
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoselective syntheses of 2-methyl-1,3-diol acetals via Re-catalyzed [1,3]-allylic alcohol transposition†
Jiaming
Liu
and
Ming
Chen
 *
*
Department of Chemistry and Biochemistry, Auburn University, Auburn, AL 36849, USA. E-mail: mzc0102@auburn.edu
First published on 6th July 2023
Abstract
Rhenium-catalyzed stereoselective transposition of allylic alcohols is reported. In the presence of 1 mol% of Re2O7, (E)- or (Z)-δ-hydroxymethyl-anti-homoallylic alcohols were converted into the acetals of 2-methyl-1,3-syn-diols with excellent diastereoselectivities. 1,3-syn-Diol acetals can also be synthesized from (E)-δ-hydroxymethyl-syn-homoallylic alcohols.
Introduction
Polyhydroxylated natural products, oxo polyene macrolide antibiotics for instance, have played an important role in modern drug discovery.1,2 Driven by a myriad of the biological activities, these natural products have garnered significant attention from the organic synthesis and chemical biology communities. One indispensible structural motif of these polyhydroxylated natural products is the stereodefined 2-methyl-1,3-diol (Fig. 1).3 Traditionally, these 2-methyl-1,3-diols are often assembled via a reiterative carbonyl crotylation approach4 or an aldol–reduction reaction sequence.5 These methods have been widely adopted in the total syntheses of polyhydroxylated natural products. Despite these advances, the development of catalytic methods that permit stereoselective syntheses of these 2-methyl-1,3-diol entities still remains an important objective in organic chemistry.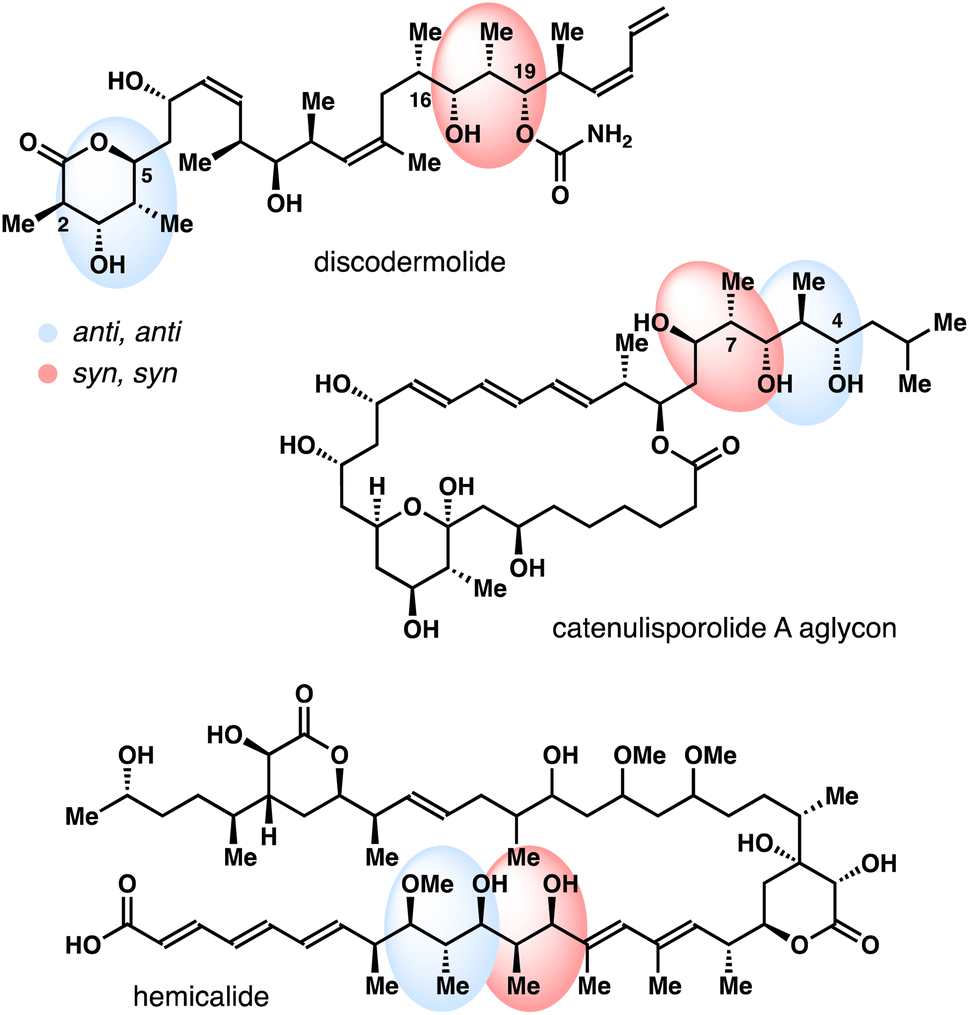 | ||
| Fig. 1 Selected natural products with 2-methyl-1,3-diols. | ||
Pioneered by the Osborn group, rhenium catalysts, such as O3ReOSiPh3, Re2O7, have been shown to promote [1,3]-allylic alcohol transposition.6,7 However, the regiochemical control of these reactions is generally poor, resulting in a thermodynamic mixture of allylic alcohols (e.g., A and B, Scheme 1). More recently, several strategies have been developed to address this limitation. For instance, the reaction can be rendered regioselective through capitalizing on alkene conjugation or steric effect.8 High regio-control can also be accomplished through capturing the alcohol group with a trapping agent, thereby shifting the equilibrium toward the desired direction.9
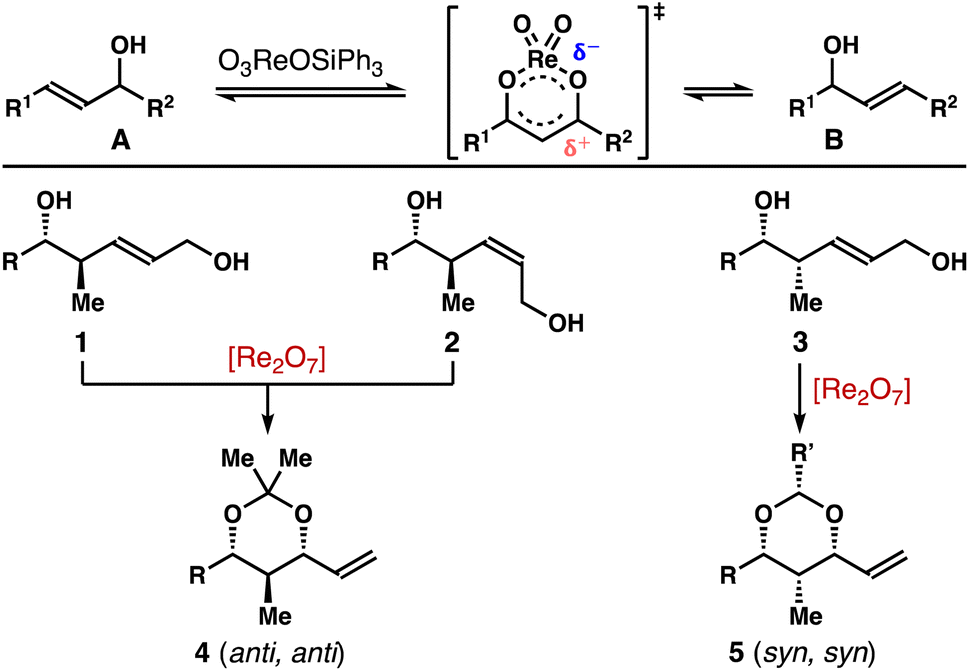 | ||
| Scheme 1 Transposition of allylic alcohols. | ||
