
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective direct Michael addition of cyanohydrin ether derivatives to enones catalyzed by chiral bis(guanidino)iminophosphorane organosuperbase†
Saikat
Das
a,
Azusa
Kondoh
 *b and
Masahiro
Terada
*a
*b and
Masahiro
Terada
*a
aDepartment of Chemistry, Graduate School of Science, Tohoku University, Aramaki, Aoba-ku, Sendai 980-8578, Japan. E-mail: mterada@tohoku.ac.jp
bResearch and Analytical Center for Giant Molecules, Graduate School of Science, Tohoku University, Aramaki, Aoba-ku, Sendai, 980-8578, Japan. E-mail: kondoha@tohoku.ac.jp
First published on 31st January 2023
Abstract
The direct use of cyanohydrin ether derivatives as less acidic pronucleophiles was achieved for the first time in the catalytic enantioselective Michael addition reaction under transition metal free conditions. Chiral bis(guanidino)iminophosphoranes as the higher order organosuperbase facilitated the intended catalytic Michael addition to enones, giving rise to the corresponding products in high yields with moderate to high diastereo- and enantioselectivities in most cases. Further elaboration of the corresponding enantioenriched product was conducted by derivatization into a lactam derivative through hydrolysis followed by cyclo-condensation.
Introduction
Cyanohydrin ether derivatives are an important class of umpolung synthons that function as an acyl anion equivalent in organic synthesis.1 The anion of the cyanohydrin ether derivatives undergoes a range of reactions with various electrophiles to form intriguing products in which the cyanohydrin unit is converted into an acyl functionality and multifunctionalized carbon frameworks having a tetrasubstituted stereogenic center are formed (Fig. 1). The formation of cyanohydrins having a tetrasubstituted stereogenic center through the addition reaction of ketones with a cyanide nucleophile is well established (Fig. 1a).2 Indeed, successful examples of enantioselective variants have been reported using a variety of chiral metal catalysts3 and a limited number of organocatalysts,4 and these have led to the development of efficient methods for the construction of a tetrasubstituted cyanohydrin unit in an enantioenriched form. However, in principle, those methods deal with the formation of a single stereogenic center and not the construction of multiple stereogenic centers in the bond formation step or the formation of multifunctionalized carbon frameworks. In contrast, the use of protected cyanohydrin derivatives as the pronucleophile has resulted in the introduction of multiple functional groups into the corresponding product and the formation of multiple-stereogenic centers, one of which is a tetrasubstituted one (Fig. 1b).5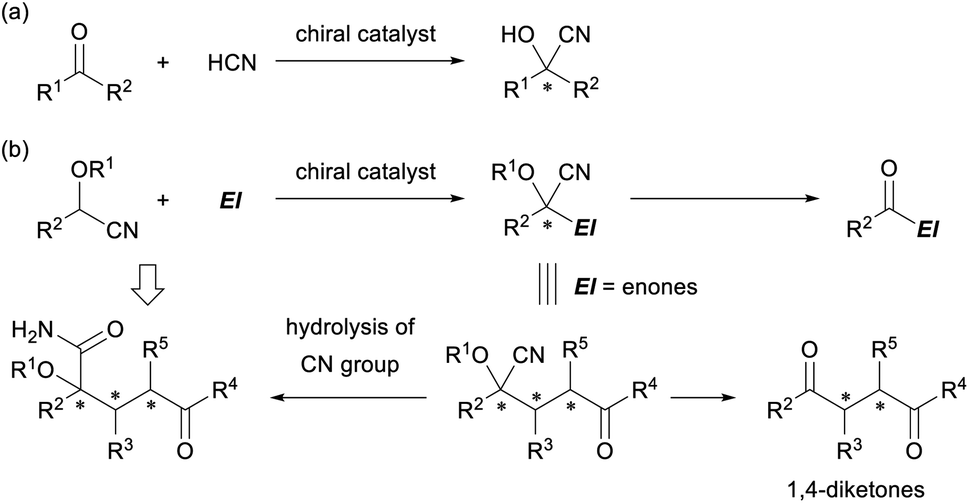 | ||
| Fig. 1 (a) Cyanohydrin formation by the reaction of ketones with the cyanide nucleophile. (b) Cyanohydrin ether derivatives as a versatile intermediate in organic synthesis. | ||
In general, the generation of an active anionic nucleophile from a cyanohydrin ether derivative requires a stoichiometric amount of a strong base, and hence marked efforts have been devoted to make the reaction catalytic. In fact, a cyanide-mediated [1,2]-Brook rearrangement of acyl silanes was employed to generate anions of cyanohydrin derivatives.6 However, catalytic enantioselective reactions have been largely unexplored despite the seminal work by the Denmark group in 2010,7 in which preformed N-silyl oxyketene imines were utilised to generate the active nucleophile catalytically. The direct use of protected cyanohydrin derivatives in a catalytic enantioselective reaction has rarely been reported8,9 despite the additional advantage of the cyanohydrin derivatives, as they can be easily derivatized into other functional groups. For instance, the retro cyanohydrin formation reaction affords a ketone derivative, and the cyano group is hydrolyzed into an amide derivative or transformed into an amine functionality through the reduction or addition of organometallic reagents. In particular, the enantioselective Michael addition of cyanohydrin ether derivatives as the pronucleophile to enones provides multifunctionalized products, and further elaboration of the products gives not only 1,4-diketones10 but also densely functionalized derivatives having a tetrasubstituted stereogenic center in an enantioenriched form (Fig. 1b).
