
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselective C–H alkylation of anisoles with olefins by cationic imidazolin-2-iminato scandium(III) alkyl complexes†
Shiyu
Wang
,
Chenhao
Zhu
,
Lichao
Ning
,
Dawei
Li
,
Xiaoming
Feng
 and
Shunxi
Dong
*
and
Shunxi
Dong
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: dongs@scu.edu.cn
First published on 21st January 2023
Abstract
A new type of rare-earth alkyl complexes supported by monoanionic imidazolin-2-iminato ligands were synthesised and structurally characterised by X-ray diffraction and NMR analyses. The utility of these imidazolin-2-iminato rare-earth alkyl complexes in organic synthesis was demonstrated by their performance in highly regioselective C–H alkylation of anisoles with olefins. With as low as 0.5 mol% catalyst loading, various anisole derivatives without ortho-substitution or 2-methyl substituted anisoles reacted with several alkenes under mild conditions, producing the corresponding ortho-Csp2-H and benzylic Csp3-H alkylation products in high yield (56 examples, 16–99% yields). Control experiments revealed that rare-earth ions, ancillary imidazolin-2-iminato ligands, and basic ligands were crucial for the above transformations. Based on deuterium-labelling experiments, reaction kinetic studies, and theoretical calculations, a possible catalytic cycle was provided to elucidate the reaction mechanism.
Introduction
Anisole and its derivatives are frequently occurring structural units in many pharmaceuticals, natural products, and functional materials.1 The development of succinct and efficient approaches for the production of anisoles and their derivatives has therefore attracted significant interest in the past few decades.2–5 In particular, the C–H alkylation of anisoles with alkenes represents one of the most atom-efficient and environmentally benign synthetic routes.2–4 Although the well-known Friedel–Crafts type reactions of anisoles with alkenes via carbocation intermediates have been extensively investigated with Lewis and Brønsted acids as the catalysts, the control of regioselectivity has been problematic; a mixture of ortho- and para-regioisomers are always concomitantly generated in the reaction process (Scheme 1a).2 The metal–organic complex-mediated C–H alkylation of anisoles with alkenes via C–H activation is an alternative pathway to obtain alkylated anisoles.3,4 However, owing to the weak interaction between late-transition metals and the ether moiety, the use of transition metal-based catalysts in such transformations is fruitless.6 In contrast, rare-earth organic complexes benefit from their unique chemical properties and have been successfully disclosed to be efficient promoters by Hou and others.3 Taking advantage of the strong oxophilicity of rare-earth metal ions and their high activity towards olefin migration insertion, Hou and co-workers first accomplished intermolecular and intramolecular C–H bond addition of anisoles to various alkenes with cationic half-sandwich yttrium(III) or scandium(III) alkyl complexes as the catalysts (cat-1 and cat-2; Scheme 1b, left).3a,c In 2019, the group of Chen extended alkene substrates to 1,5-dienes and 1,6-dienes under the influence of cationic 2-picoline-tether-half-sandwich scandium(III) alkyl catalyst (cat-3). The cyclisation/hydroarylation reaction of aromatic ethers took place successfully, delivering several anisole derivatives in good yield with high regio- and diastereoselectivity (Scheme 1b, right).3b Overall, despite such impressive advances in this field, there is still ample room for improvement in terms of new catalysts and product distribution.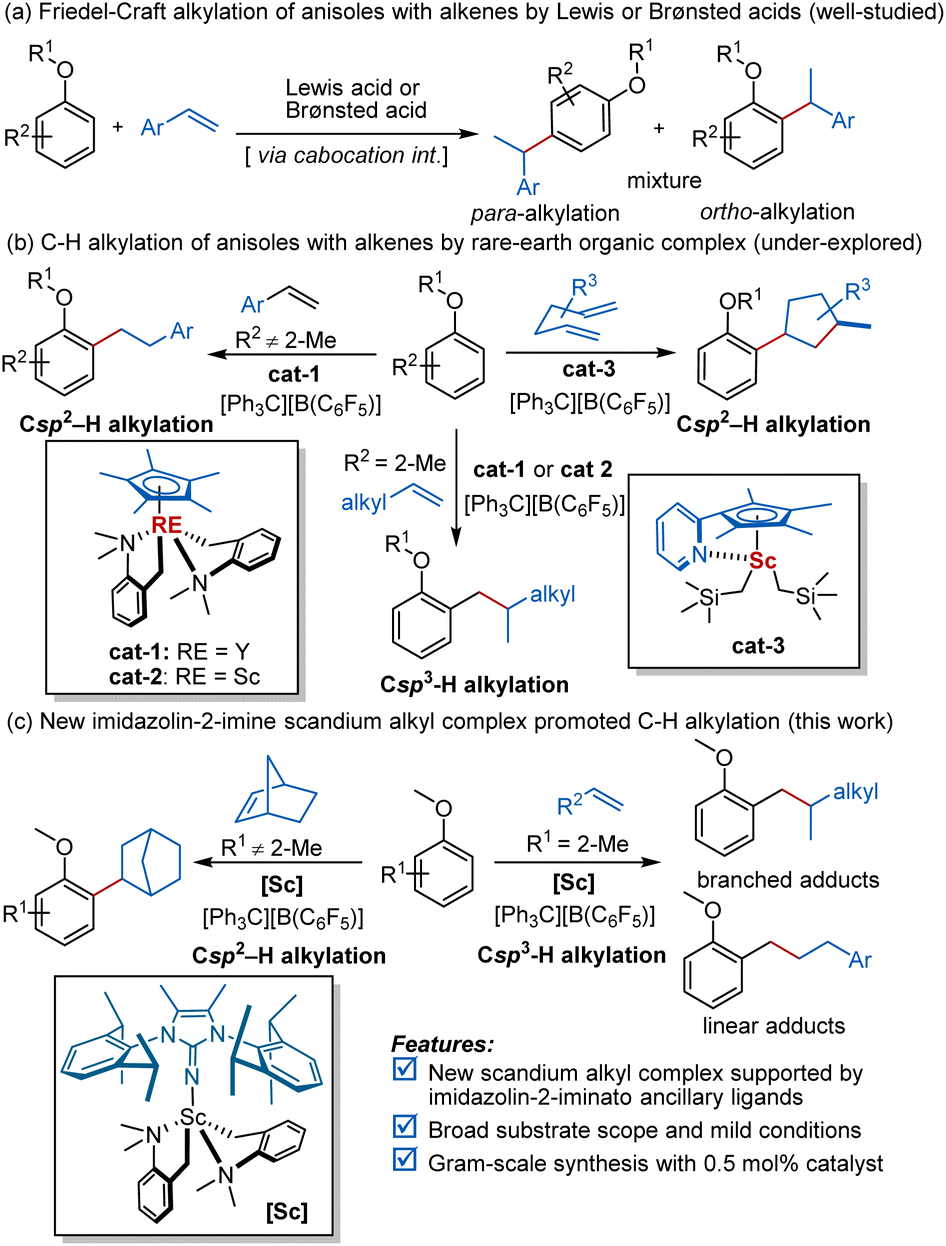 | ||
| Scheme 1 Catalytic synthesis of anisoles derivatives through C–H alkylation of anisoles with alkenes. | ||
Rare-earth organic complexes have been emerging as competent catalysts for several important organic transformations,7 including C–H functionalisation8 and polymerisation reactions.9 The development of this area, in particular C–H functionalisation, heavily relies on the use of cyclopentadienyl (Cp) and its analogues as ancillary ligands. The quest for alternatives of Cp and related aromatic ligands is of great interest and significance, but has met with limited success to date.10 Among them, N-heterocyclic iminato (NHI) ligands, for example, monoanionic imidazolin-2-iminato groups, have been successfully investigated as Cp-analogous ligands by the groups of Tamm, Inoue, Eisen, and others.11 Structurally, imidazolin-2-iminato groups are isolobally related to the Cp moiety. As shown in Scheme 2a, the two mesomeric structures of the imidazolin-2-iminato groups indicated that they can serve as strong 2σ,4π-electron N-donor ligands (Scheme 2a).12 Thanks to their strong electron donation and steric tunability, the related imidazolin-2-iminato rare-earth alkyl complexes exhibit high activity toward several reactions, including hydroamination,13 hydrosilylation,13 nucleophilic addition,14 and polymerisation.15 Nevertheless, it remains unclear whether imidazolin-2-iminato rare-earth alkyl complexes could be used in C–H activation. Motivated by the distinct selectivity and functional group tolerance frequently shown in rare-earth mediated C–H functionalisation8 and elegant work11 from Tamm's group, we envisaged that the judicious choice of rare-earth ions and basic ligands, as well as modification of imidazolin-2-iminato supporting ligands, may have the potential to achieve C–H alkylation with olefins. Herein, we wish to disclose our preliminary results along this line. An array of imidazolin-2-iminato rare-earth alkyl complexes were synthesised and structurally characterised by X-ray diffraction and NMR analyses. The cationic imidazolin-2-iminato scandium(III) alkyl complex was eventually identified as an efficient catalyst for highly regioselective C–H alkylation of anisole with ortho-Csp2-H and 2-methylanisole with Csp3-H (0.5–10 mol% catalyst loading, 56 examples, 16–99% yield). Notably, in comparison with cationic half-sandwich rare earth alkyl catalyst, a different catalytic performance was observed with these newly designed rare-earth metal complexes obtained from the reaction of 2-methylanisoles with styrenes.3a In addition, a possible catalytic cycle has been provided to understand the reaction mechanism based on deuterium-labelling experiments, reaction kinetic studies, and DFT calculations.
