
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConvergent chemoenzymatic synthesis of O-GalNAc rare cores 5, 7, 8 and their sialylated forms†
Madhusudhan Reddy
Gadi‡
a,
Congcong
Chen‡
ab,
Shumin
Bao‡
a,
Shuaishuai
Wang
 a,
Yuxi
Guo
a,
Jinghua
Han
a,
Weidong
Xiao
c and
Lei
Li
*a
a,
Yuxi
Guo
a,
Jinghua
Han
a,
Weidong
Xiao
c and
Lei
Li
*a
aDepartment of Chemistry and Center for Diagnostics & Therapeutics, Georgia State University, Atlanta, GA 30303, USA. E-mail: lli22@gsu.edu
bShandong Academy of Pharmaceutical Science, Key Laboratory of Biopharmaceuticals, Engineering Laboratory of Polysaccharide Drugs, National-Local Joint Engineering Laboratory of Polysaccharide Drugs, Jinan 250101, China
cDepartment of Pediatrics, Herman B Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202, USA
First published on 18th January 2023
Abstract
All O-GalNAc glycans are derived from 8 cores with 2 or 3 monosaccharides linked via α- or β-glycosidic bonds. While chemical and chemoenzymatic syntheses of β-linked cores 1–4 and 6 and derived glycans have been well developed, the preparation of α-linked rare cores 5, 7, and 8 is challenging due to the presence of this 1,2-cis linkage. Meanwhile, the biosynthesis and functional roles of these structures are poorly understood. Herein, we synthesize 3 α-linked rare cores with exclusive α-configuration from a versatile precursor through multifaceted chemical modulations. Efficient regioselective α2-6sialylion of the rare cores was then achieved by Photobacterium damselae α2-6sialyltransferase-catalyzed reactions. These structures, together with β-linked cores 1–4 and 6, and their sialylated forms, were fabricated into a comprehensive O-GalNAc core microarray to profile the binding of clinically important GalNAc-specific lectins. It is found that only Tn, (sialyl-)core 5, and core 7 are the binders of WFL, VVL, and SBA, while DBA only recognized (sialyl-)core 5, and Jacalin is the only lectin that binds core 8. In addition, activity assays of human α-N-acetylgalactosaminide α2-6sialyltransferases (ST6GalNAcTs) towards the cores suggested that ST6GalNAc1 may be involved in the biosynthesis of previously identified sialyl-core 5 and sialyl-core 8 glycans. In conclusion, we provide efficient routes to access α-linked O-GalNAc rare cores and derived structures, which are valuable tools for functional glycomics studies of mucin O-glycans.
Introduction
O-GalNAc glycans (also known as mucin-type O-glycans) are not only major components of the mucus that coats all non-keratinized epithelial surfaces, but are also widely expressed on other proteins. By estimation, over 80% of human secretory and cell surface proteins are decorated with O-GalNAc glycans.1 It is thus not surprising that they are associated with diverse biological processes, including cellular recognition, differentiation, signaling, adhesion, and apoptosis.2 In addition, aberrant O-GalNAc glycosylation is closely related to various diseases including tumor development.3,4 As a result, profiling this type of glycosylation5–7 and developing associated biomarkers and therapeutics8,9 have become hot topics of research.All O-GalNAc glycans initiate with a GalNAc residue α-linked to the hydroxyl group of serine (Ser), threonine (Thr), or seldomly tyrosine (Tyr).10 This initial GalNAc can then be extended with one or two additional sugar residues (Gal, GalNAc, or GlcNAc) to generate 8 core structures (Fig. 1A).11 Among these, cores 1–4 are commonly observed with relatively high abundance, whereas cores 5–8 are rare cores with restricted occurrence and low abundance.12 Structurally, the initial GalNAc of cores 1–4 and 6 are extended with β-linked residues, which can be further elongated to present various glycan epitopes.13 Cores 5, 7, and 8, on the other hand, are extended with an α-linked GalNAc or Gal residue. To date, further glycosylations of these cores other than sialylation of cores 5 and 8 have not been observed in mammals.
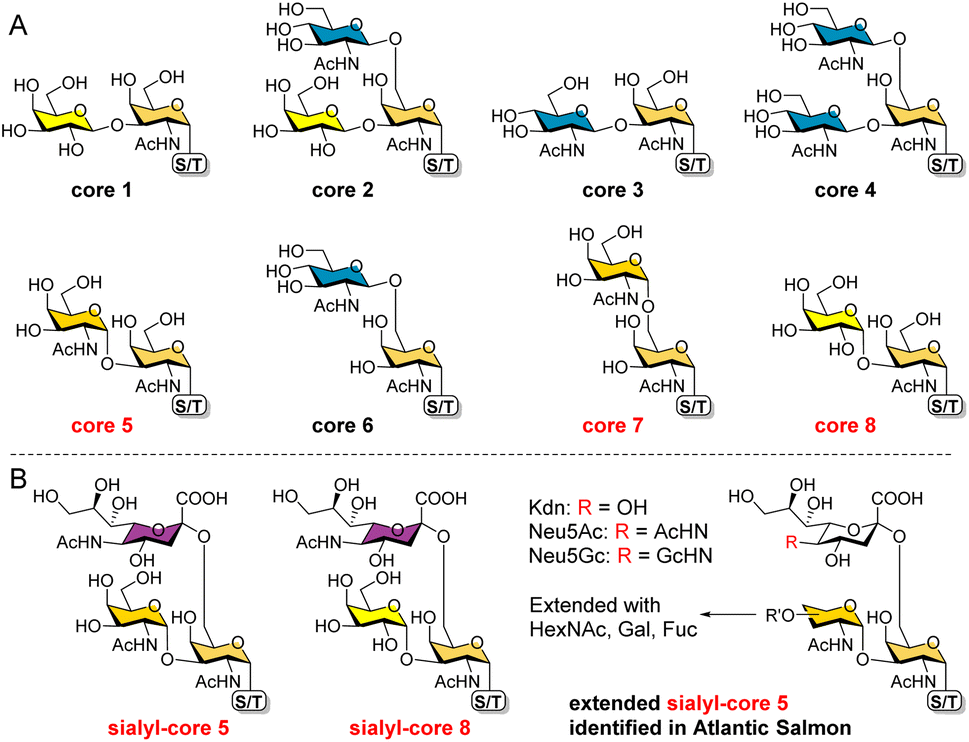 | ||
| Fig. 1 The 8 O-GalNAc glycan core structures (A) and extended rare cores 5, 7, and 8 found in mammalian systems and fish (B). | ||
In mammalian systems, core 5 (GalNAcα1-3GalNAcα) was only identified in human gastric mucins and meconium, as well as Toxoplasma gondii mucin-like glycoprotein.14,15 Its sialylated form GalNAcα1-3(Neu5Acα2-6)GalNAcα (Fig. 1B) was identified in human colon mucosa, rectal adenocarcinoma, and meconium,16–18 as well as bovine submaxillary mucin.19 In contrast, core 5 and elongated glycans (Fig. 1B) are abundant in skin mucus of fish including rainbow trout and Atlantic salmon,20–23 suggesting that the corresponding biosynthetic enzymes may be highly expressed or upregulated in such species. So far, core 7 (GalNAcα1-6GalNAcα) has only been reported in bovine submaxillary mucin, accounting for 3% of total glycans by mass,24 and sialyl-core 8 (Galα1-3(Neu5Acα2-6)GalNAcα) was solely identified in human bronchial mucin.25 Limited reports on these α-linked cores may be ascribed to their low abundance in mammals and the lack of appropriate methods to enrich/distinguish them from highly abundant cores. Furthermore, the biosynthesis and potential functions of cores 5, 7, and 8 are still unknown. Such studies require well-defined structures as standards and probes.
