suitability for both bioimaging as well as light induced Förster resonance energy transfer studies (FRET). Majority of the QD based FRET donors used so far emitted from the green region, which limits the use of acceptor molecules that emit from the yellow region and above. In other words, the unavailability of pure-blue emitting QD donors restricts the use of acceptor molecules (biomolecules, organic and inorganic chromophores) that absorb in the blue to cyan regions. Thus, there is a high demand for the demonstration of a FRET system based on a pure-blue emitting QD donor, and this forms the main focus of the present work. As a proof of concept, we have demonstrated an efficient FRET from the blue-emitting InP/ZnS QD donor to the red-emitting rhodamine B dye acceptor. This system also serves as an example for a long-range FRET process (here, long-range is in terms of wide color gamut).
A fine-control over the core size and shell thickness of InP/ZnS QDs, by optimizing the precursor amounts and the growth kinetics, enabled us to achieve a strong and stable PL in the pure-blue region. A rational combination of less-reactive metal-halides, phosphorus, and sulphur precursors was the key for achieving the uniform growth of the InP core and ZnS shell. For instance, the dual role of aminophosphine as both the precursor and reducing agent (disproportionation), coupled with the weak bond strength of iodide salts of indium and zinc was crucial in achieving the desired control over the growth kinetics of the core. The as-synthesized InP/ZnS QDs (3.5 ± 0.4 nm diameter) exhibited a first excitonic peak at ∼410 nm with the peak-to-valley ratio of 1.45, and a sharp PL peak at ∼462 nm (full-width half-maxima of ∼58 nm). The uniformity in the ZnS shell led to the proper surface modification with 11-mercaptoundecanoic acid ([−], MUA), which imparted the much-required colloidal stability to pure-blue emitting InP/ZnS QDs in water as well as in biological media. Importantly, there was only a marginal decrease in the absolute PL quantum yield (QY) of blue-emitting InP/ZnS QDs after the surface functionalization and dispersion in water (∼60% in chloroform and ∼50% in water). The blue-emitting InP/ZnS QDs retained their strong PL inside the biological cells without interfering with the fluorescence signal of commercial dyes. Moreover, the InP/ZnS QDs participated in an efficient FRET process, which expands the spectrum of aqueous QDs towards the blue region for future light harvesting studies. The presence of negative surface charges on QDs led to the formation of an electrostatically bounded donor–acceptor system consisting of a [−] pure-blue emitting InP/ZnS QD donor and a [+] rhodamine B dye (Rh B) acceptor. Installing a favourable electrostatic interaction turned out to be crucial in achieving a ∼75% efficient FRET process from InP/ZnS QDs to Rh B dye, in water. Furthermore, the process of FRET was successfully translated into the solid state as well, which proves their suitability for device level studies. Thus, the bioimaging and FRET studies showcase the potency of water-stable pure-blue emitting InP/ZnS QDs for future biological as well as light harvesting studies.
Results and discussion
Synthesis and characterization of pure-blue emitting InP/ZnS QDs
One of the main challenges in the preparation of stable blue-emitting InP/ZnS QDs was controlling the growth kinetics of the core as well as the shell. In the literature, the control over the growth kinetics of the InP core has been achieved by the appropriate choice of metal-ion precursors in combination with aminophosphine.17,33,39 For instance, the emission of InP/ZnS QDs shifts from red to green to blue regions, by changing the halide salts of In and Zn precursors from chloride to bromide to iodide, respectively.17 Likewise, the moderate disproportionation reaction of aminophosphine controls the speed of the reaction, and thus, the emission colour of InP QDs. By adopting this approach, stable blue-emitting InP/ZnS/ZnS QDs (λem ∼475 nm) have already been prepared by Sun and co-workers.33 Our first attempt was to use these blue-emitting InP/ZnS/ZnS QDs as a model system for energy transfer studies with appropriate acceptor molecules. However, the thick double ZnS shell (∼8 monolayers) prevented any photophysical interaction between the blue-emitting InP core and the acceptor molecules. Consequently, our efforts were to fine-tune the kinetics of shell growth to generate a thin layer of ZnS around the blue emitting InP core. Decreasing the reaction temperature and time of the shell formation failed to reduce the shell thickness, and the emission was still centered around 475 nm (even non-uniform shell growth was observed in some cases). Next, the ratios of Zn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S precursors were optimized to obtain a thin shell of ZnS (∼3 monolayers), which allowed the InP core to participate in an efficient FRET process with rhodamine B acceptor (vide infra). Along with reducing the shell thickness, the optimization of Zn:S precursors resulted in lowering the emission maxima of the InP/ZnS QDs to the pure-blue region (∼462 nm). To the best of our knowledge, this is the shortest wavelength reported so far for environmentally friendly blue-emitting InP/ZnS QDs in water.
:S precursors were optimized to obtain a thin shell of ZnS (∼3 monolayers), which allowed the InP core to participate in an efficient FRET process with rhodamine B acceptor (vide infra). Along with reducing the shell thickness, the optimization of Zn:S precursors resulted in lowering the emission maxima of the InP/ZnS QDs to the pure-blue region (∼462 nm). To the best of our knowledge, this is the shortest wavelength reported so far for environmentally friendly blue-emitting InP/ZnS QDs in water.