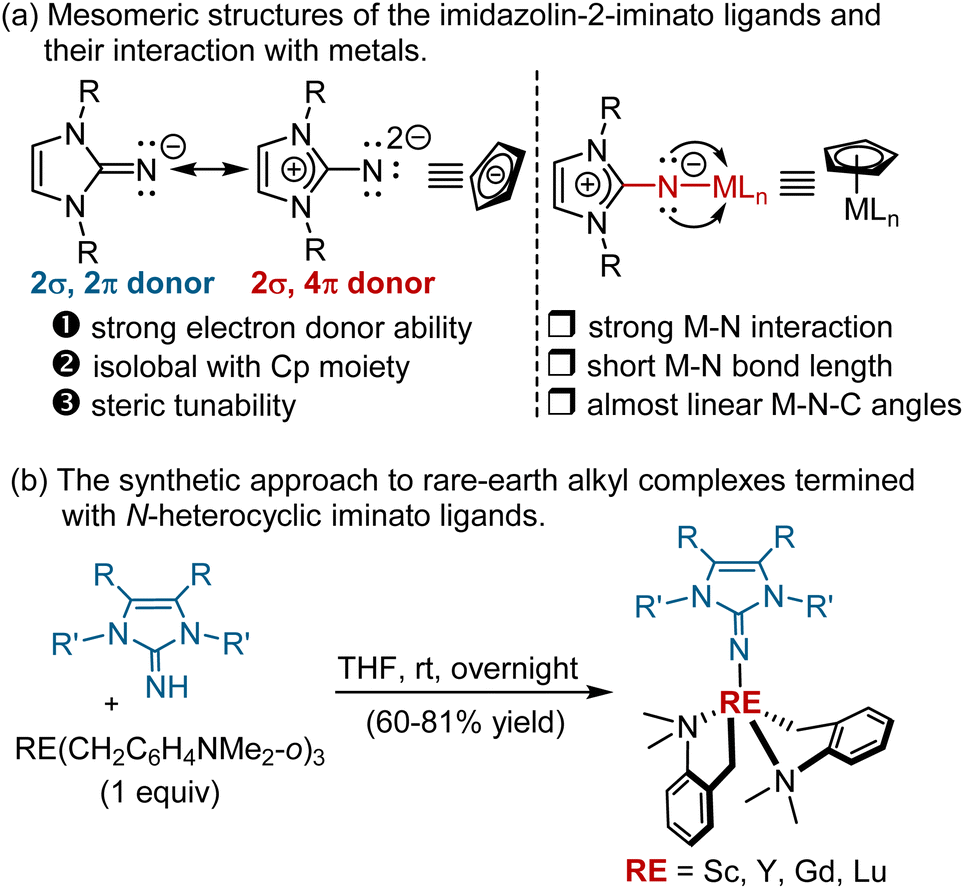 | ||
| Scheme 2 (a) The structural features of N-heterocyclic iminato ligands; and (b) synthetic method for their rare-earth alkyl complexes. | ||
Results and discussion
To validate the feasibility of the hypothesis, we first prepared a set of imidazolin-2-iminato rare-earth alkyl complexes by acid–base reaction of homoleptic tris(aminobenzyl) rare-earth complex RE(CH2C6H4NMe2-o)3 and 1 equivalent of imidazolin-2-imine16 in THF at room temperature for 12 h (Scheme 2b). The molecular structure of Sc-1, Sc-2, Y-1, and Gd-1 were established by single crystal X-ray diffraction analysis. As shown in single crystal structures, extremely short metal-nitrogen bonds [Sc–N: 1.956(4) Å, 1.971(3) Å, Y–N: 2.104(2) Å, Gd–N: 2.147(2) Å] and almost linear M-N-C angles (178.1–178.9°) were observed.17 To better understand the nature of the Sc–N bond, the bond order, localised molecular orbitals, and canonical molecular orbitals analysis were performed with Multiwfn software (see ESI† for more details). As depicted in Fig. 1a, the calculated Wiberg bond order of Sc–N is 1.36,18 indicating a strong interaction between the N atom of NHI and the scandium(III) ion. The decomposing Mayer bond order (MBO) analysis shows that one σ and two π orbitals contribute to the Sc–N bond. The molecule orbital analysis of Sc-1 also disclosed that except for the existence of one σ-bond, two p orbitals of the N atom in NHI form two π bonds with two d orbitals of the Sc ion, respectively, providing further evidence for their capability of acting as 2σ,4π-electron donors (Fig. 1b–d). The calculated ADCH charges suggested the charge donation from the NHI to the scandium(III) centre is −0.91 (for more details, see ESI†).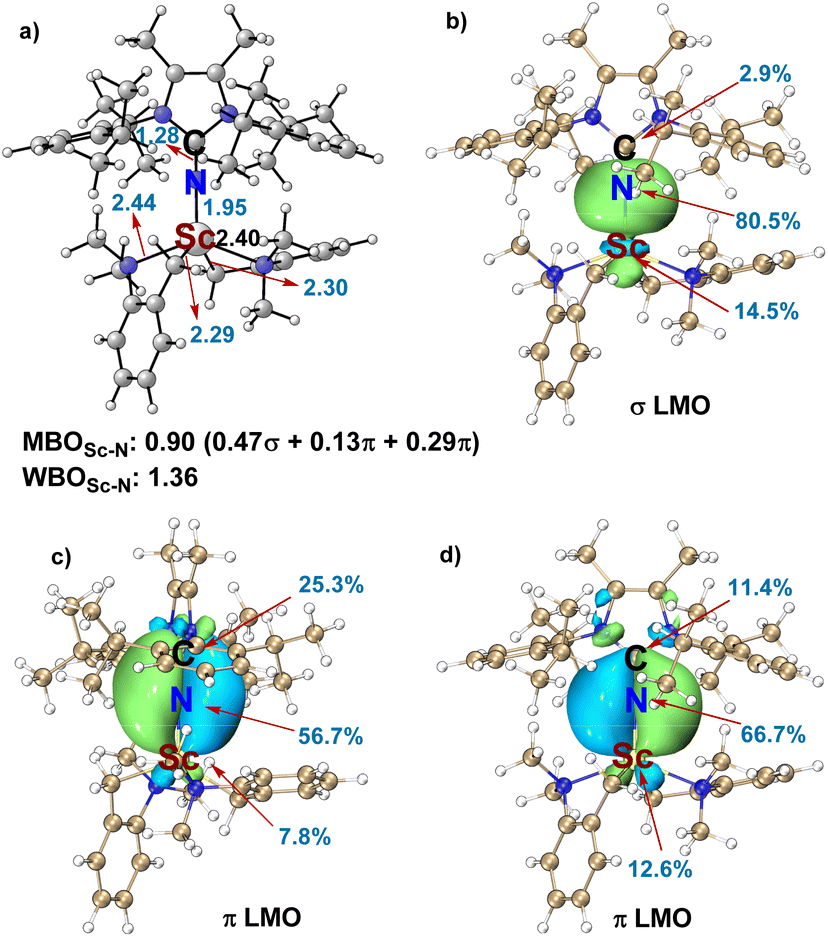 | ||
| Fig. 1 The Mayer bond order (MBO), Wiberg bond order (WBO), and the localised molecular orbitals of σ and π bond between the Sc and N atoms in the catalyst Sc-1 (isovalue = 0.02). | ||
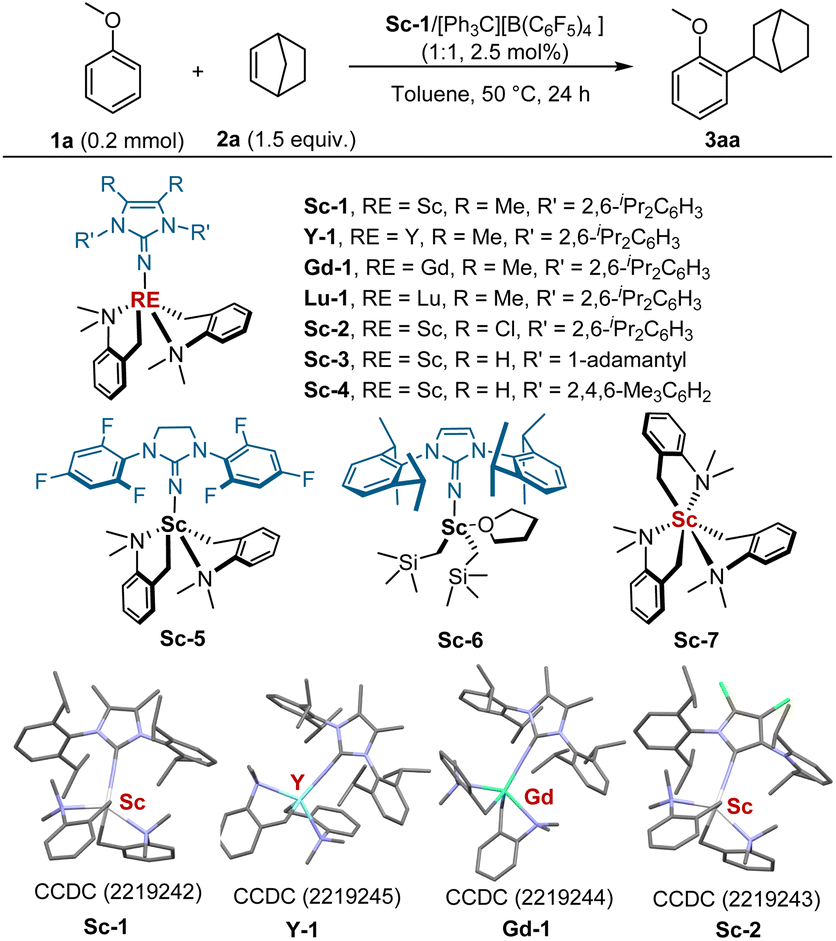
To investigate the catalytic activity of the newly designed imidazolin-2-iminato rare earth alkyl complexes, we conducted anisole Csp2-H alkylation and 2-methylanisole benzyl Csp3-H alkylation with alkenes. Initially, the reaction of methyl phenyl ether (1a) with norbornene (2a) was selected as the model reaction for optimisation. As summarised in Table 1, the rare-earth ions displayed a significant effect on the activity of catalysts. With 2.5 mol% imidazolin-2-iminato scandium bis(aminobenzyl) complex Sc-1 as the catalyst, the reaction of 1a and 2a underwent well in toluene at 50 °C for 24 h, affording the corresponding ortho-alkylation product 3aa in 99% yield (Table 1, entry 1). In stark contrast, no reaction was detected for its analogues, such as yttrium (Y-1), gadolinium (Gd-1), or lutecium (Lu-1) (entry 2). The effect of ligand structure on catalytic activity was examined. Changing 4,5-dimethyl groups on the skeleton of the imidazolin-2-imine ring to 4,5-dichloride presented comparable activity (entry 3, 99% yield). Decreasing the steric hindrance of the aromatic group on nitrogen atoms from 2,6-iPr2 to 2,4,6-Me3 or replacing 2,6-iPr2C6H3 with 1-adamantyl substitution led to no product. No reaction was observed with 1,3-bis(2,4,6-trifluorophenyl)imidazolidin-2-imine-derived Sc-5 (entry 4). Sc-6 with bis(noesilyl) [CH2Si(CH3)3] basic ligand also performed well with 99% yield (entry 5).19 Switching Sc-1 to tris(aminobenzyl) scandium(III) Sc-7 resulted in an obvious decrease in yield (entry 6). Performing the reaction without [Ph3C][B(C6F5)4] led to a full loss of activity (entry 7). Running the reaction without Sc-1 resulted in an obvious decrease in yield (entry 8). These results suggested that the cationic imidazolin-2-iminato scandium(III) alkyl complex was probably the real catalytic active