Our interest lies in the facile synthesis of O-GalNAc glycans for glycomics studies. We have recently developed an efficient modular assembly strategy to rapidly prepare diverse cores 1–4 and 6 glycans starting from a versatile precursor (Fig. 2, compound 4).26 In this work, we focused on rare cores 5, 7, 8 and their sialylated forms. Different from cores 1–4 and 6, the presence of 1,2-cis linkages in cores 5, 7, and 8 poses a major challenge in devising a synthetic route. All early attempts to synthesize core 5,27–30 sialyl-core 5,28 core 7,27,29 and core 8 (ref. 31) gave 1,2-cis (α-linkage)/1,2-trans(β-linkage) mixtures or low overall yields. An efficient and stereoselective strategy to access these rare cores and their natural sialylated forms is still lacking. Here, we report the convergent synthesis of cores 5, 7, and 8 with exclusive α-selectivity using the same precursor 4, followed by regioselective preparation of their sialylated forms using a bacterial α2-6sialyltransferase.
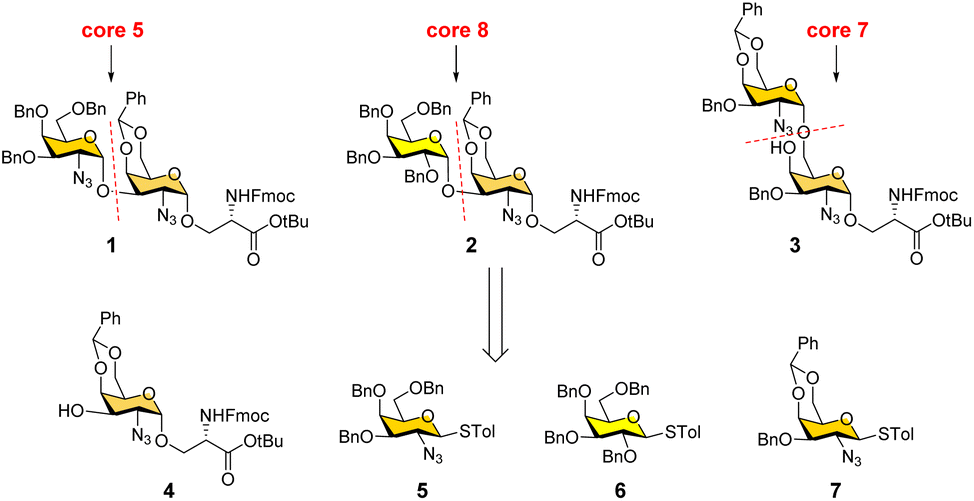 | ||
| Fig. 2 Retrosynthetic strategy for convergent synthesis of O-GalNAc cores 5, 7, and 8. | ||
Results and discussion
Convergent synthesis of α-linked rare cores 5, 7, and 8
We conceived a convergent retrosynthetic route (Fig. 2) towards cores 5, 7, and 8, which can be obtained from protected disaccharides 1, 2, and 3 respectively upon reduction of the azide group followed by global deprotection. Conversely, the protected disaccharides were planned to be synthesized by a convergent route utilizing four monosaccharide building blocks 4–7.32,33 All four building blocks were designed with a non-participating group at the C2 position to achieve exclusive α-selective glycosylation.34,35 Efficient large-scale synthesis of protected glycosyl amino acid derivative 4 was achieved previously in the synthesis of β-linked O-GalNAc cores.26Facile stereoselective synthesis of 1,2-cis linkages has been a significant challenge in the synthesis of oligosaccharides.36 The most common approach is to place a non-participating group at the C2 position of a glycosyl donor.37 However, such non-participating groups did not afford exclusive α-selectivity (data not shown). In addition, we investigated solvent-assisted glycosylation with pre-activation of the donor38 to obtain the desired stereochemical outcomes. For example, to synthesize cores 5 and 8, thioglycosyl donor 5 or 6 was first activated with p-TolSCl in diethyl ether at −78 °C using AgOTf as the promoter (Scheme 1). To the activated donor, the acceptor 4 in diethyl ether was slowly added to provide protected disaccharide 1 or 2, respectively, in very good yields with exclusive α-selectivity (Scheme 1).35,39,40 Solvent assistance from the β-side provided by diethyl ether ensured exclusive α-selectivity, whereas our initial attempts of using a mixture of dichloromethane and diethyl ether resulted in a mix of α/β-anomers. Going forward, the azide functional group on 1 and 2 was reduced to an amine with zinc and in situ protected as acetyl amide using acetic anhydride. The crude reaction mixture was then subjected to hydrogenation using 10% Pd/C under acidic conditions to afford deprotected core 5 (Scheme 1, 8) and core 8 (9). Finally, deprotection of the tBu ester using trifluoroacetic acid afforded Fmoc protected cores 5 (10) and core 8 (11) quantitively.