The as-synthesized InP/ZnS QDs exhibited a sharp first excitonic peak at ∼410 nm with a peak-to-valley ratio of 1.45, indicating a high degree of size homogeneity (Fig. 1 and S1†).25 The PL of InP/ZnS QDs was centered around the pure-blue region (∼462 nm) in chloroform, with a colour purity of ∼80% (CIE 0.16, 0.15) and a full-width half-maxima of ∼58 nm (Fig. S1 and S2†). The absolute PL QY of InP/ZnS QDs was estimated to be ∼0.6 ± 0.2 in chloroform, which is significant for this class of blue-emitting QDs (inset of Fig. 1a and S3†). Now, it is essential to disperse these blue-emitting QDs in aqueous medium to expand their scope beyond the organic medium. However, it is often difficult to retain the excellent PL properties of QDs in aqueous medium, especially for blue-emitting QDs. In this regard, a place exchange protocol was developed to replace oleylamine ligands on blue-emitting InP/ZnS QDs with 11-mercaptoundecanoic acid ([−], MUA) (Scheme 1).21,40 The transfer of the blue PL from the chloroform to the aqueous phase indicates the successful dispersion of QDs in water (Scheme 1). Zeta potential studies revealed that the surface of the QDs is negatively charged (−45.8 ± 1.8 mV), which confirms the success of the place exchange reaction (Fig. S4†). The presence of MUA ligands on the surface of blue-emitting InP/ZnS QDs was further confirmed using standard analytical techniques (see Section 2 in the ESI†) (Fig. S5–S7, Tables S1 and S2†). The thiol groups in MUA ligands bind on the surface of the ZnS shell, whereas the terminal carboxylate groups provide the much-needed dispersion in aqueous medium.
 | ||
| Fig. 1 Spectroscopic and microscopic characterization of pure-blue emitting InP/ZnS QDs. (a) The normalized UV-vis absorption and PL spectra of InP/ZnS QDs before (black) and after (olive) ligand exchange with [−] MUA ligands. The corresponding absolute PL QY data shown in the inset prove ∼85% retention of PL in water. (b) The PL decay profiles of InP/ZnS QDs before (black) and after (olive) place exchange with [−] MUA ligands. (c) A representative TEM image of [−] InP/ZnS QDs. The corresponding size distribution histogram (from ∼500 QDs) is shown in the inset. | ||
 | ||
| Scheme 1 Schematic representation of the ligand exchange process, along with the optical photographs showing the transfer of pure-blue emitting InP/ZnS QDs from the chloroform to the water layer. | ||
All the photophysical properties of pure-blue emitting InP/ZnS QDs were well-retained even after the place exchange reaction (Fig. 1a, b, S8, S9 and Table S3†). The multiexponential PL decay of blue-emitting InP/ZnS QDs was preserved in the aqueous medium, with an average lifetime of ∼212 ns (Fig. 1b). Importantly, the absolute PL QY was estimated to be 0.47 ± 0.2 in water for blue-emitting [−] InP/ZnS QDs (∼85% retention) (inset of Fig. 1a, S3 and S8†). The photostability experiment shows that the PL of the [−] InP/ZnS QD blue-emitter was preserved for at least ∼10 h under continuous illumination with a 365 nm xenon lamp (the power measured at the cuvette wall was ∼6.2 mW cm−2) (Fig. S10†). High-resolution transmission electron microscopy (HRTEM) studies confirm the formation of uniform sized [−] InP/ZnS QDs having an average diameter of 3.5 ± 0.4 nm (Fig. 1c and S11†). Furthermore, the powder X-ray diffraction (PXRD) studies confirm the zinc blend phase for the InP core (Fig. S12†).24 A large shift in the diffraction peaks toward the ZnS planes was observed for the core/shell [−] InP/ZnS QDs. The shell thickness was determined to be ∼0.85 nm (∼3 monolayers) (Fig. S12†). The average diameter of the blue-emitting [−] InP/ZnS QDs was estimated to be 3.5 ± 0.2 nm from the PXRD study, which corroborates well with the TEM data. The elemental analyses from X-ray photoelectron spectroscopy (XPS) and energy dispersive X-ray spectroscopy (EDAX) confirm the presence of all the major elements in [−] InP/ZnS QDs (see the detailed discussion in Section 2 of the ESI†) (Fig. S13–S16 and Table S4†). The valence and conduction band positions of [−] InP/ZnS QDs were estimated to be at −6.33 eV and −3.63 eV (vs. vacuum), respectively, with the combination of cyclic voltammetry and absorption studies (Fig. S17†). These values are in good agreement with literature reports on sky-blue emitting InP/ZnS/ZnS QDs.33 It is worth mentioning that the molar extinction coefficient (ε) for small InP/ZnS QDs is not available in the literature. In the present study, the ε of pure-blue emitting [−] InP/ZnS QDs was estimated to be ∼2.5 × 104 M−1 cm−1, from the combination of inductively coupled plasma-mass spectroscopy (ICP-MS), PXRD, and UV-vis absorption spectroscopy (see the detailed discussion in Section 2 of the ESI†) (Fig. S18 and Table S5†). In the literature, the ε of InP magic-sized clusters at the first excitonic peak ∼390 nm is reported to be ∼2.8 × 104 M−1 cm−1.41 Thus, the ε of pure-blue emitting [−] InP/ZnS QDs is within the range of small sized InP QDs.
