
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFree radicals promote electrocatalytic nitrogen oxidation†
Zuochao
Wang
a,
Jiao
Liu
a,
Huan
Zhao
a,
Wenxia
Xu
a,
Jiaxin
Liu
a,
Ziyi
Liu
a,
Jianping
Lai
 *a and
Lei
Wang
*ab
*a and
Lei
Wang
*ab
aState Key Laboratory of Eco-chemical Engineering, International Science and Technology Cooperation Base of Eco-chemical Engineering and Green Manufacturing, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao 266042, P. R. China. E-mail: jplai@qust.edu.cn; inorchemwl@126.com
bShandong Engineering Research Center for Marine Environment Corrosion and Safety Protection, College of Environment and Safety Engineering, Qingdao University of Science and Technology, Qingdao 266042, P. R. China
First published on 26th January 2023
Abstract
In this work, we introduce hydroxyl radicals into the electrocatalytic nitrogen oxidation reaction (NOR) for the first time. Cobalt tetroxide (Co3O4) acts not only as an electrocatalyst, but also as a nanozyme (in combination with hydrogen peroxide producing ˙OH), and can be used as a high-efficiency nitrogen oxidation reaction (NOR) electrocatalyst for environmental nitrate synthesis. Co3O4 + ˙OH shows an excellent nitrogen oxidation reaction (NOR) performance among Co3O4 catalysts in 0.1 M Na2SO4 solution. At an applied potential of 1.7 V vs. RHE, the HNO3 yield of Co3O4 + ˙OH reaches 89.35 μg h−1 mgcat−1, which is up to 7 times higher than that of Co3O4 (12.8 μg h−1 mgcat−1) and the corresponding FE is 20.4%. The TOF of Co3O4 + ˙OH at 1.7 V vs. RHE reaches 0.58 h−1, which is higher than that of Co3O4 (0.083 h−1), demonstrating that free radicals greatly enhance the intrinsic activity. Density functional theory (DFT) demonstrates that ˙OH not only can drive nitrogen adsorption, but also can decrease the energy barrier (rate-determining step) of N2 to N2OH*, thus producing great NOR activity.
Introduction
Nitrate plays an important role in promoting social progress and economic development,1–5 because it is a material for growing fertilizers and explosives.6–12 Nowadays, the industrial production of nitric acid mainly adopts high temperature (400–600 °C) and high pressure (15–25 MPa) conditions, involving a multi-step reaction Ostwald process,13–15 which is a high energy consumption and high emission production process.16–20 The electrocatalytic nitrogen oxidation reaction (NOR) to produce nitrate is clean, pollution-free and environmentally friendly,21–23 because the electrocatalytic NOR to produce nitrate only requires nitrogen and water in the environment as raw materials.24–26 At the same time, it does not discharge polluting gases and waste liquids.27–29 The electrocatalytic NOR to produce nitrate is very important to replace the Ostwald process with high energy consumption and high emission.25,30There are great challenges in the production of nitrate in an aqueous medium by the electrocatalytic NOR.31–34 The splitting of the strong N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond requires an oxidation potential in which the oxygen evolution reaction (OER) occurs readily,35,36 leading to extremely low Faraday efficiency (FE) of the NOR37–39 under ambient conditions. Therefore, it is ideal to find a catalytic system that promotes the NOR and inhibits the competing OER in neutral solution. In addition, it was previously reported that the adsorption of OH enhances the activity of the NOR, but the yield of nitrate was very low (<25 μg h−1 mgcat−1) due to the weak adsorption of N2 and a high energy barrier for the conversion of N2 to N2OH* (the rate-determining step). O2˙− radicals have been studied in the photocatalytic NOR, but no one has studied the role of free radicals in electrocatalytic nitrogen oxidation.9,35
N bond requires an oxidation potential in which the oxygen evolution reaction (OER) occurs readily,35,36 leading to extremely low Faraday efficiency (FE) of the NOR37–39 under ambient conditions. Therefore, it is ideal to find a catalytic system that promotes the NOR and inhibits the competing OER in neutral solution. In addition, it was previously reported that the adsorption of OH enhances the activity of the NOR, but the yield of nitrate was very low (<25 μg h−1 mgcat−1) due to the weak adsorption of N2 and a high energy barrier for the conversion of N2 to N2OH* (the rate-determining step). O2˙− radicals have been studied in the photocatalytic NOR, but no one has studied the role of free radicals in electrocatalytic nitrogen oxidation.9,35
In this work, we introduce hydroxyl radicals into the electrocatalytic NOR for the first time. Cobalt tetroxide (Co3O4) acts not only as an electrocatalyst, but also as a nanozyme (in combination with hydrogen peroxide producing ˙OH), and can be used as a high-efficiency NOR electrocatalyst for environmental nitrate synthesis. Co3O4 + ˙OH shows an excellent NOR performance among Co3O4 catalysts in 0.1 M Na2SO4 solution. At an applied potential of 1.7 V vs. RHE, the NO3− yield of Co3O4 + ˙OH is 7 times higher than that of Co3O4. We use ultraviolet spectroscopy and ion chromatography to determine the concentration of nitrate produced. At the same time, we also conduct 15N isotope labeling experiments by mass spectrometry to further determine the nitrogen source. The TOF of Co3O4 + ˙OH is higher than that of Co3O4, demonstrating that free radicals can greatly enhance the intrinsic activity. It is the most active non-precious metal catalyst reported so far for the NOR process (Table S1†). This catalyst also demonstrates long-term electrochemical and structural stability, and the current density did not have a tendency to decrease even in a 50 h continuous test. Density functional theory (DFT) demonstrates that ˙OH not only can drive nitrogen adsorption, but also can further lower the energy barrier for the conversion of N2 to N2OH* (the rate-determining step), generating significant NOR activity.