To establish the intended catalytic enantioselective system using cyanohydrin ether derivatives as the pronucleophile, a highly basic catalyst is strictly required because of the less acidic proton of the cyanohydrin ether derivatives.5c,11 In the recent past, several chiral organobase catalysts having different structural features have been reported;12,13 however, the low basicity of these catalysts has restricted the success of the intended methodology. To overcome this inherent limitation, recently, our group has developed chiral bis(guanidino)iminophosphoranes (M)-1 as an uncharged higher order organosuperbase, which have emerged as a new class of chiral organocatalysts.14,15 As (M)-1 have enabled the achievement of novel catalytic enantioselective transformations using several less acidic pronucleophiles, we have directed our attention to the activation of cyanohydrin ether derivatives as a less acidic pronucleophile using (M)-1. Herein we report the enantioselective direct Michael addition of protected cyanohydrin ether derivatives 2 to enones 3 catalyzed by bis(guanidino)iminophosphoranes (M)-1, which gives rise to corresponding products 4 in a diastereo- and enantioselective manner in most cases (Scheme 1). The direct use of cyanohydrin ether derivatives as less acidic pronucleophiles was accomplished for the first time in the catalytic enantioselective addition reaction under transition metal free conditions.
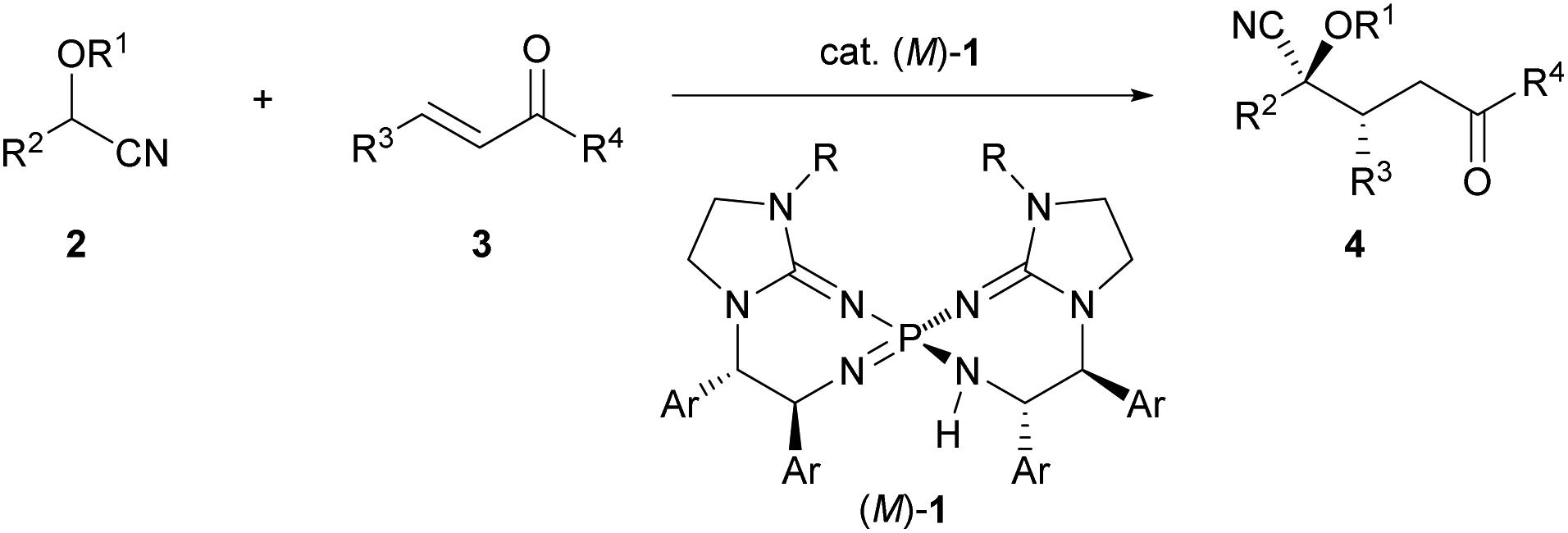 | ||
| Scheme 1 Enantioselective direct Michael addition of cyanohydrin ether derivatives to enones catalyzed by chiral bis(guanidino)iminophosphoranes (M)-1. | ||
Results and discussion
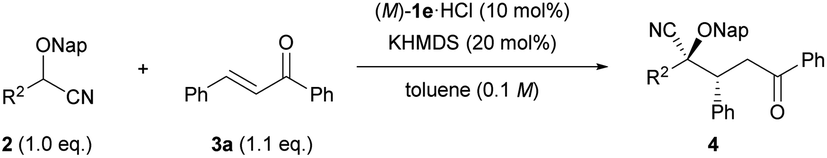
We set out to investigate our intended Michael reaction by selecting cyanohydrin benzyl ether 2a and chalcone (3a) as the model substrates (Table 1). The catalyst, which was generated in situ by treating 11 mol% (M)-1a·HBr salt with 10 mol% KHMDS (potassium hexamethyldisilazide) as the inorganic base, facilitated the reaction of 2a with 3a to afford desired product 2-(benzyloxy)-5-oxo-2,3,5-triphenylpentanenitrile (4aa) in excellent yield albeit with a low diastereomeric ratio and ee value of the major diastereomer (entry 1). In order to increase both enantio- and diastereoselectivities, we investigated the influence of substituent R attached to the nitrogen atom of the guanidine unit of (M)-1 (entries 2–4), but found that it did not exhibit any beneficial effect. Next, replacing the phenyl groups on the 7,7-membered spirocyclic ring with bulky 2-naphthyl groups, giving (M)-1e, improved the diastereo- and enantioselectivities (entry 5). Lowering the temperature from rt to −20 °C resulted in a slight enhancement of the stereoselectivity (entry 6). To further improve the stereochemical outcome, we examined the substituent effect of R1 on the cyanohydrin ethers.16 Introduction of the bulky naphthalen-2-ylmethyl group (Nap) into cyanohydrin ether 2b enhanced the ee value significantly while maintaining the diastereomeric ratio [entry 7, dr (diastereomeric ratio) = 84![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :16, 79% ee]. Lowering the reaction temperature to −40 °C had a positive influence on the stereoselectivity (entry 8, dr = 87:13, 84% ee); however, further lowering the temperature to −60 °C had an adverse effect on the yield without any marked improvement of the stereoselectivity (entry 9). We then thoroughly explored the reaction conditions to enhance the yield and the stereoselectivity. To our delight, the use of an excess amount of