
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMembrane-tethered activation design of a photosensitizer boosts systemic antitumor immunity via pyroptosis†
Pei
Lu
,
Xianjun
Liu
 ,
Xia
Chu
,
Fenglin
Wang
* and
Jian-Hui
Jiang
,
Xia
Chu
,
Fenglin
Wang
* and
Jian-Hui
Jiang
State Key Laboratory of Chemo/Bio-Sensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha, 410082, P. R. China. E-mail: fengliw@hnu.edu.cn
First published on 8th February 2023
Abstract
Pyroptotic immunogenic cell death presents an emerging targeting pathway for cancer immunotherapy. We report a novel membrane-tethered activation design of a photosensitizer (PS) that boosts systemic anti-tumor immunity to primary and distant tumors via pyroptosis induction. The membrane-tethered PS is designed by installing a new phenylbenzopyrylium PS with zwitterionic lipid anchors and a target-cleavable caging moiety. This design affords excellent membrane tethering and enzymatic activation of the PS, exerting specific phototoxicity to cancer cells and inducing effective pyroptosis. Our design demonstrates prolonged circulation, long-lasting fluorogenic imaging and persistent photodynamic therapy of immunogenic ‘cold’ tumors in vivo, eliciting potent immunity toward local and abscopal tumors via promoted maturation of dendritic cells and recruitment of cytotoxic T lymphocytes. This design affords a promising approach for enhancing systemic antitumor immunity for cancer immunotherapy.
Introduction
Immunotherapies have revolutionized the field of cancer therapy by orchestrating the host's immune system to fight against tumors. Several immunotherapies such as immune checkpoint blockade and adoptive T-cell therapy have achieved great success in the clinic.1,2 However, their efficacy is compromised by the hostile tumor microenvironments with low immunogenicity, limited infiltration of immune cells and immuno-suppressiveness.3,4 Immunogenic cell death (ICD), which can lead to release of intracellular neoantigens and damage-associated molecular patterns (DAMPs), creates the potential for reprogramming the tumor immune microenvironments.5 By activating the maturation of antigen-presenting cells and priming the cytotoxic T lymphocytes, ICD can elicit potent antitumor immunity for cancer ablation. Hence, targeting ICD pathways has become an emerging paradigm for developing effective strategies in cancer immunotherapy.Pyroptosis is a newly identified mode of ICD that is triggered by caspase-mediated cleavage of gasdermins.6,7 Cleavage of gasdermins releases their N terminal domains that translocate into the cell membrane to form pores, inducing cell swelling and membrane rupture with rapid release of intracellular contents and proinflammatory cytokines. Accumulating evidence shows that pyroptosis-mediated inflammation is able to evoke substantial antitumor immunity,8–10 which creates a new dimension for the development of ICD inducers in cancer immunotherapy. However, most of the current pyroptotic inducers are chemotherapeutic drugs that are susceptible to an increased risk of drug resistance and systemic side effects.11 There is an urgent need to develop highly specific therapeutics to induce tumor-selective pyroptotic pathways.
Photodynamic therapy (PDT) is a therapeutic modality for cancer treatment with high spatiotemporal selectivity, minimal invasiveness and low side effects.12 Either administered alone or in a combination therapy,13,14 PDT is demonstrated to stimulate effective pyroptosis of cancer cells, suggesting its potential for pyroptosis-inducing cancer immunotherapy. Very recently, novel membrane-anchoring designs of photosensitizers (PSs) have been proposed for pyroptosis induction.15,16 The first work along this direction was reported using an aggregation-induced emission PS with tunable membrane anchorage endowed by several cationic pendant groups.15 An alternative design is to utilize a membrane protein-targeting metal-complex PS that renders facile membrane anchoring.16 However, these membrane anchoring designs rely on poly-cationic groups or poorly hydrophilic moieties, which may exhibit increased non-specific adsorption and toxicity with a concurrent high clearance rate. Besides, the short-wavelength excitation and non-activatable characteristics of these PSs can compromise their efficacy due to low penetration depth and collateral phototoxicity.
Here we report a novel membrane-tethered activation design of a pyroptosis-inducing PS, which is tethered to the cell membrane using a zwitterionic lipid anchor and activated specifically by a membrane-localized enzyme overexpressed in cancer cells (Fig. 1a). The membrane-tethered activation design ensures highly specific localization of the activated PSs on the cancer cell membrane, enabling membrane-proximal generation of reactive oxygen species (ROS) and efficient induction of pyroptosis with minimal collateral phototoxicity. Elaborate engineering renders a bi-pendant zwitterionic lipid anchor for the PS (Fig. 1b), which confers excellent membrane localization with minimal penetration. The activatability of the PS by a membrane-localized enzyme exerts highly specific phototoxicity with a self-indicative fluorogenic signal toward cancer cells (Fig. 1c). Mechanistic studies reveal that the membrane-tethered activatable PS induces rapid and specific immunogenic death of cancer cells via the pyroptotic pathway. Our design demonstrates prolonged circulation, long-lasting fluorogenic imaging (>7 days) and persistent PDT of immunogenic cold tumors in vivo. Moreover, this design not only enables efficacious ablation of primary tumors, but also elicits substantial systemic immunity toward distant tumors by promoting maturation of dendritic cells, recruitment of cytotoxic T lymphocytes and production of inflammatory cytokines, converting immune ‘cold’ tumor ‘hot’. Our design thus affords a promising approach for enhancing systemic antitumor immunity and developing new combination cancer immunotherapy.