In the context of our program on asymmetric synthesis,10,11 we recently reported enantio- and stereoselective syntheses of δ-hydroxymethyl homoallylic alcohols 1, 2 and 3.12 As one venue to explore the synthetic utilities of these molecules, we were intrigued whether the allylic alcohol moiety in 1–3 can undergo [1,3]-allylic transposition to generate stereodefined 2-methyl-1,3-diols (e.g., 4, 5). Taking the inspiration from prior studies,8,9 we report herein Re-catalyzed highly stereoselective allylic transposition of alcohols 1–3 (Scheme 1). With 1 mol% Re2O7 as the catalyst, the operationally simple protocol permits stereoselective syntheses of 2-methyl-1,3-syn-diol acetals 4 (anti, anti) and 5 (syn, syn) in high yields from alcohols 1–3.13
Results and discussion
In initial experiments, alcohol 1a was treated with 1 mol% of Re2O7 and 2,2-dimethoxypropane in CH2Cl2. The reaction progress was monitored by the 1H NMR spectroscopy. We discovered that, within 10 minutes, about 80% of alcohol 1a was converted into a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5:6 mixture of diols 6a, 7a and acetal 4a. Formation of acetal 8a was not detected (panel a, Scheme 2). Full conversion of 1a was achieved in 1 h, affording a 1:0.5:7.5 mixture of 6a, 7a and 4a. After 6 h at ambient temperature, acetal 4a was isolated as the only product in 95% yield. Similar results were obtained when Z-isomer 2a was used. Next, we treated alcohol 1a with 1 mol% of Re2O7 in the absence of 2,2-dimethoxypropane (panel b, Scheme 2). After 12 h at ambient temperature, a 1:1.7 mixture of diols 6a and 7a was obtained in 41% yield, and 52% of alcohol 1a was recovered. In the case of alcohol 2a, a 1:1.6 mixture of 6a, 7a was isolated in 15% yield, and a 1:2 mixture of diols 1a, 2a was also obtained in 50% yield after 12 h, together with some unidentifiable by-products. We also treated a mixture of acetals 4a and 8a with Re2O7 catalyst (panel c, Scheme 2). After 6 h at ambient temperature, acetal 8a completely decomposed and 4a remained unchanged under the reaction conditions. Finally, when single isomer 1,3-anti-diol 7a was exposed to the reaction conditions, it was fully converted into 1,3-syn-acetal 4a. We did not observe the formation of 1,3-anti-acetal 8a. The structures of acetals 4a and 8a were assigned based on the coupling constant analyses and nOe studies. While 1,3-syn-acetal 4a adopts a chair conformation, 1,3-anti-isomer 8a prefers a twist-boat conformation, as the corresponding chair conformation is disfavoured due to the 1,3-diaxial interactions (shown in red in 8a). On the basis of these data, it is apparent that, for alcohol 1a, the equilibrium among 1a and diols 6a, 7a was established under the catalytic conditions (panel b, Scheme 2). And 1,3-syn-diol 6a reacted irreversibly with 2,2-dimethoxypropane to give acetal 4a. 1,3-anti-Diol 7a did not react to form acetonide 8a under the conditions, presumably owing to the higher energy of a twist-boat-like transition state. Diol 7a was funnelled into 6avia1a through the reversible Re-catalyzed [1,3]-allylic transposition, eventually affording acetal 4a as the sole product.