achiral inorganic base KHMDS (15–30 mol%) and 10 mol% of (M)-1e·HCl promoted the reaction efficiently,14a resulting in the significant enhancement not only of the yield but also of the enantioselectivity (entries 10–12). It is worth mentioning that despite using an excess amount of the achiral inorganic base, the enantioselectivity was improved markedly. Meanwhile, lowering the reaction temperature from −60 °C to −78 °C had a negative effect on the yield (entry 13). On the basis of our results, the optimized conditions were established as follows: use of excess KHMDS (20 mol%) and 10 mol% of (M)-1e·HCl at −60 °C gave rise to corresponding cyanohydrin derivative 4ba in excellent yield (95%) with high stereoselectivity (dr = 90:10, 92/43% ee) (entry 11). Finally, the effect of different inorganic bases was also investigated. NaHMDS and tBuOK had similar outcomes to KHMDS in terms of enantioselectivity as well as diastereoselectivity (entries 14 and 15). These results imply that the alkaline metal species and the conjugate acid of the inorganic base are not involved in the stereo determining step, namely, the carbon–carbon bond formation of the Michael addition reaction.17 On the other hand the use of LiHMDS failed to produce 4ba under the optimized conditions (entry 16) presumably because the Li cation is Lewis acidic rather than other alkaline metal cations to suppress the catalytic efficiency of (M)-1e.14a
:16, 79% ee]. Lowering the reaction temperature to −40 °C had a positive influence on the stereoselectivity (entry 8, dr = 87:13, 84% ee); however, further lowering the temperature to −60 °C had an adverse effect on the yield without any marked improvement of the stereoselectivity (entry 9). We then thoroughly explored the reaction conditions to enhance the yield and the stereoselectivity. To our delight, the use of an excess amount of achiral inorganic base KHMDS (15–30 mol%) and 10 mol% of (M)-1e·HCl promoted the reaction efficiently,14a resulting in the significant enhancement not only of the yield but also of the enantioselectivity (entries 10–12). It is worth mentioning that despite using an excess amount of the achiral inorganic base, the enantioselectivity was improved markedly. Meanwhile, lowering the reaction temperature from −60 °C to −78 °C had a negative effect on the yield (entry 13). On the basis of our results, the optimized conditions were established as follows: use of excess KHMDS (20 mol%) and 10 mol% of (M)-1e·HCl at −60 °C gave rise to corresponding cyanohydrin derivative 4ba in excellent yield (95%) with high stereoselectivity (dr = 90:10, 92/43% ee) (entry 11). Finally, the effect of different inorganic bases was also investigated. NaHMDS and tBuOK had similar outcomes to KHMDS in terms of enantioselectivity as well as diastereoselectivity (entries 14 and 15). These results imply that the alkaline metal species and the conjugate acid of the inorganic base are not involved in the stereo determining step, namely, the carbon–carbon bond formation of the Michael addition reaction.17 On the other hand the use of LiHMDS failed to produce 4ba under the optimized conditions (entry 16) presumably because the Li cation is Lewis acidic rather than other alkaline metal cations to suppress the catalytic efficiency of (M)-1e.14a
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | (M)-1·HX (mol%) | KHMDS (mol%) | 2 | Temp. (°C) | Yieldb (%) | Drb | Eec (%) |