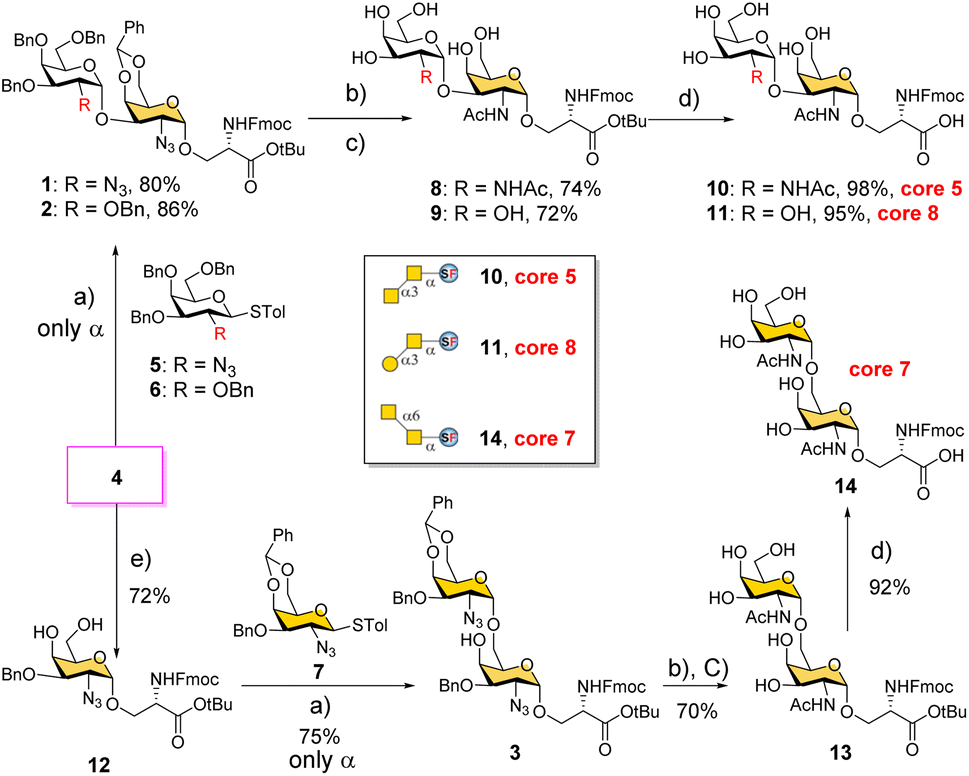 | ||
Scheme 1 Convergent synthesis of O-GalNAc glycan rare cores 5, 7, and 8 attached with a Ser residue. (a) p-TolSCl, AgOTf, −78 °C, Et2O; (b) Zn, THF, Ac2O; (c) Pd/C, H2 atm, MeOH/THF/AcOH/H2O; (d) TFA/anisole = 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; (e) (i) NaH and BnBr and (ii) PTSA and MeOH. :1; (e) (i) NaH and BnBr and (ii) PTSA and MeOH. | ||
To synthesize core 7 (Scheme 1, 14), the versatile precursor 4 was first converted to the diol 12 by protecting the C3–OH as benzyl ether and subsequent deprotection of the benzylidene acetal under acidic conditions. The higher nucleophilicity of the C6–OH over the C4–OH allowed regioselective glycosylation using glycosyl donor 5 or 7 under similar solvent-assisted glycosylation conditions for the synthesis of cores 5 and 8. However, when glycosyl donor 5 was used, a mixture of anomers (α:β = 7:3) was formed, presumably due to the higher nucleophilicity of the acceptor. We then tried glycosyl donor 7 with benzylidene protection, which afforded the disaccharide 3 in good yield (75%) with exclusive α-selectivity (Scheme 1). It is likely that the benzylidene acetal assisted in blocking the β-face attack of the acceptor 12. Finally, successive azide reduction and amine acetylation followed by global deprotection provided compound 13, which was converted to the Fmoc protected core 7 (14) under acidic conditions. Collectively, the introduction of non-participating groups at C2 of glycosyl donors and the pre-activation of donors, together with solvent-assisted glycosylation realized perfect stereoselectivity in the synthesis of α-linked O-GalNAc cores starting from the versatile acceptor 4. The overall yields for cores 5, 7, and 8 are 58% (4 steps), 33% (5 steps), and 59% (4 steps), respectively.