:1.5:6 mixture of diols 6a, 7a and acetal 4a. Formation of acetal 8a was not detected (panel a, Scheme 2). Full conversion of 1a was achieved in 1 h, affording a 1:0.5:7.5 mixture of 6a, 7a and 4a. After 6 h at ambient temperature, acetal 4a was isolated as the only product in 95% yield. Similar results were obtained when Z-isomer 2a was used. Next, we treated alcohol 1a with 1 mol% of Re2O7 in the absence of 2,2-dimethoxypropane (panel b, Scheme 2). After 12 h at ambient temperature, a 1:1.7 mixture of diols 6a and 7a was obtained in 41% yield, and 52% of alcohol 1a was recovered. In the case of alcohol 2a, a 1:1.6 mixture of 6a, 7a was isolated in 15% yield, and a 1:2 mixture of diols 1a, 2a was also obtained in 50% yield after 12 h, together with some unidentifiable by-products. We also treated a mixture of acetals 4a and 8a with Re2O7 catalyst (panel c, Scheme 2). After 6 h at ambient temperature, acetal 8a completely decomposed and 4a remained unchanged under the reaction conditions. Finally, when single isomer 1,3-anti-diol 7a was exposed to the reaction conditions, it was fully converted into 1,3-syn-acetal 4a. We did not observe the formation of 1,3-anti-acetal 8a. The structures of acetals 4a and 8a were assigned based on the coupling constant analyses and nOe studies. While 1,3-syn-acetal 4a adopts a chair conformation, 1,3-anti-isomer 8a prefers a twist-boat conformation, as the corresponding chair conformation is disfavoured due to the 1,3-diaxial interactions (shown in red in 8a). On the basis of these data, it is apparent that, for alcohol 1a, the equilibrium among 1a and diols 6a, 7a was established under the catalytic conditions (panel b, Scheme 2). And 1,3-syn-diol 6a reacted irreversibly with 2,2-dimethoxypropane to give acetal 4a. 1,3-anti-Diol 7a did not react to form acetonide 8a under the conditions, presumably owing to the higher energy of a twist-boat-like transition state. Diol 7a was funnelled into 6avia1a through the reversible Re-catalyzed [1,3]-allylic transposition, eventually affording acetal 4a as the sole product.
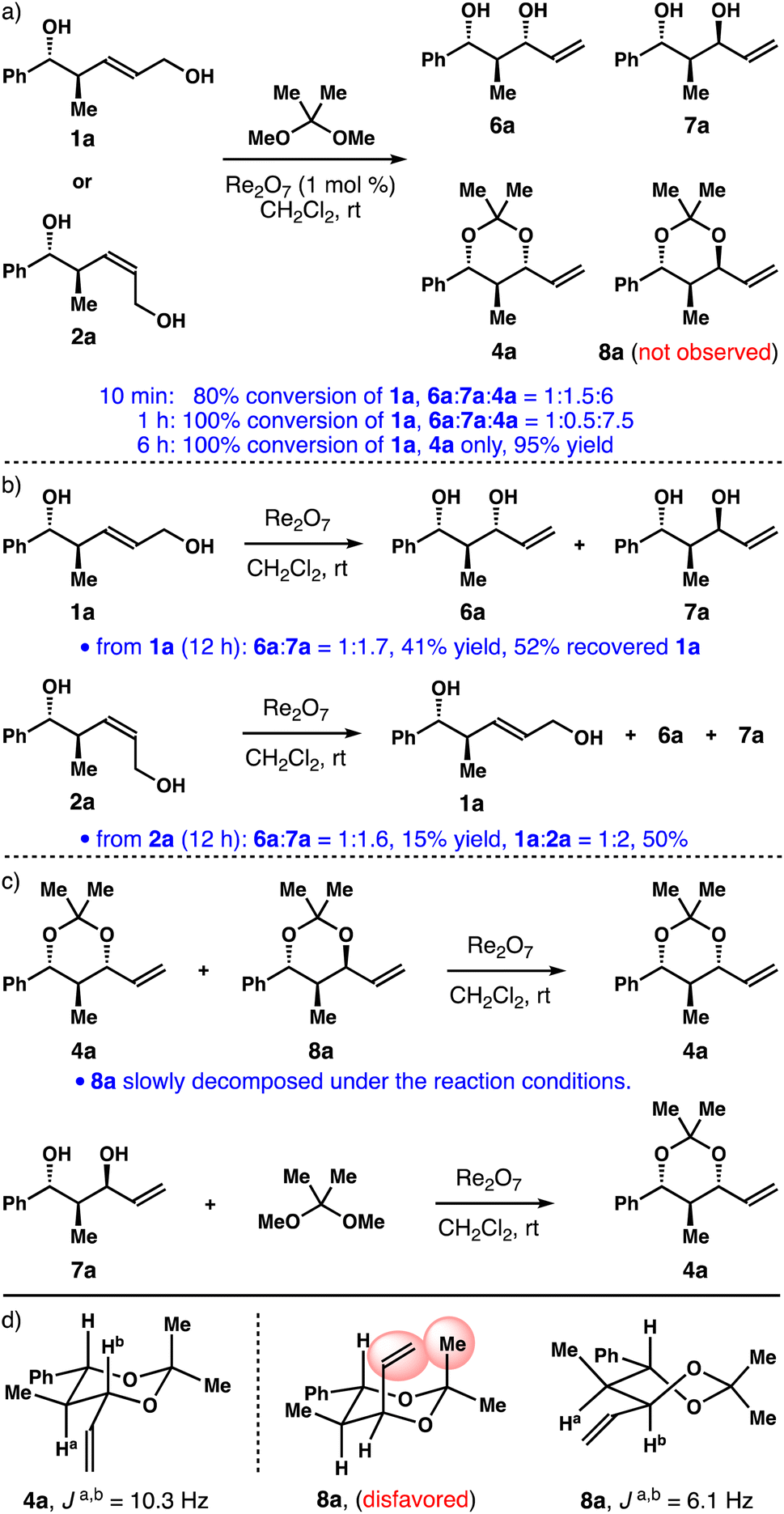 | ||
| Scheme 2 Initial studies on allylic alcohol transposition. | ||
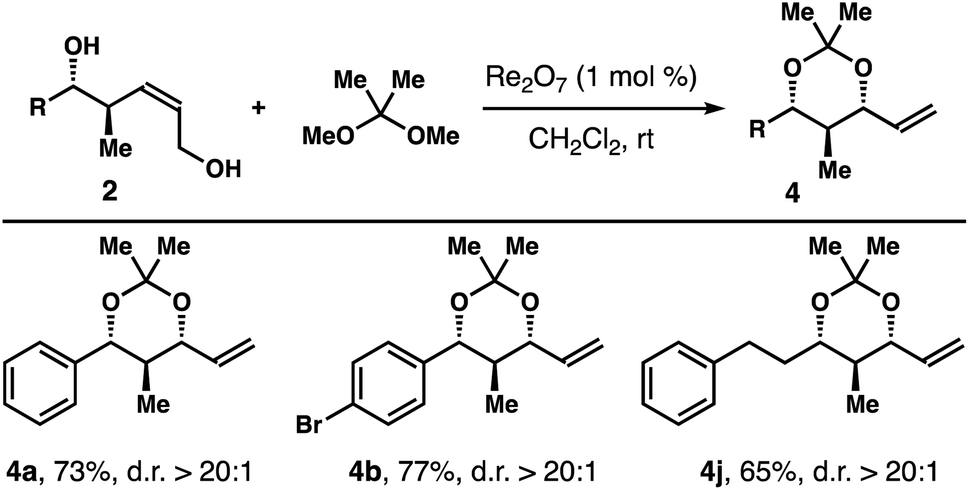
The operationally simple protocol was extended to a variety of diols 1 and the results are summarized in Table 1. Except diols with an electron-donating group at the para-position of the arene, 1h for example, the reaction worked quite well with other diols regardless the substitution patterns and steric environments on the arenes. And acetals 4a-g were obtained in 72–96% yields with excellent 1,3-syn-selectivities. The reaction tolerates an alkyne group, and acetal 4i was isolated in 66% yield with >20:1 diastereoselectivity. Diols with an aliphatic side chain also reacted under the reaction conditions to give 4j-n in 66–88% yield with >20:1 1,3-syn-selectivities. Encouraged by these results, we tested the protocol with structurally more elaborated diol substrates. Gratifyingly, the reactions proceeded smoothly to give acetals 4o–r in 64–82% yields with excellent diastereoselectivities.