| a Unless otherwise noted, all reactions were carried out using (M)-1·HX (5.0–5.5 μmol) with KHMDS (5.0–15 μmol), 2 (0.050 mmol), and 3a (0.055 mmol) in toluene (0.5 mL) at the indicated temperature for 24 h. b Yield and diastereomeric ratio were determined by crude 1H NMR spectroscopy using CH2Br2 as the internal standard. c Enantiomeric excess of 4 was determined by chiral-stationary-phase HPLC analysis. d For 6 h. e Enantiomeric excess of the major diastereomer. f For 48 h. g NaHMDS. h tBuOK. i LiHMDS. | |||||||
| 1d | 1a (11) | 10 | 2a | rt | 93 | 60:40 |
25/6 |
| 2d | 1b (11) | 10 | 2a | rt | 69 | 70:30 |
38/8 |
| 3d | 1c (11) | 10 | 2a | rt | 83 | 64:36 |
11/8 |
| 4d | 1d (11) | 10 | 2a | rt | 95 | 66:34 |
6/6 |
| 5d | 1e (11) | 10 | 2a | rt | 91 | 78:22 |
55/32 |
| 6 | 1e (11) | 10 | 2a | −20 | 75 | 84:16 |
69/26 |
| 7 | 1e (11) | 10 | 2b | −20 | 76 | 84:16 |
79e |
| 8 | 1e (11) | 10 | 2b | −40 | 89 | 87:13 |
84e |
| 9f | 1e (11) | 10 | 2b | −60 | 53 | 89:11 |
83e |
| 10 | 1e (10) | 15 | 2b | −60 | 88 | 90:10 |
90e |
| 11 | 1e (10) | 20 | 2b | −60 | 95 | 90:10 |
92/43 |
| 12 | 1e (10) | 30 | 2b | −60 | 85 | 90:10 |
90e |
| 13 | 1e (10) | 20 | 2b | −78 | 66 | 90:10 |
90e |
| 14 | 1e (10) | 20g | 2b | −60 | 96 | 87:13 |
90e |
| 15 | 1e (10) | 20h | 2b | −60 | 84 | 90:10 |
90e |
| 16 | 1e (10) | 20i | 2b | −60 | Trace | — | — |

The scope and limitations of this reaction were evaluated under the optimized conditions. Initially, the generality of the reaction was tested using a series of cyanohydrin ether derivatives 2 with diverse steric and electronic demands on the aryl ring R2 (Table 2). Substrate 2c having a para-tolyl group was well tolerated under the optimized reaction conditions, affording corresponding product 4ca in good yield with high enantioselectivity (entry 1). The use of substrate 2d, in which an electron-donating methoxy group is introduced at the para-position of the aryl group, retarded the reaction; an elevated temperature (−20 °C) was required to afford 4da in good yield, albeit with modest dr and ee values (entry 2). The reaction of substrates 2e and 2f having electron-withdrawing groups such as chloro and bromo proceeded smoothly to afford 4ea and 4fa, respectively, in excellent yields with fairly good enantioselectivities (entries 3 and 4). Substitution of a 4-trifluoromethyl group at the para-position, as in the case of substrate 2g, accelerated the reaction, albeit with moderate enantioselectivity (entry 5). Desirable results were obtained by the introduction of a methyl group and an electron-withdrawing chloro group at the meta-position of the aryl group, 2h and 2i, respectively (entries 6 and 7). In contrast, an ortho-tolyl group failed to afford any product even at room temperature (entry 8), presumably because of the steric congestion around the nucleophilic site. As expected, aliphatic cyanohydrin ether derivative 2k was not suitable even when the reaction was performed at the elevated reaction temperature because of its inherent low acidity.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 2 | R2 | Temp. (°C) | Time (h) | Yieldb (%) | Drc | Eed (%) |
| a Unless otherwise noted, all reactions were carried out using (M)-1·HX (5.0 μmol) with KHMDS (10.0 μmol), 2 (0.050 mmol), and 3a (0.055 mmol) in toluene (0.5 mL) at the indicated temperature. b Yield of the isolated products. c Diastereomeric ratio was determined by crude 1H NMR spectroscopy using CH2Br2 as the internal standard. d Enantiomeric excess of the major diastereomer was indicated and was determined by chiral-stationary-phase HPLC analysis. e No reaction. | |||||||
| 1 | 2c | 4-MeC6H4 | −60 | 48 | 72 | 85:15 |
88 |
| 2 | 2d | 4-MeOC6H4 | −20 | 48 | 81 | 71:29 |
73 |