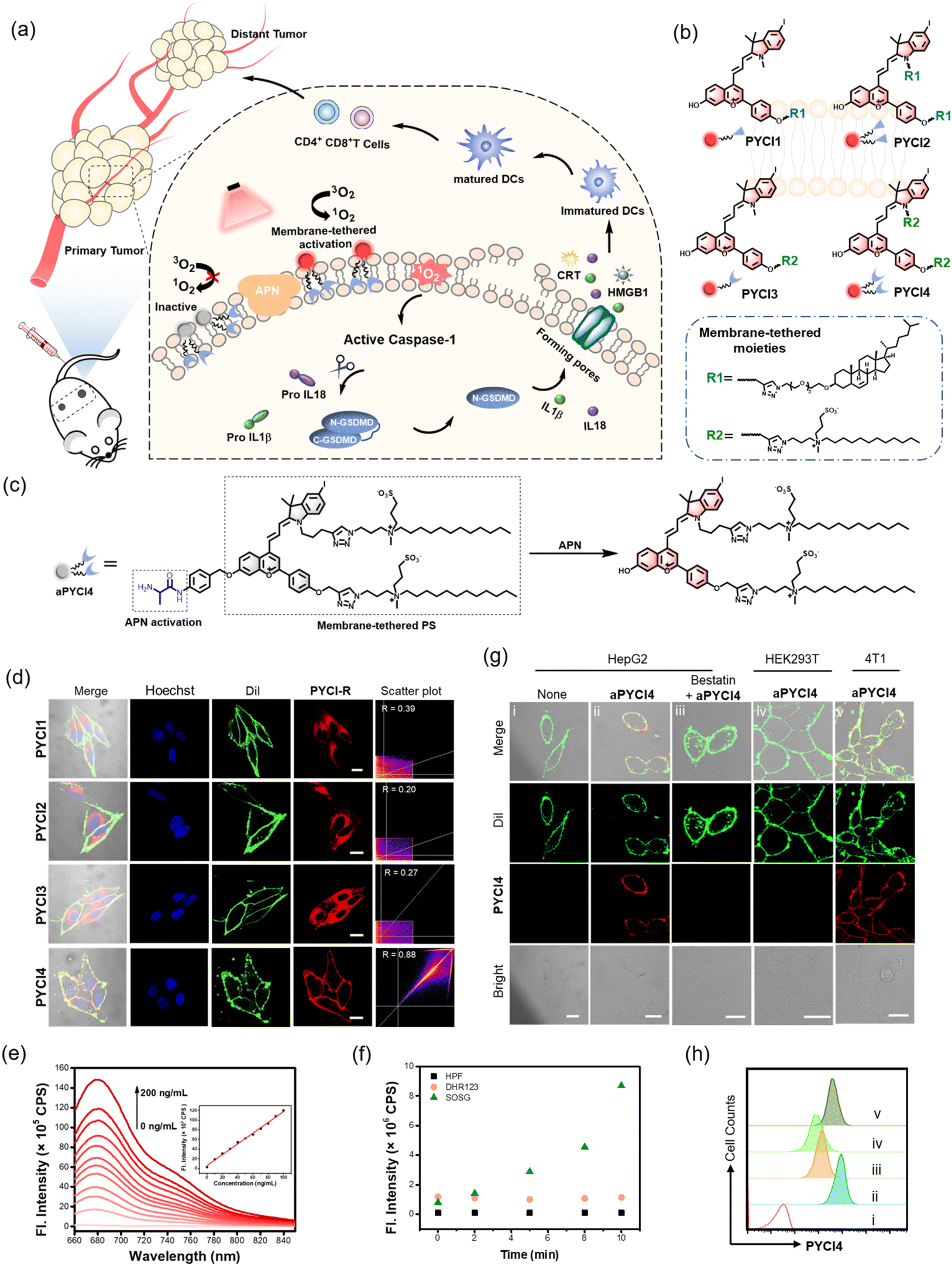 | ||
| Fig. 1 Membrane-tethered activation design and characterizations of PSs. (a) Schematic illustration of the membrane-tethered activation design for inducing pyroptosis with potent systemic anti-tumor immunity. (b) Structures for the four PSs, PYCI1, PYCI2, PYCI3 and PYCI4. (c) Schematic diagram for the structure of aPYCI4 and its reaction mechanism toward APN. (d) CLSM images for HepG2 cells incubated with PSs (2.5 μM, 30 min), membrane tracker Dil (2 μg mL−1, 15 min) and Hoechst 33342 (2 μg mL−1, 10 min), and scatter plots for the correlation between the fluorescence for PSs and Dil. Scale bar: 20 μm. (e) Fluorescence spectra for aPYCI (2.5 μM) in the presence of varying concentrations of APN (0–200 ng mL−1) for 1 h. Inset: linear relationship between fluorescence intensities at 680 nm and various concentrations of APN (0–100 ng mL−1). (f) Fluorescence intensities of HPF (2.5 μM) at 516 nm, DHR123 (2.5 μM) at 536 nm and SOSG (2.5 μM) at 525 nm for aPYCI4 (2.5 μM) incubated with APN (100 ng mL−1) and irradiated for different times. (g) CLSM images for cells treated under different conditions. Scale bar: 20 μm. (h) Flow cytometry profiles for the corresponding cells in (g). | ||
Results and discussion
Membrane-tethered activation design of PSs
The rationale of membrane-tethered activation design is to tether a PS to the cell membrane which is activatable by a membrane-localized enzyme. For noninvasive deep-tissue PDT, we designed a new PS, PYCI, by extending a phenylbenzopyrylium moiety with a conjugated iodinated indole (Fig. 1b and Scheme S1†), which was anticipated to exhibit near-infrared (NIR) absorption and efficient ROS generation. To facilitate derivation of the PS with the alkyne–azide click chemistry, alkyne groups were introduced to the phenylbenzopyrylium and/or indole moieties. To endow the PS with membrane-anchoring ability, we exploited two pendant options using cholesterol or lipid derivation (Fig. 1b and Scheme S2†).17,18 These highly lipophilic pendants were inclined to anchor in the membrane, affording the possibility for membrane-tethering engineering of various PSs. Furthermore, we engineered a specific zwitterionic lipid,19 because this highly hydrophilic moiety could minimize membrane penetration. Four derivatives of the PS were obtained with one or two cholesterol pendants (PYCI1 and PYCI2) and one or two zwitterionic lipid pendants (PYCI3 and PYCI4). The successful synthesis of PYCI and PYCI1-4 was characterized by 1H NMR, 13C NMR and ESI-MS.The ability of the PS derivatives, PYCI1-4, to localize on the plasma membrane was investigated using confocal laser scanning microscopy (CLSM) imaging. HepG2 cells treated with PYCI4 exhibited bright fluorescence on the cell membrane, which perfectly colocalized with that obtained with Dil, a lipophilic membrane indicator (Fig. 1d). By contrast, the cells incubated with PYCI1-3 still displayed evident fluorescence signals in the cytosol. This result demonstrated that two zwitterionic lipids endowed excellent membrane-anchoring ability. Although cholesterol moieties had been documented in facilitating membrane anchoring of DNA structures,17PYCI1-2 failed to localize on the cell membrane, which might be ascribed to the hydrophobicity of the PS. PYCI3 with a single zwitterionic lipid derivation also failed to target the cell membrane. Further assays using other cell lines including A549, MCF-7 and HeLa confirmed the ideal membrane-anchoring characteristic of PYCI4 (Fig. S1†). This result suggested that an optimized number of zwitterionic lipid pendants afforded a viable approach for effective membrane-anchoring.