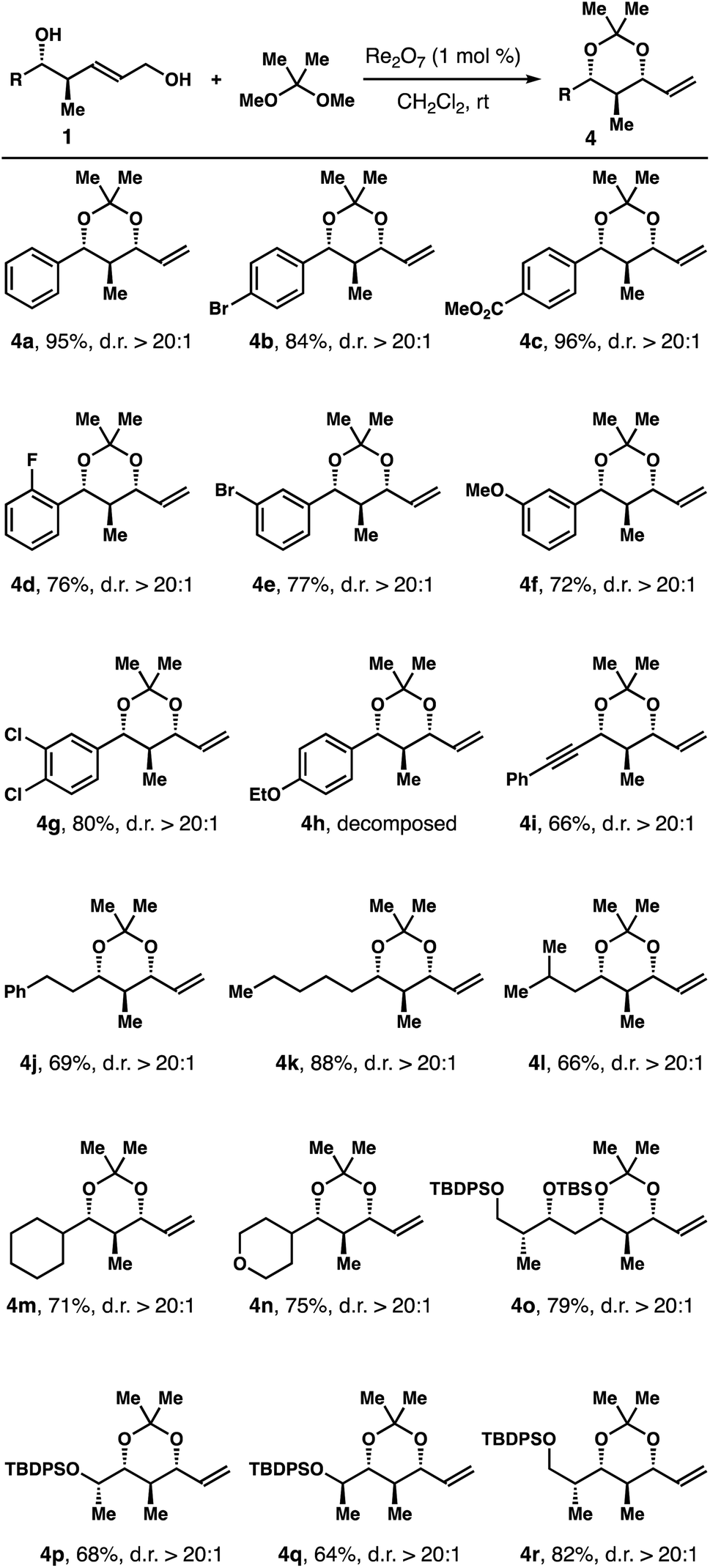
Table 2 summarizes the results of reactions with a few (Z)-alcohols 2 under the same conditions. As anticipated, 1,3-syn-isomers 4a, 4b, and 4j were obtained in 65–77% yields with >20:1 diastereoselectivities.