| 3 | 2e | 4-ClC6H4 | −60 | 4 | 99 | 87:13 |
87 |
| 4 | 2f | 4-BrC6H4 | −60 | 2 | 90 | 85:15 |
83 |
| 5 | 2g | 4-CF3C6H4 | −60 | 1 | 88 | 62:38 |
73 |
| 6 | 2h | 3-MeC6H4 | −60 | 24 | 83 | 85:15 |
88 |
| 7 | 2i | 3-ClC6H4 | −60 | 2 | 89 | 87:13 |
85 |
| 8 | 2j | 2-MeC6H4 | rt | 24 | NRe | — | — |
| 9 | 2k | Cyclohexyl | rt | 24 | NRe | — | — |
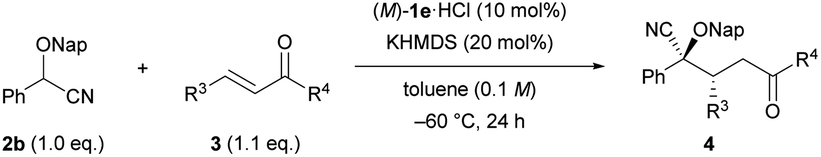
Next, we turned our attention to Michael acceptors in the reaction of 2-(naphthalen-2-ylmethoxy)-2-phenylacetonitrile (2b) (Table 3). First, the substituent effect on the aryl ring R4 adjacent to the keto group was examined. Substrates having an electron-donating substituent at the para-position of the aromatic ring (e.g., Me and OMe) (3b and 3c) were well suited for the reaction, affording products with high diastereo- and enantioselectivities (entries 1 and 2). However, the reaction of the substrate with the methoxy group was extremely sluggish compared with that of the methyl group containing substrate (entry 2 vs. 1), and 4bc was afforded in moderate yield (entry 2). Chalcone derivative 3d having an electron-withdrawing para-chloro group also participated in this reaction with equal efficiency to the reaction using 3b (entry 3 vs. 1), and the absolute configuration of 4bd was unambiguously determined to be (2S,3R) by single-crystal X-ray diffraction analysis.18 The reaction of 3e having a para-4-trifluoromethyl group afforded product 4be in moderate yield with a slight decrease in enantioselectivity (entry 4). The introduction of a methyl group at the meta-position of the aryl group, as in the case of 3f, led to a relatively high stereochemical outcome (entry 5). In contrast, a methyl group, which was introduced at the ortho-position of the aryl group, caused a negative influence on both dr and ee values (entry 6). It is presumed that the ortho-substituent broke the conjugation between the carbonyl group and the o-tolyl group, resulting in a marked conformational change around the site of the hydrogen bonding interaction between the catalyst and the carbonyl oxygen. Substrate 3h having a thiophene-2-yl group was compatible with the reaction conditions, affording product 4bh in good yield with moderate enantioselectivity (entry 7). Next, the effect of the aryl substituent R3 at the β-position was investigated. Substrates 3i–k having such electron-donating groups as methyl and methoxy groups as well as an electron-withdrawing chloro group at the para-position of the aryl group (entries 8–10) smoothly afforded corresponding products 4bi–4bk in high yields with good stereoselectivities. Unfortunately, substrate 3l having a 4-trifluoromethyl group resulted in a product with low diastereo- and enantioselectivities (entry 11). Substrate 3m having a meta-methyl group at the aryl ring underwent the reaction without any problem (entry 12). The introduction of a substituent at the ortho-position retarded the reaction (entry 13) and had a deleterious effect on both yield and enantioselectivity presumably because of the steric congestion around the reaction site. Further investigation of the Michael acceptors,19 such as β-alkyl substituted enones and cinnamaldehyde, instead of chalcone derivatives, displayed negative results. The use of β-alkyl substituted enones resulted in only low conversion of 2b even at room temperature. On the other hand, in the reaction of cinnamaldehyde, a mixture of 1,2- and 1,4-addition products was formed.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 3 | R3 | R4 | Yieldb (%) | Drc | Eed (%) |