The photophysical properties of QDs depend on the quality of the crystals formed, which is governed by the kinetics of nucleation and crystallization growth processes. In the present study, the less reactive aminophosphine and weak In/Zn–I bond strength led to the balance between the nucleation and growth rates.33,39 As a result, the size of the core InP QDs was controlled below 2 nm. Furthermore, a fine control over the shell growth and thickness was achieved, which led to the unusually high photophysical performance of blue-emitting InP/ZnS QDs. This was achieved by optimizing the amount of Zn- and S-precursors and growing the ZnS shell at a high temperature (details are provided in the ESI†). The consequence of having a small InP core size and a thin ZnS shell is as follows. The energy difference between the small InP core (∼1.8 nm) and ZnS shell is not significant enough to form a typical type-I core–shell QDs. Instead, they form a quasi-type-II core–shell QDs.36 As the shell thickness is less, the excitons will be mainly confined within the InP core.36,42 As a result, the PL QY will be high, and the PL maxima will be centered around the band edge emission of the core QDs (∼462 nm in the present study). A thicker shell will lead to the delocalization of the electron wave function to the shell, which compromises the photophysical properties of the QDs.42 Furthermore, the uniformity in the ZnS shell and the robust nature of the core–shell QD allowed for the transfer of blue-emitting InP/ZnS QDs to the aqueous medium (via place exchange reaction), without compromise in their photophysical properties.
Thus, the blue-emitting InP/ZnS QDs exhibit all the fundamental characteristics that a material should possess for studying the applied properties. Accordingly, the InP QD based pure-blue emitters were used in multicolour bioimaging and resonance energy transfer studies, as representative examples, to prove their potency for future biological and light harvesting applications.
Biocompatibility and cellular bioimaging studies
First, the compatibility of blue-emitting [−] InP/ZnS QDs as bioimaging agents was tested. The colloidal stability as well as the PL of [−] InP/ZnS QDs was well-retained in buffer and biological media for a long time (∼30 h) (Fig. S19†). Next, the cytotoxicity of [−] InP/ZnS QDs was tested in HeLa cell lines using the MTT assay.21 Up to ∼95% cell viability was exhibited by [−] InP/ZnS QDs till ∼120 μg mL−1 concentration (∼2 μM), beyond which a mild toxicity was observed (∼75% cell viability at ∼300 μg mL−1) (Fig. 2a and S20†). It can be concluded that the blue-emitting [−] InP/ZnS QDs are less cytotoxic compared to the traditional Cd based QDs.21 The less toxic metal ions and anionic charges displayed on the surface of the QDs prevent their unwarranted interactions with a multitude of cytosolic proteins and other biomolecules inside the cells, thereby preserving the cellular viability. Inspired by the excellent cell viability data, detailed imaging experiments were then carried out. As seen in Fig. S21,† the PL signals emanating from the cells intensified as the concentration of the [−] InP/ZnS QD blue-emitter was increased. The observed images were then quantified, and the resultant corrected total cell fluorescence (CTCF) values were coherent with the physical inspection (Fig. 2b and S21†). Co-staining experiments with the commercially available red Lysotracker dye confirm that the cellular uptake of blue-emitting [−] InP/ZnS QDs is via the endocytosis pathway (details are provided in Section 3 of the ESI†) (Fig. S22†). | ||
| Fig. 2 Biocompatibility and bioimaging studies with pure-blue emitting [−] InP/ZnS QDs. (a) MTT assay showing the viability of HeLa cells incubated with different concentrations of [−] InP/ZnS QDs for ∼72 h. (b) Representative confocal images of HeLa cells recorded, after internalizing with 0.70 μM [−] InP/ZnS QDs for ∼7 h. (c) Multicolour imaging of pure-blue emitting InP/ZnS QDs with PHD and SYTO-Deep Red commercial dyes. | ||
The narrow absorption and emission features of blue-emitting [−] InP/ZnS QDs motivated us to check their compatibility with other commercially available fluorophores. For this, the cells were treated with blue-emitting [−] InP/ZnS QDs for a period of 6 hours, followed by the sequential staining of the nuclei as well as the actin and filaments with commercially available SYTO-dye and phalloidin green (PHD), respectively. The cells were visualized using Zeiss confocal microscopy under simultaneous excitations with 3 different laser sources (405 nm, 488 nm, and 633 nm). The QDs and the commercial markers fluoresce individually at their respective excitation wavelengths. The presence of QDs did not interfere with the fluorescence signals of the other two dyes (Fig. 2c). Thus, the blue-emitting [−] InP/ZnS QDs displayed excellent multicolour bioimaging abilities, which are essential for any optical probes in cellular imaging studies.