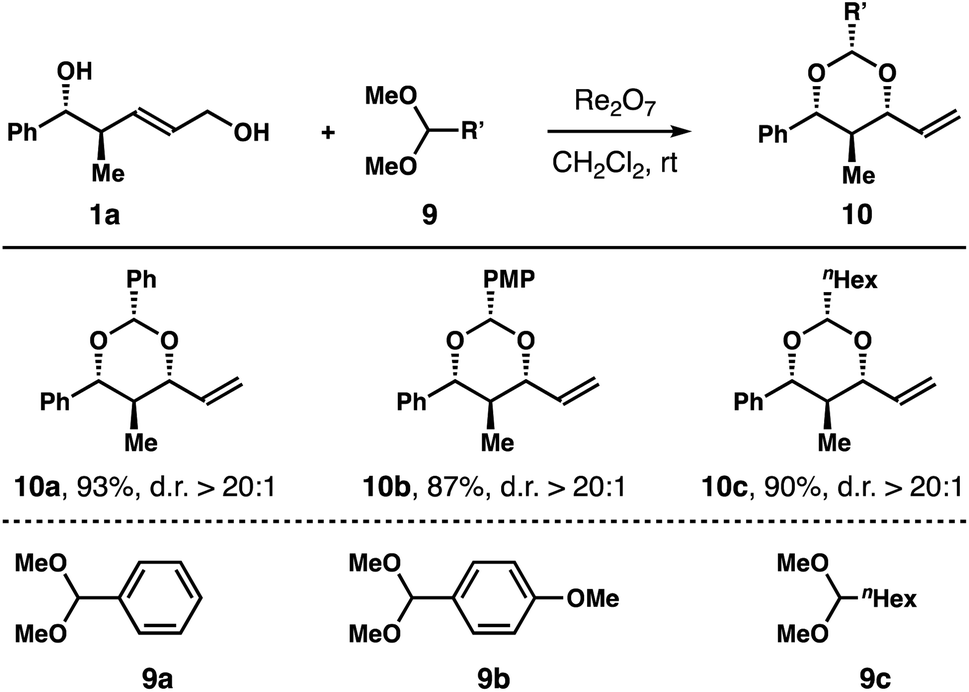
In addition to 2,2-dimethoxypropane, we also conducted reactions of diol 1a with dimethyl acetals 9a–c. As shown in Table 3, the reactions afforded products 10a–c in 87–93% yields with excellent 1,3-syn-selectivities.
Next, we considered the reactions with (E)-syn-homoallylic alcohols 3 under the standard reaction conditions. Intriguingly, when R is a phenyl group in 3, the reaction mostly led to the decomposition of the starting diol, only a small amount of acetal product was obtained (∼20%). For substrate 3a bearing an aliphatic side chain (R = PhCH2CH2), 70% of starting diol 3a was consumed in 1.5 h. And a 1:1 mixture of diols 11a and 12a was obtained in 18% yield, together with a 9:1 mixture of acetals 13a and 14a in 45% isolated yield (panel a, Scheme 3). When the reaction was conducted at ambient temperature for 12 h, 3a was fully consumed, and acetal 13a was isolated in 73% yield with 15:1 syn-selectivity (panel b, Scheme 3). When diol 3a was subjected to the reaction conditions in the absence of 2,2-dimethoxypropane for 12 h, a 1:1 mixture of 11a and 12a was obtained in 45% yield, and remaining 3a was recovered in 50% yield (panel c, Scheme 3). On the basis of these data, we conclude that an analogous reaction pathway to anti-diols 1 is operational for the syn counterpart 3a. Similar to the cases of 4a and 8a in Scheme 2, 1,3-syn-isomer 13a adopts a chair conformation and 1,3-anti-isomer 14a prefers a twist-boat conformation (panel d, Scheme 3).
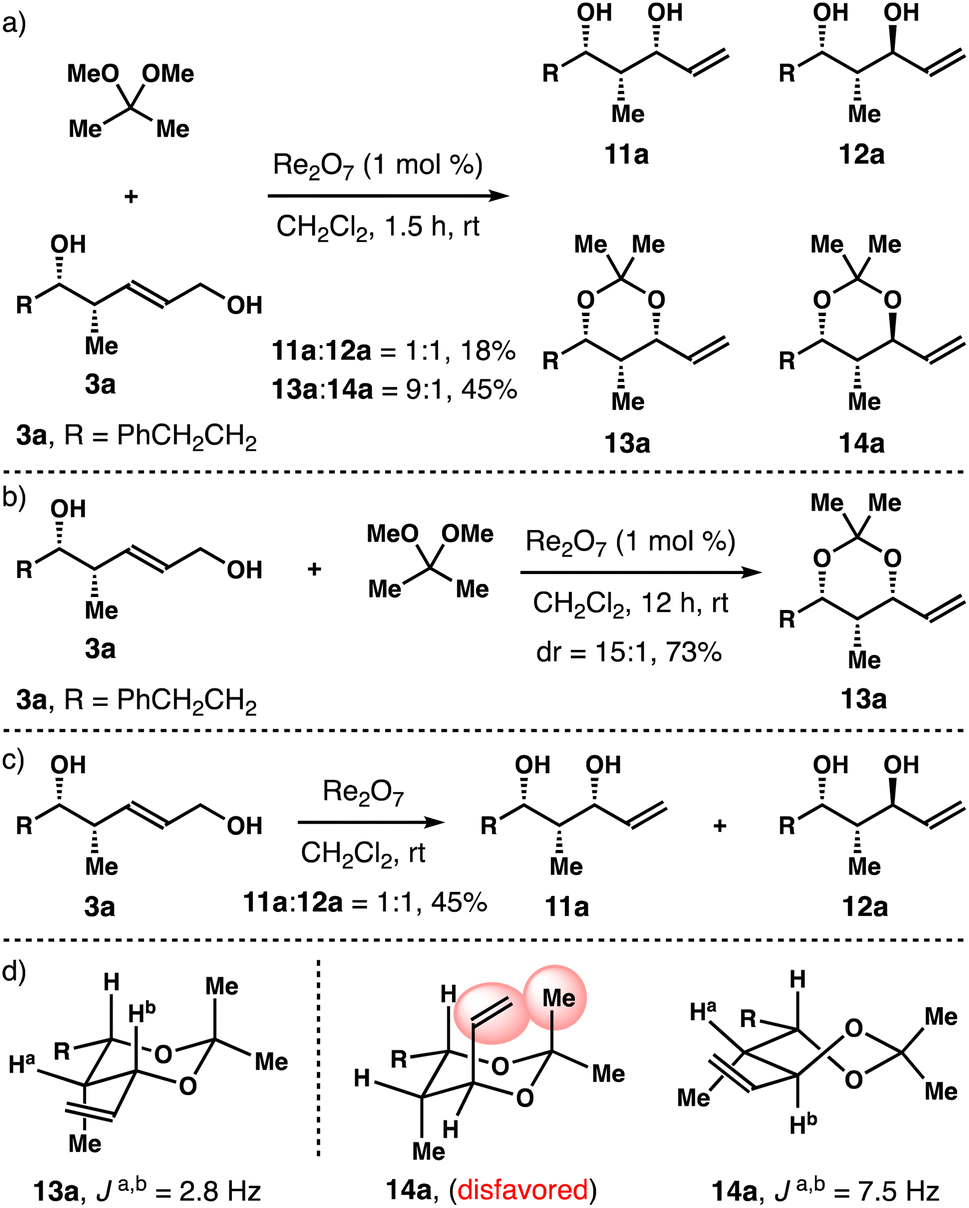 | ||
| Scheme 3 Rhenium-catalyzed stereoselective allylic alcohol transposition studies with diol 3a. | ||
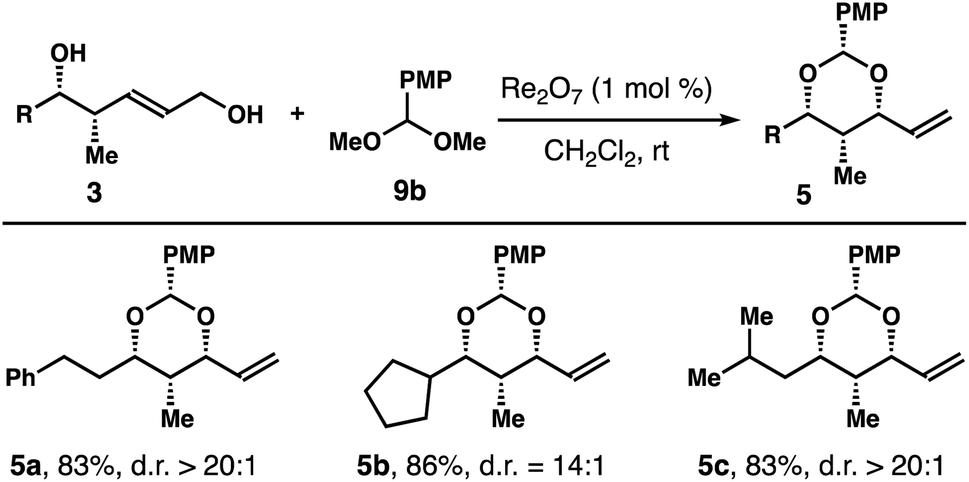