| a Unless otherwise noted, all reactions were carried out using (M)-1·HX (5.0 μmol) with KHMDS (10.0 μmol), 2b (0.050 mmol), and 3 (0.055 mmol) in toluene (0.5 mL) at −60 °C for 24 h. b Yield of isolated products. c Diastereomeric ratio was determined by crude 1H NMR spectroscopy using CH2Br2 as the internal standard. d Enantiomeric excess of the major diastereomer was indicated and was determined by chiral-stationary-phase HPLC analysis. e For 72 h. f For 48 h. | ||||||
| 1 | 3b | Ph | 4-MeC6H4 | 75 | 96:4 |
92 |
| 2e | 3c | Ph | 4-MeOC6H4 | 55 | 91:9 |
89 |
| 3 | 3d | Ph | 4-ClC6H4 | 86 | 91:9 |
92 |
| 4e | 3e | Ph | 4-CF3C6H4 | 63 | 80:20 |
80 |
| 5 | 3f | Ph | 3-MeC6H4 | 80 | 88:12 |
89 |
| 6e | 3g | Ph | 2-MeC6H4 | 73 | 68:32 |
41 |
| 7 | 3h | Ph | Thiophen-2-yl | 78 | 83:17 |
79 |
| 8 | 3i | 4-MeC6H4 | Ph | 92 | 91:9 |
87 |
| 9e | 3j | 4-MeOC6H4 | Ph | 98 | 92:8 |
91 |
| 10 | 3k | 4-ClC6H4 | Ph | 91 | 82:18 |
86 |
| 11 | 3l | 4-CF3C6H4 | Ph | 93 | 64:36 |
76 |
| 12 | 3m | 3-MeC6H4 | Ph | 97 | 90:10 |
90 |
| 13f | 3n | 2-MeC6H4 | Ph | 73 | 93:7 |
68 |
In order to highlight the synthetic importance of the present enantioselective reaction, we further carried out the derivatization of the product (Scheme 2). Rather than forming a ketone through the retro cyanohydrin formation reaction (Fig. 1b, right bottom), our primary aim was to convert the cyano group into another functionality as an alternative to the conventional catalytic Stetter reactions.10 Towards this goal, the hydrolysis of the cyano group of Michael product 4ba was conducted by taking advantage of the characteristics of the multifunctionalized product formed in the present reaction. Enantioenriched Michael product 4ba (92% ee) was treated with platinum catalyst 5 at 80 °C in an ethanol/water mixture.20
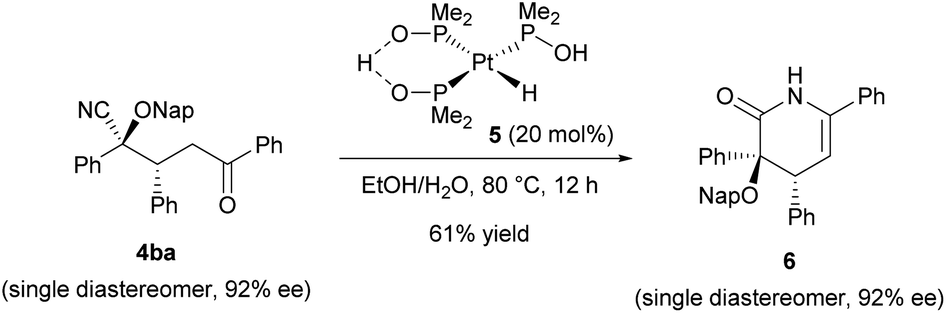 | ||
| Scheme 2 Derivatization of product 4. | ||
However, the corresponding amide derivative was obtained as a mixture with lactam 6 and recovered 4ba when the reaction time was short (for 5 h). Prolonging the reaction (for 12 h) resulted in the exclusive formation of lactam 6 in an acceptable yield without any loss of enantiomeric purity.
Conclusions
We have developed an enantioselective direct Michael addition reaction of cyanohydrin ether derivatives as less acidic pronucleophiles using chiral bis(guanidino)iminophosphoranes as the higher order organosuperbase catalyst. It is worth mentioning that the less acidic cyanohydrin ether derivatives were directly utilised for the first time in the catalytic enantioselective Michael addition reaction under transition metal free conditions. The corresponding Michael addition products were formed in high yields with moderate to high diastereo- and enantioselectivities in most cases. The usefulness of this methodology was demonstrated by the derivatization of the corresponding multifunctionalized product into a lactam derivative through hydrolysis followed by cyclo-condensation. Further mechanistic studies of the catalytic cycles in detail and the origin of the stereocontrol are ongoing in our laboratory.Data availability
The datasets supporting this article, i.e., additional experimental results, experimental procedures, characterization data, and crystallographic data for 4bd, have been uploaded as part of the ESI.†Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by a Grant-in-Aid for Scientific Research from the JSPS (No. JP16H06354 and JP22H04969).Notes and references
- (a) G. Stork and L. Maldonado, J. Am. Chem. Soc., 1971, 93, 5286–5287 CrossRef CAS; (b) J. D. Albright, Tetrahedron, 1983, 39, 3207–3233 CrossRef CAS.