Enzymatic synthesis of sialylated cores 5, 7, and 8
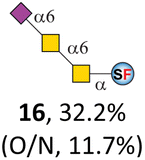
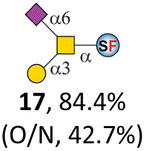
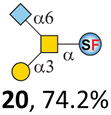
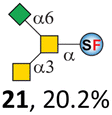
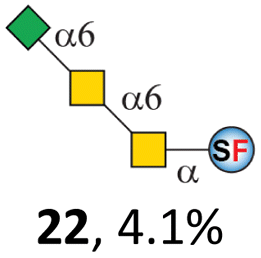
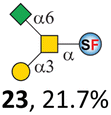
To date, the only identified modification on α-linked O-GalNAc cores in mammalian systems is α2-6sialylation on the initial GalNAc of core 5 or 8.16,17,25 However, glycosyltransferases responsible for the sialylation, as well as for the biosynthesis of cores 5, 7, and 8 are yet unknown. To generate their sialylated forms, we tested the activity of α2-6sialyltransfease from Photobacterium damselae (Pd26ST)41 towards synthesized cores 5, 7, and 8 (ESI†), as Pd26ST has a broad acceptor specificity, which recognizes distal, internal, and reducing end Gal and GalNAc residues.41,42 Reactions were performed at 37 °C for 1 h or overnight. As expected, core 7 was mono-sialylated as it has a single exposed C6–OH on the distal GalNAc residue (a new mass spectrometry (MS) peak at m/z = 1023.3874 [M–H]− and a new HPLC peak at 15.46 min, ESI†). Interestingly, the conversion rate of core 7 to its sialylated form showed a positive correlation to the concentration of the acceptor, which is <1% (1 mM of core 7), 16.6% (5 mM of core 7), and 32.2% (10 mM of core 7) (Table 1).| Donor | Concentration of core | Acceptor | ||
|---|---|---|---|---|
| 10 (core 5) | 14 (core 7) | 11 (core 8) | ||
| Product | ||||
| a Reactions were performed in 20 μL systems in 100 mM Tris–HCl (pH 8.0), containing varying concentrations of cores (1, 5, and 10 mM), donors CMP–sialic acid (2 equivalents), and 80 μg of purified Pd26ST. All reactions were incubated at 37 °C for 1 h unless otherwise stated. Conversion of cores is monitored by HPLC. b One-pot two-enzyme system was used to generate CMP–Kdn in situ (ESI). O/N, overnight reaction. | ||||
| CMP–Neu5Ac | 10 mM |
|
|
|
| CMP–Neu5Ac | 5 mM | 15, 62.1% | 16, 16.6% | 17, 67.0% |
| CMP–Neu5Ac | 1 mM | 15, 15.3% | 16, <1% | 17, 11.7% |
| CMP–Neu5Gc | 10 mM |
|
|
|
| CMP–Kdnb | 10 mM |
|
|
|

|
||||
Surprisingly, for cores 5 and 8, only MS peaks corresponding to mono-sialylated core 5 (m/z = 1023.3695, [M–H]−) and core 8 (m/z = 982.3695, [M–H]−) were observed in Pd26ST-catalyzed reactions. Meanwhile, HPLC analyses of the reactions showed a single new peak (TR = 14.87 min for core 5 and TR = 15.07 min for core 8) corresponding to the products in both reactions, with high conversion rates of 85.5% and 84.4%, respectively (ESI†). Note that elongated reaction times did not result in di-sialylated forms, instead a significantly lower conversion rate (49.0% and 42.7% for core 5 and core 8 respectively) was observed (Table 1, ESI†), indicating that sialylated forms of cores 5 and 8 may be labile under the reaction conditions. Nevertheless, these results suggested that one specific Gal/GalNAc residue in cores 5 and 8 was sialylated. These sialylated core 5 and core 8 were then synthesized in very good yields (92%, 7 mg for core 5; 90%, 6 mg for core 8) and purified for NMR characterization. The 2D-HMBC NMR spectra (ESI†) of both products showed a positive correlation between the anomeric C2 of Neu5Ac and the C6 of GalNAc at the reducing end, revealing that only the initial GalNAc was α2-6sialylated in both cases. These results confirmed that the sialylated cores are natural sialyl-core 5 and sialyl-core 8 identified in mammalian systems.16,17,25 Such a strict regioselectivity may stem from the distorted structures of cores 5 and 8 where the C6–OH of the distal GalNAc/Gal residue is adjacent to and thus masked by the proximal C2–N-acetyl of the initial GalNAc residue (Fig. S1†). Similar to those observed in core 7 reactions, the conversion rates of cores 5 and 8 again showed positive correlations to the concentration of acceptor cores (Table 1). These results may be explained by high Km values of Pd26ST towards α-linked GalNAc residues.43
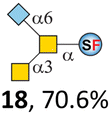
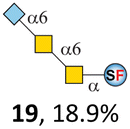
N-Glycolylneuraminic acid (Neu5Gc) and deaminated sialic acid (Kdn) are two other common sialic acid forms found in eukaryotes besides Neu5Ac.44 Sialyl-core 5 structures with Neu5Gc and Kdn were previously identified in salmon.21,22 We performed activity assay of Pd26ST using CMP–Neu5Gc and CMP–Kdn as donors and cores 5, 7, and 8 as acceptors (Table 1, ESI†). Pd26ST efficiently catalyzed the sialylation of cores 5 and 8 using CMP–Neu5Gc as the donor (70.6% and 74.2%). The conversion rate of core 7 (18.9%) is low but comparable to that using CMP–Neu5Ac as the donor (32.2%). On the other hand, the conversion rates are much lower towards Kdn, which may be partially due to the use of a one-pot multi-enzyme system to generate the donor CMP–Kdn in situ (ESI†) instead of pure CMP–Kdn. Nevertheless, mg scale reactions with excess amounts of Pd26ST afforded Neu5Ac and Kdn modified cores 5, 7, and 8 in good overall yields. The compounds were purified by HPLC and analyzed by HPLC, MS, and/or NMR (ESI†).