Table 4 summarizes the results of reactions with several alcohols 3 with an aliphatic side chain. In these experiments, acetal 9b was utilized to minimize the volatility of the reaction products. Under the standard reaction conditions, acetals 5a–c were isolated in 83–86% yields with high 1,3-syn-selectivities.
A few structurally more complex diols 15a–c were subjected to the reaction conditions in the presence of 9b (Scheme 4). The reaction with 15a produced acetal 16 in 72% yield with >20:1 diastereoselectivity. For substrate 15b, the product was inseparable from the aldehyde by-product derived from 9b. After treating the reaction mixture with TBAF, alcohol 17 was obtained in 62% yield with 18:1 dr. For substrate 15c, partial deprotection of the silyl ether was observed during the course of the reaction. After prolonged reaction time (24 h) with 3 equiv. of 9b, bis-acetal 18 was isolated in 70% yield with >20:1 diastereoselectivity.
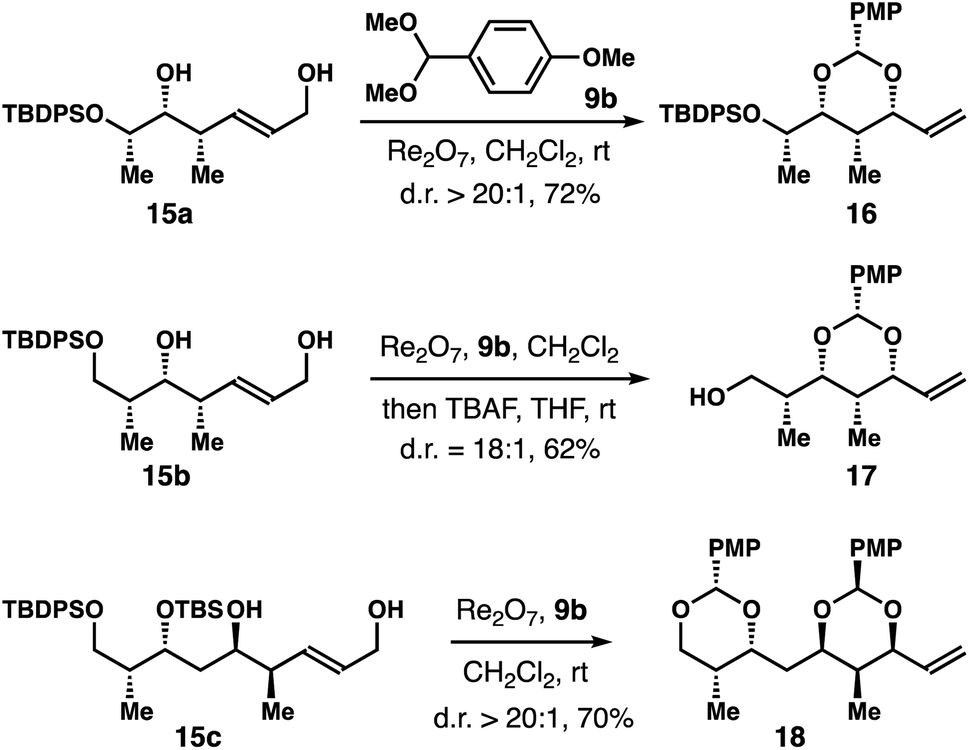 | ||
| Scheme 4 Rhenium-catalyzed stereoselective allylic alcohol transposition with structurally complex diols 15. | ||
The results from stereochemically rich diol substrates in Table 1 (4o–r) and Scheme 4 are worth noting, as the products could be utilized as valuable intermediates for the synthesis of polyhydroxylated natural products. For example, the relative configuration of the four contiguous stereocenters in acetal 4r nicely matches the C2–C5 fragment of discodermolide and the C4–C7 fragment of catenulisporolide A aglycon (Fig. 1). The relative configuration of the four contiguous stereocenters in acetal ent-17 matches the C16–C19 fragment of discodermolide and the C2–C5 fragment of erythronolide B.