To demonstrate the membrane-tethered activation design, we chose aminopeptidase N (APN), a membrane-localized metalloproteinase overexpressed in various cancer cells.20–23 We caged the hydroxyl group of PYCI4 using the alanine substrate of APN through a self-immolative linker, p-aminobenzyl alcohol, to obtain an activatable PS, aPYCI4 (Fig. 1c). Fluorescence and absorption spectra analysis showed that aPYCI4 exhibited very weak fluorescence with a blue-shifted absorption peak at 636 nm (Fig. S2†). After incubation with APN, the absorption peak was red-shifted to 651 nm with an intense fluorescence response at 680 nm. The red-shifted NIR absorption and fluorogenic properties after APN-mediated reaction verified the activatability of the designed PS. Moreover, the fluorescence peak at 680 nm dynamically increased with increasing concentrations of APN in the range from 0 to 200 ng mL−1 (Fig. 1e). A linear correlation was obtained in the concentration range of 0–100 ng mL−1 with an estimated limit of detection of 0.30 ng mL−1. Time-dependent measurements showed gradually increased fluorescence intensities till completion at ∼100 min after addition of APN (Fig. S3†). ESI-MS analysis testified the activation mechanism via APN-mediated cleavage followed by a self-immolative decaging reaction (Fig. S4†). In addition, selectivity studies revealed that aPYCI4 was specifically activated by APN with negligible fluorescence enhancements in the presence of other biologically relevant substances (Fig. S5†). Moreover, the fluorescence of aPYCI4 was dramatically decreased for APN pretreated with a specific APN inhibitor bestatin.24 This result confirmed the specific activation of aPYCI4 by APN.
We anticipated that the heavy-atom effect facilitated the intersystem crossing transition to a triplet state, rendering PYCI an efficient PS for ROS generation. We next explored whether aPYCI4 afforded a potential activatable PS in response to APN. We chose 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA), which was oxidized to the emissive dichlorofluorescein (DCF), as a fluorogenic ROS indicator. Negligible green fluorescence was observed from the indicator in the presence of aPYCI4 after NIR irradiation (Fig. S6†). NIR irradiation of aPYCI4 in the presence of APN induced strong green fluorescence for the ROS indicator, while the presence of APN inhibitor bestatin almost inhibited the fluorescence enhancement for the ROS indicator (Fig. S7†). This result suggested that aPYCI4 was an activatable PS specifically responsive to APN. Additionally, we found that the fluorescence for DCFH-DA dynamically increased as the irradiation time increased, indicating an irradiation dose dependent ROS generation process. The type of ROS was further determined using a selective singlet oxygen trap molecule (singlet oxygen sensor green, SOSG), a specific OH˙ indicator (hydroxyl phenyl fluorescein, HPF) and a specific fluorogenic O2˙− indicator (dihydrorhodamine 123, DHR123). Substantial fluorescence enhancement was only obtained with SOSG upon irradiation of APN-activated aPYCI4 (Fig. 1f and S8†), implying a dominant type-II PDT process for the activated PS. In addition, the yield of singlet oxygen for PYCI4 was determined to be 22% using methylene blue as a reference (Fig. S9†). We also found that PYCI4 exhibited photostability comparable to cyanine 5 (Fig. S10†). Together, these results demonstrated the successful development of our activatable type-II PS specifically responsive to APN.
Live cell study of membrane-tethered activation for the PS
We investigated whether membrane-tethered activation of the caged PS aPYCI4 was achieved on live cells. APN-overexpressed HepG2 cells,23 after treatment with aPYCI4, were found to deliver intense fluorescence on the plasma membrane with low fluorescence in the cytoplasm. The CLSM image showed highly colocalized fluorescence with a membrane indicator Dil (Fig. 1g and S11†). Likewise, 4T1 cells with positive APN expression23 displayed intense fluorescence selectively on the membrane upon incubation with aPYCI4. By contrast, fluorescence activation from aPYCI4 dramatically reduced for HepG2 cells pretreated with the APN inhibitor. A control using HEK-293T cells with negative APN expression exhibited negligible fluorescence. Flow cytometry showed a consistent result with the CLSM images (Fig. 1h). This finding demonstrated the specific membrane-tethered activation of aPYCI4 by APN. On the other hand, we synthesized a control activatable PS (aPYCI) with a sulfonic acid moiety in place of the membrane-tethering pendant (Scheme S2†). As anticipated, HepG2 cells incubated with aPYCI displayed strong fluorescence predominantly localized in the cytosol, which was remarkably decreased for the cells pretreated with the APN inhibitor (Fig. S12†).We further inspected the ability of our membrane-tethered activatable PS for ROS production in live cells. We chose DCFH-DA as the fluorogenic ROS indicator. CLSM imaging showed that there was bright green fluorescence for DCF in aPYCI4-treated HepG2 cells upon irradiation with a 640 nm laser, and the fluorescence dramatically decreased for cells pretreated with the APN inhibitor (Fig. 2a). A control experiment using cells only irradiated with a 640 nm laser or merely treated with aPYCI4 delivered negligible fluorescence for DCF, indicating no production of ROS. Flow cytometry analysis showed a consistent result (Fig. 2b and c). The type of ROS was examined using the ROS indicator DCFH-DA in combination with selective ROS quenchers. The DCF fluorescence largely decreased for HepG2 cells pretreated with NaN3, a singlet oxygen quencher, suggesting that singlet oxygen was the predominant ROS produced for aPYCI4-treated cells upon irradiation (Fig. 2d and S13†). By contrast, The DCF fluorescence remained almost unchanged for HepG2 cells pretreated with tiron, a superoxide anion scavenger, or D-mannitol, a hydroxyl quencher. Further CLSM imaging showed that aPYCI4-treated HepG2 cells exhibited bright fluorescence using SOSG as an intracellular singlet oxygen indicator, and this fluorescence was dramatically reduced for cells pretreated with the APN inhibitor (Fig. 2e and S14†). Likewise, we found that the control activatable PS aPYCI could also generate singlet oxygen in HepG2 cells (Fig. S15†).
 | ||
| Fig. 2 Cell-specific toxicity for membrane-tethered activation PDT design. (a) CLSM imaging of DCF fluorescence for HepG2 cells under different treatments: (i) cells irradiated with light only (10 mW cm−2, 10 min); (ii) cells treated with aPYCI4 (2.5 μM) for 1 h; (iii) cells treated with aPYCI4 (2.5 μM, 1 h) and irradiated with a 640 nm laser (10 mW cm−2,10 min); (iv) cells pre-treated with bestatin (100 μM, 2 h), treated with aPYCI4 (2.5 μM, 1 h) and irradiated with a 640 nm laser (10 mW cm−2,10 min). Scale bar: 20 μm. Flow cytometry profiles (b) and quantitative analysis (c) of the corresponding cells in (a). MFI: Mean fluorescence intensity. (d) Flow cytometry profiles of DCF fluorescence for HepG2 cells pretreated with different ROS quenchers and DCFH-DA (10 μM) under irradiation. (e) CLSM images of SOSG fluorescence for HepG2 cells under different treatments. Scale bar: 20 μm. (f) Cytotoxicity of various cell lines treated with aPYCI4 (2.5 μM, 1 h) with or without light irradiation. (g) Fluorescence imaging of calcein AM and PI for aPYC4-treated HepG2 cells under different treatments. Scale bar: 50 μm. Flow cytometry profiles (h) and quantitative analysis for PI + population (i) for HepG2 cells costained with Annexin V-FITC/PI under different treatments. (c) and (i) Statistical comparison was made by conducting the two-tailed t-test (****p < 0.0001, **p < 0.01, *p < 0.05, n = 3). | ||
Next, the ability of our membrane-tethered PS to exert cell-specific phototoxicity was interrogated. We found that both aPYCI4 and aPYCI exhibited negligible dark cytotoxicity towards different cell lines such as APN-positive HepG2 and 4T1 cancer cells and APN-negative HEK-293T normal cells (Fig. 2f and S16†). In contrast, the cell viability dramatically decreased for aPYCI4 treated 4T1 and HepG2 cells with 50% cell viability obtained at aPYCI4 concentrations of 1.3 μM and 1.7 μM, respectively. Furthermore, irradiation of HEK-293T cells treated with aPYCI4 of varying concentrations did not show appreciable toxicity. This result verified the selective phototoxicity for the membrane-tethered PS toward APN-positive cells. Likewise, we observed specific cytotoxicity for the cell-internalized control PS aPYCI to APN-positive HeLa and 4T1 cells with higher concentrations (4.2 and 4.7 μM) needed to achieve 50% cell viability (Fig. S17†). This finding suggested a higher efficacy for the membrane-tethering PS than the cell-internalized PS.