species, and the presence of imidazolin-2-iminato ligand was crucial for the high activity observed. Next, other reaction parameters were studied (for more details, see ESI†). It was found that the reaction took place well in n-hexane, and 86% yield was obtained (entry 9). The coordinative solvents, for example, THF, inhibited the reaction (entry 10). When the reaction was carried out at a higher temperature (70 °C), an excellent yield was obtained (entry 11). A further decrease in temperature to 40 °C led to reduced yield (entry 12, 88% yield). Performing the reaction with 2 mol% of catalyst provided a slightly lower yield (91% vs. 99%). Further decrease in the catalyst loading to 1 mol% led to severe erosion in yield (entries 14, 47% yield).
|
|
||
|---|---|---|
| Entry | Deviations | Yield (%)b |
a Standard conditions: Sc-1/[Ph3C][B(C6F5)4] (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2.5 mol%), 1a (0.20 mmol), 2a (0.30 mmol) in toluene at 50 °C for 24 h.
b Yield was determined by 1H NMR with C2H2Br4 as an internal standard. Yield in brackets refers to isolated yield. :1, 2.5 mol%), 1a (0.20 mmol), 2a (0.30 mmol) in toluene at 50 °C for 24 h.
b Yield was determined by 1H NMR with C2H2Br4 as an internal standard. Yield in brackets refers to isolated yield.
|
||
| 1 | None | 99 (99) |
| 2 | 10 mol% Y-1, Gd-1 or Lu-1 | NR |
| 3 | 10 mol% Sc-2 | 99 |
| 4 | 10 mol% Sc-3, Sc-4 or Sc-5 | NR |
| 5 | 10 mol% Sc-6 | 99 |
| 6 | 10 mol% Sc-7 | 37 |
| 7 | No [Ph3C][B(C6F5)4] | NR |
| 8 | No Sc-1 | 30 |
| 9 | n-hexane | 86 |
| 10 | THF | NR |
| 11 | 70 °C | 94 (94) |
| 12 | 40 °C | 88 |
| 13 | 2 mol% Sc-1 | 91 (90) |
| 14 | 1 mol% Sc-1 | 47 (45) |
With the optimum reaction conditions in hand, we assessed the reactions of norbornene with various anisoles in the presence of Sc-1 (Scheme 3). Anisoles with different substituents at the para- and meta-position of the phenyl ring were all compatible in the current system, yielding the related ortho-alkylation products 3aa–3ia in 86–99% yields. Halide atoms (F, Cl, Br, I) at the para-position of the phenyl ring were retained after the reaction (3ea–3ha), which is different from the late-transition metals catalytic system.20 The reaction of the substrate with the 4-SMe group exclusively took place at the ortho-position of OMe in 99% yield (3ja), which was probably due to the strong oxophilicity of rare-earth metals. The anisoles bearing vinyl or allylic substitutions were well-tolerated as well, affording the corresponding adducts 3ka and 3la in 97 and 99% yields, respectively. In cases of meta-methyl anisole and 2-methoxynaphthalene, which have two different ortho-C–H bonds, the alkylation selectively occurred at less sterically congested positions (3ca and 3ma, 99% yields for both). For the reaction of anisole with the 4-OMe group, the increase in catalyst loading (5 mol% and prolonged reaction time 36 h) were necessary to get good results, and the NMR spectra indicated that the dialkylation adduct 3na was obtained (99% yield). Carrying out the reaction with 1 equivalent of norbornene led to the formation of monoalkylation product 3oa in 99% yield. The trimethylsilyl group at para-position also performed well with 99% yield (3pa). When 4-methoxy-N,N-dimethylaniline was used as the reaction partner, monoalkyation (3qa, 29%) and dialkylation adducts (3ra, 55%) were obtained. 2,3-Dihydrobenzofuran transformed into the product 3sa smoothly with 10 mol% catalyst loading (80% yield). Unfortunately, the reactions of methyl phenyl ether with other alkenes, such as styrene, 1-hexene, 1-octene, and vinyl trimethylsilane, were sluggish, and only low yields were obtained (16–23% yields). When other alkyl phenyl ethers, for instance, ethyl phenyl ether, were subjected to standard conditions, no reaction occurred at all (see ESI, Page 11 for more details†).