Further product derivatization studies are summarized in Scheme 5. Acetal 4a underwent an alkene metathesis reaction with acrylate 19 to furnish α,β-unsaturated ester 20 in 72% yield with >20:1 E-selectivity. Hydroboration–oxidation of the alkene group of 4a gave primary alcohol 21 in 71% yield. Dess–Martin oxidation of alcohol 21 afforded aldehyde 22 in 68% yield.14 Aldehyde 22 could participate in a variety of transformations. For instance, Wittig olefination of aldehyde 22 with reagent 23 delivered α,β-unsaturated ester 24 in 89% yield with >20:1 E-selectivity.
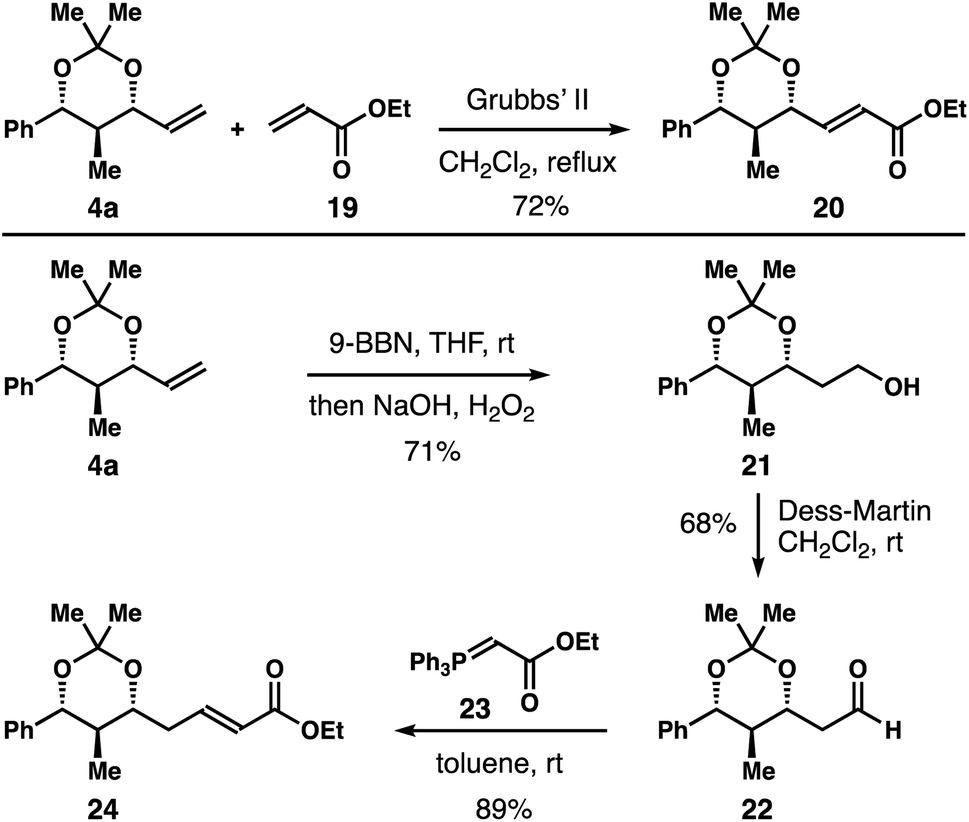 | ||
| Scheme 5 Product derivatization. | ||
Conclusions
In summary, we developed a Re-catalyzed allylic alcohol transposition protocol to convert diols 1–3 into the acetals of 2-methyl-1,3-syn-diols with excellent diastereoselectivities. In addition to 2,2-dimethoxypropane, several other acetals can also be utilized in the reactions. Importantly, reactions with a variety of structurally complex allylic alcohols worked well under the standard conditions, affording acetal intermediates that are synthetically valuable for the construction of polyhydroxylated natural products.Data availability
All the data have been included in the ESI.†Author contributions
The concept of the manuscript is conceived by Ming Chen, and Jiaming Liu conducted all the experiments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support provided by the National Science Foundation (CAREER Award CHE-2042353) is gratefully acknowledged.References
- (a) S. D. Rychnovsky, Chem. Rev., 1995, 95, 2021 CrossRef CAS; (b) K. S. Madden, F. A. Mosa and A. Whiting, Org. Biomol. Chem., 2014, 12, 7877 RSC.
- (a) H. C. Kwon, C. A. Kauffman, P. R. Jensen and W. Fenical, J. Am. Chem. Soc., 2006, 128, 1622 CrossRef CAS PubMed; (b) H. C. Kwon, C. A. Kauffman, P. R. Jensen and W. Fenical, J. Org. Chem., 2009, 74, 675 CrossRef CAS PubMed; (c) Y. Morishita, H. Zhang, T. Taniguchi, K. Mori and T. Asai, Org. Lett., 2019, 21, 4788 CrossRef CAS PubMed; (d) P. Shi, Y. Li, J. Zhu, Y. Shen and H. Wang, J. Nat. Prod., 2021, 84, 1924 CrossRef CAS PubMed.
- (a) C. Schneider, Angew. Chem., Int. Ed., 1998, 37, 1375 CrossRef CAS; (b) S. E. Bode, M. Wolberg and M. Müller, Synthesis, 2006, 2006, 557 CrossRef.