Mechanistic studies of membrane-tethered activation mediated cell death
We further determined the type of cell death induced by our membrane-tethered activation PDT design. Initial evaluation of cell death was performed using two fluorescence staining experiments, live/dead assay with calcein AM/FITC and propidium iodide (PI) and apoptosis/necrosis assay with Annexin V-FITC and PI. We found that APN-positive HepG2 cells exhibited bright PI fluorescence upon incubation with aPYCI4 followed by laser irradiation, and the cell death was substantially inhibited for cells pretreated with the APN inhibitor (Fig. 2g). Negligible cell death was observed for APN-negative normal HEK-293T cells. Quantitative flow cytometry analysis using Annexin V-FITC and PI showed that aPYCI4-treated HepG2 cells upon irradiation underwent a dominant necrosis pathway (89.8%, PI+) (Fig. 2h and i). CLSM imaging showed a consistent result (Fig. S18†). Interestingly, cells treated with the cell-internalized PS aPYCI under irradiation exhibited a much lower efficiency in inducing cell necrosis (34.9%, PI+). In addition, HEK293T cells treated with the membrane-tethered PS PYCI4 exhibited substantial cell death upon laser irradiation, indicating that PYCI4 without caging caused side effects to healthy cells (Fig. S19†). Together, these results revealed that the membrane-tethered activation design conferred higher PDT efficiency than the cell-internalized PS toward APN-positive cancer cells through a dominant necrosis mechanism.As the PS aPYCI4 was tethered on the cell membrane, we hypothesized that the localized production of ROS could cause lipid peroxidation on the cell membrane, which was suggested to augment pyroptosis.25 Recent membrane-anchoring PDT studies also revealed a pyroptosis pathway.15,16 Hence, we explored whether our membrane-tethered activation design of aPYCI4 induced pyroptosis of APN-positive cancer cells. As anticipated, bubble-like protrusions around aPYCI4-treated HepG2 cells were observed upon irradiation with a 640 nm laser (Fig. 3a). As the irradiation time increased up to 15 min, more bubbles appeared around the cells, typical of pyroptotic cells. Moreover, the activated fluorescence profiles from the PS on the cell membrane showed characteristics of discontinuity after irradiation, indicating obvious membrane rupture. In contrast, the cell-internalized activatable PS aPYCI, under laser irradiation, showed remarkable morphological transitions but with no bubble-like protrusions. The different morphological changes during cell death induced by aPYCI4 and aPYCI suggested that membrane-tethered activation was essential for pyroptosis. An additional control using cells not treated with aPYCI4 showed no appreciable changes in morphology or membrane rupture upon irradiation (Fig. S20†). Furthermore, a lactate dehydrogenase (LDH) release assay revealed that aPYCI4-treated HepG2 cells showed increased amounts of LDH release upon irradiation, whereas cells irradiated with NIR light alone or treated with the cell-internalized PS aPYCI and irradiated only showed insignificant LDH release (Fig. 3b). Besides, we observed increased LDH release with prolonged irradiation time for aPYCI4-treated cells (Fig. 3c).
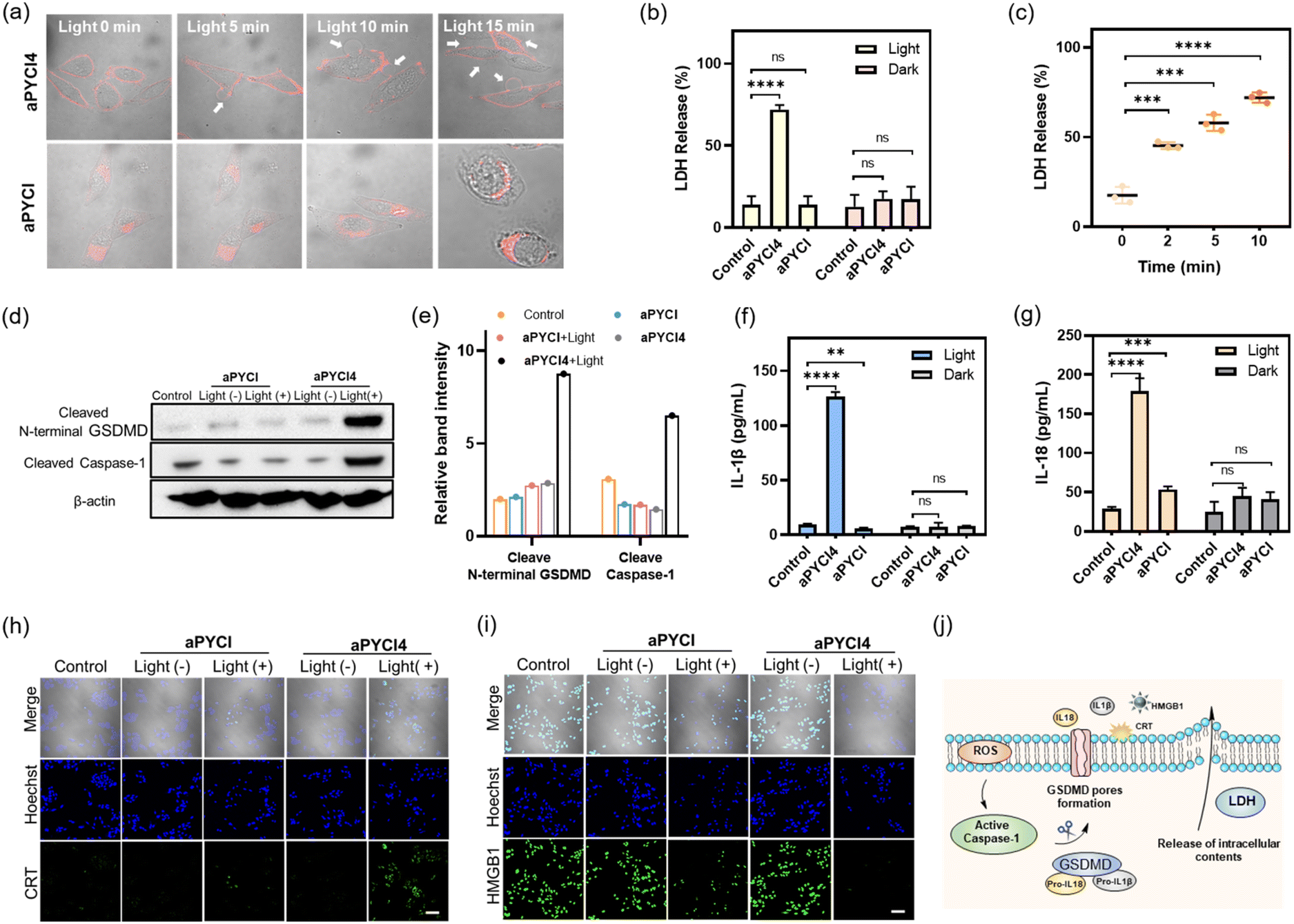 | ||
| Fig. 3 (a) Merged images for HepG2 cells treated with aPYCI4 (2.5 μM) or aPYCI (2.5 μM) and irradiated with a 640 nm laser (10 mW cm−2) for different time periods. White arrows indicate bubble-like protrusions. Scale bar: 20 μm. (b) LDH release for HepG2 cells under different treatments. (c) LDH release for aPYCI4-treated HepG2 cells after irradiation for different time periods. Western blot (d) and quantitative analysis (e) of N-terminal GSDMD and cleaved caspase-1 in HepG2 cells under different treatments. Levels of secreted IL-1β (f) and IL-18 (g) in HepG2 cells under different treatments determined by ELISA. CLSM images for CRT (h) and HMGB1 (i) in HepG2 cells under different treatments (Hoechst 33342, Ex: 405 nm, Em: 430–470 nm; CRT and HMGB1, Ex: 488 nm, Em: 500–550 nm). Scale bar: 100 μm. (j) Schematic illustration of membrane-tethered PDT for inducing pyroptosis and ICD. (c), (f) and (g) Statistical comparison was made by conducting the two-tailed t-test (****p < 0.0001, ***p < 0.001, **p < 0.01, n.s., not significant, n = 3). | ||
Next, we verified the molecular mechanism of pyroptosis induced by our membrane-tethered PDT. One of the distinct pathways for pyroptosis was the activated cleavage of caspase 1 and GSDMD.6,7 As anticipated, western blotting (WB) analysis displayed intense bands for cleaved caspase 1 and N-terminal GSDMD for aPYCI4-treated HepG2 cells after irradiation (Fig. 3d and e). No appreciable N-terminal GSDMD with a basal cleavage of caspase 1 was obtained in control experiments for cells without irradiation or treated with the cell-internalized PS aPYCI under irradiation. This result gave direct evidence that our membrane-tethered PDT was critical for pyroptosis induction.