ST6GalNAc1 is responsible for the biosynthesis of sialyl-core 5/8
Humans have 6 α-N-acetylgalactosaminide α2-6sialyltransferases (ST6GalNAcTs) that catalyze the α2-6siaylation of the initial GalNAc residue of O-GalNAc cores. Among these, ST6GalNAc1 and ST6GalNAc2 are classified into one subfamily and exhibit activity to Tn, T (core 1), and 3′-sialyl-T (Neu5Acα2-3Galβ1-3GalNAcα) antigens in vitro.45In vivo, ST6GalNAc1 and ST6GalNAc2 were thought to be responsible for the biosynthesis of sialyl-Tn (Neu5Acα2-6GalNAcα) and 6-sialyl-T (Galβ1-3(Neu5Acα2-6)GalNAcα) antigens, respectively.45 The other subfamily contains ST6GalNAc3–6, which recognizes Neu5Acα2-3Galβ1-3GalNAc. While ST6GalNAc4 prefers O-glycan (3′-sialyl-T) to generate disialyl-T (Neu5Acα2-3Galβ1-3(Neu5Acα2-6)GalNAcα),46 the other 3 members effectively synthesize gangliosides.45 To test whether any of these GTs may be involved in the biosynthesis of sialyl-cores 5 and 8, we carried out activity assay of human ST6GalNAcs towards synthesized rare cores, with core 1 as a positive control (Table 2).| Enzyme | Acceptor | ||||
|---|---|---|---|---|---|
| Core 1 | 10 (core 5) | 14 (core 7) | 11 (core 8) | ||
| Product | |||||
| a Reactions were carried out in a 10 μL system in 100 mM MES buffer (pH 7.0), containing 0.1 mM acceptor, 10 mM CMP–Neu5Ac, and 2 μg of enzymes. Reactions were incubated at 37 °C for 48 hours. b GTs with a N-terminal GFP-tag expressed in CHO cell lines were obtained from Glyco Expression Technologies, Inc. (Athens, GA). c ST6GalNAc4 with a C-terminal His6-tag expressed in a mouse myeloma cell line was obtained from R&D Systems. ND, not detected. | |||||
| ST6GalNAc1b | 6-Sialyl-T | ND | 15 | 16 | 17 |
| 21.2% | 47.5% | 9.4% | 33.8% | ||
| ST6GalNAc2b | 6-Sialyl-T | ND | ND | ND | ND |
| 4.3% | |||||
| ST6GalNAc4b | 6-Sialyl-T | Disialyl-T | ND | ND | ND |
| 11.8% | 52.9% | ||||
| ST6GalNAc4c | 6-Sialyl-T | Disialyl-T | ND | ND | ND |
| 58.0% | 3.7% | ||||
| ST6GalNAc5b | 6-Sialyl-T | Disialyl-T | ND | ND | ND |
| 2.4% | 3.9% | ||||
| ST6GalNAc6b | 6-Sialyl-T | Disialyl-T | ND | ND | ND |
| 6.2% | 7.1% | ||||
As expected, both ST6GalNAc1 and ST6GalNAc2 catalyzed the sialylation of core 1 to generate 6-sialyl-T, despite low conversion rates of 21.2% and 4.3%, respectively. This may be explained by the possible low activity of ST6GalNAcs to Fmoc-protected glyco-amino acids, as parallel reactions using a MUC1 glycopeptide bearing core 1 gave excellent yields (Fig. S2†). Most surprisingly, HPLC profiles of reactions catalyzed by ST6GalNAc4–6 showed two new peaks, one (TR = 14.48 min) corresponding to 6-sialyl-T as expected, while the other one (TR = 11.45 min) corresponding to disialyl-T (Neu5Acα2-3Galβ1-3(Neu5Acα2-6)GalNAcα) (Fig. S3†). The conversion rate of the ST6GalNAc4-catalyzed reaction was much higher than those of ST6GalNAc5–6 catalyzed ones, with 11.8% of 6-sialyl-T and 52.9% of disialyl-T (Table 2, Fig. S3†). Controversially, ST6GalNAc4 bearing a His6-tag yielded 58.0% of 6-sialyl-T but only 3.9% disialyl-T. Such disparate activities may result from fused tags (N-terminal GFP vs. C-terminal His6) and glycosylation patterns derived from expression cell lines (CHO vs. mouse myeloma). Nevertheless, both 6-sialyl-T and disialyl-T were observed in all reactions catalyzed by ST6GalNAc4–6, suggesting that they could recognize core 1 (T antigen) besides their preferred substrate 3′-sialyl-T,46 and surprisingly may also possess weak α2-3sialyltransferase activity (at least in vitro) to sialylate T or 6-sialyl-T antigens, which subsequently converted to disialyl-T.