Light induced resonance energy transfer studies
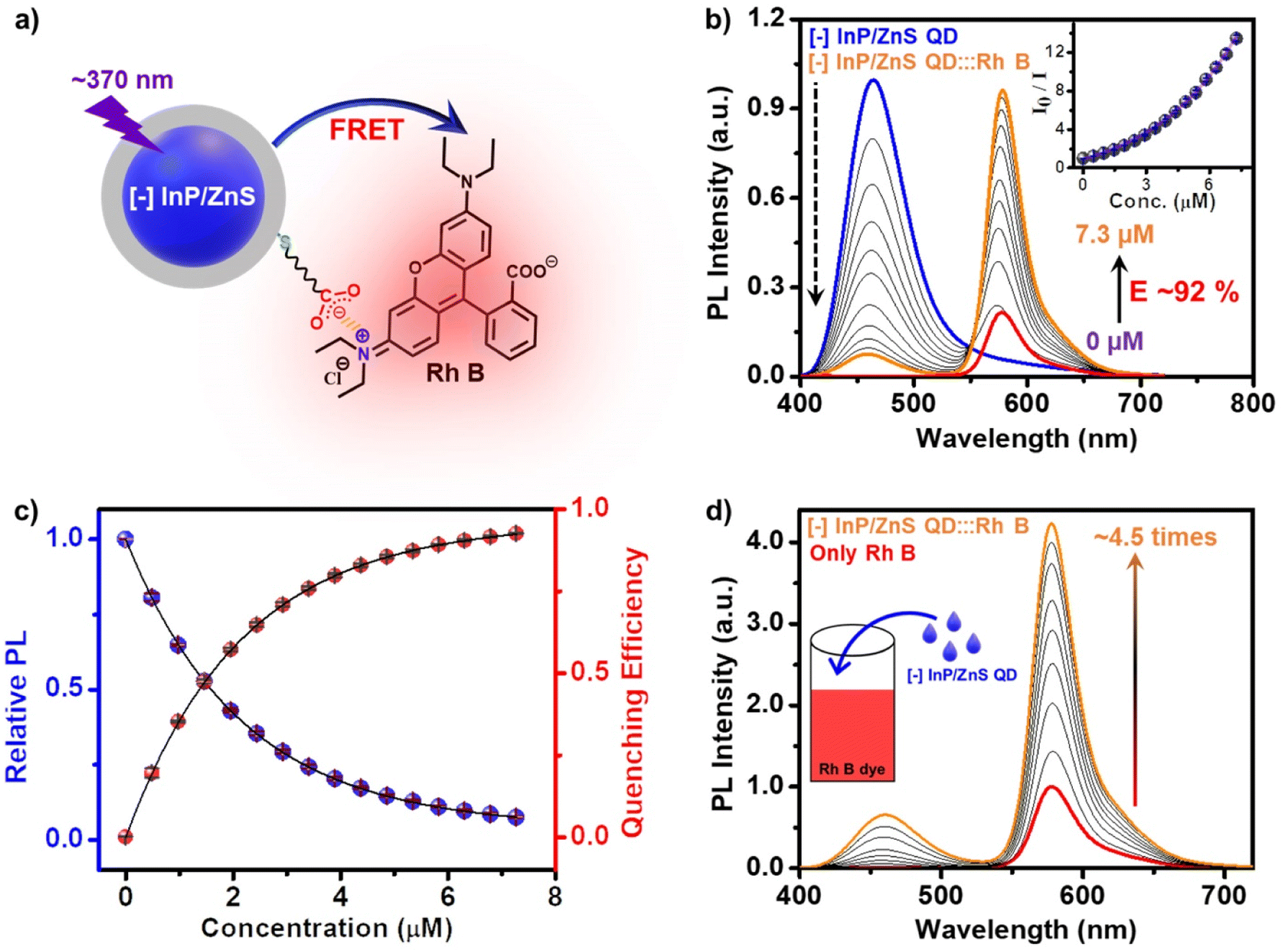 | ||
| Fig. 3 Steady-state FRET studies in the [−] InP/ZnS QD:::Rh B donor–acceptor system. (a) Schematic representation of the electrostatically driven FRET process from the pure-blue emitting [−] InP/ZnS QD donor to the Rh B acceptor dye. (b) The PL spectral changes in [−] InP/ZnS QDs upon addition of varying concentrations of Rh B dye. The inset shows the corresponding Stern–Volmer plot. (c) A saturation in both the relative PL intensity of donor [−] InP/ZnS QDs and the quenching efficiency was observed after ∼6.5 μM addition of the acceptor Rh B dye. (d) Reverse addition experiment: a steady enhancement in the PL of ∼7.5 μM acceptor Rh B dye was observed, upon addition of varying concentrations of the [−] InP/ZnS QD donor. The excitation wavelength was 370 nm in all these studies. | ||
In another set of studies, reverse addition experiments were performed to rule out the interference from the direct excitation of Rh B dye acceptors. Here, the PL of the acceptor Rh B dye (∼7.5 μM) was monitored upon successive addition of the donor QDs (4 μL each of ∼418 μM QD). A steady increase in the PL of Rh B dye was observed (∼4.5 times, I/I0 = 4.5) in the presence of ∼6 μM of InP/ZnS QDs (Fig. 3d), which further confirms an efficient energy transfer process from the QDs to the Rh B dye. Likewise, detailed excitation spectroscopy studies reaffirm the involvement of an efficient energy transfer process in the [−] InP/ZnS QD:::Rh B dye donor–acceptor system (see Section 5 in the ESI†) (Fig. S25†). A high efficiency in the energy transfer process (∼92%) proves a strong interaction between the blue-emitting [−] InP/ZnS QDs and Rh B dye. The presence of opposite charges on both InP/ZnS QDs and Rh B point towards the involvement of electrostatic attraction in achieving an efficient PL quenching in the [−] InP/ZnS QD:::Rh B dye donor–acceptor system. Accordingly, PL quenching experiments were performed in buffer to weaken the electrostatic interactions in the [−] InP/ZnS QD:::Rh B dye donor–acceptor system.54,55 An increase in the ionic strength of the medium will weaken the electrostatic attraction due to the screening of charges on both the donor and the acceptor.55 As predicted, the PL enhancement for the acceptor Rh B dye showed a substantial decrease from 5.5 to 3.4 times, as the ionic strength of the medium was increased from 0.05 mM (basic water, pH ∼10) to 0.85 M (5× PBS) (see details in Section 5 of the ESI†) (Fig. S26†). This confirms an electrostatically driven energy transfer from the InP/ZnS QD donor to the Rh B dye acceptor.