It is recognized that pyroptosis can induce inflammation and cause the release of inflammatory cytokines.26,27 We then determined the concentrations of two typical cytokines, IL-1β and IL-18, in the cell media for our membrane-tethered PDT treated cells. There was negligible production of IL-1β and IL-18 for the control cells after irradiation, whereas their concentrations were elevated by 13.5- and 6.2- fold, respectively, after the membrane-tethered PDT (Fig. 3f and g). In contrast, only a slight increase of IL-18 secretion with negligible IL-1β production was obtained for the cells treated with the internalized PS aPYCI under irradiation. Together, these results demonstrated that our membrane-tethered PDT design afforded high efficiency in inducing cell pyroptosis. We further investigated whether our membrane-tethered PDT could induce ICD using immunological staining of two critical ICD indicators of DAMPs, high mobility group box 1 (HMGB1) and calreticulin (CRT). The images showed that aPYCI4-treated HepG2 cells induced substantial extracellular release of HMGB1 and translocation of CRT to the cell surface after irradiation, implying that the membrane-tethered PDT was potent in activating ICD (Fig. 3h, i and S21†). In addition, cells treated with the internalized PS aPYCI under irradiation mediated, to a less extent, the efflux or exposure of HMGB1 and CRT. Together, these results demonstrated that our membrane-tethered PDT triggered pyroptosis via activation of caspase 1, cleavage of GSDMD and release of inflammatory IL-18 and IL-1β, accompanied by the release of DAMPs (Fig. 3j).
In vivo tumor imaging and anti-tumor therapy
Our membrane-tethered activation of the PS aPYCI4 held the potential for fluorogenic imaging of tumors in vivo. To investigate it, a tumor-bearing mice model was established using 4T1 cells with over-expression of APN by subcutaneous injection to the left hind leg of Balb/C mice. The tumor-bearing mice were randomly divided into two groups and administered PBS and aPYCI4, via tail-vein injection, respectively, followed by fluorescence image collection at different time points. For aPYCI4-injected tumor-bearing mice, appreciable fluorescence signals appeared at 8 h post-injection, which gradually increased and reached the maximum at ∼24 h (Fig. 4a and b). Moreover, the fluorescence signals remained nearly constant for 5 days with only slight decrease on the 7th day. Ex vivo fluorescence images for dissected organs and tumors on day 7 confirmed strong fluorescence signals from the tumors (Fig. 4c and S22†). Further blood circulation analysis showed that the PS PYCI4 had a half-life of 2.06 h (Fig. 4d), which was much longer than those of current small-molecule based probes (usually <1 h).28–31 This long blood circulation time could be ascribed to its binding with serum proteins (Fig. S23†). On the other hand, the tumor-bearing mice treated with the control PS (no membrane anchor) aPYCI also delivered maximal fluorescence signals at ∼24 h with an inferior signal-to-background ratio to that obtained with aPYCI4 (Fig. S24†). By contrast, the fluorescence signals became remarkably decreased on day 2. This result demonstrated that our membrane-tethered activatable PS aPYCI4 enabled long-lasting fluorogenic imaging of APN-overexpressed tumors in vivo. This long-lasting fluorescence imaging for aPYCI4 was ascribed to its excellent membrane anchoring in tumors, implying its potential for persistent PDT. | ||
| Fig. 4 In vivo tumor imaging and anti-tumor therapy. (a) In vivo fluorescence imaging for 4T1 tumor-bearing mice injected with aPYCI4 (100 μM, 100 μL) via the tail-vein at different time intervals. (b) Average fluorescence intensities from the tumor regions as a function of time. (c) Ex vivo fluorescence intensities of various organs and tumor from 4T1 tumor-bearing mice intravenously injected with aPYCI4 (100 μM, 100 μL) on the 7th day. (n = 3). (d) Blood circulation for PS PYCI4 (100 μM, 100 μL) in healthy BALB/c mice (n = 3). Inset: linear correlation between the logarithm of PS concentrations and time. (e) Schematic illustration of timeline for anti-tumor therapy. (f) Growth curves of tumors in 4T1 tumor-bearing mice under different treatments (n = 5). (g) Photographs of dissected tumors at the end of treatments. (h) Box plot for weights of dissected tumors at the end of treatments. (n = 5). (i) Weight curves for 4T1 tumor-bearing mice under different treatments (n = 5). (j) H&E staining and immunofluorescence imaging of tumors for 4T1 tumor-bearing mice under different treatments. Blue: fluorescence from DAPI; green fluorescence indicates CD4+ T cells and red fluorescence indicates CD8+ T cells. (b–d), (f), (h) and (i) data are presented as mean ± s.d. Statistical comparison was made by conducting the two-tailed t-test (****p < 0.0001, ***p < 0.001, **p < 0.01, n.s., not significant). | ||