- For selected reviews, see: (a) R. J. H. Gregory, Chem. Rev., 1999, 99, 3649–3682 CrossRef CAS PubMed; (b) J.-M. Brunel and I. P. Holmes, Angew. Chem., Int. Ed., 2004, 43, 2752–2778 CrossRef CAS PubMed; (c) W. Wang, X. Liu, L. Lin and X. Feng, Eur. J. Org. Chem., 2010, 2010, 4751–4769 CrossRef; (d) N. Kurono and T. Ohkuma, ACS Catal., 2016, 6, 989–1023 CrossRef CAS; (e) P. Bracco, H. Busch, J. von Langermann and U. Hanefeld, Org. Biomol. Chem., 2016, 14, 6375–6389 RSC.
- For selected reviews, see: (a) M. North, D. L. Usanov and C. Young, Chem. Rev., 2008, 108, 5146–5226 CrossRef CAS PubMed; (b) H.-u. H. Khan, K. I. Kureshy, S. H. R. Abdi, S. Agrawal and R. V. Jasra, Coord. Chem. Rev., 2008, 252, 593–623 CrossRef; (c) H. Pellissier, Adv. Synth. Catal., 2015, 357, 857–882 CrossRef CAS; (d) W.-B. Wu, J.-S. Yu and J. Zhou, ACS Catal., 2020, 10, 7668–7690 CrossRef CAS; (e) F.-X. Chen and X. Feng, Curr. Org. Synth., 2006, 3, 77–97 CrossRef CAS.
- For selected examples, see: (a) S.-K. Tian, R. Hong and L. Deng, J. Am. Chem. Soc., 2003, 125, 9900–9901 CrossRef CAS PubMed; (b) D. E. Fuerst and E. N. Jacobsen, J. Am. Chem. Soc., 2005, 127, 8964–8965 CrossRef CAS PubMed; (c) B. Qin, X. Liu, J. Sin, K. Zheng, H. Zhao and X. Feng, J. Org. Chem., 2007, 72, 2374–2378 CrossRef CAS PubMed; (d) Q.-W. Yu, L.-P. Wu, T.-C. Kang, J. Xie, F. Sha and X.-Y. Wu, Eur. J. Org. Chem., 2018, 2018, 3992–3996 CrossRef CAS; (e) H. Zhou, Y. Zhou, M. Leutzsch, Y. Li, C. K. De, D.-J. Cheng and B. List, Nature, 2022, 605, 84–90 CrossRef CAS PubMed; (f) S.-K. Tian and L. Deng, J. Am. Chem. Soc., 2001, 123, 6195–6196 CrossRef CAS PubMed; (g) Y. Ogura, M. Akakura, A. Sakakura and K. Ishihara, Angew. Chem., Int. Ed., 2013, 52, 8299–8303 CrossRef CAS PubMed.
- (a) T. Schrader, Chem.–Eur. J., 1997, 3, 1273–1282 CrossRef CAS; (b) S. Yoshida, T. Yamanaka, T. Miyake, Y. Moritani, H. Ohmizu and T. Iwasaki, Tetrahedron, 1997, 53, 9585–9598 CrossRef CAS; (c) T. Nanjo, X. Zhang, Y. Tokuhiro and Y. Takemoto, ACS Catal., 2019, 9, 10087–10092 CrossRef CAS.
- (a) D. A. Nicewicz, C. M. Yates and J. S. Johnson, Angew. Chem., Int. Ed., 2004, 43, 2652–2655 CrossRef CAS PubMed; (b) D. A. Nicewicz, C. M. Yates and J. S. Johnson, J. Org. Chem., 2004, 69, 6548–6555 CrossRef CAS PubMed; (c) X. Linghu, J. R. Potnick and J. S. Johnson, J. Am. Chem. Soc., 2004, 126, 3070–3071 CrossRef PubMed.
- S. E. Denmark and T. W. Wilson, Nat. Chem., 2010, 2, 937–943 CrossRef CAS PubMed.
- For organocatalytic SN2′ reactions of cyanohydrin ether derivatives, see: Z. Zhuang, F. Pan, J.-G. Fu, J.-M. Chen and W.-W. Liao, Org. Lett., 2011, 13, 6164–6167 CrossRef CAS PubMed.
- For enantioselective organocatalytic reactions of α-trifluoromethylthionitriles as an analogue of cyanohydrin derivatives, see: L. Xu, H. Wang, C. Zheng and G. Zhao, Adv. Synth. Catal., 2017, 359, 2942–2948 CrossRef CAS.