 | ||
| Fig. 4 Time-resolved FRET studies in [−] InP/ZnS:::Rh B dye donor–acceptor system. (a) PL decay profiles of [−] InP/ZnS QDs in the presence (orange) and absence (blue) of ∼7.5 μM Rh B dye (the excitation wavelength was 370 nm). (b) The corresponding Stern–Volmer plot based on relative PL lifetime values of InP/ZnS QDs. (c) A plot of FRET efficiency (estimated from lifetime quenching) vs. temperature. Negligible effect of temperature on the quenching efficiency confirms the process of resonance energy transfer in the [−] InP/ZnS:::Rh B dye donor–acceptor system. (d) Table summarizing various quenching parameters estimated based on FRET formalism: the spectral overlap integral [J(λ)]; Förster radius (R0); FRET efficiency (E); rate of energy transfer (kT); quenching volume (V); quenching radius (rq), and bimolecular quenching constant (ka). | ||
The ultimate proof for the FRET in [−] InP/ZnS QD:::Rh B dye donor–acceptor system was obtained from the temperature dependent PL lifetime studies. Typically, the PL lifetime increases upon decreasing the temperature, since the non-radiative recombination channels will be inhibited because of the suppression of phonon vibrations.57 On the other hand, FRET is a non-radiative process which will be independent of the temperature, at a given donor–acceptor distance.54,57 Thus, performing temperature dependent PL lifetime studies is a common practice to confirm the process of FRET in a donor–acceptor system.54,57 In the present study as well, a linear increase was observed in the individual PL lifetime of the [−] InP/ZnS QD donor and [−] InP/ZnS QD:::Rh B complex as the temperature was lowered (Fig. S29†). In contrast, negligible changes were observed in the efficiency of PL lifetime quenching as a function of temperature, which confirms the prominent involvement of the non-radiative resonance energy transfer process in the PL quenching of InP/ZnS QD by the Rh B dye (Fig. 4c, S30 and Table S7†). Accordingly, different energy transfer parameters were calculated for the [−] InP/ZnS QD:::Rh B dye donor–acceptor system using the FRET formalism, as summarized in Fig. 4d (details of each parameter calculation are given in Section 5 of the ESI†).