Inspired by its tumor-specific activation and long-lasting retention, we explored the possibility of our membrane-tethered activatable PS aPYCI4 for tumor therapy. We chose the 4T1 tumor model as it is known to be immunogenically cold. BALB/c mice with tumor sizes of ∼100 mm3 were randomly divided into four groups through once tail-vein injection with PBS (group I and II), and aPYCI4 (group III and IV), respectively (Fig. 4e). The tumor regions of mice in group II and IV were irradiated with a 640 nm laser every other day for 4 days. The mice weight and tumor sizes were monitored. We observed that the mice treated with aPYCI4 plus radiation (group IV) showed remarkably decreased tumor growth rates as compared to the treated mice in other groups (Fig. 4f), demonstrating the high anti-tumor efficacy for our membrane-tethered PDT. Photographs of tumors dissected on day 14 confirmed the favorable PDT efficacy in group IV with a tumor growth inhibition rate of 81% using the average size of tumors from the mice in group I as a reference (Fig. 4g and h). For all treated mice, the body weights remained stable (Fig. 4i). Meanwhile, hematoxylin and eosin (H&E) staining of major organs including heart, liver, spleen, lung, and kidneys showed no abnormal cell morphology or tissue lesions (Fig. S25†), implying low toxicity of our membrane-tethered PDT strategy to normal organs. Further H&E staining showed that tumor slices in group IV exhibited the highest levels of cell death (Fig. 4j). Interestingly, immunofluorescence analysis of the dissected tumor slices revealed intratumoral accumulation of CD8+ and CD4+ T cells in group IV, indicating augmented anti-tumor immunity in the tumors. Together, these results demonstrated the potential of membrane-tethered activation design for developing efficacious PDT treatments to tumors even with a single dose.
Systemic anti-tumor immunity for distant tumor treatment
Considering that the membrane-tethered PDT was a potent inducer of cell pyroptosis that could boost anti-tumor immunity, we anticipated that our strategy had the potential for treatment of distant tumors. To investigate it, the Balb/C mice bearing primary 4T1 tumors on the left legs were randomly divided into five groups and administered once via tail-vein injection with PBS (group I), aPYCI (II and III) and aPYCI4 (group IV and V) and the primary tumors of mice in group I, III and IV were irradiated every other day for 4 days (Fig. 5a). Distant tumors were implanted by subcutaneously inoculating 4T1 cells to the right legs 24 h after the first irradiation. We observed that the sizes of both tumors from mice in group I–IV showed continuous increase. Notably, the negligible tumor growth inhibition of tumors treated with the control PS (with no membrane anchoring) aPYCI in group III was attributed to the fact that the PS had a short retention time and a low intratumoral concentration after day 2, affording ineffective PDT toward the tumors. In contrast, the mice in group V under PDT treatment with the membrane-tethered PS remarkably suppressed the growth of both primary and distant tumors (Fig. 5b, c and S26†). The growth inhibition rate for the primary and distant tumors was estimated to be 69% and 80%, respectively, using the average size of tumors from the mice in group I as a reference (Fig. 5d). H&E staining of the dissected distant tumor slices verified higher percentages of cell death in group V than in other control groups (Fig. S27†). In addition, the body weights of all the mice were stable and H&E staining of major organs showed no appreciable morphological variations (Fig. 5e and S28†). Together, these results showed that our membrane-tethered PDT allowed efficacious treatment of distant tumors.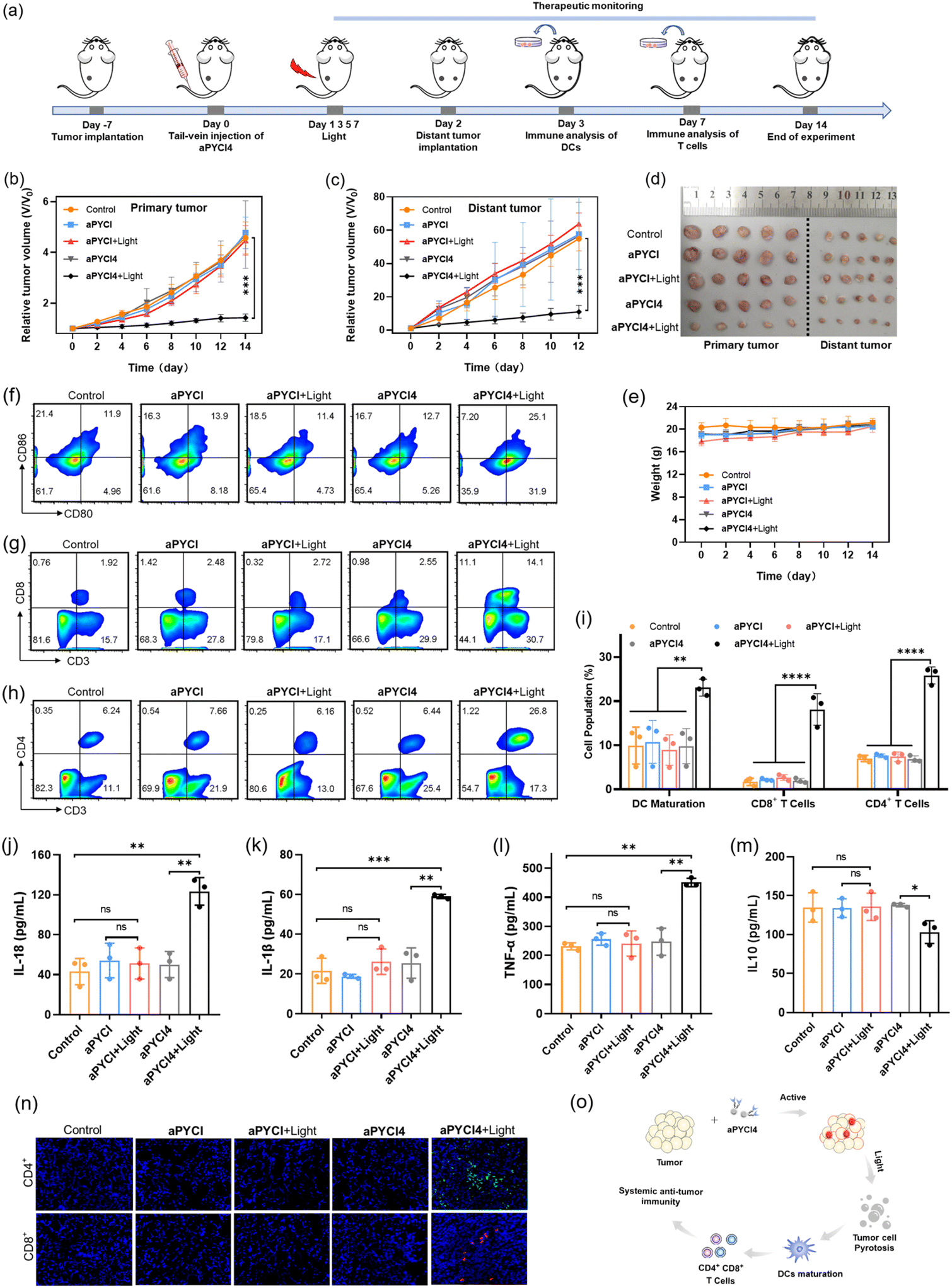 | ||
| Fig. 5 Systemic anti-tumor immunity for distant tumor treatment. (a) Schematic illustration of the timeline for distant tumor treatment and analysis. Growth curves of primary tumors (b) and distant tumors (c) in 4T1 tumor-bearing mice under different treatments (n = 5). (d) Photographs of dissected tumors at the end of treatments. (e) Weight curves for 4T1 tumor-bearing mice under different treatments (n = 5). (f) Flow cytometry profiles for CD80+ CD86+ cells gating on CD11c+ cells in primary tumors under different treatments. Flow cytometry profiles of CD3+CD8+ cells (g) and CD3+CD4+ cells (h) gating on CD3+ cells in primary tumors of 4T1 tumor-bearing mice under different treatments on day 3. (i) Quantification of matured DCs, CD8+ and CD4+ T cells. Levels of IL-18 (j), IL-1β (k), TFN-α (l) and IL10 (m) in serum from 4T1 tumor-bearing mice under different treatments on day 3. (n) Immunofluorescence imaging of distant tumors. Green fluorescence indicates CD4+ T cells and red fluorescence indicates CD8+ T cells. (o) Schematic illustration for membrane-tethered PDT with systemic anti-tumor immunity. Data are presented as mean ± s.d. Statistical comparison was made by conducting the two-tailed t-test (****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05, n.s., not significant, n = 3). | ||