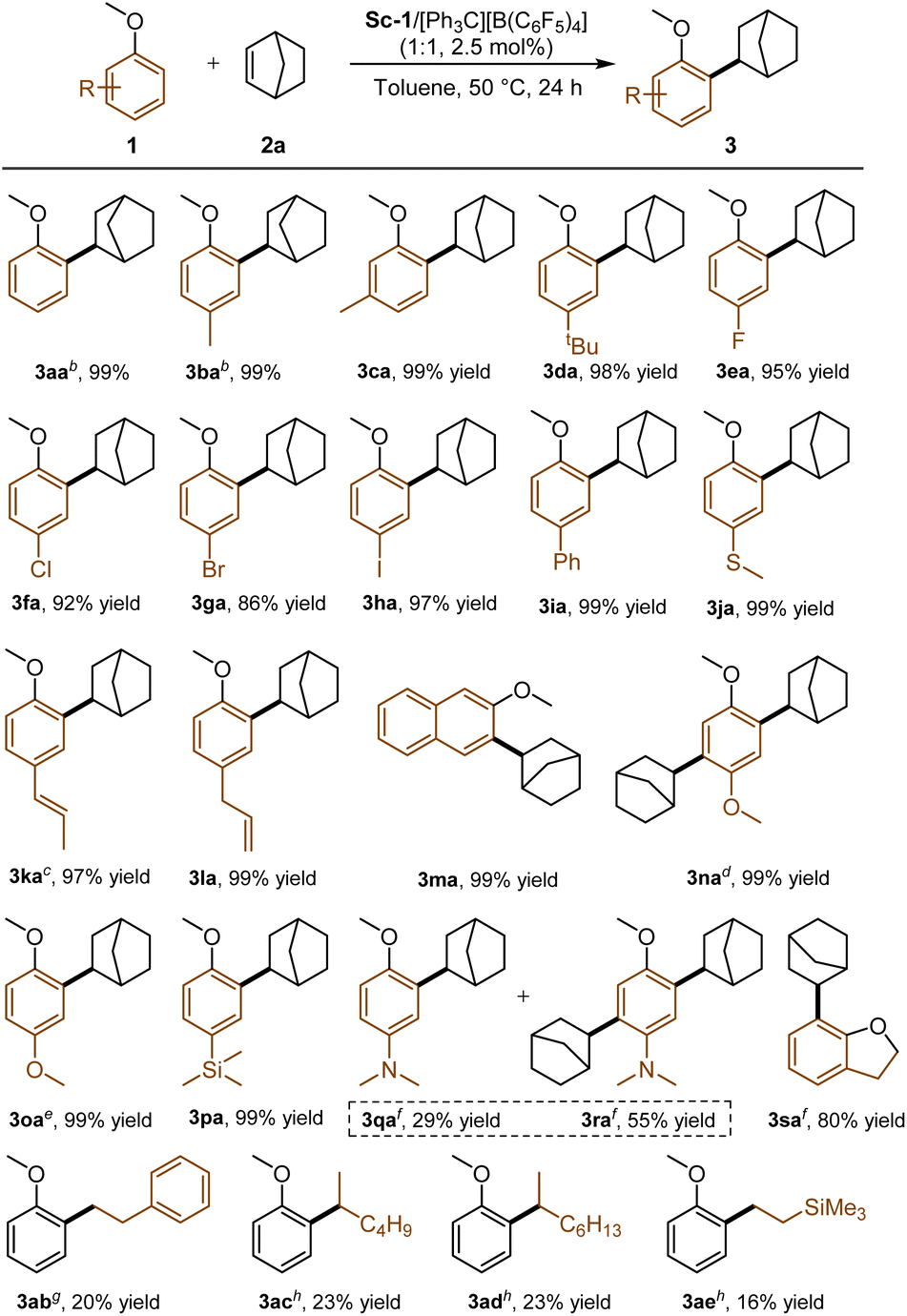 | ||
| Scheme 3 Substrate scope of anisoles. aUnless otherwise noted, all reactions were performed with Sc-1/[Ph3C][B(C6F5)4] (1:1, 2.5 mol%), 1 (0.20 mmol), 2a (0.60 mmol) in toluene (0.5 mL) at 50 °C for 24 h. b2a (0.30 mmol). c5 mol% catalyst. d5 mol% catalyst, 36 h. e5 mol% catalyst, 1 (0.20 mmol), 2a (0.20 mmol), 36 h. f10 mol% catalyst, 1 (0.20 mmol), 2a (1.20 mmol), 100 °C. g10 mol% catalyst, 1a (0.30 mmol), 2 (0.20 mmol). h10 mol% catalyst, 1a (0.20 mmol), 2 (2.00 mmol). | ||
Encouraged by the above results, we extended the substrates to 2-methyl substituted anisoles. As illustrated in Scheme 4, with 2 mol% of Sc-1, 2-methyl substituted anisole (4a) reacted with 1-octene effectively at 70 °C for 24 h, and the desired benzylic Csp3-H alkylation product 6aa was isolated in 94% yield (for detailed optimisation, see ESI, Pages 12 and 13†). The scope of 2-methylanisoles was then investigated. 2-Methyl-substituted anisoles with an additional methyl substituted at 3-, 4-, or 6-positions of the phenyl ring were amenable to the reaction, yielding branched C–H alkylation products 6ba–6da in 91–96% yields. The reactions of halide atoms substituted substrates were sluggish, and higher catalyst loading (10 mol%) was used. From fluorine to iodine, the isolated yield decreased gradually (6ea–6ha, 99 → 47%). In the case of 4-OMe substituted one, benzylic Csp3-H alkylation preferred to take place rather than ortho-Csp2-H alkylation (6ia, 94% yield), which was different from the previous report by Hou and co-workers.3a The use of norbornene instead of 1-octene led to a mixture of benzylic Csp3-H alkylation adduct 6ja and dialkylation product 6jb (ca. 1:9 ratio). Performing the reaction with 1.5 equivalents of 1,4-dimethoxy-2-methylbenzene provided 6ja as the sole product in 99% yield.
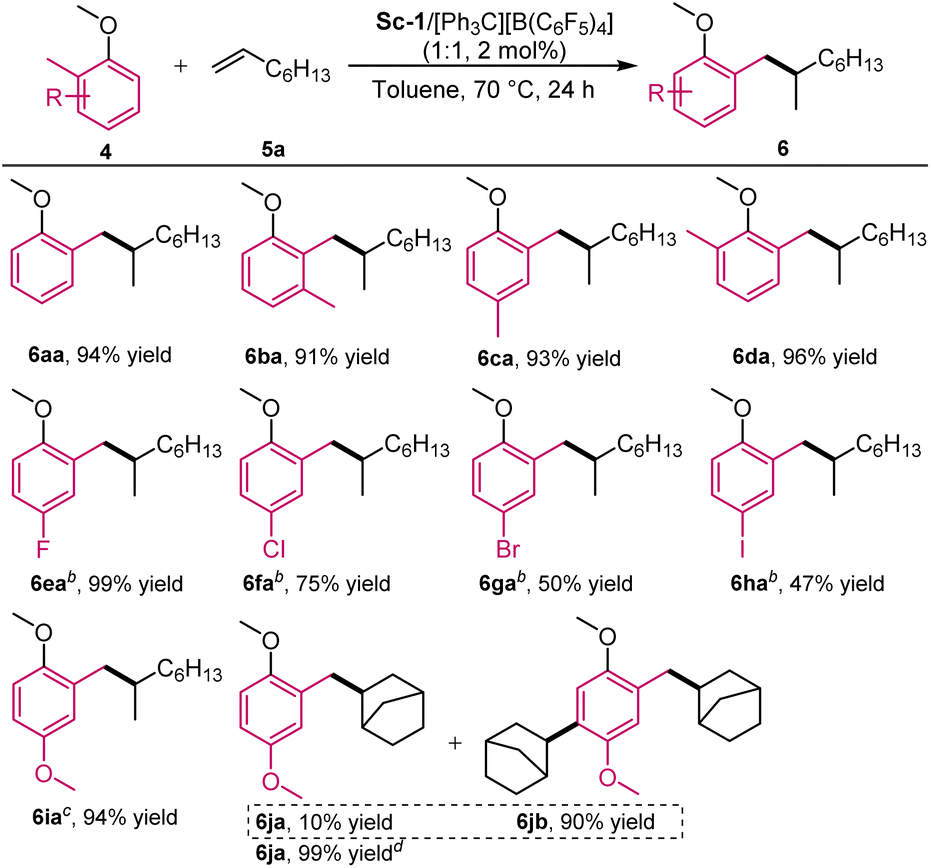 | ||
| Scheme 4 Substrate scope of 2-methylanisole. aUnless otherwise noted, all reactions were performed with Sc-1/[Ph3C][B(C6F5)4] (1:1, 2.0 mol%), 4 (0.20 mmol), 5a (0.60 mmol) in toluene (0.5 mL) at 70 °C for 24 h. The yield of isolated product. b10 mol% catalyst. c5 mol% of catalyst. dCarried out with 4a (0.30 mmol), norbornene 2a (0.20 mmol). | ||
Next, diverse alkenes were evaluated with 2-methyl substituted anisole (4a). Alkyl-substituted alkenes, such as 1-hexene, allyltrimethylsilane, and norbornene, were all suitable, affording branched benzyl Csp3-H alkylation products (6ab, 6ad, and 6ae) in 91–99% yields (Scheme 5). Notably, the use of vinyltrimethylsilane generated the linear product 6ac in 68% yield due to the ability of silicon to aid the stabilisation of negative charge.21 In the case of 1,5-hexadiene, continuous 2,1-insertion resulted in hydroalkylation and cyclisation product 6af in 99% yield with >98 cis. Vinyl cyclohexane was subjected to the reaction, branched product 6ag was produced in 81% yield. The present reaction was applicable for allyl cyclohexane, allylbenzene, and 2-allylnaphthalene as well, giving the corresponding products 6ah–6aj in 72–99% yields. In Hou's work,3a no reaction of 2-methylanisole with styrene occurred in the presence of a half-sandwich rare-earth complex. To our delight, styrene and its derivatives were feasible in the current reaction system, and linear products were generated as the major products with a variable amount of branched adducts.21 As shown in Scheme 5, the bottom part, ortho-, and meta-methyl substituted styrenes provided higher yield than para-substituted ones due to the formation of less amount of branched by-products (85 and 81% yield vs. 75% yield). Relatively lower yields (6aq: 42% yield; 6ar: 68% yield) were afforded for the reaction of styrenes with fluoro or bromine groups than those of other alkyl groups. Olefins derived from estradiol also transformed into the related product 6as in moderate yield (63%).