 | ||
| Fig. 5 FRET studies in the solid-state. (a) Steady-state and (b) time-resolved PL of [−] InP/ZnS QDs in the absence (blue) and presence (orange) of Rh B dye, in 1% agarose gel. The inset of (a) shows the true colour PL image of pure-blue emitting [−] InP/ZnS QDs and [−] InP/ZnS QD:::Rh B complex agarose film. (c) Temporal evolution of FRET in the solid state: true colour PL images of [−] InP/ZnS QDs (first row), 1:0.3 molar ratio of QDs:dye (2nd row), 1:0.6 molar ratio of QDs:dye (3rd row), 1:1.3 molar ratio of QDs:dye (4th row), and only Rh B dye (5th row), as a function of drying. The first and the third columns correspond to the liquid drop (0 h) and solid film (∼12 h), respectively. The donor–acceptor distance decreases as drying proceeds, resulting in an increase in the FRET efficiency. Consequently, the colour of the droplet shifts towards the red region corresponding to the Rh B dye (rows 2–4). First row: the blue PL of the InP/ZnS QDs shifts towards the cyan colour upon complete drying, indicating inter-QD interactions. Last row: the background of the last image was intentionally changed to blue to improve the visibility of the dried droplet. | ||
Finally, a temporal evolution of the FRET process was visualized via the droplet-evaporation method. In a typical experiment, droplets (∼10 μL) of different donor:acceptor (D:A) ratios were placed on a Teflon-coated glass slide, and the PL colour of the droplet was monitored in a UV chamber as a function of time/evaporation. At the initial stage, the droplet showed a PL colour corresponding to the mixing of donor and acceptor colours (such as magenta, pink, and orange corresponding to the D:A of 1:0.3, 1:0.6, and 1:1.3, respectively). As the time proceeds, the drying of the droplet occurs, and the colour of the droplet shifts towards red (corresponding to the Rh B dye) on complete evaporation of water (Fig. 5c). This PL colour change of the droplets can be explained based on Förster formalism.43 The efficiency of the FRET process is extremely sensitive towards the distance between the donor and the acceptor (E α 1/r6, where r is the donor–acceptor distance).43 In the present study, the evaporation of the droplet decreases the D–A distance, resulting in an increase in the FRET efficiency. This will be accompanied by a temporal colour change towards the PL of the Rh B acceptor, which once again ascertains the involvement of an efficient FRET process in the [−] InP/ZnS QD:::Rh B dye donor–acceptor system.
Conclusions
Our work introduces environmentally friendly InP/ZnS QDs to the family of pure-blue emitting luminescent materials. A fine control over the growth kinetics of the core as well as the shell was crucial in achieving a stable PL in blue-emitting InP/ZnS QDs (PL QY ∼60% at 462 nm). The retention of the strong PL of blue-emitting InP/ZnS QDs in the aqueous medium enabled their use in diverse areas including cellular bioimaging and light harvesting. Cytotoxicity and bioimaging experiments showcase the potency of blue-emitting InP/ZnS QDs as optical probes for multicolour cellular imaging studies. As part of the light harvesting, an efficient FRET process from blue-emitting [−] InP/ZnS QDs to Rh B dye was demonstrated in water. Detailed steady-state and time-resolved photophysical studies show that the non-linear Stern–Volmer (SV) plots fit well with the Perrin formalism and DDQ model, which further confirms a multi-layer assembly of Rh B acceptor molecules in the quenching radius. The large values of bimolecular quenching constant (∼1.24 × 1012 M−1 s−1) and quenching radius (∼54 nm) reveal the involvement of electrostatic attraction between the oppositely charged InP/ZnS QDs and Rh B dyes. This turned out to be a crucial factor in achieving an efficient FRET process in blue-emitting [−] InP/ZnS QD:::Rh B dye donor–acceptor system. The process of FRET was demonstrated in the solid state as well, in a temporal fashion, which enhances the prospects of blue-emitting InP/ZnS QDs for device-level applications. The successful demonstration of a FRET process based on blue-emitting InP/ZnS QDs opens up various possibilities in energy transfer studies with a wide range of acceptors emitting from the cyan region onwards. A logical extension will be to check the suitability of blue-emitting InP/ZnS QDs in photocatalysis and photovoltaics, to establish them as an efficient light harvester. Looking forward, the blue-emitting InP/ZnS QDs developed in this work can potentially be an alternative to the existing luminescent materials for both biological as well as energy applications.Data availability
All the experimental data were provided in the main text and/or ESI.†Author contributions
P. Roy carried out the QD synthesis and performed all the characterization and energy transfer studies. M. Virmani performed the cytotoxicity and bioimaging experiments. Prof. P. P. Pillai conceived and designed the project. The manuscript was written through contributions of all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from IISER Pune and DST SERB India (Grant No. CRG/2019/003960). The authors thank Prof. M. Jayakannan for the useful discussion on the cytotoxicity and bioimaging experiments. The authors also thank Mr Joy Chatterjee for helping with the measurements of absolute PL QY. P. R. thanks MoE/PMRF and M. V. thanks MoE for PhD fellowships.Notes and references
- S. Nakamura, Angew. Chem., Int. Ed., 2015, 54, 7770–7788 CrossRef CAS PubMed.
- Z. Shen, P. E. Burrows, V. Bulović, S. R. Forrest and M. E. Thompson, Science, 1997, 276, 2009–2011 CrossRef CAS.
- Y. Wang, I. Fedin, H. Zhang and D. V. Talapin, Science, 2017, 357, 385–388 CrossRef CAS PubMed.
- R. Xie, D. Battaglia and X. Peng, J. Am. Chem. Soc., 2007, 129, 15432–15433 CrossRef CAS PubMed.
- A. Wolf, V. Lesnyak, N. Gaponik and A. Eychmüller, J. Phys. Chem. Lett., 2012, 3, 2188–2193 CrossRef CAS PubMed.
- L. E. Brus, J. Chem. Phys., 1984, 80, 4403 CrossRef CAS.