Activity assay of these ST6GalNAcs towards rare cores revealed that only ST6GalNAc1 is active towards α-linked cores 5 and 8, generating sialyl-core 5 and sialyl-core 8 with moderate conversion rates of 47.5% and 33.8%. Interestingly, ST6GalNAc1 also sialylated core 7 with a low conversion rate of 9.4% (ESI†). These results double-confirmed that ST6GalNAc1 has a broad substrate specificity.46 None of other ST6GalNAcTs showed activity towards the 3 α-linked rare cores. ST6GalNAc3 was not tested due to unavailability and fails in heterogeneous expression attempts. Nevertheless, our results indicated that it is highly likely that ST6GalNAc1 rather than other ST6GalNAcTs is involved in the biosynthesis of sialyl-core 5 and 8. Accordingly, putative biosynthetic routes for these glycans were proposed (Fig. S4†).
O-GalNAc core glycan microarray assay
With herein prepared rare cores 5, 7, and 8, previously prepared Tn antigen, cores 1–4 and 6,26 and their α2-6sialylated forms, we fabricated an O-GalNAc core glycan microarray (Fig. 3A) as a tool to study the recognition of a set of GalNAc- and core O-glycan-specific lectins, including Wisteria floribunda lectin (WFL), Vicia villosa lectin (VVL), soybean agglutinin (SBA), Dolichos biflorus agglutinin (DBA), Jacalin, and peanut agglutinin (PNA). These lectins have been widely applied to probe O-GalNAc glycans in research and clinical settings. For example, WFL was used as a serum biomarker probe in various disease.47 PNA and jacalin were used as tools for cancer diagnosis and O-glycopeptide capturing.48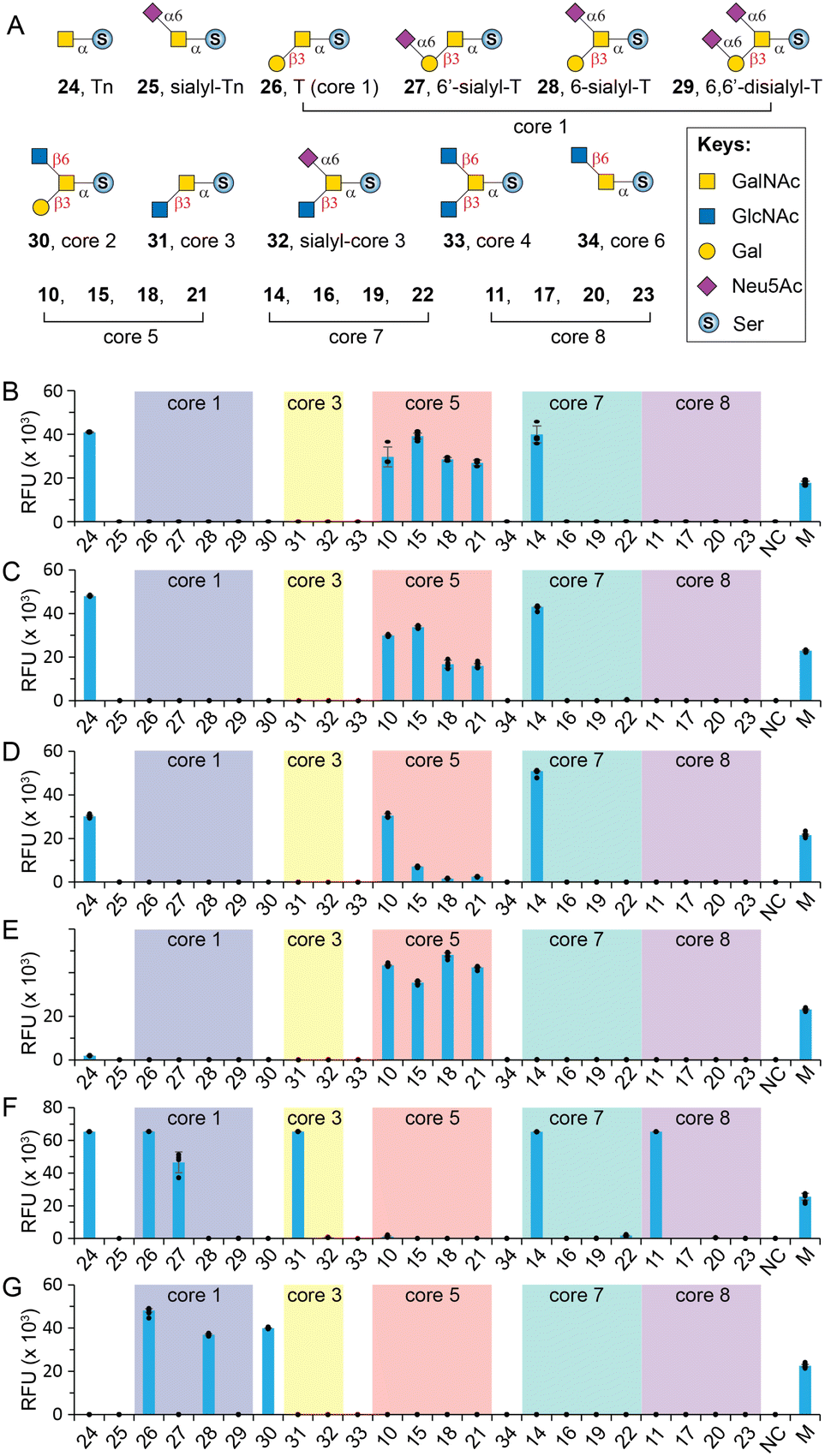 | ||
| Fig. 3 Glycan microarray analysis of GalNAc-specific lectins. (A) O-GalNAc glycan structures printed on the array; (B) WFL; (C) VVL; (D) SBA; (E) DBA; (F) Jacalin; (G) PNA. M = marker (0.01 mg mL−1 Alexs647-conjugated anti-human IgG); NC = printing buffer negative control. The individual data points are shown as dots. Data are presented as mean values. Error bars represent standard deviation. | ||
Our results showed that WFL (Fig. 3B) and VVL (Fig. 3C) strongly bind all structures with a terminal unmodified GalNAc residue, including Tn antigen (24), core 5 (10), sialyl-core 5 (15, 18, and 21), and core 7 (14), concordant with previous reports.49 Modifications on either C3–OH (e.g., cores 1, 3, and 8) or C6–OH (e.g., sialyl-T, core 6, and sialyl-core 7) of GalNAc completely abolished the bindings. SBA has a similar binding profile towards the O-glycan core array and prefers terminal GalNAc (Fig. 3D). Unlike WFL and VVL, α2-6sialylation of proximal residues (e.g., sialyl-core 5) significantly diminished bindings (Fig. 3D).
DBA has been used as a probe for terminal α-GalNAc residues and to bind blood group A antigen. However, Forssman antigen (GalNAcα1-3GalNAcβ) was identified to be the best binder of DBA, whereas only weak binding was observed to other α-GalNAc terminal glycans.49,50 In consonanance with this, we found that DBA very weakly binds or didn't bind Tn (24) or core 7 (14) with a terminal α-GalNAc residue (Fig. 3E). In contrast, it strongly binds core 5 (10) that contains an α-linked Forssman disaccharide (GalNAcα1-3GalNAcα). Surprisingly, DBA well tolerated α2-6sialylation on the initial GalNAc residue (15, 18, 21) (Fig. 3E). These results identified GalNAcα1-3GalNAc disaccharide instead of the β-linked Forssman antigen as the minimum binding motif of DBA.