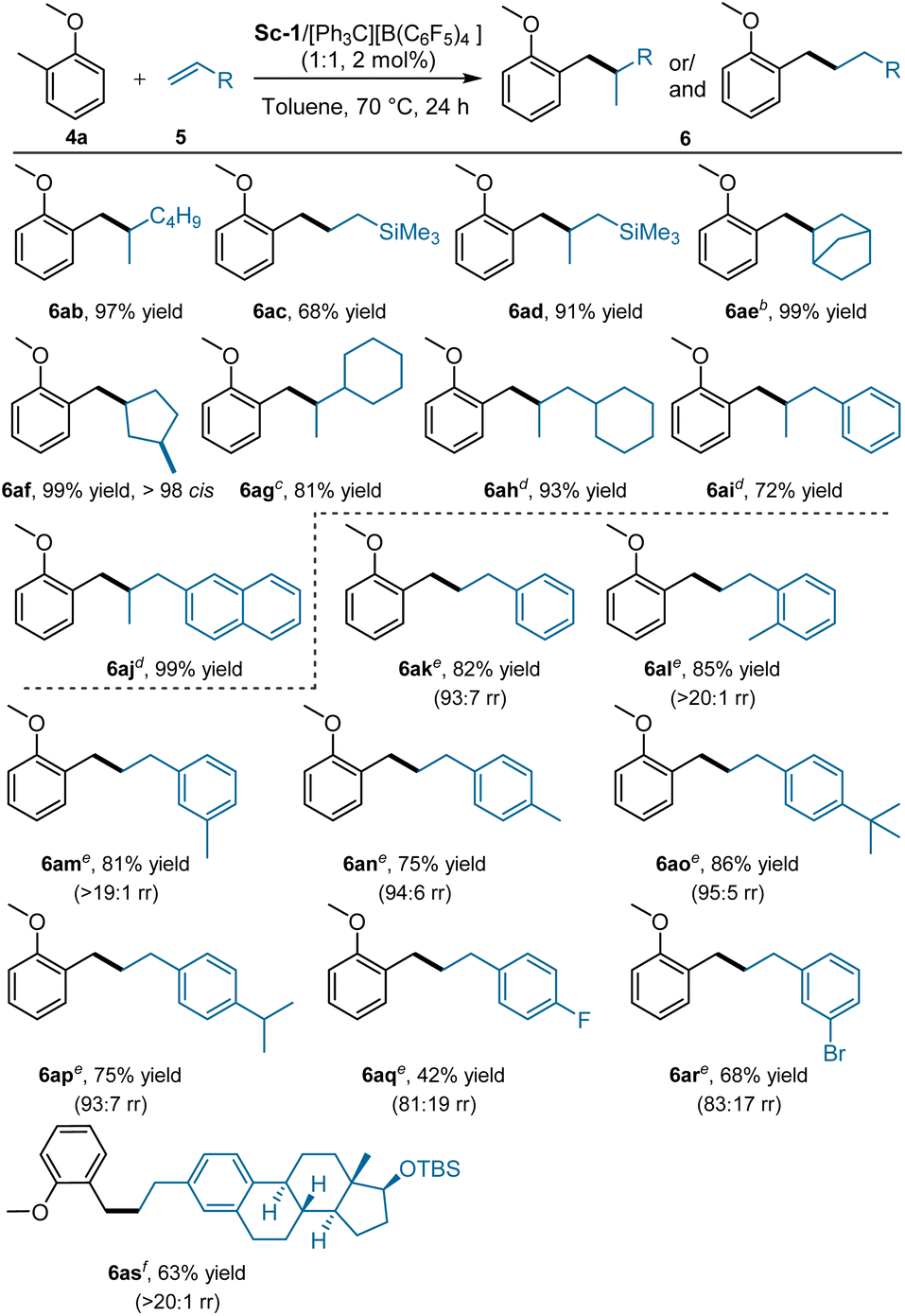 | ||
| Scheme 5 Substrate scope of alkenes. aUnless otherwise noted, all reactions were performed with Sc-1/[Ph3C][B(C6F5)4] (1:1, 2 mol%), 4a (0.20 mmol), 5 (0.60 mmol) in toluene (0.5 mL) at 70 °C for 24 h. The yield of isolated product. b0.5 mol% catalyst. c5 mol% catalyst. d10 mol% catalyst. eRun with 10 mol% catalyst, 4a (0.10 mmol), 5 (0.15 mmol). Yield of isolated linear product. fCarried out with 10 mol% catalyst, 4a (0.15 mmol), 5 (0.10 mmol). | ||
In addition, sulfides were feasible as well in the current reaction system.22 As shown in Scheme 6, high yields (9aa: 99% yield; 9ba: 99% yield) were obtained for the reaction of cyclohexyl(methyl)sulfane and methyl(pentyl)sulfane with norbornene. Vinyl cyclohexane converted into the related branched product 9ab with 72% yield. The reaction of styrene afforded the linear product 9ac in low yield (29%).
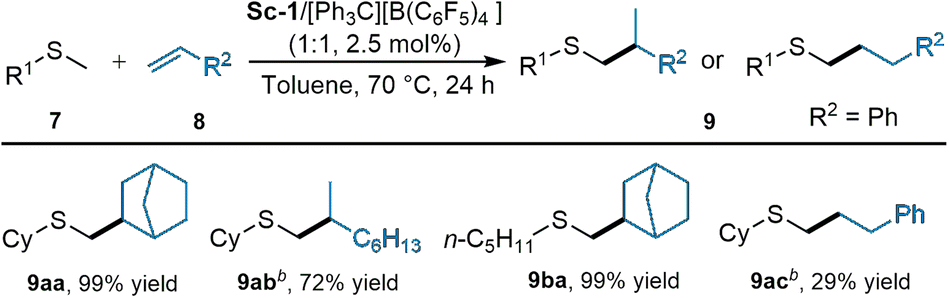 | ||
| Scheme 6 Substrate scope of sulfides. aUnless otherwise noted, all reactions were performed with Sc-1/[Ph3C][B(C6F5)4] (1:1, 2.5 mol%), 7 (0.20 mmol), 8 (0.60 mmol) in toluene (0.5 mL) at 70 °C for 24 h. The yield of isolated product. b10 mol% catalyst, 7a (1.0 mmol), 8 (0.20 mmol). | ||
To further demonstrate the synthetic utility of this protocol, a gram–scale reaction was conducted. As shown in Scheme 7, the reaction of 5 mmol of 4a with 1.5 equivalents of 2a in the presence of 0.5 mol% of Sc-1 proceeded smoothly, furnishing the corresponding benzyl Csp3-H alkylation product 6ae in 99% isolated yield (1.08 g) (Scheme 7).