- (a) H. Lachance and D. G. Hall, Org. React., 2009, 73, 1 Search PubMed; (b) M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2011, 111, 7774 CrossRef CAS PubMed; (c) M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2013, 113, 5595 CrossRef CAS PubMed.
- (a) E. M. Carreira, in Modern Carbonyl Chemistry, ed. J. Otera, Wiley-VCH, Weinheim, 2000, p. 227 Search PubMed; (b) B. Schetter and R. Mahrwald, Angew. Chem., Int. Ed., 2006, 45, 7506 CrossRef CAS PubMed.
- I. Volchkov and D. Lee, Chem. Soc. Rev., 2014, 43, 4381 RSC.
- S. Bellemin-Laponnaz, H. Gisie, J. P. Le Ny and J. A. Osborn, Angew. Chem., Int. Ed. Engl., 1997, 36, 976 CrossRef CAS.
- (a) C. Morrill and R. H. Grubbs, J. Am. Chem. Soc., 2005, 127, 2842 CrossRef CAS PubMed; (b) C. Morrill, G. L. Beutner and R. H. Grubbs, J. Org. Chem., 2006, 71, 7813 CrossRef CAS PubMed.
- (a) E. C. Hansen and D. Lee, J. Am. Chem. Soc., 2006, 128, 8142 CrossRef CAS PubMed; (b) A. T. Herrmann, T. Saito, C. E. Stivala, J. Tom and A. Zakarian, J. Am. Chem. Soc., 2010, 132, 5962 CrossRef CAS PubMed; (c) I. Volchkov, S. Park and D. Lee, Org. Lett., 2011, 13, 3530 CrossRef CAS PubMed; (d) Y. Xie and P. E. Floreancig, Chem. Sci., 2011, 2, 2423 RSC; (e) Y. Xie and P. E. Floreancig, Angew. Chem., Int. Ed., 2013, 52, 625 CrossRef CAS PubMed; (f) Y. Xie and P. E. Floreancig, Angew. Chem., Int. Ed., 2014, 53, 4926 CrossRef CAS PubMed; (g) T. M. Rohrs, Q. Qin and P. E. Floreancig, Angew. Chem., Int. Ed., 2017, 56, 10900 CrossRef CAS PubMed; (h) A. H. Asari and P. E. Floreancig, Angew. Chem., Int. Ed., 2020, 59, 6622 CrossRef CAS PubMed; (i) J.-M. I. A. Lawrence and P. E. Floreancig, Org. Lett., 2020, 22, 9513 CrossRef CAS PubMed.
- (a) M. Wang, S. Gao and M. Chen, Org. Lett., 2019, 21, 2151 CrossRef CAS PubMed; (b) S. Gao, J. Chen and M. Chen, Chem. Sci., 2019, 10, 3637 RSC; (c) S. Gao and M. Chen, Chem. Sci., 2019, 10, 7554 RSC; (d) S. Gao and M. Chen, Chem. Commun., 2019, 55, 11199 RSC; (e) J. Chen, S. Gao, J. D. Gorden and M. Chen, Org. Lett., 2019, 21, 4638 CrossRef CAS PubMed; (f) J. Chen, S. Gao and M. Chen, Org. Lett., 2019, 21, 9893 CrossRef CAS PubMed; (g) J. Liu and M. Chen, Chem. Commun., 2021, 57, 10799 RSC; (h) J. Liu, B. Su and M. Chen, Org. Lett., 2021, 23, 6035 CrossRef CAS PubMed; (i) Z. Zhang, J. Liu, S. Gao, B. Su and M. Chen, J. Org. Chem., 2023, 88, 3288 CrossRef CAS PubMed.
- (a) M. Wang, S. Khan, E. Miliordos and M. Chen, Org. Lett., 2018, 20, 3810 CrossRef CAS PubMed; (b) M. Wang, S. Khan, E. Miliordos and M. Chen, Adv. Synth. Catal., 2018, 360, 4634 CrossRef CAS; (c) S. Gao and M. Chen, Org. Lett., 2018, 20, 6174 CrossRef CAS PubMed; (d) S. Gao and M. Chen, Org. Lett., 2020, 22, 400 CrossRef CAS PubMed; (e) J. Chen and M. Chen, Org. Lett., 2020, 22, 7321 CrossRef CAS PubMed; (f) J. Liu and M. Chen, Org. Lett., 2020, 22, 8967 CrossRef CAS PubMed; (g) S. Gao, M. Duan, K. N. Houk and M. Chen, Angew. Chem., Int. Ed., 2020, 59, 10540 CrossRef CAS PubMed; (h) S. Gao, M. Duan, Q. Shao, K. N. Houk and M. Chen, J. Am. Chem. Soc., 2020, 142, 18355 CrossRef CAS PubMed; (i) J. Chen, E. Miliordos and M. Chen, Angew. Chem., Int. Ed., 2021, 60, 840 CrossRef CAS PubMed; (j) S. Gao, M. Duan, L. R. Andreola, P. Yu, S. E. Wheeler, K. N. Houk and M. Chen, Angew. Chem., Int. Ed., 2022, 61, e202208908 CAS.
- (a) S. Gao, M. Duan, J. Liu, P. Yu, K. N. Houk and M. Chen, Angew. Chem., Int. Ed., 2021, 60, 24096 CrossRef CAS PubMed; (b) S. Gao, J. Liu and M. Chen, Chem. Sci., 2021, 12, 13398 RSC; (c) J. Liu, S. Gao and M. Chen, Org. Lett., 2021, 23, 7808 CrossRef CAS PubMed; (d) J. Liu, S. Gao and M. Chen, Org. Lett., 2021, 23, 9451 CrossRef CAS PubMed.
- H. Y. Cho and J. P. Morken, J. Am. Chem. Soc., 2010, 132, 7576 CrossRef CAS PubMed.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc07059f |
| This journal is © The Royal Society of Chemistry 2023 |