We then investigated the mechanism of our membrane-tethered PDT strategy for distant tumor suppression. We first assessed the PDT-induced reprogramming of immune microenvironments in the primary tumors. Flow cytometry analysis showed that the percentages of CD80+CD86+ dendritic cells in the primary tumors were significantly elevated after PDT treatment in group V (25.1%), whereas those in group I–IV were only 11.9%, 13.9%, 11.4% and 12.7%, respectively (Fig. 5f). This result suggested that membrane-tethered PDT facilitated the maturation of dendric cells with the potential of enhanced antigen presentation. Moreover, the percentages of intratumoral CD3+CD8+ T cells substantially increased in group V (14.1%), whereas those in group I–IV were only 1.92%, 2.48%, 2.72% and 2.55%, respectively (Fig. 5g). Recruitment of intratumoral CD3+CD4+ T cells was also remarkably augmented in group V, and the percentages were 26.8% in group V versus group I–IV (6.24%, 7.66%, 6.16% and 6.44%, respectively) (Fig. 5h). Quantitative analysis of DC maturation, CD4+ and CD8+ T cells showed significant differences between group V and group I–IV (Fig. 5i). These results revealed that our membrane-tethered PDT was capable of converting immune ‘cold’ tumor ‘hot’ by promoting maturation of dendritic cells and recruitment of cytotoxic T lymphocytes.
We further explored whether the membrane-tethered PDT treatment could induce systemic inflammatory responses. The levels of several typical cytokines including interleukin (IL)-18, IL-1β, tumor necrosis factor α (TNF-α) and IL10 in the peripheral blood of treated mice were determined using enzyme-linked immunosorbent assay (ELISA). We found that the levels of inflammatory cytokines including IL-18, IL-1β and TNF-α were increased by ∼2.9-, 2.7- and 2.0-fold, respectively, for group V as compared to those from group I (Fig. 5j–l). Moreover, the concentration of a typical immunosuppressive cytokine IL-10 was decreased by ∼1.3-fold in group V (Fig. 5m). Notably, the concentrations of cytokines in the peripheral blood of mice under PDT treatment with the cell-internalized PS in group IV were close to the levels in group I, verifying that membrane-tethered PDT was crucial for eliciting systemic anti-tumor immunity. Furthermore, we examined the abscopal anti-tumor immunity of our PDT strategy through an immunofluorescence staining experiment for the slices of the distant tumors. The images revealed substantial accumulation of CD8+ and CD4+ T cells in the distant tumors in group V, whereas negligible recruitment of CD8+ and CD4+ T cells was obtained in other control groups (Fig. 5n), confirming the abscopal anti-tumor immunity. This result revealed that the growth inhibition for distant tumors was due to the anti-tumor immunity in enhancing systemic inflammatory responses and recruiting cytotoxic T lymphocytes in the distant tumors (Fig. 5o).
Conclusions
We developed a new design of membrane-tethered activation for PSs that enabled long-lasting fluorogenic imaging and boosted potent systemic anti-tumor immunity for cancer therapy. In vitro and live cell analysis demonstrated that our design exhibited excellent membrane localization, evoked efficient singlet oxygen production, and elicited specific phototoxicity to tumor cells via pyroptotic ICD. The membrane-tethered PS demonstrated prolonged blood circulation, long-lasting fluorogenic imaging and persistent PDT of tumors in vivo. The membrane-proximal PDT not only exerted excellent primary tumor ablation, but also efficiently suppressed the growth of distant tumors by turning immunogenic ‘cold’ tumors ‘hot’ and augmenting systemic antitumor immunity. The membrane-tethered activation design could afford a useful approach for boosting systemic antitumor immunity and developing new combination cancer immunotherapy.Data availability
Main experimental data are provided within the main text and the ESI.† Additional data are available from the corresponding author upon reasonable request.Author contributions
F. Wang and J. H. Jiang conceived the project and wrote the manuscript. X. Chu edited the manuscript. P. Lu synthesized the compounds and collected experimental data. X. Liu participated in in vitro characterization assays. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research Program (2019YFA0905800), National Natural Science Foundation of China (22274040, 22090053) and Hunan Provincial Science Fund for Distinguished Young Scholars (2022JJ10004).Notes and references
- A. D. Waldman, J. M. Fritz and M. J. Lenardo, Nat. Rev. Immunol., 2020, 20, 651–668 CrossRef CAS PubMed.