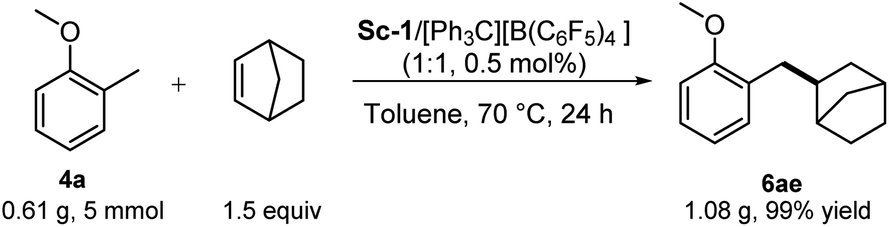 | ||
| Scheme 7 Gram-scale synthesis of 6ae. | ||
To gain further insights into the mechanism of Csp2-H alkylation of anisole and benzyl Csp3-H alkylation of 2-methylanisole, a series of deuterium-labelling and kinetic studies were carried out. Intramolecular KIE experiment of deuterium-labelled 4-methyl[2-D]anisole (1b-D) with norbornene (2a) showed a significant kinetic isotope effect (KIE) value of 5.2 (Scheme 8a), indicating that Csp2-H cleavage of anisole might be involved in the turnover limiting step. In Hou's work, an intramolecular kinetic isotopic effect was not observed (kH/KD = 1) in the reaction with styrene.3a Two sets of intermolecular KIE experiments of 2-methylanisole and 1-methoxy-2-(methyl-d3)benzene react with norbornene or 1-octene mixed in 1:1:3 molar ratio showed a significant kinetic isotope effect (KIE) value of 4.5 and 5.7, respectively (Scheme 8b and 8d). Two sets of parallel reactions were conducted and KIE values of 7.3 and 2.8 were observed (Scheme 8c and 8e). The above outcomes suggested that benzyl Csp3-H cleavage of 2-methylanisole might be involved in the turnover-limiting step as well. In addition, we measured the rate concentration dependences of 2-methylanisole (4a), 1-octene (5a), and catalyst precursor Sc-1. It was found that the reaction showed a first-order rate dependence on the concentration of 4a and the catalyst. Interestingly, the concentration of 5a also affected the reaction rate, an approximate first-order rate (0.75) dependence on the 5a concentration was observed. Overall, the reaction rate was affected by the concentration of 2-methylanisole (4a), 1-octene (5a), and the catalyst precursor Sc-1.
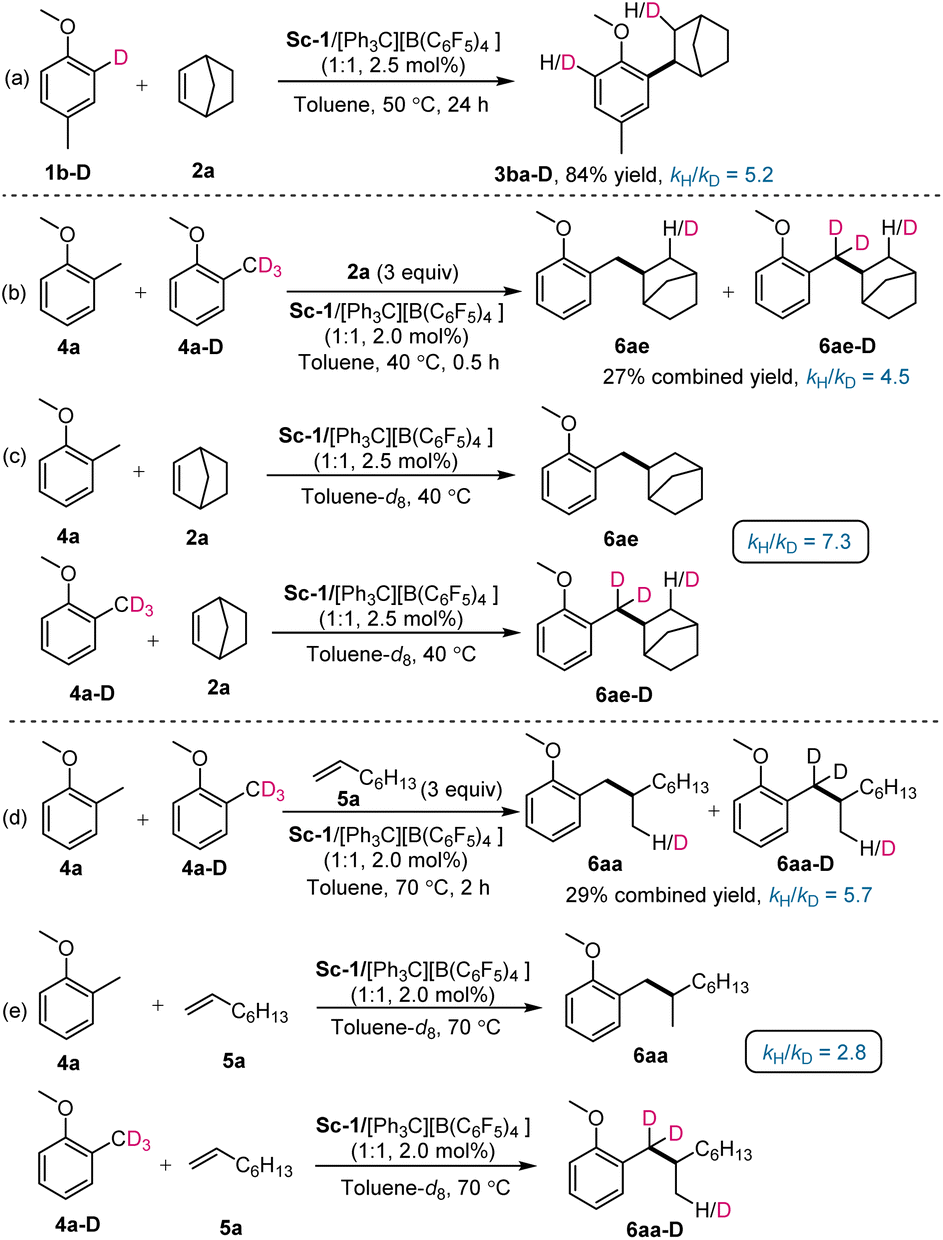 | ||
| Scheme 8 Deuterium-labelling experiments. | ||
To understand the reaction mechanism, theoretical calculations were performed by using Sc-1 in the formation 6aa. The detailed results, including Gibbs free energy profiles and structures, are summarised in Fig. 2. Initially, the catalyst precursor Sc-1 reacted with [Ph3C][B(C6F5)4] to generate the corresponding cationic scandium alkyl species, which was confirmed by 1H NMR analysis.23 The coordination of the methoxy group of 4a to scandium(III) provided the cationic scandium species, followed by selective deprotonation at benzylic position afforded the five-membered metallacycle intermediate INT1 through σ-bond metathesis, which was thought to be the real catalytic active species. According to the energy profile, the pathway involving another 2-methylanisole viaTS1 (9.8 kcal mol−1) was more favoured than the other pathway viaTS1' (15.2 kcal mol−1).24 Then, the simultaneous coordination of INT1 with another 4a and 1-octene (5a) furnished INT2, which subsequently underwent 1,2-insertion of 5a into the Sc–C bond in INT2 to give rise to intermediate INT3via the transition state TS1. This process needed to overcome an energy barrier of 9.8 kcal mol−1. In contrast, the 2,1-insertion of 1-octene into the Sc–C bond in INT2 was unfavoured in terms of the energy barrier (12.0 kcal mol−1vs. 9.8 kcal mol−1). In comparison, the DFT calculation suggested that the energy barrier of 1,2-insertion of styrene into the Sc–C bond was slightly higher than that of 2,1-insertion (10.9 kcal mol−1vs. 11.6 kcal mol−1, see ESI† for more details). The above results provide a rational explanation for the regioselectivity observed in experimental studies in Schemes 4 and 5. Upon activating the benzylic Csp3-H bond of another molecule 4a, intermediate INT4 was generated with an activation barrier of 12.6 kcal mol−1. Finally, INT4 liberated the desired product 6aa and regenerated catalytic species INT1. Although the turnover limiting step was identified to be the Csp3-H activation step, the activation barriers viaTS1 and TS2 were similar. Therefore, the concentration of alkene substrate had an obvious effect on the reaction rate as well, which was in agreement with kinetic results. In addition, the calculated KIE value (kH/kD) was 3.7, which was consistent with the KIE experimental result.
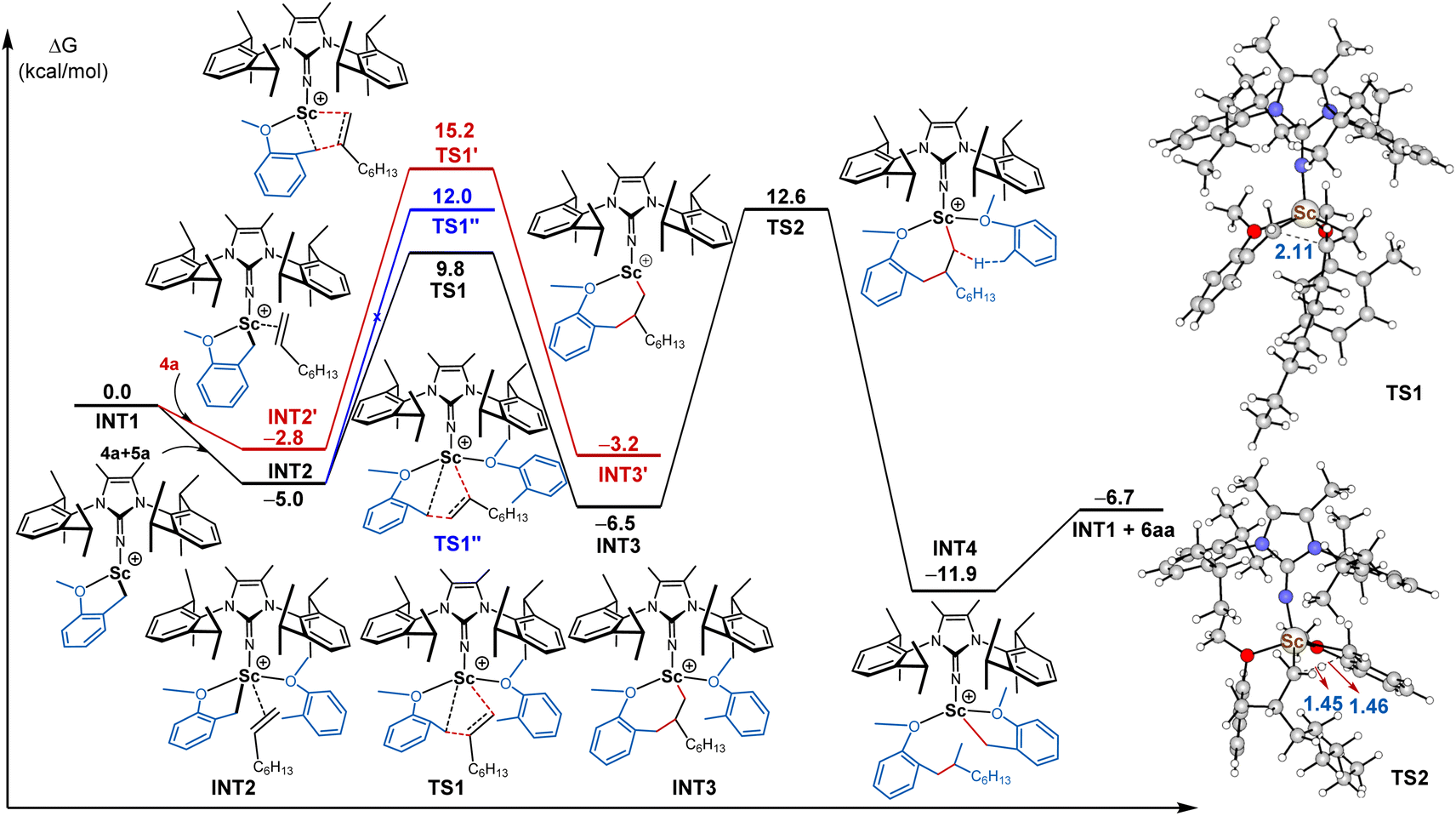 | ||
| Fig. 2 Energy profile for the regioselective benzylic Csp3-H alkylation of 4a with 5a. | ||
Next, the reaction of 1,4-dimethoxy-2-methylbenzene (4i) with 1-octene (5a) was studied by DFT calculations to further clarify the regioselectivity (see ESI† for more details). The results indicated that the Csp3-H activation pathway is more favourable than the Csp2-H activation pathway for the 1,4-dimethoxy-2-methylbenzene kinetically and thermodynamically. Therefore, only the Csp3-H alkylation product was obtained in the current reaction system.
Conclusions
A new type of imidazolin-2-iminato rare earth alkyl complexes was prepared and successfully employed as competent catalysts for C–H alkylation of anisoles and 2-methylanisoles with various alkenes. A series of anisole derivatives were obtained under mild conditions with high regioselectivity and yield. The experimental results indicated that the activation of the benzylic Csp3-H bond is preferred to that of ortho-Csp2-H in an aromatic ring. Rare-earth ions, ancillary imidazolin-2-iminato ligands, base ligands, and borate play a significant role in the titled transformations. Based on deuterium-labelling experiments, reaction kinetic studies, and DFT calculations, a catalytic cycle along with possible working modes were provided under the reaction mechanism. The development of chiral imidazolin-2-iminato rare earth alkyl complexes and their further utility in other related C–H alkylation reactions are in progress.25Data availability
The data supporting this article has been uploaded as part of the ESI.†Author contributions
S. Y. Wang performed experiments and prepared the ESI† and paper. C. H. Zhu repeated some experiments. L. C. Ning carried out the DFT calculations. D. W. Li helped with crystal growth. X. M. Feng and S. X. Dong supervised the project and polished the manuscript and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (No. 92056107, 22271199, 92256303 and 21890723) and Sichuan University (2020SCUNL204).Notes and references
- For selected examples of valuable molecules containing anisole moiety, see: (a) S. Shiotani, T. Kometani, K. Mitsuhashi, T. Nozawa, A. Kurobe and O. Futsukaichi, J. Med. Chem., 1976, 19, 803 CrossRef CAS PubMed; (b) T. Pacher, C. Seger, D. Engelmeier, S. Vajrodaya, O. Hofer and H. Greger, J. Nat. Prod., 2002, 65, 820 CrossRef CAS PubMed; (c) L. Garrido, E. Zubía, M. J. Ortega and J. Salvá, J. Org. Chem., 2003, 68, 293 CrossRef CAS PubMed; (d) M. A. Grundl and D. Trauner, Org. Lett., 2006, 8, 23 CrossRef CAS PubMed; (e) T. Zhou, Q. Shi, K. F. Bastow and K.-H. Lee, J. Med. Chem., 2010, 53, 8700 CrossRef CAS PubMed; (f) R. Gaspari, A. E. Prota, K. Bargsten, A. Cavalli and M. O. Steinmetz, Chem, 2017, 2, 102 CrossRef CAS; (g) Z.-S. Liu, G.-G. Qian, Q.-W. Gao, P. Wang, H.-G. Cheng, Q. Wei, Q. Liu and Q.-H. Zhou, ACS Catal., 2018, 8, 4783 CrossRef CAS; (h) G. A. Pakora, J. Mpika and D. Kone, Environ. Sci. Pollut. Res., 2018, 25, 29901 CrossRef CAS PubMed; (i) L. Chen, M. P. Pu, S. Y. Li, X. P. Sang, X. H. Liu, Y. D. Wu and X. M. Feng, J. Am. Chem. Soc., 2021, 143, 19091 CrossRef CAS PubMed; (j) Y. Li, S. Xin, R. Weng, X. H. Liu and X. M. Feng, Chem. Sci., 2022, 13, 8871 RSC; (k) Y. Xu, H. K. Wang, Z. Yang, Y. Q. Zhou, Y. B. Liu and X. M. Feng, Chem, 2022, 8, 2011 CrossRef CAS.
- For selected examples of hydroalkylation of anisole via Friedel-Craft type reactions, see: (a) H.-B. Sun, B. Li, R.-M. Hua and Y.-W. Yin, Eur. J. Org. Chem., 2006, 18, 4231 CrossRef; (b) S. Y. Lee, A. V. Gale and C. C. Eichman, Org. Lett., 2016, 18, 5034 CrossRef CAS PubMed; (c) R. K. Nandi, F. Ratsch, R. Beaud, R. Guillot, C. Kouklovskya and G. Vincent, Chem. Commun., 2016, 52, 5328 RSC; (d) J. N. Bentley and C. B. Caputo, Organometallics, 2018, 37, 3654 CrossRef CAS; (e) C. D. T. Nielsen, A. J. P. White, D. Sale, J. Bures and A. C. Spivey, J. Org. Chem., 2019, 84, 14965 CrossRef CAS PubMed.
- (a) J. Oyamada and Z. Hou, Angew. Chem., Int. Ed., 2012, 51, 12828 CrossRef CAS PubMed; (b) B. Tang, X.-Y. Hu, C.-L. Liu, T. Jiang, F. Alam and Y.-H. Chen, ACS Catal., 2019, 9, 599 CrossRef CAS; (c) A. Mishra, P. Wu, X.-F. Cong, M. Nishiura, G. Luo and Z. Hou, ACS Catal., 2022, 12, 12973 CrossRef CAS.
- L.-X. Zhao, P. Deng, X. Gong, X.-H. Kang and J.-H. Cheng, ACS Catal., 2022, 12, 7877 CrossRef CAS.