- T. Vossmeyer, L. Katsikas, M. Giersig, I. G. Popovic, K. Diesner, A. Chemseddine, A. Eychmueller and H. Weller, J. Phys. Chem., 1994, 98, 7665–7673 CrossRef CAS.
- N. Hildebrandt, C. M. Spillmann, W. R. Algar, T. Pons, M. H. Stewart, E. Oh, K. Susumu, S. A. Díaz, J. B. Delehanty and I. L. Medintzy, Chem. Rev., 2017, 117, 536–711 CrossRef CAS PubMed.
- I. L. Medintz and H. Mattoussi, Phys. Chem. Chem. Phys., 2009, 11, 17–45 RSC.
- S. Sarkar, R. Bose, S. Jana, N. R. Jana and N. Pradhan, J. Phys. Chem. Lett., 2010, 1, 636–640 CrossRef CAS.
- M. Wu, P. Mukherjee, D. N. Lamont and D. H. Waldeck, J. Phys. Chem. C, 2010, 114, 5751–5759 CrossRef CAS.
- J. M. Pietryga, Y.-S. Park, J. Lim, A. F. Fidler, W. K. Bae, S. Brovelli and V. I. Klimov, Chem. Rev., 2016, 116, 10513–10622 CrossRef CAS PubMed.
- I. N. Chakraborty, P. Roy, A. Rao, G. Devatha, S. Roy and P. P. Pillai, J. Mater. Chem. A, 2021, 9, 7422–7457 RSC.
- V. Jain, S. Roy, P. Roy and P. P. Pillai, Chem. Mater., 2022, 34, 7579–7597 CrossRef CAS.
- L. Li and P. Reiss, J. Am. Chem. Soc., 2008, 130, 11588–11589 CrossRef CAS PubMed.
- A. Thomas, P. V. Nair and K. G. Thomas, J. Phys. Chem. C, 2014, 118, 3838–3845 CrossRef CAS.
- M. D. Tessier, D. Dupont, K. De Nolf, J. De Roo and Z. Hens, Chem. Mater., 2015, 27, 4893–4898 CrossRef CAS.
- D. C. Gary, M. W. Terban, S. J. L. Billinge and B. M. Cossairt, Chem. Mater., 2015, 27, 1432–1441 CrossRef CAS.
- G. Xu, S. Zeng, B. Zhang, M. T. Swihart, K.-T. Yong and P. N. Prasad, Chem. Rev., 2016, 116, 12234–12327 CrossRef CAS PubMed.
- S. Tamang, C. Lincheneau, Y. Hermans, S. Jeong and P. Reiss, Chem. Mater., 2016, 28, 2491–2506 CrossRef CAS.
- G. Devatha, S. Roy, A. Rao, A. Mallick, S. Basu and P. P. Pillai, Chem. Sci., 2017, 8, 3879–3884 RSC.
- S. Thirunavukkuarasu, A. George, A. Thomas, A. Thomas, V. Vijayan and K. G. Thomas, J. Phys. Chem. C, 2018, 122, 14168–14176 CrossRef CAS.
- G. Devatha, A. Rao, S. Roy and P. P. Pillai, ACS Energy Lett., 2019, 4, 1710–1716 CrossRef CAS.
- Y. Li, X. Hou, X. Dai, Z. Yao, L. Lv, Y. Jin and X. Peng, J. Am. Chem. Soc., 2019, 141, 6448–6452 CrossRef CAS PubMed.
- Y.-H. Won, O. Cho, T. Kim, D.-Y. Chung, T. Kim, H. Chung, H. Jang, J. Lee, D. Kim and E. Jang, Nature, 2019, 575, 634–638 CrossRef CAS PubMed.
- G. Devatha, P. Roy, A. Rao, S. Roy and P. P. Pillai, J. Phys. Chem. Lett., 2020, 11, 4099–4106 CrossRef CAS PubMed.
- H. A. Nguyen, F. Y. Dou, N. Park, S. Wu, H. Sarsito, B. Diakubama, H. Larson, E. Nishiwaki, M. Homer, M. Cash and B. M. Cossairt, Chem. Mater., 2022, 34, 6296–6311 CrossRef CAS.
- A. Cros-Gagneux, F. Delpech, C. Nayral, A. Cornejo, Y. Coppel and B. Chaudret, J. Am. Chem. Soc., 2010, 132, 18147–18157 CrossRef CAS PubMed.
- K. Lim, H. S. Jang and K. Woo, Nanotechnology, 2012, 23, 485609 CrossRef PubMed.
- W. Shen, H. Tang, X. Yang, Z. Cao, T. Cheng, X. Wang, Z. Tan, J. You and Z. Deng, J. Mater. Chem. C, 2017, 5, 8243–8249 RSC.
- F. Huang, C. Bi, R. Guo, C. Zheng, J. Ning and J. Tian, J. Phys. Chem. Lett., 2019, 10, 6720–6726 CrossRef CAS PubMed.
- W. Lee, C. Lee, B. Kim, Y. Choi and H. Chae, Nanomaterials, 2020, 10, 2171 CrossRef CAS PubMed.