- J. Galon and D. Bruni, Nat. Rev. Drug Discovery, 2019, 18, 197–218 CrossRef CAS.
- P. Sharma, S. Hu-Lieskovan, J. A. Wargo and A. Ribas, Cell, 2017, 168, 707–723 CrossRef CAS PubMed.
- S. I. Grivennikov, F. R. Greten and M. Karin, Cell, 2010, 140, 883–899 CrossRef CAS PubMed.
- G. Kroemer, C. Galassi, L. Zitvogel and L. Galluzzi, Nat. Immunol., 2022, 23, 487–500 CrossRef CAS PubMed.
- P. Broz, P. Pelegrin and F. Shao, Nat. Rev. Immunol., 2020, 20, 143–157 CrossRef CAS PubMed.
- J. Ding, K. Wang, W. Liu, Y. She, Q. Sun, J. Shi, H. Sun, D. C. Wang and F. Shao, Nature, 2016, 535, 111–116 CrossRef CAS PubMed.
- Q. Wang, Y. Wang, J. Ding, C. Wang, X. Zhou, W. Gao, H. Huang, F. Shao and Z. Liu, Nature, 2020, 579, 421–426 CrossRef CAS PubMed.
- Z. Zhang, Y. Zhang, S. Xia, Q. Kong, S. Li, X. Liu, C. Junqueira, K. F. Meza-Sosa, T. M. Y. Mok, J. Ansara, S. Sengupta, Y. Yao, H. Wu and J. Lieberman, Nature, 2020, 579, 415–420 CrossRef CAS PubMed.
- B. Chen, Y. Yan, Y. Yang, G. Cao, X. Wang, Y. Wang, F. Wan, Q. Yin, Z. Wang, Y. Li, L. Wang, B. Xu, F. You, Q. Zhang and Y. Wang, Nat. Nanotechnol., 2022, 17, 788–798 CrossRef CAS PubMed.
- Y. Wang, W. Gao, X. Shi, J. Ding, W. Liu, H. He, K. Wang and F. Shao, Nature, 2017, 547, 99–103 CrossRef CAS PubMed.
- X. Zhao, J. Liu, J. Fan, H. Chao and X. Peng, Chem. Soc. Rev., 2021, 50, 4185–4219 RSC.
- D. Wu, S. Wang, G. Yu and X. Chen, Angew. Chem., Int. Ed., 2021, 60, 8018–8034 CrossRef CAS PubMed.
- J. W. Kleinovink, P. B. van Driel, T. J. Snoeks, N. Prokopi, M. F. Fransen, L. J. Cruz, L. Mezzanotte, A. Chan, C. W. Lowik and F. Ossendorp, Clin. Cancer Res., 2016, 22, 1459–1468 CrossRef CAS PubMed.
- M. Wu, X. Liu, H. Chen, Y. Duan, J. Liu, Y. Pan and B. Liu, Angew. Chem., Int. Ed., 2021, 60, 9093–9098 CrossRef CAS PubMed.
- X. Su, W. J. Wang, Q. Cao, H. Zhang, B. Liu, Y. Ling, X. Zhou and Z. W. Mao, Angew. Chem., Int. Ed., 2022, 61, e202115800 CAS.
- M. You, Y. Lyu, D. Han, L. Qiu, Q. Liu, T. Chen, C. Sam Wu, L. Peng, L. Zhang, G. Bao and W. Tan, Nat. Nanotechnol., 2017, 12, 453–459 CrossRef CAS PubMed.
- V. V. Shynkar, A. S. Klymchenko, C. Kunzelmann, G. Duportail, C. D. Muller, A. P. Demchenko, J.-M. Freyssinet and Y. Mely, J. Am. Chem. Soc., 2007, 129, 2187–2193 CrossRef CAS PubMed.
- O. A. Kucherak, S. Oncul, Z. Darwich, D. A. Yushchenko, Y. Arntz, P. Didier, Y. Mely and A. S. Klymchenko, J. Am. Chem. Soc., 2010, 132, 4907–4916 CrossRef CAS.
- L. Chen, Y. L. Lin, G. Peng and F. Li, Proc. Natl. Acad. Sci., 2012, 109, 17966–17971 CrossRef CAS.
- J. Huang, Y. Jiang, J. Li, S. He, J. Huang and K. Pu, Angew. Chem., Int. Ed., 2020, 59, 4415–4420 CrossRef CAS PubMed.
- H. Li, Q. Yao, W. Sun, K. Shao, Y. Lu, J. Chung, D. Kim, J. Fan, S. Long, J. Du, Y. Li, J. Wang, J. Yoon and X. Peng, J. Am. Chem. Soc., 2020, 142, 6381–6389 CrossRef CAS PubMed.
- H. Li, Y. Li, Q. Yao, J. Fan, W. Sun, S. Long, K. Shao, J. Du, J. Wang and X. Peng, Chem. Sci., 2019, 10, 1619–1625 RSC.
- J. Shang, X. Zhang, Z. He, S. Shen, D. Liu, W. Shi and H. Ma, Angew. Chem., Int. Ed., 2022, 61, e202205043 CAS.
- D. Zhivaki and J. C. Kagan, Nat. Rev. Immunol., 2022, 22, 322–330 CrossRef CAS PubMed.
- J. Shi, Y. Zhao, K. Wang, X. Shi, Y. Wang, H. Huang, Y. Zhuang, T. Cai, F. Wang and F. Shao, Nature, 2015, 526, 660–665 CrossRef CAS PubMed.
- P. Broz and V. M. Dixit, Nat. Rev. Immunol., 2016, 16, 407–420 CrossRef CAS PubMed.
- P. Cheng and K. Pu, Nat. Rev. Mater., 2021, 6, 1095–1113 CrossRef CAS.
- C. Yang, H. Wang, S. Yokomizo, M. Hickey, H. Chang, H. Kang, T. Fukuda, M. Y. Song, S. Y. Lee, J. W. Park, K. Bao and H. S. Choi, Angew. Chem., Int. Ed., 2021, 60, 13847–13852 CrossRef CAS PubMed.
- K. S. de Valk, H. J. Handgraaf, M. M. Deken, B. G. Sibinga Mulder, A. R. Valentijn, A. G. Terwisscha van Scheltinga, J. Kuil, M. J. van Esdonk, J. Vuijk, R. F. Bevers, K. C. Peeters, F. A. Holman, J. V. Frangioni, J. Burggraaf and A. L. Vahrmeijer, Nat. Commun., 2019, 10, 3118 CrossRef PubMed.
- H. S. Choi, S. L. Gibbs, J. H. Lee, S. H. Kim, Y. Ashitate, F. Liu, H. Hyun, G. Park, Y. Xie, S. Bae, M. Henary and J. V. Frangioni, Nat. Biotechnol., 2013, 31, 148–153 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc07044h |
| This journal is © The Royal Society of Chemistry 2023 |