- For the enantioselective intermolecular Stetter reaction for the formation of 1,4-diketones and their derivatives, see: (a) D. Enders, J. Han and A. Henseler, Chem. Commun., 2008, 2008, 3989–3991 RSC; (b) D. Enders and J. Han, Synthesis, 2008, 2008, 3864–3868 CrossRef; (c) Q. Liu, S. Perreault and T. Rovis, J. Am. Chem. Soc., 2008, 130, 14066–14067 CrossRef CAS PubMed; (d) T. Jousseaume, N. E. Wurz and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 1410–1414 CrossRef CAS PubMed; (e) X. Fang, X. Chen, H. Lv and Y. R. Chi, Angew. Chem., Int. Ed., 2011, 50, 11782–11785 CrossRef CAS PubMed; (f) M. R. Khalkhali, M. M. D. Wilde and M. Gravel, Org. Lett., 2021, 23, 155–159 CrossRef PubMed.
- (a) K. S. Yang, A. E. Nibbs, Y. E. Türkmen and V. H. Rawal, J. Am. Chem. Soc., 2013, 135, 16050–16053 CrossRef CAS PubMed; (b) K. S. Yang and V. H. Rawal, J. Am. Chem. Soc., 2014, 136, 16148–16151 CrossRef CAS PubMed; (c) Y. Tokuhiro, K. Yoshikawa, S. Murayama, T. Nanjo and Y. Takemoto, ACS Catal., 2022, 12, 5292–5304 CrossRef CAS.
- (a) H. Krawczyk, M. Dzięgielewski, D. Deredas, A. Albrecht and Ł. Albrecht, Chem.–Eur. J., 2015, 21, 10268–10277 CrossRef CAS PubMed; (b) B. Teng, W. C. Lim and C.-H. Tan, Synlett, 2017, 28, 1272–1277 CrossRef CAS; (c) S. Dong, X. Feng and X. Liu, Chem. Soc. Rev., 2018, 47, 8525–8540 RSC; (d) Y.-H. Wang, Z.-Y. Cao, Q.-H. Li, G.-Q. Lin, J. Zhou and P. Tian, Angew. Chem., Int. Ed., 2020, 59, 8004–8014 CrossRef CAS PubMed; (e) A. Kondoh and M. Terada, Bull. Chem. Soc. Jpn., 2021, 94, 339–356 CrossRef CAS.
- For reviews on organobase catalysis, see: (a) C. Palomo, M. Oiarbide and R. López, Chem. Soc. Rev., 2009, 38, 632–653 RSC; (b) T. Ishikawa, Superbases for Organic Synthesis, John Wiley & Sons, Chippenham, 2009 CrossRef; (c) K. Nagasawa, Y. Sohtome, R. P. Singh, L. Deng, H. B. Jang, J. S. Oh and C. E. Song, Science of Synthesis: Asymmetric Organocatalysis 2: Brønsted Base and Acid Catalysis, and Additional Topics, ed. K. Maruoka, Thieme, Stuttgart, 2012, pp. 1–168 Search PubMed.
- (a) T. Takeda and M. Terada, J. Am. Chem. Soc., 2013, 135, 15306–15309 CrossRef CAS PubMed; (b) T. Takeda and M. Terada, Aust. J. Chem., 2014, 67, 1124–1128 CrossRef CAS; (c) A. Kondoh, M. Oishi, T. Takeda and M. Terada, Angew. Chem., Int. Ed., 2015, 54, 15836–15839 CrossRef CAS PubMed; (d) T. Takeda, A. Kondoh and M. Terada, Angew. Chem., Int. Ed., 2016, 55, 4734–4737 CrossRef CAS PubMed; (e) Q. Hu, A. Kondoh and M. Terada, Chem. Sci., 2018, 9, 4348–4351 RSC; (f) S. Das, Q. Hu, A. Kondoh and M. Terada, Angew. Chem., Int. Ed., 2021, 60, 1417–1422 CrossRef CAS PubMed.
- (a) A. Kondoh, M. Oishi, H. Tezuka and M. Terada, Angew. Chem., Int. Ed., 2020, 59, 7472–7477 CrossRef CAS PubMed; (b) A. Kondoh, S. Ishikawa and M. Terada, J. Am. Chem. Soc., 2020, 142, 3724–3728 CrossRef CAS PubMed.
- The introduction of other electron-withdrawing groups, such as benzoyl, benzenesulfonyl, and diphenylphosphinyl, as an alcohol protective group of the cyanohydrin resulted in a marked detrimental effect. In all cases only traces of the desired product were formed. On the other hand, the trimethylsilyl group was deprotected during the course of the reaction.
- These results also suggest that an excess amount of inorganic base does not involve in the stereo-determining step.
- CCDC 2076236 (4bd) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre (see the ESI† for details).
- The reaction of α,β-unsaturated esters did not afford the desired Michael addition products.
- (a) T. Ghaffar and A. W. Parkins, Tetrahedron Lett., 1995, 36, 8657–8660 CrossRef CAS; (b) For a selected review, see: R. García-Álvarez, P. Crochet and V. Cadierno, Green Chem., 2013, 15, 46–66 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental results, experimental procedures, and characterization data. CCDC 2076236. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc07065k |
| This journal is © The Royal Society of Chemistry 2023 |