- For selected examples, see: (a) J. Oyamada, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2011, 50, 10720 CrossRef CAS PubMed; (b) P. Ricci, K. Krämer and L. Larrosa, J. Am. Chem. Soc., 2014, 136, 18082 CrossRef CAS PubMed; (c) D. A. Frasco, S. Mukherjee, R. D. Sommer, C. M. Perry, N. S. Lambic, K. A. Abboud, E. Jakubikova and E. A. Ison, Organometallics, 2016, 35, 2435 CrossRef CAS; (d) C. Xue, Y. Luo, H.-L. Teng, Y.-H. Ma, M. Nishiura and Z. Hou, ACS Catal., 2018, 8, 5017 CrossRef CAS; (e) W. Xu, X. Cong, K. An, S.-J. Lou, Z. Li, M. Nishiura, T. Murahashi and Z. Hou, Angew. Chem., Int. Ed., 2022, 61, e202210624 CAS.
- R. Mandal, B. Garai and B. Sundararaju, ACS Catal., 2022, 12, 3452 CrossRef CAS.
- For the reviews on the application of rare-earth organic complexes in organic synthesis, see: (a) X. Zhang and J. Jiang, in Rare Earth Coordination Chemistry, ed. C. Huang, Wiley-VCH, Weinheim, 2010, ch. 4, p. 138 Search PubMed; (b) M. Zimmermann and R. Anwander, Chem. Rev., 2010, 110, 6194 CrossRef CAS PubMed; (c) R. Waterman, Organometallics, 2013, 32, 7249 CrossRef CAS; (d) K. R. D. Johnsona and P. G. Hayes, Chem. Soc. Rev., 2013, 42, 1947 RSC; (e) M. Nishiura, F. Guo and Z. Hou, Acc. Chem. Res., 2015, 48, 2209 CrossRef CAS PubMed; (f) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192 CrossRef CAS PubMed.
- For the reviews on C–H functionalization promoted by rare-earth alkyl complexes, see: (a) H. Tsurugi, K. Yamamoto, H. Nagae, H. Kanekoa and K. Mashima, Dalton Trans., 2014, 43, 2331 RSC; (b) J. Hannedouche and E. Schulz, Organometallics, 2018, 37, 4313 CrossRef CAS; (c) Q. Sun, X. Xu and X. Xu, ChemCatChem, 2022, e202201083 CAS; (d) M. Inoue, H. Tsurugi and K. Mashima, Coord. Chem. Rev., 2022, 473, 214810 CrossRef CAS; (e) K. Seth, Org. Chem. Front., 2022, 9, 3102 RSC.
- For selected examples of polymerization reactions promoted by rare-earth alkyl complexes, see: (a) Y. Matsuo, K. Mashima and K. Tani, Organometallics, 2001, 20, 3510 CrossRef CAS; (b) Z. Hou, in Multimetallic Catalysts in Organic Synthesis, eds M. Shibasaki and Y. Yamamoto, WILEY-VCH, Weinheim, 2004, ch. 6, p. 143 Search PubMed; (c) K. Beckerle and J. Okuda, in Syndiotactic Polystyrene, ed. J. Schellenberg, WILEY-VCH, Weinheim, 2009, ch. 7, p. 125 Search PubMed; (d) Z.-B. Jian and D.-M. Cui, Dalton Trans., 2012, 41, 2367 RSC; (e) P. Xu, Y. Yao and X. Xu, Chem. – Eur. J., 2017, 23, 1263 CrossRef CAS PubMed; (f) Y. Yang, H. Wang, L. Huang, M. Nishiura, Y. Higaki and Z. Hou, Angew. Chem., Int. Ed., 2021, 60, 26192 CrossRef CAS PubMed.
- (a) Y. Chen, D. Song, J. Li, X. Hu, X. Bi, T. Jiang and Z. Hou, ChemCatChem, 2018, 10, 159 CrossRef CAS; (b) A. Kundu, M. Inoue, H. Nagae, H. Tsurugi and K. Mashima, J. Am. Chem. Soc., 2018, 140, 7332 CrossRef CAS PubMed; (c) B. Tang, X.-Y. Hu, C.-L. Liu, T. Jiang, F. Alam and Y.-H. Chen, ACS Catal., 2019, 9, 599 CrossRef CAS; (d) J. Su, Y. Zhou and X. Xu, Org. Biomol. Chem., 2019, 17, 2013 RSC; (e) J.-H. Su, Y.-C. Luo and X. Xu, Chem. Commun., 2021, 57, 3688 RSC.
- For the reviews on N-heterocyclic iminato ligands, see: (a) A. Trambitas, T. Panda and M. Tamm, Z. Anorg. Allg. Chem., 2010, 636, 2156 CrossRef CAS; (b) X. Wu and M. Tamm, Coord. Chem. Rev., 2014, 260, 116 CrossRef CAS; (c) T. Ochiai, D. Franz and S. Inoue, Chem. Soc. Rev., 2016, 45, 6327 RSC; (d) A. Doddi, M. Peters and M. Tamm, Chem. Rev., 2019, 119, 6994 CrossRef CAS PubMed; (e) S. Revathi, P. Raja, S. Saha, M. S. Eisen and T. Ghatak, Chem. Commun., 2021, 57, 5483 RSC.
- T. K. Panda, S. Randoll, C. G. Hrib, P. G. Jones, T. Bannenberg and M. Tamm, Chem. Commun., 2007, 5007 RSC.
- A. G. Trambitas, T. K. Panda, J. Jenter, P. W. Roesky, C. Daniliuc, C. G. Hrib, P. G. Jones and M. Tamm, Inorg. Chem., 2010, 49, 2435 CrossRef CAS PubMed.
- (a) B. D. Stubbert and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 4253 CrossRef CAS PubMed; (b) S. D. Wobser and T. J. Marks, Organometallics, 2013, 32, 2517 CrossRef CAS; (c) S. Saha and M. S. Eisen, ACS Catal., 2019, 9, 5947 CrossRef CAS.
- (a) E. Barnea, D. Moradove, J. C. Berthet, M. Ephritikhine and M. S. Eisen, Organometallics, 2006, 25, 320 CrossRef CAS; (b) W.-P. Zhao, B.-B. Wang, B. Dong, H. Liu, Y.-M. Hu, M. S. Eisen and X.-Q. Zhang, Organometallics, 2020, 39, 3983 CrossRef CAS.
- Imidazolin-2-imines are guanidine derivatives, which have been employed as an important type of ligands and catalysts in organometallic chemistry of rare-earth elements and organic synthesis. For selected reviews, see: (a) F. T. Edelmann, Chem. Soc. Rev., 2012, 41, 7657 RSC; (b) S. X. Dong, X. M. Feng and X. H. Liu, Chem. Soc. Rev., 2018, 47, 8525 RSC; (c) H.-C. Chou, D. Leow and C.-H. Tan, Chem.–Asian J., 2019, 14, 3803 CrossRef CAS PubMed.
- For a detailed comparison with related rare–earth alkyl complexes supported by imidazolin-2-iminato ligands, see ESI,† Page 34 for more details..
- The calculated Wiberg bond order of Sc–Nimido is 1.32 in Chen’s work, see: (a) E. Lu, Y.-X. Li and Y.-F. Chen, Chem. Commun., 2010, 46, 4469 RSC; (b) E. Lu, J.-X. Chu and Y.-F. Chen, Acc. Chem. Res., 2018, 51, 557 CrossRef CAS PubMed.
- In the following investigation, Sc-1 was superior than Sc-2 and Sc-6 in the reactions of 2-methyl anisoles with styrenes. therefore, Sc-1 was selected as the optimal catalyst for the examination of substrate scope..
- S. Gatard, R. Çelenligil-Çetin, C.-Y. Guo, B. M. Foxman and O. V. Ozerov, J. Am. Chem. Soc., 2006, 128, 2808 CrossRef CAS PubMed.
- F. Liu, G. Luo, Z. Hou and Y. Luo, Organometallics, 2017, 36, 1557 CrossRef CAS.
- Y. Luo, Y.-H. Ma and Z. Hou, J. Am. Chem. Soc., 2018, 140, 114 CrossRef CAS PubMed.
- For more details, see ESI,† Page 15–18..
- Y. Zhou, P. Wu, F.-S. Cao, L. Shi, N. Zhang, Z.-G. Xue and G. Luo, RSC Adv., 2022, 12, 13593 RSC.
- S. J. Wang, C. F. Zhang, D. Li, Y. Q. Zhou, Z. S. Su, X. M. Feng and S. X. Dong, Sci. China: Chem., 2023, 66, 147 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C{1H} and 19F{1H} NMR. X-ray crystallographic data. CCDC [2219241–2219245]. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06725k |
| This journal is © The Royal Society of Chemistry 2023 |