Jacalin is generally considered a T-antigen binder, but several reports concluded that the lectin primarily binds C3–OH substituted GalNAcα motifs.26,49,51 It also binds Tn antigen but substitution at the C6 of core GalNAc is not tolerated according to Consortium for Functional Glycomics (CFG) microarray data.49 Consistent with these, our results (Fig. 3F) showed strong binding to Tn (24), T (26), 6′-sialyl-T (27), core 3 (31), and core 8 (11) but not sialyl-core 8 (17, 20, and 23). In addition, Jacalin didn't recognize core 5, further confirming that this Forssman antigen-like structure is not a binder.49 Interestingly, Jacalin showed comparable strong binding to core 7 (14) that contains an α1-6GalNAc modification on the initial GalNAc, against previous conclusions.26,49,51 It is possible that the binding site of Jacalin on core 7 is the distal α-GalNAc instead of the initial α-GalNAc. Collectively, O-GalNAc glycan recognized by Jacalin include Tn, core 1, core 3, core 8, their extended structures without C6-modification on the core GalNAc, and core 7. In contrast, PNA is a strict T-antigen binder (Fig. 3G), which recognizes the Galβ1-3GalNAc motif that is devoid of any modification on the Gal residue (26, 28, and 30).49
Conclusions
In summary, we developed a convergent chemoenzymatic strategy to synthesize O-GalNAc rare cores 5, 7, and 8 containing the challenging 1,2-cis linkage, and their sialylated forms. The strategy enabled exclusive α-stereoselectivity through the introduction of non-participating groups at C2 of glycosyl donors, the pre-activation of donors, and solvent-assisted glycosylation. The use of a bacterial α2-6sialyltransferase then enabled efficient and regioselective synthesis of natural sialyl-core 5, sialyl-core 8, and their derivatives. Together with a previous report,26 we have achieved the chemoenzymatic synthesis of all O-GalNAc cores and derived structures starting from one versatile precursor (protected glyco-amino acid 4). Such a convergent strategy could significantly expedite O-glycan synthesis. In addition, our in vitro activity assay of human ST6GalNAcs indicated that ST6GalNAc1 might be involved in the biosynthesis of sialylated rare cores. Finally, glycan microarray assays revealed that core 5 and sialyl-core 5 are good binders of WFL, VVL, and DBA, while core 7 but not sialyl forms are binders of WFL, VVL, SBA and Jacalin. None of the screened lectins bind core 8 except for Jacalin. Collectively, the convergent synthetic strategy, synthesized rare core structures, and the unique O-GalNAc core glycan microarray provide indispensable tools and probes to study the biosynthesis and structure-function relationships of O-glycans.Data availability
All associated experimental data can be found in ESI.†Author contributions
L. L. and M. R. G. conceived the project; M. R. G. performed chemical synthesis and NMR characterization; C. C. and S. B. performed enzymatic synthesis, compound purification, enzyme activity assay, and microarray fabrication and assay. S. W., J. H., Y. G., and W. X. helped in purification and MS analysis. L. L., M. R. G., and C. C. wrote the manuscript which was edited and approved by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by United States National Institutes of Health (NIH) awards (R44GM123820 & U54HL142019). S. B. is thankful for the support from the MBD Fellowship at Georgia State University. C. C. is thankful for the support from the China Postdoctoral Science Foundation (2021M691976 and 2021TQ0197) and Shandong Province Postdoctoral Innovative Talent Support Program.Notes and references
- C. Steentoft, S. Y. Vakhrushev, H. J. Joshi, Y. Kong, M. B. Vester-Christensen, K. T. Schjoldager, K. Lavrsen, S. Dabelsteen, N. B. Pedersen, L. Marcos-Silva, R. Gupta, E. P. Bennett, U. Mandel, S. Brunak, H. H. Wandall, S. B. Levery and H. Clausen, EMBO J., 2013, 32, 1478–1488 CrossRef CAS PubMed.
- S. Pinzon Martin, P. H. Seeberger and D. Varon Silva, Front. Chem., 2019, 7, 710 CrossRef PubMed.
- C. Reily, T. J. Stewart, M. B. Renfrow and J. Novak, Nat. Rev. Nephrol., 2019, 15, 346–366 CrossRef PubMed.
- I. Bagdonaite, E. M. H. Pallesen, M. I. Nielsen, E. P. Bennett and H. H. Wandall, Adv. Exp. Med. Biol., 2021, 1325, 25–60 CrossRef PubMed.
- S. A. Malaker, N. M. Riley, D. J. Shon, K. Pedram, V. Krishnan, O. Dorigo and C. R. Bertozzi, Nat. Commun., 2022, 13, 3542 CrossRef CAS PubMed.
- D. Ince, T. M. Lucas and S. A. Malaker, Curr. Opin. Chem. Biol., 2022, 69, 102174 CrossRef CAS PubMed.
- M. R. Kudelka, A. Antonopoulos, Y. Wang, D. M. Duong, X. Song, N. T. Seyfried, A. Dell, S. M. Haslam, R. D. Cummings and T. Ju, Nat. Methods, 2016, 13, 81–86 CrossRef CAS PubMed.
- N. Martinez-Saez, J. M. Peregrina and F. Corzana, Chem. Soc. Rev., 2017, 46, 7154–7175 RSC.
- N. Stergiou, M. Urschbach, A. Gabba, E. Schmitt, H. Kunz and P. Besenius, Chem. Rec., 2021, 21, 3313–3331 CrossRef CAS PubMed.
- A. Halim, G. Brinkmalm, U. Ruetschi, A. Westman-Brinkmalm, E. Portelius, H. Zetterberg, K. Blennow, G. Larson and J. Nilsson, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11848–11853 CrossRef CAS PubMed.
- H. C. Hang and C. R. Bertozzi, Bioorg. Med. Chem., 2005, 13, 5021–5034 CrossRef CAS PubMed.
- F. G. Hanisch, W. Chai, J. R. Rosankiewicz, A. M. Lawson, M. S. Stoll and T. Feizi, Eur. J. Biochem., 1993, 217, 645–655 CrossRef CAS PubMed.
- I. Brockhausen, H. H. Wandall, K. G. T. Hagen and P. Stanley, in Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, D. Mohnen, T. Kinoshita, N. H. Packer, J. H. Prestegard, R. L. Schnaar and P. H. Seeberger, Cold Spring Harbor, NY, 2022 Search PubMed.
- E. F. Hounsell, A. M. Lawson, J. Feeney, H. C. Gooi, N. J. Pickering, M. S. Stoll, S. C. Lui and T. Feizi, Eur. J. Biochem., 1985, 148, 367–377 CrossRef CAS PubMed.
- C. Jin, D. T. Kenny, E. C. Skoog, M. Padra, B. Adamczyk, V. Vitizeva, A. Thorell, V. Venkatakrishnan, S. K. Linden and N. G. Karlsson, Mol. Cell. Proteomics, 2017, 16, 759–769 CrossRef PubMed.
- A. Kurosaka, H. Nakajima, I. Funakoshi, M. Matsuyama, T. Nagayo and I. Yamashina, J. Biol. Chem., 1983, 258, 11594–11598 CrossRef CAS PubMed.
- C. Capon, Y. Leroy, J. M. Wieruszeski, G. Ricart, G. Strecker, J. Montreuil and B. Fournet, Eur. J. Biochem., 1989, 182, 139–152 CrossRef CAS PubMed.
- K. Madunic, O. A. Mayboroda, T. Zhang, J. Weber, G. J. Boons, H. Morreau, R. van Vlierberghe, T. van Wezel, G. S. M. Lageveen-Kammeijer and M. Wuhrer, Theranostics, 2022, 12, 4498–4512 CrossRef CAS PubMed.
- A. V. Savage, C. M. Donoghue, S. M. D'Arcy, C. A. Koeleman and D. H. van den Eijnden, Eur. J. Biochem., 1990, 192, 427–432 CrossRef CAS PubMed.