- W. Zhang, S. Ding, W. Zhuang, D. Wu, P. Liu, X. Qu, H. Liu, H. Z. Wu, K. Wang and X. W. Sun, Adv. Funct. Mater., 2020, 30, 2005303 CrossRef CAS.
- Z. Cui, S. Mei, Z. D. Yang, S. Qin, Z. Xiong, B. Yang, H. He, R. Bao, Y. Qiu, Y. Chen, E. Zhang, F. Xie, G. Xing and R. Guo, Small, 2022, 18, 2108120 CrossRef CAS PubMed.
- H. Zhang, X. Ma, Q. Lin, Z. Zeng, H. Wang, L. S. Li, H. Shen, Y. Jia and Z. Du, J. Phys. Chem. Lett., 2020, 11, 960–967 CrossRef CAS PubMed.
- S. Rakshit, B. Cohen, M. Gutiérrez, A. O. El-Ballouli and A. Douhal, ACS Appl. Mater. Interfaces, 2023, 15, 3099–3111 CrossRef CAS PubMed.
- H. B. Jalali, R. Melikov, S. Sadeghi and S. Nizamoglu, J. Phys. Chem. C, 2018, 122, 11616–11622 CrossRef PubMed.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef CAS PubMed.
- M. D. Tessier, K. De Nolf, D. Dupont, D. Sinnaeve, J. De Roo and Z. Hens, J. Am. Chem. Soc., 2016, 138, 5923–5929 CrossRef CAS PubMed.
- D. V. Talapin, A. L. Rogach, I. Mekis, S. Haubold, A. Kornowski, M. Haase and H. Weller, Colloids Surf., A, 2002, 202, 145–154 CrossRef CAS.
- R. P. Brown, M. J. Gallagher, D. H. Fairbrother and Z. Rosenzweig, Langmuir, 2018, 34, 13924–13934 CrossRef CAS PubMed.
- Y. Jia, J. Chen, K. Wu, A. Kaledin, D. G. Musaev, Z. Xie and T. Lian, Chem. Sci., 2016, 7, 4125–4133 RSC.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 1999 Search PubMed.
- A. Thomas, K. Sandeep, S. M. Somasundaran and K. G. Thomas, ACS Energy Lett., 2018, 3, 2368–2375 CrossRef CAS.
- R. Wargnier, A. V. Baranov, V. G. Maslov, V. Stsiapura, M. Artemyev, M. Pluot, A. Sukhanova and I. Nabiev, Nano Lett., 2004, 4, 451–457 CrossRef CAS.
- T. Franzl, A. Shavel, A. L. Rogach, N. Gaponik, T. A. Klar, A. Eychmüller and J. Feldmann, Small, 2005, 1, 392–395 CrossRef CAS PubMed.
- K. E. Sapsford, L. Berti and I. L. Medintz, Angew. Chem., Int. Ed., 2006, 45, 4562–4588 CrossRef CAS PubMed.
- R. Freeman, B. Willner and I. Willner, J. Phys. Chem. Lett., 2011, 2, 2667–2677 CrossRef CAS.
- R. T. Cheriya, J. Joy, A. P. Alex, A. Shaji and M. Hariharan, J. Phys. Chem. C, 2012, 116, 12489–12498 CrossRef CAS.
- K. F. Chou and A. M. Dennis, Sensors, 2015, 15, 13288–13325 CrossRef PubMed.
- X. Ji, W. Wang and H. Mattoussi, Nano Today, 2016, 11, 98–121 CrossRef CAS.
- A. Sarkar, S. Dhiman, A. Chalishazar and S. J. George, Angew. Chem., Int. Ed., 2017, 56, 13767–13771 CrossRef CAS PubMed.
- M. S. Kodaimati, S. Lian, G. C. Schatz and E. A. Weiss, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8290–8295 CrossRef CAS PubMed.
- P. Roy, G. Devatha, S. Roy, A. Rao and P. P. Pillai, J. Phys. Chem. Lett., 2020, 11, 5354–5360 CrossRef CAS PubMed.
- C. He, T. D. Nguyen, K. Edme, M. Olvera de la Cruz and E. A. Weiss, J. Am. Chem. Soc., 2017, 139, 10126–10132 CrossRef CAS PubMed.
- B. Zelent, J. Kuśba, I. Gryczynski, M. L. Johnson and J. R. Lakowicz, J. Phys. Chem., 1996, 100, 18592–18602 CrossRef CAS PubMed.
- E. Mutlugun, P. L. Hernandez-Martinez, C. Eroglu, Y. Coskun, T. Erdem, V. K. Sharma, E. Unal, S. K. Panda, S. G. Hickey, N. Gaponik, A. Eychmüller and H. V. Demir, Nano Lett., 2012, 12, 3986–3993 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental methods, synthesis, and characterization of QDs, stability studies, bioimaging, uptake mechanism, proof of electrostatics and calculation of various parameters involved in the FRET study. See DOI: https://doi.org/10.1039/d3sc00164d |
| This journal is © The Royal Society of Chemistry 2023 |