Results and discussion
Preparation and characterization of catalysts
Here, we used a simple co-precipitation and subsequent annealing way to synthesize Co3O4 NBs, as shown in Fig. 1a. A scanning electron microscope (SEM) and transmission electron microscope (TEM) were applied to detect the morphology of the catalyst. Firstly, highly uniform and small size (about 500 nm) Co–Co Prussian blue analogue (PBA) nanocubes (NCs)40 were obtained by a co-precipitation strategy at room temperature (Fig. S1a†). From Fig. S1b,† we can know that the X-ray diffraction (XRD) of the Co–Co PBA corresponded well to crystalline Co3[Co(CN)6]2·xH2O (PDF#77-1161).41 Secondly, the as-synthesized Co–Co PBA NCs were calcined at 350 °C in air for 2 h. Fig. 1b shows the XRD pattern of the catalyst, the peak of which matched greatly with the card of Co3O4 (PDF#98-006-9366). After thermal treatment, the PBA NCs were transformed into concave Co3O4 NBs, and the basic morphology remains the same, but the size is slightly smaller (Fig. 1c). As obviously shown in Fig. 1d and S2–S4,† the TEM energy-dispersive X-ray spectroscopy (TEM-EDX) and SEM-EDX elemental mapping images showed that Co and O elements are uniformly distributed on the catalyst. The high-resolution TEM (HRTEM, Fig. S5†) images demonstrated that the Co3O4 NBs are uniform and the lattice spacing is 0.243 nm, which corresponds to the (311) face of the Co3O4 NBs.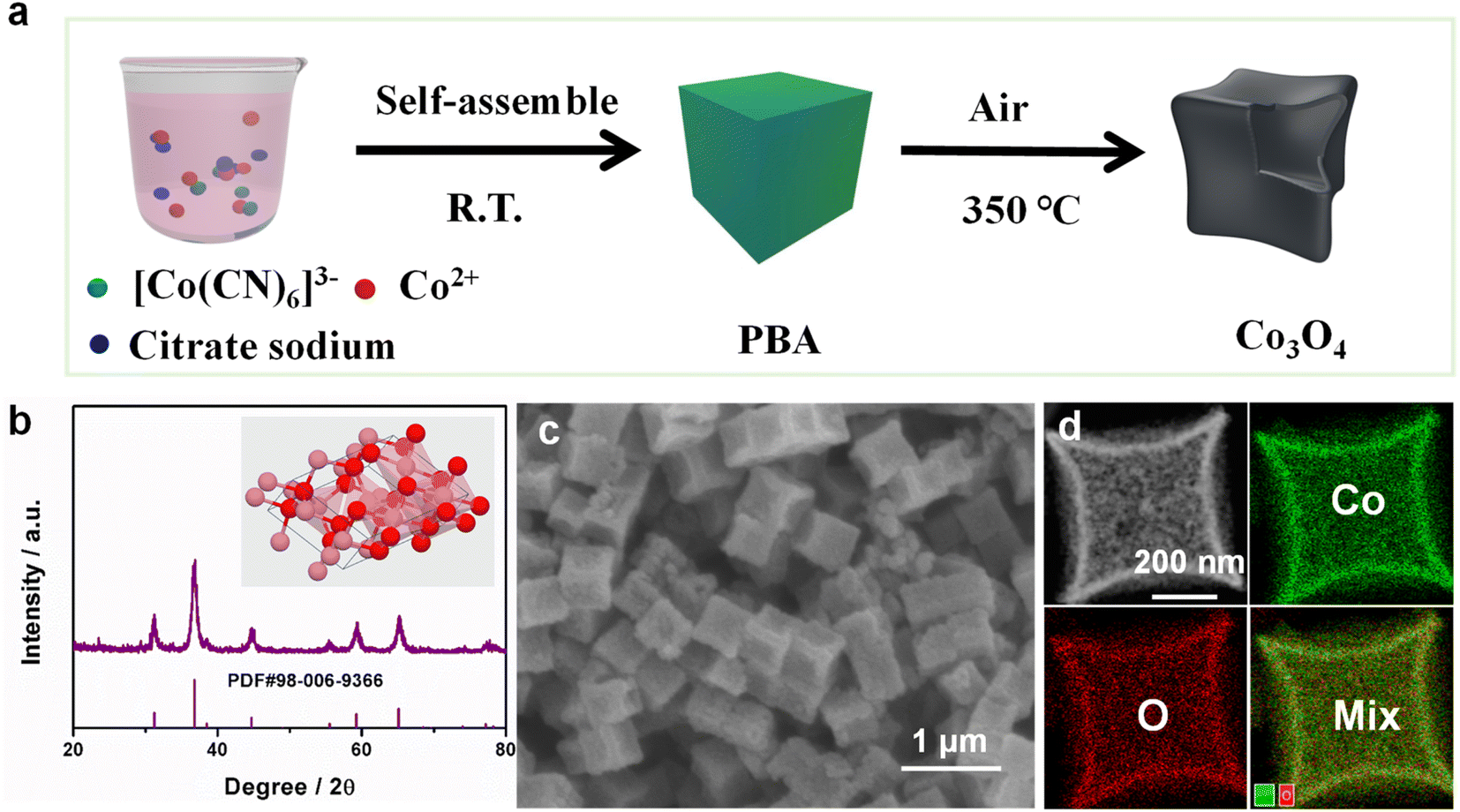 | ||
| Fig. 1 (a) Schematic illustration of the formation of Co3O4 NBs. (b) The X-ray diffraction (XRD) pattern of Co3O4 NBs. (c) SEM image of Co3O4 NBs. (d) TEM elemental mapping images of Co3O4 NBs. | ||
In addition, X-ray photoelectron spectroscopy (XPS) was used to study the valence electron states and electronic structures. As shown in Fig. S6,† the XPS survey spectrum shows that the Co3O4 NBs are composed of Co and O elements. This result is consistent with TEM-EDX and SEM-EDX spectroscopy. As obvious from Fig. S7a,† the Co 2p spectrum represented two main peaks, which belong to Co 2p3/2 and Co 2p1/2, respectively.42 The O 1s spectrum is composed of three species of oxygen, which can be attributed to the Co–O band, the defect sites (oxygen vacancies), and the absorbed oxygen, respectively (Fig. S7b†).43 From Fig. S8,† we can apparently know that the Brunauer–Emmett–Teller (BET) surface area and the pore volume of Co3O4 NBs are 51.06 m2 g−1 and 0.14 cm3 g−1. This further indicates that Co3O4 NBs with a porous structure were successfully synthesized.
To investigate the formation of free groups on the catalyst surface, a range of methods are used to detect oxygen species, including UV-vis, electron spin resonance (ESR) spectroscopy and fluorescence. Fig. 2a shows the process of generating reactive oxygen species including ˙OH free radicals and ˙O2H free radicals by nanozymes. As shown in Fig. 2b, surface Co2+ species first catalyze the reduction of H2O2 to ˙OH radicals, and play a key role in the subsequent TMB oxidation discoloration and generation of ˙O2H radicals and Co3+ regeneration.44 As displayed in Fig. 2c, the Co3O4 + TMB + H2O2 group has a distinct absorption peak compared to the other groups, indicating that Co3O4 has significant catalase-mimicking activities. At the same time, the obvious turquoise oxidation product (oxTMB) in the inset of Fig. 2c further indicates the great catalytic performance of Co3O4.45 From Fig. 2d, we can know that the Co3O4/H2O2 mixture yield ˙OH was obtained by reacting with terephthalic acid (TA). Under these conditions, TA is oxidized by ˙OH to form the fluorescent product TAOH. Fig. S9† depicts the effect of the mixture of H2O2 and Co3O4 on the adsorption spectrum of MB. H2O2/Co3O4 promoted the formation of ˙OH in the system, and the adsorption spectrum of MB + H2O2 was significantly reduced. In addition, it was observed that ˙OH and ˙O2H captured by DMPO agent had a pair of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1 and six resonance characteristic peaks (Fig. 2e and f), which also confirmed that Co3O4 produced ˙OH and ˙O2H during a catalytic reaction similar to peroxidase mimics.46
:2:2:1 and six resonance characteristic peaks (Fig. 2e and f), which also confirmed that Co3O4 produced ˙OH and ˙O2H during a catalytic reaction similar to peroxidase mimics.46
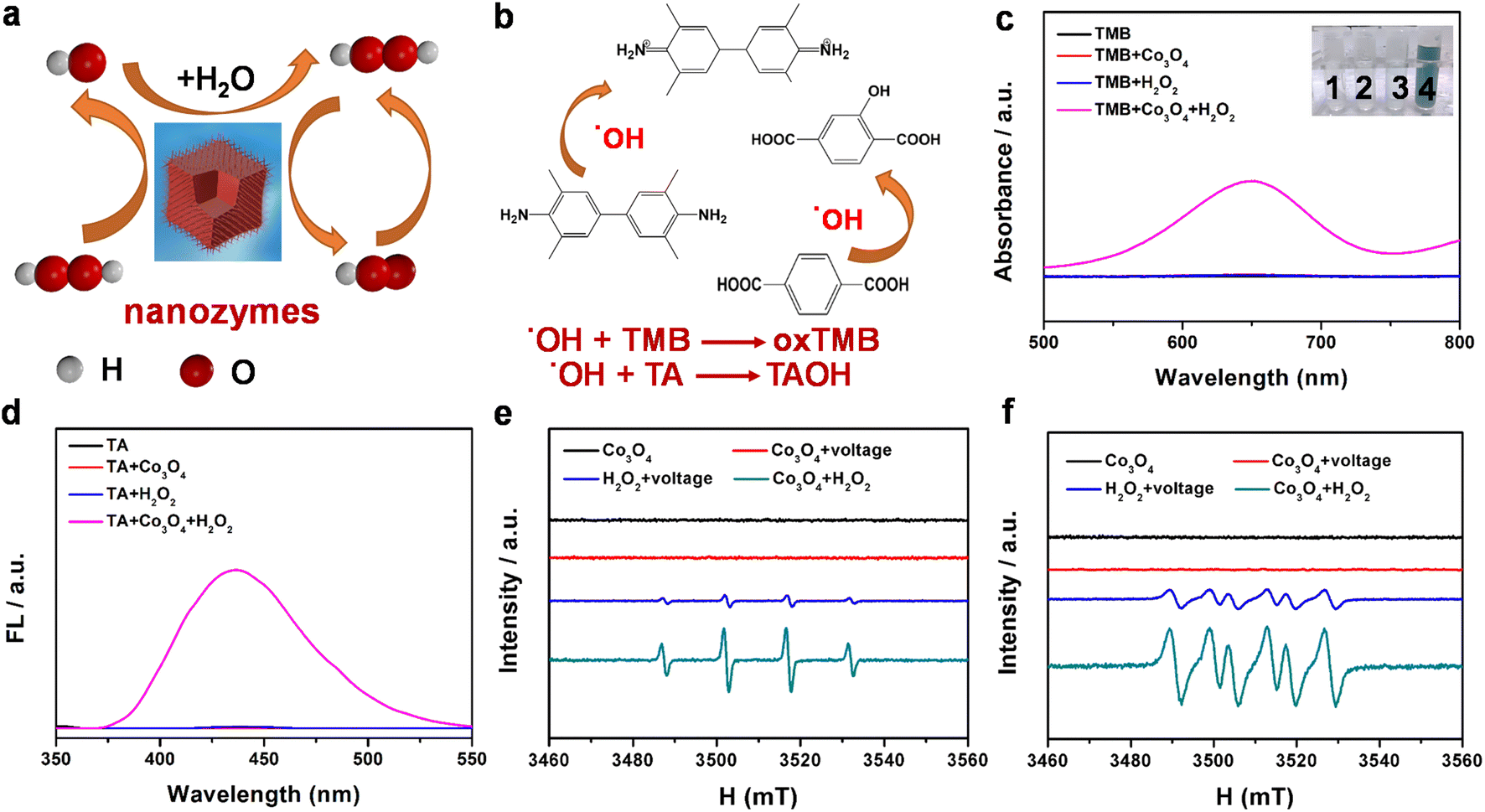 | ||
| Fig. 2 (a) Process of generating reactive oxygen species by nanozymes. (b) Reaction of ˙OH free radicals produced by nanozymes with TMB and TA. (c) The UV-vis spectra and visible color changes of TMB in various reaction systems: (1) TMB, (2) TMB + Co3O4, (3) TMB + H2O2, and (4) TMB + Co3O4 + H2O2. (d) Probing ˙OH formation through the fluorescence spectrum of the adduct of ˙OH with TA. (e) Probing ˙OH production through the electron spin resonance (ESR) spectrum of the adduct of ˙OH with DMPO. (f) Probing ˙O2H production through the ESR spectrum of the adduct of ˙O2H with DMPO. | ||
Electrocatalytic performance for the NOR
To evaluate the electrocatalytic NOR activity of nanozymes under ambient conditions, a series of electrochemical tests were performed in 0.1 M Na2SO4 solution with H2O2 (the contrast experiment is to test in 0.1 M Na2SO4 without adding H2O2 (Fig. S11†)). Before the test, the third exclusion measurement was conducted on H2O2, and no nitrogenous substances such as ammonia were found, so as to exclude the source of N in H2O2 (Fig. S12†). A Co3O4/H2O2 mixture could produce ˙OH in the electrolyte. Fig. 3a shows a schematic diagram of the electrocatalytic NOR, while Fig. S10† displays a picture of the actual device tested. The linear sweep voltammetry (LSV) curves of Co3O4 + ˙OH showed an obvious enhancement under a N2-saturated electrolyte compared to an Ar-saturated sample, indicating the possible electro-oxidation of N2 (Fig. 3b). Then, the possible oxidation product of nitrate was measured and quantified based on the standard method by using ultraviolet-visible (UV-Vis) spectrophotometry (Fig. S13 and S14†) and anion chromatography (Fig. S15†). From 1.5 to 1.9 V vs. RHE, the yield of nitrate and FEs displayed a volcanic shaped curve (Fig. 3c and S16†). At an applied potential of 1.7 V vs. RHE, the NO3− yield of nanozymes reaches 89.35 μg h−1 mgcat−1, which is up to 7 times higher than that of Co3O4 (12.8 μg h−1 mgcat−1) without H2O2. The FE reached 20.4% and 1.23% at 1.7 V vs. RHE for nanozymes and Co3O4 NBs, respectively (Fig. S17†). It is the most effective non-precious metal catalyst reported so far for the NOR process (Fig. 3d and Table S1†). As shown in Fig. S18,† we can know that the TOF of the nanozymes catalyst at 1.7 V vs. RHE reaches 0.58 h−1, which is better than that of Co3O4 (0.083 h−1), demonstrating that the nanozyme has a great intrinsic activity.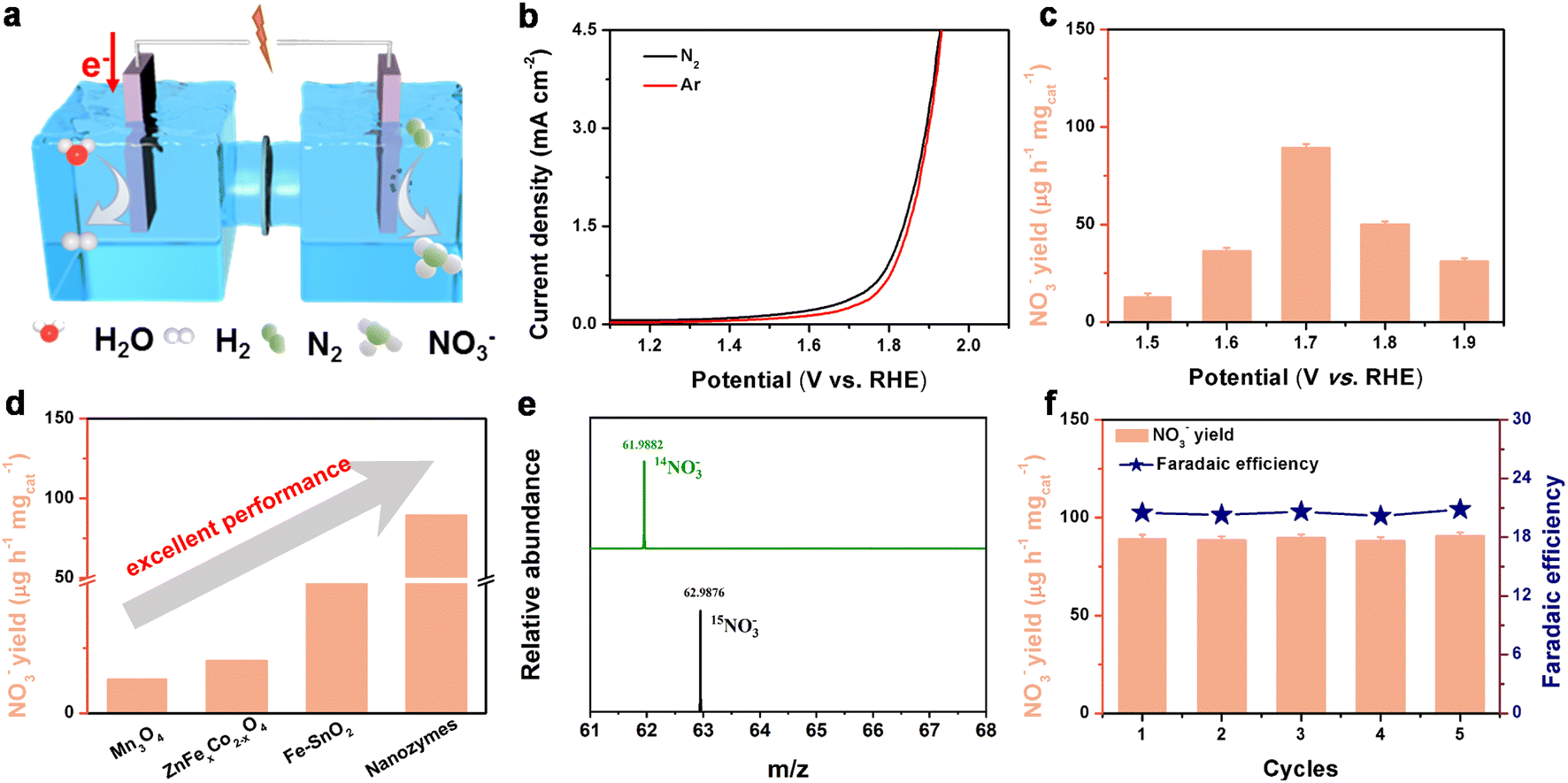 | ||
| Fig. 3 (a) Schematic diagram of Co3O4 + ˙OH for NO3− production. (b) Linear sweep voltammetric (LSV) curves of Co3O4 + ˙OH in N2- and Ar-saturated 0.1 M Na2SO4 solutions. (c) NO3− production of Co3O4 + ˙OH at various potentials in N2-saturated 0.1 M Na2SO4 electrolytes. (d) Comparison of NO3− production of Co3O4 + ˙OH with different reported non-noble metal catalysts for the NOR. (e) MS spectra of both 14NO3− and 15NO3− generated from the NOR tests (at 1.7 V vs. RHE) used as a N2 source with 14N2 or 15N2. (f) Recycling measurement of Co3O4 + ˙OH at 1.7 V vs. RHE. | ||
We experimentally demonstrate the importance of various sources of pollution and show how to remove unstable nitrogenous compounds present in N2 gas and how to make isotopic measurements with 15N2 gas to reduce pollution. In order to examine the N sources of the product in the NOR of nanozymes, some control experiments were performed (Fig. S19†), including experiments with a pure carbon paper (CP) electrode in 0.1 M Na2SO4 solution at 1.7 V vs. RHE, a nanozyme electrode in 0.1 M Na2SO4 solution at an open circuit and an electrode in an Ar-saturated electrolyte after electrolysis for 10 h. In order to exclude other possible contamination, the active test resulted in NO3− levels comparable to the control test. These sources include Nafion, which is used in some N2 oxidation experiments as a separation membrane or electrolyte (Fig. S20†). NO3− was not detected in these experiments, which ensured that the N source of NO3− came from the NOR of the nanozymes. Meanwhile, a 15N isotopic labeling experiment was also performed to examine the N source of NO3− detected. Here, we present such a rigorous procedure to reliably detect the electrooxidation of N2 to NO3− by using 15N2. In the 15N2-saturated electrolyte experiment, 15NO3− was obtained with a molecular weight of 62.9884, compared with 61.988 for 14NO3− in the 14N2-saturated electrolyte (Fig. 3e). These results suggest that the detected NO3− was completely derived from the electrochemical oxidation of N2. To further quantify the yield of NO3−, ion chromatography was used. It is found that the NO3− yield calculated by ion chromatography is close to the NO3− yield calculated by the colorimetric method (Fig. S20†).
The stability of catalysts is not only a vital factor for practical applications, but also critical to evaluate the NOR activity. As shown in Fig. S22 and S23,† this catalyst also demonstrates long-term electrochemical and structural stability, and the current density did not have a tendency to decrease even in a 50 h continuous test. Besides, it can be seen from Fig. 3f that the NO3− yield rate and FE show slight fluctuations for 5 reduplicate tests at 1.7 V (vs. RHE). Most importantly, the results of the cycling experiments clearly show that the nanozyme catalyst exhibits no appreciable decline in the NO3− yield and FEs after the five continuous cycles. Furthermore, it is distinct from the SEM (Fig. S24a†) and TEM images (Fig. S24b†) that the catalyst maintains the original structure after stability testing. XRD and XPS (Fig. S25 and S26†) analysis showed that the position and intensity of peaks did not change significantly compared with the initial results, which further indicated that the stability of the catalyst was impressive.
DFT was used to explore the specific mechanisms of ˙OH free radicals in the NOR process. At first, the N2 adsorption capacity of catalysts was studied using the N2 Temperature Programmed Desorption (N2-TPD) way. We can see in Fig. S26† that the high signal of chemisorption of nanozyme indicates that the adsorption of N2 is higher. From Fig. 4a, we can know the structural model of Co3O4. Subsequently, the energy barriers for the conversion of N2 to N2OH* (the rate-determining step) can be obtained from Fig. 4b. And it was found that the introduction of the ˙OH free radical (Co3O4/H2O2 system) decreases the energy barrier substantially and accelerates the reaction (Fig. S28 and S29†). Fig. 4c depicts the entire path diagram of the electrocatalytic NOR. Nitrogen molecules are first absorbed and oxidized to form *NNOH intermediates. NN key fracture occurred in the second step (the rate-determining step), which is suitable for *N2OH. The *N intermediate formed evolves into *NO. The *NO intermediate is continuously combined with ˙OH in the electrolyte, and the final nitrate product is formed. DFT demonstrates that ˙OH not only can drive nitrogen adsorption, but also can further decrease the energy barrier for the conversion of N2 to N2OH* (the rate-determining step), leading to great NOR activity.
 | ||
| Fig. 4 (a) The top and side views of Co3O4. (b) Reaction free energy diagram of the N2 electrooxidation process on Co3O4 and Co3O4 + ˙OH. (c) Simplistic mechanism of NO3− production on the surface of catalysts. | ||
Conclusions
In short, we introduce hydroxyl radicals into the electrocatalytic NOR for the first time. Co3O4 acts not only as an electrocatalyst, but also as a nanozyme (in combination with hydrogen peroxide producing ˙OH), and can be used as an efficient NOR electrocatalyst for environmental nitrate synthesis. Co3O4 + ˙OH shows an excellent NOR performance among Co3O4 catalysts in 0.1 M Na2SO4 solution. At an applied potential of 1.7 V vs. RHE, the NO3− yield of Co3O4 + ˙OH reaches 89.35 μg h−1 mgcat−1, which is 7 times that of Co3O4 (12.8 μg h−1 mgcat−1) and the corresponding FE is 20.4%. We use ultraviolet spectroscopy and ion chromatography to determine the concentration of nitrate produced. At the same time, we also conduct 15N isotope labeling experiments by mass spectrometry to further determine the nitrogen source. It is the most effective non-precious metal catalyst reported so far for the NOR process. This catalyst also demonstrates long-term electrochemical and structural stability, and the current density did not have a tendency to decrease even in a 50 h continuous test. The TOF of the Co3O4 + ˙OH catalyst at 1.7 V vs. RHE reaches 0.58 h−1, which is better than that of Co3O4 (0.083 h−1), demonstrating that free radicals can greatly enhance the intrinsic activity of catalysts. DFT demonstrates that ˙OH not only can drive nitrogen adsorption, but also can decrease the energy barrier (rate-determining step) of N2 to N2OH*, thus producing great NOR activity. Therefore, the next direction of development is that an electric Fenton can be used to generate more free radicals to promote the NOR.Experimental
Reagents and materials
Cobalt acetate ((H3CO2)2Co, Shanghai Macklin Biochemical Technology Co., Ltd, China, 98%). Sodium citrate dihydrate (C6H5Na3O7·2H2O, Shanghai Aladdin Biochemical Technology Co., Ltd, China, 99%). Sodium fluoride (K3[Co(CN)6], Shanghai Aladdin Biochemical Technology Co., Ltd, China, 99%). Sodium sulfate (Na2SO4·H2O, 99%) were purchased from Aladdin. Nafion solution (5%) was purchase from Sigma Aldrich. Ethanol (C2H6O) was purchased from Sinopharm Chemical Reagent Co., Ltd. Ultrapure water (Millipore Milli-Q grade) with a resistivity of 18.2 MΩ was used in all the experiments.Preparation of Co–Co PBA NCs
In a typical synthesis, 0.6 mmol of cobalt acetate and 0.9 mmol of sodium citrate were dissolved in 20 mL of deionized (DI) water to form solution A. At the same time, 0.4 mmol of potassium hexacyanocobaltate(III) was dissolved in 20 mL of DI water to form solution B. Then, solutions A and B were mixed together under magnetic stirring for 1 min. The obtained mixed solution was aged for 12 h at room temperature. Then, the obtained black gelatinous solutions were collected by centrifugation at 8000 rpm for 3 min, and washed 3 times with DI and ethanol. Finally, the precipitates were dried at 70 °C overnight.Preparation of Co3O4 NBs
The as-prepared PBA NCs were annealed at 350 °C for 2 h with a heating temperature rate of 2 °C min−1 in air.Materials characterization
X-ray diffraction (XRD) analysis was used to examine the composition of the as-synthesized samples on an X'Pert PRO MPD. Scanning electron microscopy (SEM) measurement was collected on a Hitachi, S-8200 to investigate the structure and morphology of the samples. Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) measurements were performed on a JEM-2100UHR operating at 200 kV. Electron spin resonance (ESR) was performed using a German Bruker A-300 and a magnetic field strength (0-9500) Gauss. The 1H NMR spectrum was obtained on a Bruker 500 with a Probe TXI at room temperature of 25 °C using a 3 mm tube. The electrolyte after electrolysis was collected, lyophilized and further dissolved in 1.0 M HCl solution (D2O/H2O mixed solution). The IC data were collected by using an IC (863 Basic IC Plus, Metrohm, Switzerland) equipped with a Metrosep C Supp 4-250/4.0 column. N2-TPD of N2 experiments was conducted on a Quantachrome Chem BET Pulsar TPR/TPD.Electrochemical measurements
All the electrochemical performances of the as-synthesized samples were determined on an electrochemical workstation (CHI 760E). A typical H-type electrolytic cell divided by a proton-exchange membrane (Nafion 117) was used. Except for special instructions, all potentials were recorded against the RHE. The potentials against the saturated calomel electrode (Ag/AgCl) were translated to those against the RHE using the following equation: E (vs. RHE) = E (vs. Ag/AgCl) + 0.197 + 0.059 × pH. All the polarization curves were steady lines after many cycles and the current density was normalized to the geometric surface area.Author contributions
L. W. and J. L. supervised the research. J. L. conceived the research. J. L. and Z. W. designed the experiments. Z. W. performed most of the experiments and data analysis. Z. W., J. L. and H. Z. prepared the electrodes and helped with electrochemical measurements. W. X. helped analyze physical characterization data. J. L. and Z. L. helped answer some questions. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22001143 and 52072197), Youth Innovation and Technology Foundation of Shandong Higher Education Institutions, China (2019KJC004), Outstanding Youth Foundation of Shandong Province, China (ZR2019JQ14), Taishan Scholar Young Talent Program (tsqn201909114 and tsqn201909123), Natural Science Foundation of Shandong Province (ZR2020YQ34), Major Scientific and Technological Innovation Project (2019JZZY020405), and Major Basic Research Program of Natural Science Foundation of Shandong Province under Grant (ZR2020ZD09), the 111 Project of China (Grant No. D20017).Notes and references
- F. Y. Chen, Z. Y. Wu, S. Gupta, D. J. Rivera, S. V. Lambeets, S. Pecaut, J. Y. T. Kim, P. Zhu, Y. Z. Finfrock, D. M. Meira, G. King, G. Gao, W. Xu, D. A. Cullen, H. Zhou, Y. Han, D. E. Perea, C. L. Muhich and H. Wang, Nat. Nanotechnol., 2022, 17, 759–767 CrossRef CAS PubMed.
- J. Yang, H. Qi, A. Li, X. Liu, X. Yang, S. Zhang, Q. Zhao, Q. Jiang, Y. Su, L. Zhang, J.-F. Li, Z.-Q. Tian, W. Liu, A. Wang and T. Zhang, J. Am. Chem. Soc., 2022, 144, 12062–12071 CrossRef CAS PubMed.
- H. He, H.-K. Li, Q.-Q. Zhu, C.-P. Li, Z. Zhang and M. Du, Appl. Catal., B, 2022, 316, 121673 CrossRef CAS.
- E. Contreras, R. Nixon, C. Litts, W. Zhang, F. M. Alcorn and P. K. Jain, J. Am. Chem. Soc., 2022, 144, 10743–10751 CrossRef CAS PubMed.
- J. Cai, Y. Wei, A. Cao, J. Huang, Z. Jiang, S. Lu and S.-Q. Zang, Appl. Catal., B, 2022, 316, 121683 CrossRef CAS.
- H. Iriawan, S. Z. Andersen, X. Zhang, B. M. Comer, J. Barrio, P. Chen, A. J. Medford, I. E. L. Stephens, I. Chorkendorff and Y. Shao-Horn, Nat. Rev. Methods Primers, 2021, 1, 56 CrossRef CAS.
- T. Li, S. Han, C. Cheng, Y. Wang, X. Du, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202204541 CAS.
- S. Han, C. Wang, Y. Wang, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 4474–4478 CrossRef CAS PubMed.
- M. Kuang, Y. Wang, W. Fang, H. Tan, M. Chen, J. Yao, C. Liu, J. Xu, K. Zhou and Q. Yan, Adv. Mater., 2020, 32, 2002189 CrossRef CAS PubMed.
- Y. Lin, J. Liang, H. Li, L. Zhang, T. Mou, T. Li, L. Yue, Y. Ji, Q. Liu, Y. Luo, N. Li, B. Tang, Q. Wu, M. S. Hamdy, D. Ma and X. Sun, Mater. Today Phys., 2022, 22, 100611 CrossRef CAS.
- Y. Li, C. Cheng, S. Han, Y. Huang, X. Du, B. Zhang and Y. Yu, ACS Energy Lett., 2022, 7, 1187–1194 CrossRef CAS.
- M. Jiang, J. Su, X. Song, P. Zhang, M. Zhu, L. Qin, Z. Tie, J. L. Zuo and Z. Jin, Nano Lett., 2022, 22, 2529–2537 CrossRef CAS PubMed.
- B. H. R. Suryanto, H.-L. Du, D. Wang, J. Chen, A. N. Simonov and D. R. MacFarlane, Nat. Catal., 2019, 2, 290–296 CrossRef CAS.
- Y. Wang, C. Wang, M. Li, Y. Yu and B. Zhang, Chem. Soc. Rev., 2021, 50, 6720–6733 RSC.
- J. Shi, C. Wang, R. Yang, F. Chen, N. Meng, Y. Yu and B. Zhang, Sci. China: Chem., 2021, 64, 1493–1497 CrossRef CAS.
- T. Li, S. Han, C. Wang, Y. Huang, Y. Wang, Y. Yu and B. Zhang, ACS Catal., 2021, 11, 14032–14037 CrossRef CAS.
- J. Choi, H.-L. Du, C. K. Nguyen, B. H. R. Suryanto, A. N. Simonov and D. R. MacFarlane, ACS Energy Lett., 2020, 5, 2095–2097 CrossRef CAS.
- X. Zhang, C. Wang, Y. Guo, B. Zhang, Y. Wang and Y. Yu, J. Mater. Chem. A, 2022, 10, 6448–6453 RSC.
- J. Wang, C. Cai, Y. Wang, X. Yang, D. Wu, Y. Zhu, M. Li, M. Gu and M. Shao, ACS Catal., 2021, 11, 15135–15140 CrossRef CAS.
- Y. Ling, Q. Ma, Y. Yu and B. Zhang, Trans. Tianjin Univ., 2021, 27, 180–200 CrossRef CAS.
- C. Dai, Y. Sun, G. Chen, A. C. Fisher and Z. Xu, Angew. Chem., Int. Ed., 2020, 59, 9418–9422 CrossRef CAS PubMed.
- Y. Ren, C. Yu, L. Wang, X. Tan, Z. Wang, Q. Wei, Y. Zhang and J. Qiu, J. Am. Chem. Soc., 2022, 144, 10193–10200 CrossRef CAS PubMed.
- Z. Li, Z. Ma, J. Liang, Y. Ren, T. Li, S. Xu, Q. Liu, N. Li, B. Tang, Y. Liu, S. Gao, A. A. Alshehri, D. Ma, Y. Luo, Q. Wu and X. Sun, Mater. Today Phys., 2022, 22, 100586 CrossRef CAS.
- Y. Zhang, F. Du, R. Wang, X. Ling, X. Wang, Q. Shen, Y. Xiong, T. Li, Y. Zhou and Z. Zou, J. Mater. Chem. A, 2021, 9, 17442–17450 RSC.
- Y. Wang, T. Li, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2021, 61, e202115409 Search PubMed.
- A. J. Medford and M. C. Hatzell, ACS Catal., 2017, 7, 2624–2643 CrossRef CAS.
- Y. Wang, Y. Yu, R. Jia, C. Zhang and B. Zhang, Natl. Sci. Rev., 2019, 6, 730–738 CrossRef CAS PubMed.
- W. Fang, C. Du, M. Kuang, M. Chen, W. Huang, H. Ren, J. Xu, A. Feldhoff and Q. Yan, Chem. Commun., 2020, 56, 5779–5782 RSC.
- Z. Y. Wu, M. Karamad, X. Yong, Q. Huang, D. A. Cullen, P. Zhu, C. Xia, Q. Xiao, M. Shakouri, F. Y. Chen, J. Y. T. Kim, Y. Xia, K. Heck, Y. Hu, M. S. Wong, Q. Li, I. Gates, S. Siahrostami and H. Wang, Nat. Commun., 2021, 12, 2870 CrossRef CAS PubMed.
- S. Andersen, V. Čolić, S. Yang, J. Schwalbe, A. Nielander, J. McEnaney, K. Enemark, J. Baker, A. Singh, B. Rohr, M. Statt, S. Blair, S. Mezzavilla, J. Kibsgaard, P. Vesborg, M. Cargnello, S. Bent, T. Jaramillo, I. Stephens, J. Nørskov and I. Chorkendorff, Nature, 2019, 570, 504–508 CrossRef CAS PubMed.
- B. M. Hoffman, D. Lukoyanov, Z. Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed.
- M. D. V. Rosca, M. T. de Groot, M. T. M. Koper and M. T. M. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef.
- J. Chen, R. Crooks, L. Seefeldt, K. Bren, R. Bullock, M. Darensbourg, P. Holland, B. Hoffman, M. Janik, A. Jones, M. Kanatzidis, P. King, K. Lancaster, S. Lymar, P. Pfromm, W. Schneider and R. Schrock, Science, 2018, 360, 873 CAS.
- S. Garcia-Segura, M. Lanzarini-Lopes, K. Hristovski and P. Westerhoff, Appl. Catal., B, 2018, 236, 546–568 CrossRef CAS.
- S. J. Yuan, J. J. Chen, Z. Q. Lin, W. W. Li, G. P. Sheng and H. Q. Yu, Nat. Commun., 2013, 4, 2249 CrossRef PubMed.
- U. B. Shahid, K. Siddharth and M. Shao, Curr. Opin. Electrochem., 2021, 30, 100790 CrossRef CAS.
- B. S. Patil, Q. Wang, V. Hessel and J. Lang, Catal. Today, 2015, 256, 49–66 CrossRef CAS.
- Y. Liu, M. Cheng, Z. He, B. Gu, C. Xiao, T. Zhou, Z. Guo, J. Liu, H. He, B. Ye, B. Pan and Y. Xie, Angew. Chem., Int. Ed., 2019, 58, 731–735 CrossRef CAS PubMed.
- S. D. Minteer, P. Christopher and S. Linic, ACS Energy Lett., 2018, 4, 163–166 CrossRef.
- L. Deng, Z. Yang, L. Tan, L. Zeng, Y. Zhu and L. Guo, Adv. Mater., 2018, 30, 1802510 CrossRef PubMed.
- W. Sun, Z. Wei, J. Qi, L. Kang, J. Li, J. Xie, B. Tang and Y. Xie, Chin. J. Catal., 2021, 39, 2347–2353 CAS.
- D. Zhang, H. Zhao, X. Wu, Y. Deng, Z. Wang, Y. Han, H. Li, Y. Shi, X. Chen, S. Li, J. Lai, B. Huang and L. Wang, Adv. Funct. Mater., 2020, 31, 2006939 CrossRef.
- W. Zhong, C. Yang, J. Wu, W. Xu, R. Zhao, H. Xiang, K. Shen, Q. Zhang and X. Li, Chem. Eng. J., 2022, 436, 134813 CrossRef CAS.
- Y. Tao, E. Ju, J. Ren and X. Qu, Adv. Mater., 2015, 27, 1097–1104 CrossRef CAS PubMed.
- M. M. Pan, Y. Ouyang, Y. L. Song, L. Q. Si, M. Jiang, X. Yu, L. Xu and I. Willner, Small, 2022, 18, 2200548 CrossRef CAS PubMed.
- J. Cai, T. Niu, P. Shi and G. Zhao, Small, 2019, 15, 1900153 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06599a |
| This journal is © The Royal Society of Chemistry 2023 |