- T. Sumi, Y. Hama, H. Nakagawa, D. Maruyama and M. Asakawa, Fish Physiol. Biochem., 2001, 25, 11–17 CrossRef CAS.
- C. Jin, J. T. Padra, K. Sundell, H. Sundh, N. G. Karlsson and S. K. Linden, J. Proteome Res., 2015, 14, 3239–3251 CrossRef CAS PubMed.
- J. T. Padra, H. Sundh, K. Sundell, V. Venkatakrishnan, C. Jin, T. Samuelsson, N. G. Karlsson and S. K. Linden, Infect. Immun., 2017, 85, e00189 CrossRef CAS PubMed.
- K. A. Thomsson, J. Benktander, M. P. Quintana-Hayashi, S. Sharba and S. K. Lindén, Fish Shellfish Immunol., 2022, 131, 349–357 CrossRef CAS PubMed.
- W. G. Chai, E. F. Hounsell, G. C. Cashmore, J. R. Rosankiewicz, C. J. Bauer, J. Feeney, T. Feizi and A. M. Lawson, Eur. J. Biochem., 1992, 203, 257–268 CrossRef CAS PubMed.
- H. van Halbeek, A. M. Strang, M. Lhermitte, H. Rahmoune, G. Lamblin and P. Roussel, Glycobiology, 1994, 4, 203–219 CrossRef CAS PubMed.
- S. Wang, C. Chen, M. R. Gadi, V. Saikam, D. Liu, H. Zhu, R. Bollag, K. Liu, X. Chen, F. Wang, P. G. Wang, P. Ling, W. Guan and L. Li, Nat. Commun., 2021, 12, 3573 CrossRef CAS PubMed.
- S. Rio-Anneheim, H. Paulsen, M. Meldal and K. Bock, J. Chem. Soc., Perkin Trans. 1, 1995, 8, 1071–1081 RSC.
- D. Qiu and R. R. Koganty, Tetrahedron Lett., 1997, 38, 961–964 CrossRef CAS.
- K. Kakita, T. Tsuda, N. Suzuki, S. Nakamura, H. Nambu and S. Hashimoto, Tetrahedron, 2012, 68, 5005–5017 CrossRef CAS.
- M. Wakao, T. Miyahara, K. Iiboshi, N. Hashiguchi, N. Masunaga and Y. Suda, Carbohydr. Res., 2022, 516, 108565 CrossRef CAS PubMed.
- M. Maemura, A. Ohgaki, Y. Nakahara, H. Hojo and Y. Nakahara, Biosci., Biotechnol., Biochem., 2005, 69, 1575–1583 CrossRef CAS PubMed.
- X.-S. Ye and C.-H. Wong, J. Org. Chem., 2000, 65, 2410–2431 CrossRef CAS PubMed.
- S.-R. Lu, Y.-H. Lai, J.-H. Chen, C.-Y. Liu and K.-K. T. Mong, Angew. Chem., Int. Ed. Engl., 2011, 50, 7315–7320 CrossRef CAS PubMed.
- T. Nukada, A. Berces, M. Z. Zgierski and D. M. Whitfield, J. Am. Chem. Soc., 1998, 120, 13291–13295 CrossRef CAS.
- D. Crich, Acc. Chem. Res., 2010, 43, 1144–1153 CrossRef CAS PubMed.
- S. S. Nigudkar and A. V. Demchenko, Chem. Sci., 2015, 6, 2687–2704 RSC.
- R. A. Mensink and T. J. Boltje, Eur. J. Chem., 2017, 23, 17637–17653 CrossRef CAS PubMed.
- G. Wasonga, Y. Zeng and X. Huang, Sci. China: Chem., 2011, 54, 66–73 CrossRef CAS PubMed.
- P. O. Adero, H. Amarasekara, P. Wen, L. Bohé and D. Crich, Chem. Rev., 2018, 118, 8242–8284 CrossRef CAS PubMed.
- A. Kafle, J. Liu and L. Cui, Can. J. Chem., 2016, 94, 894–901 CrossRef CAS.
- H. Yu, S. Huang, H. Chokhawala, M. Sun, H. Zheng and X. Chen, Angew. Chem., Int. Ed. Engl., 2006, 45, 3938–3944 CrossRef CAS PubMed.
- N. Lu, J. Ye, J. Cheng, A. Sasmal, C. C. Liu, W. Yao, J. Yan, N. Khan, W. Yi, A. Varki and H. Cao, J. Am. Chem. Soc., 2019, 141, 4547–4552 CrossRef CAS PubMed.
- L. Ding, H. Yu, K. Lau, Y. Li, S. Muthana, J. Wang and X. Chen, Chem. Commun., 2011, 47, 8691–8693 RSC.
- X. Chen and A. Varki, ACS Chem. Biol., 2010, 5, 163–176 CrossRef CAS PubMed.
- N. Taniguchi, K. Honke, M. Fukuda, H. Narimatsu, Y. Yamaguchi and T. Angata, Handbook of glycosyltransferases and related genes, Springer, 2014 Search PubMed.
- L. Wen, D. Liu, Y. Zheng, K. Huang, X. Cao, J. Song and P. G. Wang, ACS Cent. Sci., 2018, 4, 451–457 CrossRef CAS PubMed.
- H. Narimatsu and T. Sato, Expert Rev. Proteomics, 2018, 15, 183–190 CrossRef CAS PubMed.
- G. Poiroux, A. Barre, E. J. M. van Damme, H. Benoist and P. Rouge, Int. J. Mol. Sci., 2017, 18, 1232 CrossRef PubMed.
- D. Bojar, L. Meche, G. Meng, W. Eng, D. F. Smith, R. D. Cummings and L. K. Mahal, ACS Chem. Biol., 2022, 17, 2993–3012 CrossRef CAS PubMed.
- D. A. Baker, S. Sugii, E. A. Kabat, R. M. Ratcliffe, P. Hermentin and R. U. Lemieux, Biochemistry, 1983, 22, 2741–2750 CrossRef CAS PubMed.
- K. Tachibana, S. Nakamura, H. Wang, H. Iwasaki, K. Tachibana, K. Maebara, L. Cheng, J. Hirabayashi and H. Narimatsu, Glycobiology, 2006, 16, 46–53 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06925c |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |